DOI:
10.1039/C8QI01098F
(Research Article)
Inorg. Chem. Front., 2019,
6, 126-136
Enhanced UV-Vis-NIR activated photocatalytic activity from Fe3+-doped BiOBr:Yb3+/Er3+ upconversion nanoplates: synergistic effect and mechanism insight†
Received
11th October 2018
, Accepted 14th November 2018
First published on 14th November 2018
Abstract
Upconversion (UC) materials are recognized as promising candidates to harvest solar energy for photocatalysis. In this work, a simple strategy for simultaneously enhancing the UC luminescence and photocatalytic efficiency of BiOBr:Yb3+/Er3+ nanoplates through Fe3+ ion doping is reported. Compared to the Fe3+-free sample, the UC emission intensity was significantly enhanced through tailoring of the crystal symmetry by Fe3+ ions. Experiment and DFT calculations reveal that the introduction of Fe3+ ions resulted in the formation of an impurity energy level, extending to the light absorption region. As expected, the Fe3+-doped BiOBr:Yb3+/Er3+ nanoplates exhibit a wide photoresponse from the UV to NIR regions, good stability, and obviously enhanced photocatalytic activities compared with the BiOBr:Yb3+/Er3+ nanoplates in the degradation of RhB. The boosted photocatalytic activity can be attributed to the synergic effect of the efficient utilization of UC luminescence and Fe3+ doping, where Fe3+ doping could improve the light harvesting capacity, enhance the separation efficiency of electron and hole (e−/h+) pairs, and promote the production of highly oxidative species. This work not only provides a promising system for the efficient utilization of solar light, but also offers a feasible guideline for the further design of broad-spectrum active photocatalysts.
1. Introduction
Today, environmental pollution and the energy crisis are two significant challenges for human society. Photocatalysis is considered as one of the most valuable and promising approaches to tackle these problems, as it can directly utilize solar light.1 Recently, 2D materials have been recognized as a new kind of potential material to improve the photocatalytic efficiency, which provides the possibility to reduce the recombination of electron and hole (e−/h+) pairs via shortening the migration distance and increasing the activity of interfacial reactions, as well as large specific surface areas.1–6 Among various 2D materials, BiOBr, a promising photocatalyst, has attracted particular attention due to its unique layered structure and catalytic properties.7–9 However, pure BiOBr is still not satisfactory because of its poor absorption range, which can only be activated by ultraviolet (UV) and part of visible (Vis) light,10 resulting in limited solar energy utilization. Although many effective ways have been developed to extend the light absorption range of BiOBr, such as doping,11 oxygen vacancies,12 semiconductor recombination,13 and so on, the main absorption is still limited to the Vis light region. However, the near-infrared (NIR) light, which occupies ∼50% of the solar spectrum, has still not been exploited. As a result, the photocatalytic activity of BiOBr still could not achieve the requirements of practical applications. Therefore, it is an urgent need to seek an approach to utilize the NIR light, in order to improve the photocatalytic efficiency and promote its practical applications.
In order to fully utilize solar light, upconversion (UC) materials are potential candidates for this task, which can serve as a medium for converting longer wavelength light (NIR) to shorter wavelength light (UV and Vis),14 which is then reabsorbed by UV or Vis-responsive photocatalysts.15–20 In recent years, the UC luminescence properties of rear-earth (RE3+) doped BiOBr have been reported, such as Yb3+/Er3+,21 Yb3+/Tm3+,22 Yb3+/Ho3+,23etc., which showed efficient UC luminescence. However, these studies only focus on the UC luminescence properties and their NIR photocatalytic performances are rarely reported. In 2018, Liang et al.24 reported that BiOBr:Yb3+,Er3+/g-C3N4 exhibited satisfactory photocatalytic activity for the degradation of RhB under Vis and NIR light irradiation due to the UC effect of Er3+ and the construction of the p–n junction. Unfortunately, their insufficient UC intensity and the high recombination rate of electrons (e−) and holes (h+) strongly impede their practical application.
To overcome the aforementioned drawbacks, doping transition metal ions is an effective strategy, which not only enhances UC luminescence,25 but also modifies the band structure and consequently extends the absorption range of photocatalysts, as well as suppresses the recombination of e−/h+ pairs.26 Among various transition metals ions, Fe3+ has been recognized as an appropriate dopant for Bi-based photocatalysts. Chala et al.27 reported that Fe loaded BiVO4 improved the photocatalytic activity for MB degradation under visible light compared to pure BiVO4, which resulted from effectively extending the absorption range to the visible region. Mi et al.28 and Yuan et al.29 reported that the Fe-modified BiOCl and BiOBr can enhance the visible light photocatalytic performance, respectively, due to the enhanced photogenerated carrier separation efficiency. Recently, Liu et al.30 have shown that the Er3+/Fe3+ co-doped porous Bi5O7I microspheres exhibited enhanced photocatalytic activity under visible light, but the Fe3+-doping effects on the UC emissions had not been investigated. What's more, doping Fe3+ ions also could enhance UC emissions, such as Fe3+ doping in NaGdF4:Yb/Er,25 NaYF4:Yb/Er,31 and β-NaYbF4:Tm32 nanocrystals. However, there is no report on the improvement of the UC luminescence of BiOBr:Yb3+,Er3+ by introducing Fe3+ ions, not to mention their NIR photocatalytic performances.
Inspired by the previous work, our objective is to prepare Fe3+-doped BiOBr:Yb3+/Er3+ nanoplates as a highly efficient UV-Vis-NIR active photocatalyst. In this work, it is found that Fe3+ ion doping could improve UC emission and photocatalytic activity in the RhB degradation of BiOBr:Yb3+/Er3+ nanoplates under UV, Vis, NIR and full spectrum light irradiation. The reasons for the enhanced photocatalytic performance are discussed in detail, and the related photocatalytic mechanisms are proposed.
2. Results and discussion
2.1. Structure and morphology
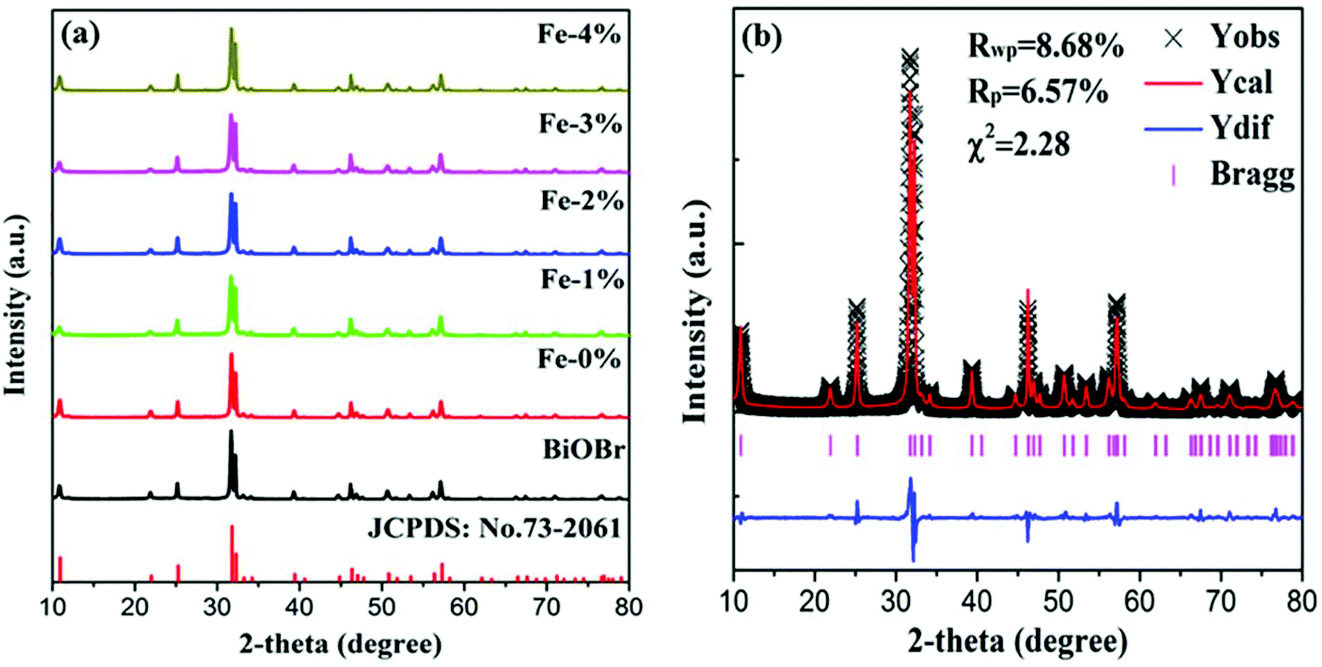
Fig. 1(a) shows the XRD patterns of pure BiOBr and BiOBr:10%Yb3+/1%Er3+ (BYE) nanoplates doped with different Fe3+ (0, 1, 2, 3, and 4%) concentrations. It is found that all diffraction peaks of the products are well indexed to the tetragonal phase BiOBr (JCPDS no. 73-2061) and no other diffraction peaks are observed, indicating the successful incorporation of Yb3+, Er3+, and Fe3+ into the BiOBr host. To reveal the subtle differences caused by Fe3+ doping, a selected region of diffraction peaks in the range of 31.4–32.6 is studied, as shown in Fig. S1.† Compared with the Fe3+-free sample, the diffraction peaks show a slight shift to a high angle with increasing Fe3+ ion concentration, which indicates that Fe3+ ions can be doped into the host lattice through substitution. Because the radius of the Fe3+ ion (0.79 Å) is smaller than the radius of the Bi3+ ion (1.03 Å),33 substituting the Bi3+ ion with this smaller Fe3+ ion can induce the shrinking of the host lattice. In order to obtain a deeper insight into the influence of Fe3+ doping on the phase structure, the representative Rietveld XRD refinement of BiOBr:10% Yb3+/1% Er3+/4% Fe3+ (BYE-4% Fe) is performed by using the GSAS program, as shown in Fig. 1(b). The Rietveld refinement result shows that the measured diffraction peaks are well in agreement with the calculated one, and the fitting result is found to be Rwp = 8.68%, Rp = 6.57%, and χ2 = 2.23, which indicates that the synthesized BYE-4% Fe nanoplates have a pure tetragonal phase with a P4/nmm (no. 129) space group.
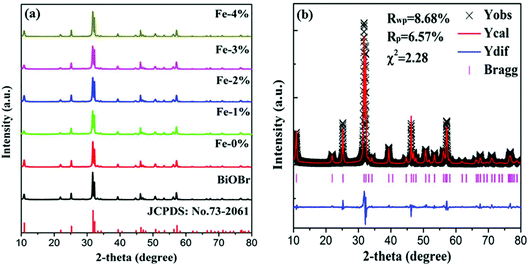 |
| | Fig. 1 (a) XRD patterns of pure BiOBr and BYE nanoplates doped with different Fe3+ concentrations; (b) Rietveld XRD refinement for the BYE-4% Fe sample. | |
SEM and TEM images characterize the morphologies of the prepared samples. As shown in Fig. 2(a) and (b), the pure BiOBr and BYE-0% Fe samples present a large number of nanoplates with the side length in the range of 200–300 nm and a thickness of ∼40 nm, which indicates that the doping of Yb3+/Er3+ ions does not change the morphology. When Fe3+ ions are introduced, the samples also appear with a sheet morphology, but the particle size of the samples become smaller, which is evident in Fig. 2(c–f). The SEM images indicate that Fe3+ doping has obvious influence on the particle size of BiOBr. To further verify the change of particle size, the surface areas of these samples are measured using the BET method, and are calculated to be 4.64, 5.77, 16.22, 17.34, 15.01, and 15.22 m2 g−1 for BiOBr, BYE-0% Fe, BYE-1% Fe, BYE-2% Fe, BYE-3% Fe, and BYE-4% Fe, respectively (Fig. S2† and Table 1), which is consistent with the particle size variation in the SEM images. The TEM images of the BYE-4% Fe sample shown in Fig. 2(g and h) further confirm its sheet morphology. From the HRTEM image [Fig. 2(i)], clear lattice fringes can be found, and the distance of the lattice fringes is 0.273 nm, which is consistent with the (110) plane of the tetragonal BiOBr. Fig. S3† represents the EDX analysis of BYE-4% Fe. From the EDX spectrum, it is clear that Bi, Br, O, Er, Yb and Fe elements are present in the sample. The results confirm that Yb3+, Er3+, and Fe3+ ions are effectively incorporated into the BiOBr host lattice.
 |
| | Fig. 2 SEM images of (a) BiOBr; (b) BYE-0% Fe; (c) BYE-1% Fe; (d) BYE-2% Fe; (e) BYE-3% Fe; (f) BYE-4% Fe; (g, h) TEM images and (i) HRTEM image of BYE-4% Fe. | |
Table 1 The kinetics of the degradation reaction under different forms of light source irradiation
| Sample |
S
BET (m2 g−1) |
Degradation rate |
| Vis (min−1) |
NIR (h−1) |
Full (min−1) |
| BiOBr |
4.64 |
0.0107 |
0.0087 |
0.0435 |
| Fe-0% |
5.77 |
0.0151 |
0.2776 |
0.0494 |
| Fe-1% |
16.22 |
0.0202 |
0.3435 |
0.0509 |
| Fe-2% |
17.34 |
0.0264 |
0.3649 |
0.0523 |
| Fe-3% |
15.01 |
0.0298 |
0.4207 |
0.0692 |
| Fe-4% |
15.22 |
0.0243 |
0.3328 |
0.0597 |
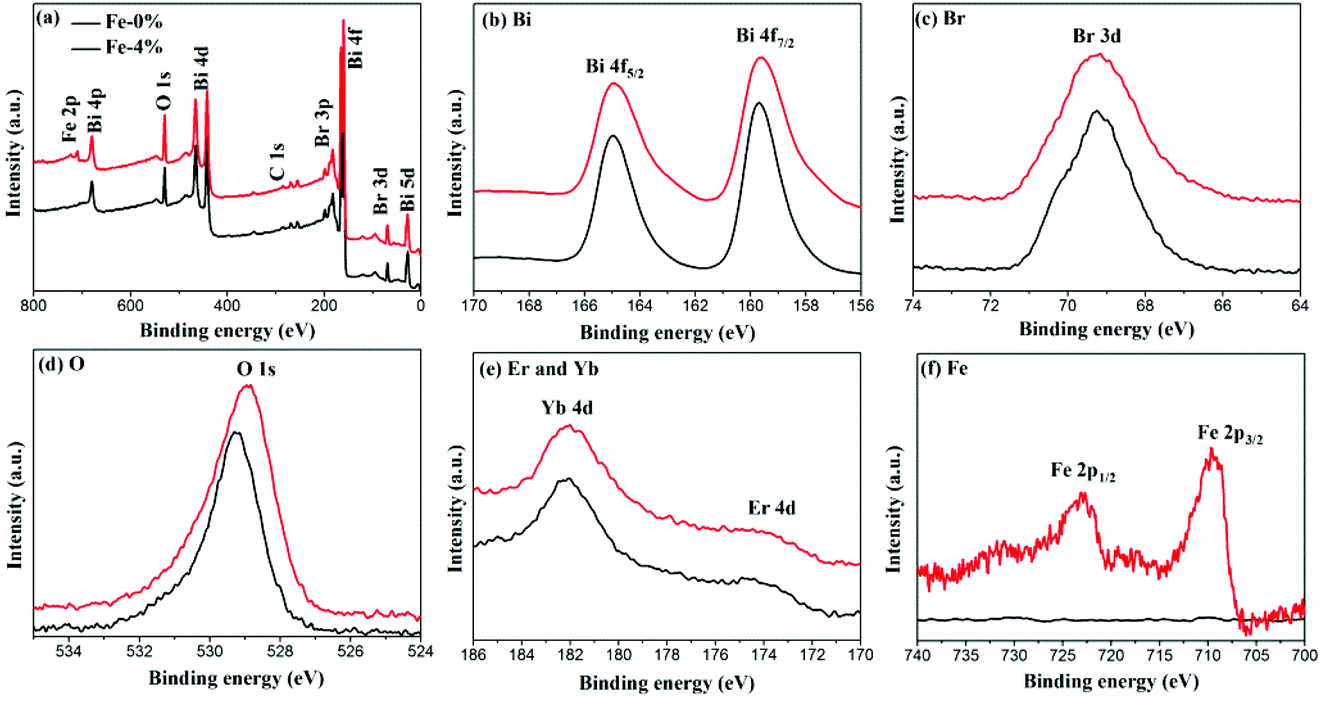
The composition and surface chemical states of BYE-0% Fe and BYE-4% Fe are analyzed by XPS. Fig. 3(a) shows the survey spectrum which depicts the presence of Bi, Br, O, Fe, Yb and Er elements. Fig. 3(b) displays the high-resolution Bi 4f spectra; two peaks at 164.9 and 159.5 eV are identified as Bi 4f7/2 and Bi 4f5/2, respectively.29 The peak located at 5 69.2 eV is ascribed to Br 3d in BiOBr.34 As shown in Fig. 3(d), the O 1s peak of BYE-0% Fe located at 529.2 eV is assigned to the Bi–O bond.29 Compared with BYE-0% Fe, the O 1s peak of BYE-4% Fe is shifted to a lower binding energy, which may be attributed to the weakened hybridization between Bi 6s and O 2p, leading to the decrease of the binding energy of Bi–O bond.35 The Yb 4d and Er 4d peaks [Fig. 3(e)] can be observed at 182.1 and 172.6 eV, respectively, which is consistent with the previous reports.25 The peaks around 709.2 and 722.8 eV are ascribed to Fe 2p3/2 and Fe 2p1/2 of the Fe3+ ions, respectively.30 From the above results, it can be further confirmed that the Fe3+, Yb3+ and Er3+ ions are successfully incorporated into the BiOBr host.
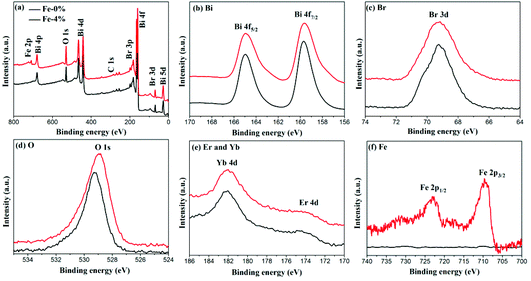 |
| | Fig. 3 XPS spectra of BYE-0% Fe and BYE-4% Fe. (a) Full survey spectrum; (b) Bi; (c) Br; (d) O; (e) Er; Yb; and (f) Fe. | |
2.2. Optical properties
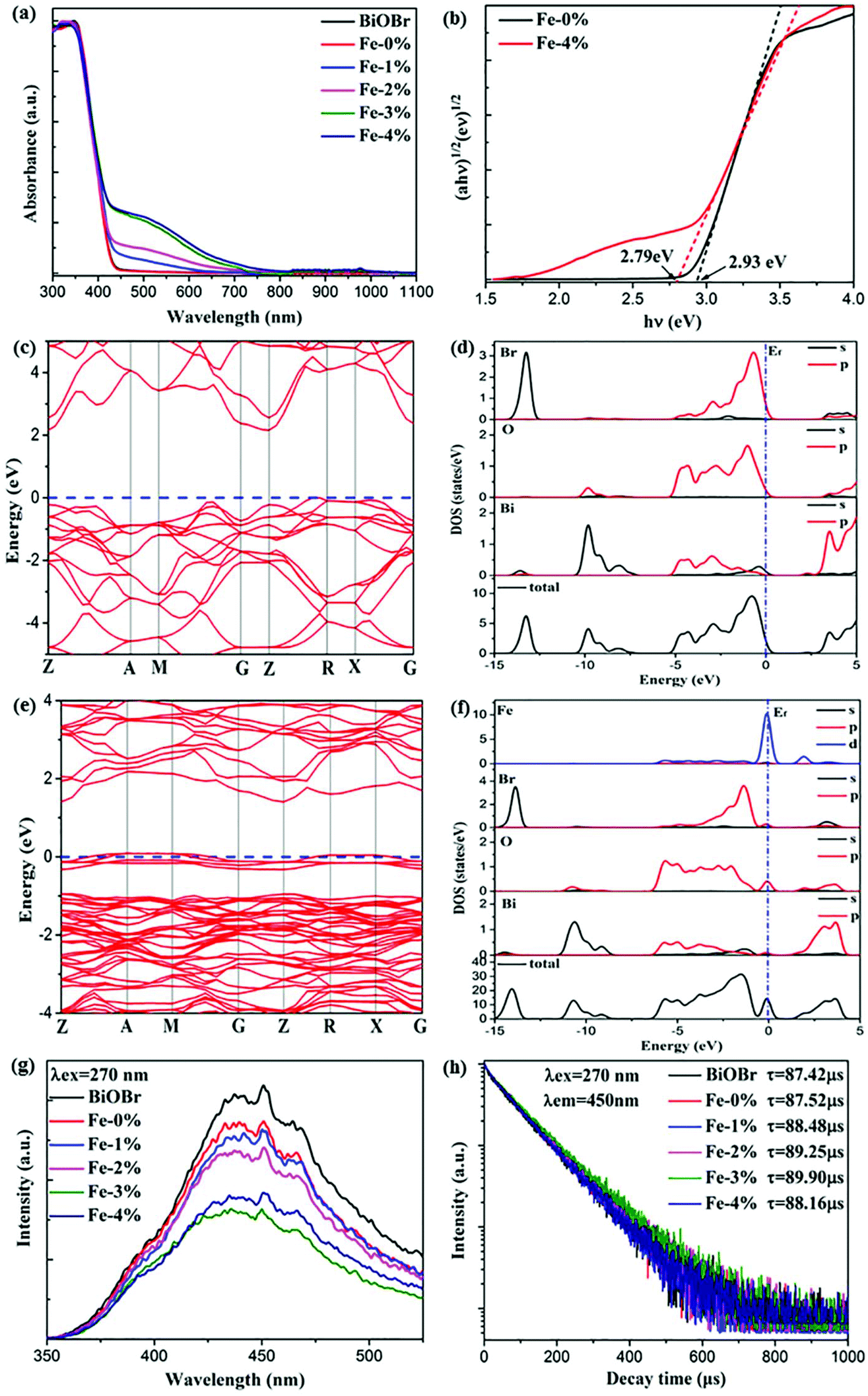
It is acknowledged that Fe3+ doping plays an important role in the Vis light harvesting ability, which would further affect the photocatalytic properties.26 Therefore, the UV–Vis–NIR absorption spectra of all samples are recorded. As can be seen in Fig. 4(a), the absorption edges display a slight red shift with increasing Fe3+ concentration. The corresponding optical band gap energy (Eg) was calculated to be 2.93 and 2.79 eV for BYE-0% Fe and BYE-4% Fe, respectively [Fig. 4(b)]. However, it is worth noting that Fe3+ doping greatly increased light absorption in the Vis light region. There are two explanations for this phenomenon: (1) surface doping, which can generate a doping energy level in the band gap and result in increased absorption of the additional tail without changing the intrinsic optical absorption edge.36,37 (2) The increase in the Vis light absorption may be due to the d–d transition of Fe3+ ions (2T2g → 2A2g, 2T1g) or the charge transfer transition between the interacting Fe3+ ions via the conduction band (Fe3+ + Fe3+ → Fe4+ + Fe2+).26 To confirm the origin of the increase in Vis light absorption, the effect of Li+ or Zn2+ ion dopants on the absorption spectra is investigated for comparison. It shows that Vis light absorption is almost not affected by single or co-doping Li+ and Zn2+ ions (Fig. S4†). Therefore, we can conclude that the increased Vis light absorptions are attributed to the transition of Fe3+ ions. The enhanced Vis light absorption is beneficial to the photocatalytic activity under Vis light irradiation.
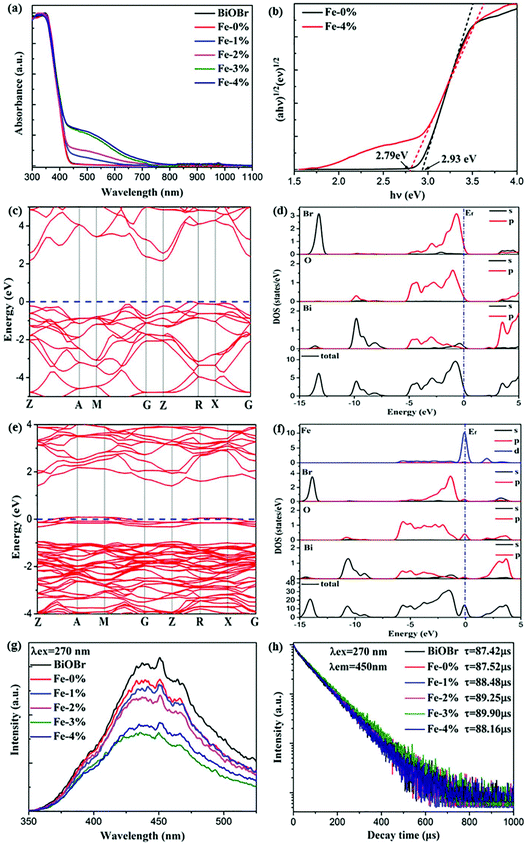 |
| | Fig. 4 (a) UV–Vis–NIR absorption spectra of pure BiOBr and BYE nanoplates doped with different Fe3+ ion concentrations; (b) plots of (αhv)1/2vs. hν of BYE-0% Fe and BYE-4% Fe; calculated electronic energy band structures and DOS spectra of (c, d) pure BiOBr and (e, f) Fe–BiOBr; (g) PL spectra and (h) lifetime decay curves of the samples under excitation at a wavelength of 270 nm. | |
To determine the effect of Fe3+ doping on the absorption spectra of BiOBr, the electronic structures of pure BiOBr and Fe3+-doped BiOBr are calculated by first-principles density functional theory (DFT). The band structure of pure BiOBr is calculated and presented in Fig. 4(c), where the valence band maximum (VBM) locates between Z → R points while the conduction band minimum (CBM) locates at the Z point, which implies that BiOBr is an indirect band gap semiconductor. The calculated band gap is 2.19 eV, which is smaller than the experimental value, owing to the well-known GGA-PBE underestimation.13,38Fig. 4(d) shows the detailed density of state (DOS) spectra of pure BiOBr. It can be seen that the VBM is mainly contributed from the Br 4p and O 2p orbitals, whereas the CBM is mainly derived from the Bi 6p orbitals, which is consistent with the results of previous studies.39 Compared to pure BiOBr, the doping of Fe3+ can effectively narrow the band gap of BiOBr (1.39 eV) by generating an impurity energy level within the band gap of pure BiOBr [Fig. 4(e)]. The band gap decrease will induce more Vis light absorption, which is consistent with the result of the UV-Vis-NIR spectra in Fig. 4(a). From the detailed DOS spectra [Fig. 4(f)], it can be seen that the VBM is still mainly derived from the Br 4p and O 2p orbitals. The CBM is mainly composed of the Bi 6p and Fe 3d orbitals, while the new impurity energy band is mainly contributed by the Fe 3d orbitals and a few O 2p orbitals. This result shows that the doped Fe3+ can induce the O 2p orbitals to generate the impurity energy level in the band gap of BiOBr to promote the photogenerated electron–hole (e−/h+) pair separation, which is beneficial to the photocatalytic performance.
To confirm the charge transfer and recombination processes of e−/h+ pairs, the photoluminescence (PL) spectra of the samples under excitation at a wavelength of 270 nm are recorded, as shown in Fig. 4(g). A broad emission peak located at about 440 nm is observed. As is known, a weak PL intensity means a lower recombination rate of the photogenerated e−/h+ pairs.15,40 The result shows that the PL intensity of the sample decreased upon introducing Fe3+, illustrating that Fe3+ doping could effectively inhibit the recombination of e−/h+ pairs. In addition, the production and transfer of e−/h+ pairs are also studied by using time-resolved PL decay. The decay curves of the samples are measured and presented in Fig. 4(h). All curves conform to a double-exponential fit:31
| | I(t) = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) exp(−t/τ1) + A2exp(−t/τ2) | (1) |
where
I is the luminescence intensity, A
1 and A
2 are constants,
t is the time, and
τ1 and
τ2 refer to the rapid and slow components of decay time, respectively. The average decay times (
τ) can be calculated by:
| | | τ = (A1τ12 + A2τ22)/(A1τ1 + A2τ2) | (2) |
Obviously, compared with the other samples, BYE-3% Fe exhibits the longest lifetime of photogenerated charges, indicating that the recombination rate of e−/h+ pairs is significantly inhibited,40,41 which is consistent with the PL spectra. These results clearly illustrate that Fe3+ ion doping favors the separation of photogenerated e−/h+ pairs, which may lead to excellent photocatalytic activity.
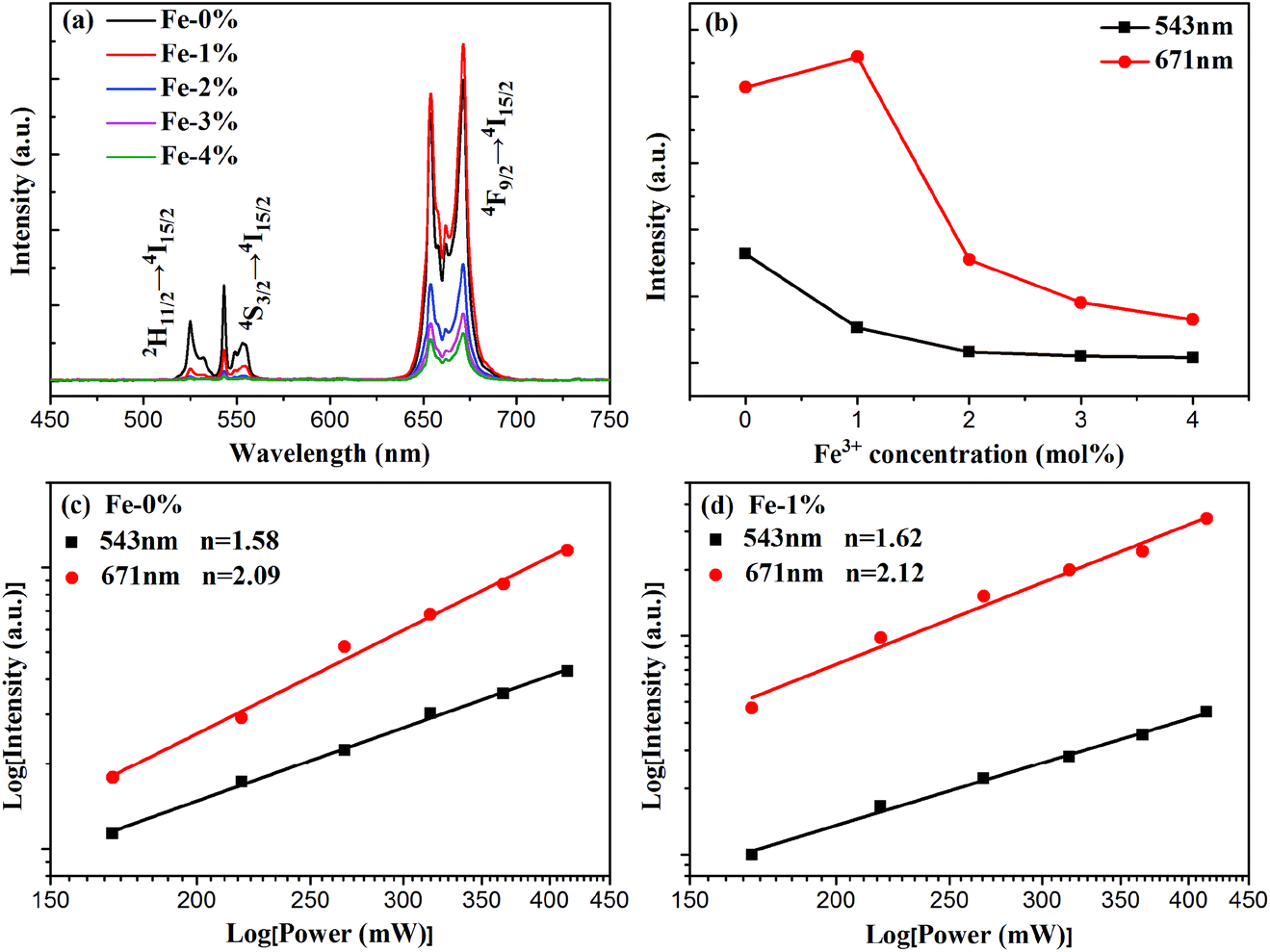
To analyze the effect of Fe3+ ion concentration on the UC emission behaviors of the samples, the UC emission spectra of BYE nanoplates with different Fe3+ ion concentrations are recorded under excitation at a wavelength of 980 nm and the results are shown in Fig. 5. As illustrated in Fig. 5(a), all samples exhibit three main emission peaks centered at 523, 543, and 671 nm, which are ascribed to the 2H11/2 → 4I15/2, 4S3/2 → 4I15/2, and 4F9/2 → 4I15/2 transitions of Er3+ ions, respectively. Clearly, the UC emission intensities are drastically changed with the increase in the Fe3+ concentration. As is shown in Fig. 5(b), the emission intensity of red luminescence increases first and then decreases with the increment of Fe3+ concentrations, while the green one continues to decrease. The enhancement of red UC luminescence is caused by the tailored local crystal field symmetry around the Er3+ ions.25 To confirm our hypothesis, the experiments taking Eu3+ as the structural probe,42,43 to investigate the modification of symmetry around RE3+ ions induced by Fe3+ doping, is also conducted. As shown in Fig. S5,† the intensity ratio of 5D0 → 7F2/5D0 → 7F1 gradually increases from 3.13 to 3.41 with the increase of the Fe3+ content. This result indicates that Fe3+ doping can tailor the local crystal field symmetry, which causes enhanced UC luminescence. According to the analysis of the absorption spectra [Fig. 4(a) and S6†], the green and red emission can be reabsorbed by the Fe3+-doped BYE nanoplates, which results in the decrease of UC emissions with the introduction of Fe3+ ions. To further confirm that the green and red emission decrease is associated with the reabsorption process, the UC luminescence of the BYE nanoplates single-doped or co-doped with Li+ and Zn2+ ions is measured. By contrast, the green and red emission intensity exhibits an upward tendency, as shown in Fig. S7,† because the absorption edge of less than 450 nm is not enough to absorb the longer wavelength emissions, such as green and red emission. Therefore, we can speculate that the green and red UC emission energy can probably be transferred to the BiOBr host. The time resolved spectra also support the energy transfer from UC emission to BiOBr. The decay curves are fitted using the double-exponential function and the average lifetimes are calculated for the 4S3/2 (543 nm) and 4F9/2 (671 nm) levels of Er3+ ions (Fig. S8†). One can see that the average lifetimes of the 4S3/2 and 4F9/2 levels decrease significantly with increasing Fe3+ ion concentration, which further confirms the efficient energy transfer from UC emission to the BiOBr host.44 Therefore, Fe3+-doped BYE nanoplates should be a promising candidate to achieve efficient photocatalytic activity under NIR light irradiation.
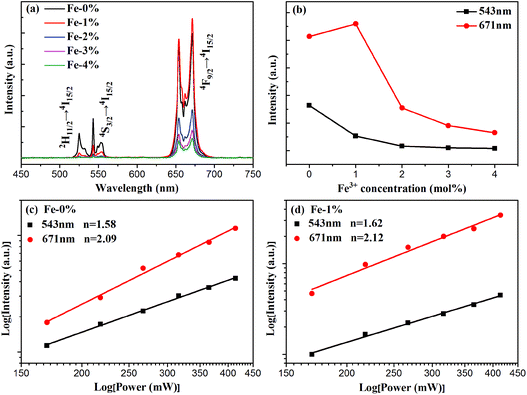 |
| | Fig. 5 (a) UC spectra and (b) the emission intensities of green and red luminescence of BYE nanoplates with different Fe3+ concentrations under excitation at a wavelength of 980 nm; power dependence of the UC emission of (c) BYE and (d) BYE-1% Fe. | |
For UC luminescence processes, the UC intensity IUC depends on the pumping power density Ip according to the following: IUC ∝ InP,31 where n is the number of pumping photons emitted per UC photon. The corresponding double logarithmic plot centered at 543 and 671 nm is displayed for BYE-0% Fe and BYE-1% Fe nanoplates in Fig. 5(c) and (d). The fitted slopes are 1.58 and 2.09 for BYE-0% Fe nanoplates, indicating the two-photon UC process. After Fe3+ ion doping, the n values are 1.62 and 2.12. The results illustrate that Fe3+ ion doping does not affect the UC process of BYE nanoplates.
2.3. Photocatalytic activity
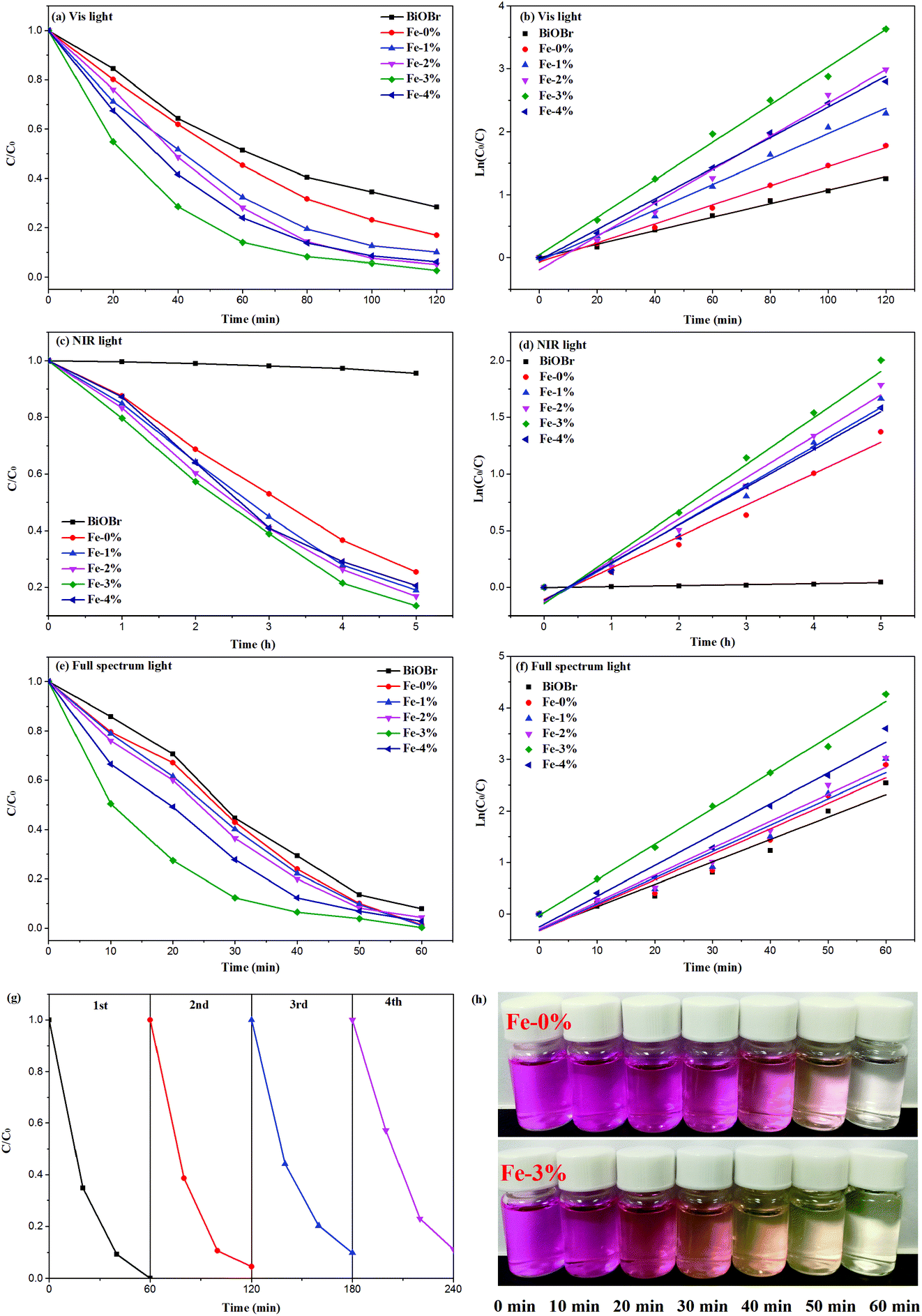
According to the above results, Fe3+ doping results in an increase of the Vis light absorption of BYE nanoplates, which should improve the Vis light photocatalytic activity. Therefore, the photocatalytic performance of the BYEF (BiOBr:Yb3+/Er3+/Fe3+) nanoplates is firstly evaluated by the degradation of RhB under Vis light irradiation. As shown in Fig. 6(a), under Vis light irradiation after 2 h, the degradation efficiency of RhB for pure BiOBr and BYE-0% Fe is 72% and 83%, respectively. With the increase of the Fe3+ ion concentration, the photocatalytic activity of BYE nanoplates gradually increases and then decreases, and the RhB degradation activity decreases in the following order: BYE-3% Fe > BYE-2% Fe > BYE-4% Fe > BYE-1% Fe > BYE-0% Fe > BiOBr. Furthermore, the corresponding kinetic constants are calculated by applying a first-order model: ln(C0/C) = kt,17 where k is the rate constant, C0 is the initial concentration of RhB and C is the concentration of RhB at different reaction times. As shown in Fig. 6(b) and Table 1, the reaction rate constant k is 0.0107, 0.0151, 0.0202, 0.0264, 0.0298 and 0.0243 min−1 for pure BiOBr, BYE-0% Fe, BYE-1% Fe, BYE-2% Fe, BYE-3% Fe and BYE-4% Fe, respectively. It is found that BYE-3% Fe has the highest degradation rate, which is about 2 times higher than that of BYE-0% Fe nanoplates. The result confirms that the photocatalytic activity of BYE nanoplates is significantly enhanced through Fe3+ ion doping. Under UV light irradiation, the degradation rule is consistent with the degradation curves of Vis light irradiation (Fig. S9†).
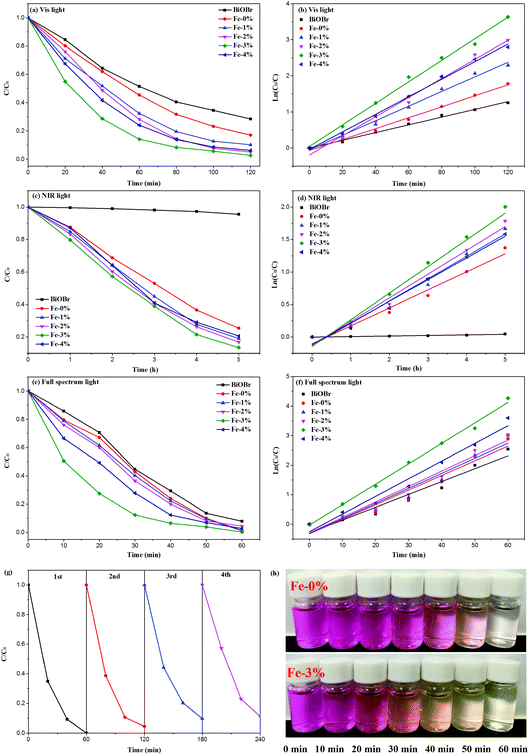 |
| | Fig. 6 Photocatalytic degradation of RhB and kinetics rates over different photocatalysts under (a, b) Vis, (c, d) NIR, and (e, f) full spectrum light irradiation; (g) recycling performance of BYE-3% Fe nanoplates under full spectrum light irradiation; (h) photographic image of the temporal evolution of the RhB solution over BYE-0% Fe and BYE-3% Fe under full spectrum light irradiation. | |
In order to improve the utilization efficiency of solar light, the photocatalytic properties of the samples under NIR light irradiation is shown in Fig. 6(c). Obviously, pure BiOBr shows almost no photocatalytic activity, because it cannot absorb NIR light. After Yb3+/Er3+ doping, the UV UC emission peaks centered at 380 and 410 nm can be observed (Fig. S10†), which are assigned to the transitions of 4G11/2 → 4I15/2 and 2H9/2 → 4I15/2 of Er3+, which can induce excitation across the band gap generating e− and h+. Therefore, the generated e− and h+ should be able to photodegrade RhB under the NIR (980 nm) irradiation. As expected, with the introduction of Fe3+ ions, the degradation performance is further improved, and the degradation rate of BYE-3% Fe reached about 86% after 5 h of NIR light irradiation; the reason will be discussed later. The photocatalytic degradation efficiency follows the order: BYE-3% Fe > BYE-2% Fe > BYE-1% Fe > BYE-4% Fe > BYE-0% Fe > BiOBr. The rate constants of all samples are presented in Fig. 6(d) and Table 1. The highest degradation rate of the BYE-3% Fe sample (0.4207 h−1) is 1.5 times higher than that of BYE-0% Fe (0.2776 h−1). Note that, in comparison with the NIR photoactivities of other RE3+ doped upconverting materials, as listed in Table 2, the BYE-3% Fe nanoplates exhibit a relatively high photodegradation ability under NIR light, implying that the Fe3+-doped BiOBr:Yb3+/Er3+ nanoplates have potential applications as NIR-activated photocatalysts.
Table 2 Comparison of dye degradation between our material and similar reported materials in the literature
| Photocatalyst |
Dyes |
Degradation rate |
Ref. |
| UV (min−1) |
Vis (min−1) |
NIR (h−1) |
Solar (min−1) |
| β-NaYF4: Yb3+, Er3+@TiO2–Ag6Si2O7 |
MB |
0.5050 |
0.0098 |
0.1191 |
1.8805 |
15
|
| β-NaYF4:Yb3+,Tm3+@SiO2@TiO2 |
RhB |
— |
— |
0.3624 |
— |
16
|
| β-NaYF4:Yb3+,Tm3+@SnO2@Ag |
RhB |
0.0342 |
0.0229 |
0.5757 |
0.2700 |
19
|
| BiOBr:Yb3+,Er3+/g-C3N4 |
RhB |
— |
0.0264 |
0.3649 |
0.0523 |
24
|
| Bi2WO6:Yb3+/Ho3+ |
RhB |
— |
0.1007 |
— |
— |
45
|
| BiPO4:Yb3+,Tm3+/BiVO4 nanocomposite |
MB |
— |
0.0107 |
0.0415 |
0.0471 |
46
|
| BiVO4/CaF2:Er3+, Tm3+,Yb3+ |
MO |
— |
— |
0.0113 |
0.036 |
47
|
| BiVO4:Er3+, Tm3+,Yb3+ |
MB |
|
0.0130 |
0.1656 |
— |
48
|
| BiOBr:Yb3+/Er3+/Fe3+ |
RhB |
— |
0.0298 |
0.4098 |
0.0692 |
This work |
As a step toward practical applications, the photocatalytic activity under full spectrum light irradiation has been further investigated. Under full spectrum light irradiation, the BYE-3% Fe nanoplates show the best photocatalytic activity, and almost completely degrade RhB within 1 h [Fig. 6(e)]. The corresponding rate constant k is presented in Fig. 6(f) and Table 1. For different forms of light irradiation, the degradation efficiency is in the following order: full spectrum light > Vis > NIR. Additionally, the stability of the photocatalyst is of great importance for practical applications. Therefore, the stability of BYE-3% Fe nanoplates is further investigated by recycling the photocatalysis of RhB under full spectrum light irradiation [Fig. 6(g)]. After four cycles, BYE-3% Fe shows a slight decrease in photocatalytic efficiency due to the loss of the catalyst in the recovery process, but the photocatalytic activity remains high, which implies that the BYEF nanoplates can be an excellent catalyst for practical applications.
2.4. Mechanism study
To understand the enhancement of the photocatalytic activity of the Fe3+-doped BiOBr:Yb3+/Er3+ nanoplates, several factors have to be taken into consideration, such as the specific surface area, photogenerated charge separation and utilization efficiency of sunlight. Generally, a high specific surface area is helpful in providing more surface active sites, resulting in higher photocatalytic activity.30 From Fig. S2† and Table 1, it can be seen that the specific surface area of the samples follows the order of BYE-2% Fe > BYE-1% Fe > BYE-4% Fe > BYE-3% Fe > BYE-0% Fe > BiOBr, which is not consistent with the photocatalytic activity (BYE-3% Fe > BYE-4% Fe > BYE-2% Fe > BYE-1% Fe > BYE-0% Fe > BiOBr). This result indicates that the specific surface area is not the main factor responsible for the enhanced photocatalytic activity of Fe3+-doped BYE nanoplates.
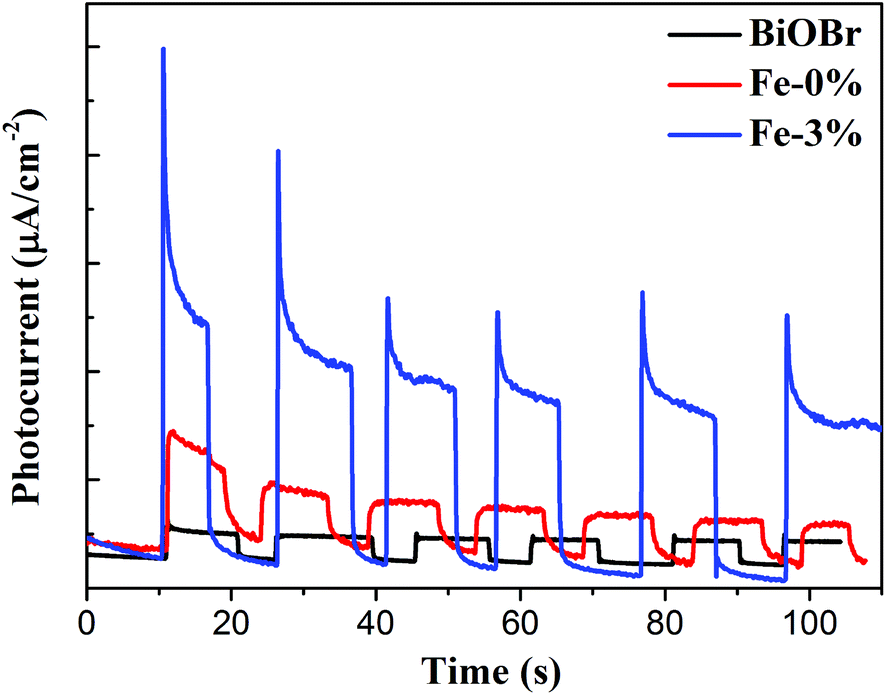
The above PL result suggests that Fe3+ doping can effectively reduce the recombination of e−/h+ pairs. To further understand the improved electron production and transfer, the transient photocurrent responses of pure BiOBr, BYE-0% Fe and BYE-3% Fe under full spectrum light irradiation are presented in Fig. 7. As expected, the photocurrent generated by BYE-3% Fe is obviously stronger than that of the other samples, which indicates that BYE-3% Fe could lead to the generation of more charge carriers. The enhanced photocurrent confirms that the Fe3+ ions could promote the separation of e−/h+ pairs, and hence reduce the recombination rate, which leads to enhanced photocatalytic activity.
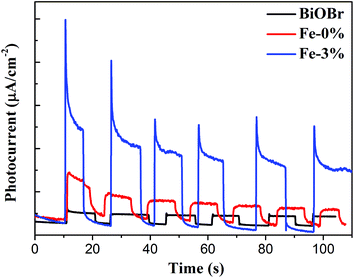 |
| | Fig. 7 Transient photocurrent response of pure BiOBr, BYE-0% Fe and BYE-3% Fe nanoplates under full spectrum light irradiation. | |
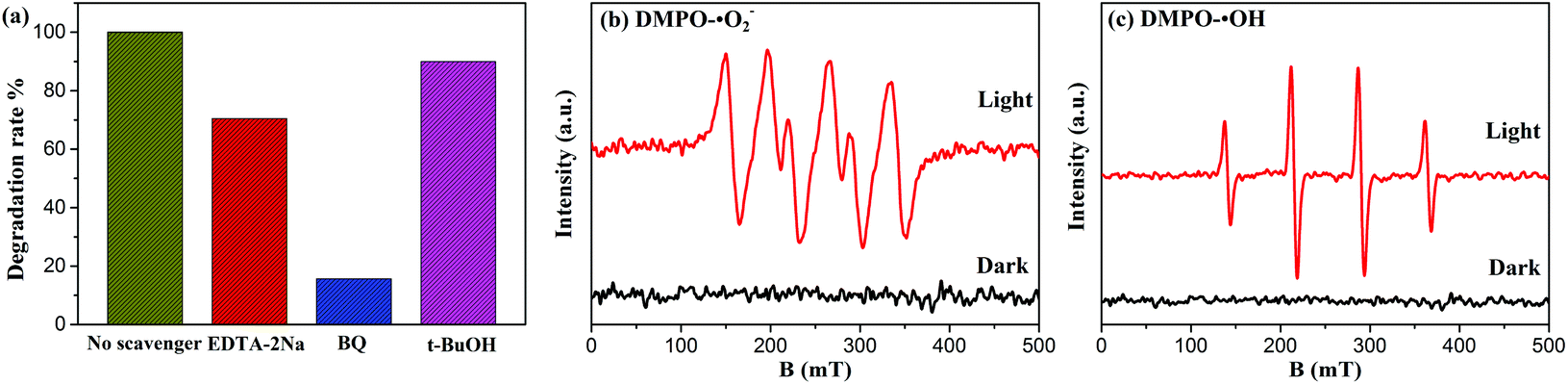
To obtain deeper insight into the intrinsic photocatalytic mechanism, the active species trapping experiment is performed. EDTA-2Na, t-BuOH, and BQ are used as scavengers for trapping photogenerated holes (h+), hydroxyl radicals (˙OH), and superoxide radicals (˙O2–), respectively.15,17 As shown in Fig. 8(a), under full spectrum light irradiation, the degradation efficiency of BYE-3% Fe is significantly restrained after the addition of BQ, indicating that the ˙O2– radical species has the dominant role in the photocatalytic process. By adding EDTA-2Na, the degradation efficiency is also reduced, suggesting that h+ species participated in the photocatalytic process but did not play a predominant role. However, the presence of t-BuOH has a slight influence on the catalytic efficiency, indicating that ˙OH is not the main reactive species in the reaction process. The above results demonstrated that the photocatalytic process is mainly governed by direct h+ and ˙O2– oxidation reactions, and ˙OH plays a secondary role. Meanwhile, the ESR spin-trap technique is employed to further identify the generation of active species of BYE-3% Fe nanoplates. As shown in Fig. 8(b and c), under the irradiation of full spectrum light for 10 min, DMPO-˙O2– and DMPO-˙OH signals are observed, which indicates that ˙O2– and ˙OH are produced during the photocatalytic process, and this is well consistent with the trapping experimental results.
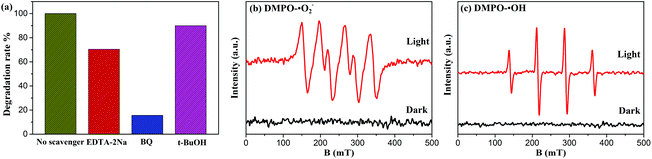 |
| | Fig. 8 (a) Effects of scavengers on the degradation of RhB for BYE-3% Fe nanoplates; ESR spectra (b) of DMPO-˙O2– and (c) DMPO-˙OH adducts of BYE-3% Fe nanoplates in the dark and under full spectrum light irradiation. | |
To further investigate the generation process of ˙O2– and ˙OH, analysis of the valence band (VB) and conduction band (CB) positions on BYE is necessary. It has been reported that the VB and CB positions could be calculated as follows:13
where
EVBM represents the VB edge potential and
ECBM is the CB edge potential;
X is the electronegativity of the semiconductor;
Ee is the energy of free electrons on the hydrogen scale (about 4.5 eV), and
Eg is the band gap energy of the semiconductor [2.93 eV;
Fig. 4(b)]. According to
eqn (3) and (4), the
ECBM of BYE was −1.345 eV, as shown in Fig. S11,
† which is more negative than the potential of the ˙O
2–/O
2 couple (−0.33 eV
vs. NHE),
15,49 and thus facilitates the dissolved O
2 molecule to generate ˙O
2–. Theoretically, h
+ cannot efficiently oxidize OH
− groups to ˙OH radicals, because the
EVBM (1.585 eV) is less positive than the potential of the ˙OH/OH
− couple (∼2.38 V
vs. NHE).
49 However, the trapping experimental and ESR results show the coexistence of the ˙OH in the photocatalytic process. This may be because Fe
3+ doping could promote the production of ˙OH radicals;
28 the detailed explanations will be discussed later.
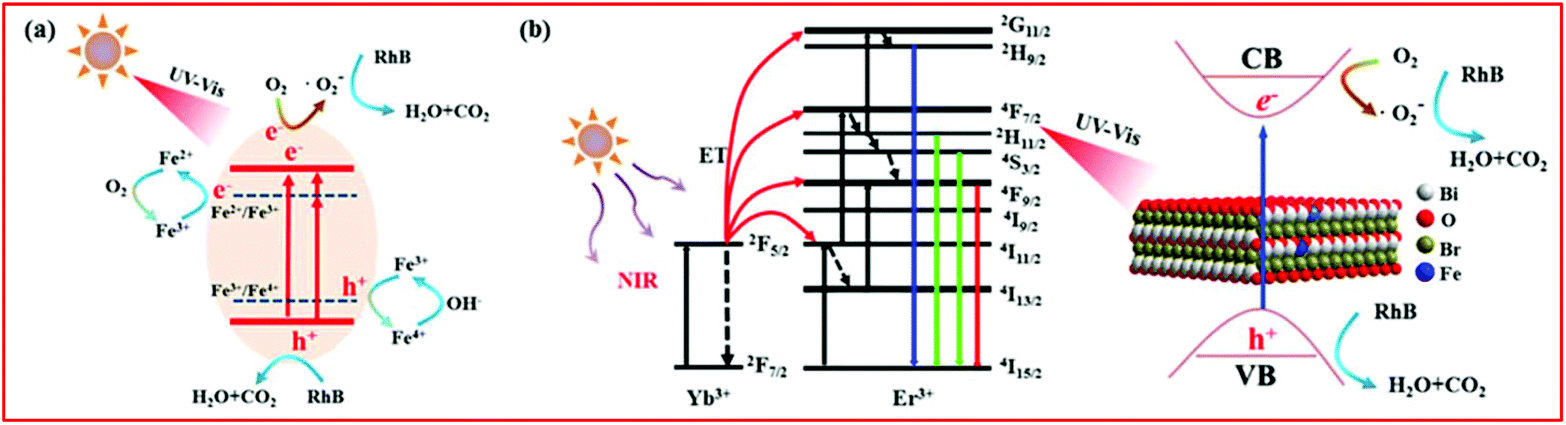
According to the above experimental results and discussion, the enhanced photocatalytic activity of BiOBr:Yb3+/Er3+/Fe3+ nanoplates could involve different mechanisms under different forms of light irradiation, as depicted in Fig. 9. Under UV and Vis light illumination [Fig. 9(a)], the electrons (e−) and holes (h+) are generated in the CB and VB of BiOBr, respectively. The CB electrons are scavenged by the oxygen adsorbed on the surface of BiOBr to produce ˙O2–, playing a dominant role in RhB degradation. Simultaneously, the VB holes can directly degrade RhB. Moreover, as depicted in the UV–Vis absorption spectra, Fe3+ doping can form an impurity level between the VB and CB of BiOBr, which increases the light absorption region. Therefore, much more electrons can be easily excited by photons with a lower energy, which can efficiently improve the photocatalytic activity. In addition, the electrons and holes can be trapped by Fe3+, which can turn into Fe2+ and Fe4+ ions.26,39,50,51 However, Fe2+ and Fe4+ ions are relatively unstable than Fe3+ ions, since they have the half-filled 3d5 electronic configuration.26,39,50,51 Therefore, Fe2+ ions are oxidized to Fe3+ ions through transferring electrons to the absorbed O2 molecule on the surface of BiOBr, and then the adsorbed O2 is reduced to ˙O2–, which can degrade RhB. Similarly, Fe4+ ions can also be reduced to Fe3+ ions by releasing electrons, while the OH− groups on the surface of BiOBr can convert into ˙OH.39,50 This explains the coexistence of ˙OH in the photocatalytic process. The above process can be described as follows:
| | | Fe2+ + O2 → Fe3+ + ˙O2– | (9) |
| | | Fe4+ + OH− → Fe3+ + ˙OH | (10) |
| | | ˙O2– or h+ + RhB → CO2 + H2O | (11) |
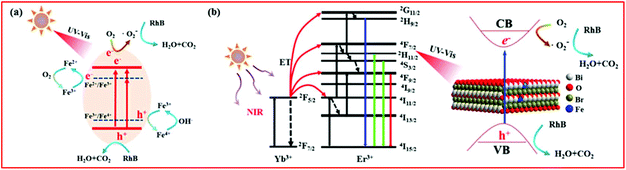 |
| | Fig. 9 The plausible photocatalytic mechanism of BiOBr:Yb3+/Er3+/Fe3+ nanoplates under (a) UV-Vis and (b) NIR irradiation. | |
Based on the UC spectra, the mechanism for NIR-activated photocatalysis is illustrated in Fig. 9(b). As for the Yb3+/Er3+ co-doped system under excitation at a wavelength of 980 nm, the Yb3+ ion acts as a sensitizer to absorb 980 nm photons, and the Yb3+ ions can transfer energy to Er3+ ions. Through the energy transfer (ET) UC process, the Er3+ ions are excited to the 4G11/2, 2H9/2, 2H11/2/4S3/2 and 4F9/2 levels. After that, the excited Er3+ ions fall to the ground state: 4G11/2 → 4I15/2, 2H9/2 → 4I15/2, 2H11/2 → 4I15/2, 4S3/2 → 4I15/2 and 4F9/2 → 4I15/2, leading to emission at 380, 411, 525, 545 and 671 nm, respectively. According to the absorption spectra, the generated UV and Vis light of the Er3+ ion can be reabsorbed by the BiOBr nanoplates. Under the excitation of these UC emissions, the activated BiOBr produces e− and h+ in the CB and VB bands, respectively, by following the same pathway described for the UV and Vis light illumination. The same as under the Vis light irradiation, BYE-3% Fe exhibits the highest photocatalytic activity under NIR irradiation, which is attributed to the enhanced UC emission and increase in the light absorption region by Fe3+ doping. As a result, more UC luminescence could participate in catalytic reactions, which helps to improve the NIR photocatalytic activity. Under the full spectrum light, all the abovementioned processes can actually take place. However, when the Fe3+ ion doping concentration is too high, recombination centers are formed for the photogenerated e− and h+,52 which can reduce the photocatalytic activity.
3. Conclusion
In summary, Fe3+-doped BiOBr:Yb3+/Er3+ nanoplates are successfully prepared by a simple solvothermal process. The introduction of Fe3+ ions can enhance the UC emission intensity through tailoring the crystal symmetry. The Fe3+-doped BiOBr:Yb3+/Er3+ nanoplates exhibit enhanced photocatalytic activity in the degradation of RhB under UV, Vis, NIR and full spectrum light irradiation compared to BiOBr:Yb3+/Er3+ and pure BiOBr nanoplates. In particular, the BiOBr:Yb3+/Er3+/3% Fe3+ nanoplates show the highest photocatalytic activity for the degradation of RhB under Vis (0.0298 min−1), NIR (0.4207 h−1) and full spectrum light (0.0692 min−1), which is about 1.5 times higher than that of BiOBr:Yb3+/Er3+ nanoplates. The enhanced photocatalytic activity of BiOBr:Yb3+/Er3+/Fe3+ can be attributed to the synergic effect of the efficient utilization of UC luminescence and Fe3+ doping. The Fe3+-doped BiOBr could form an impurity energy level in the band gap, resulting in expanding the light absorption region, enhancing the separation efficiency of e−/h+ pairs and promoting the production of highly oxidative species. Moreover, stability tests indicate good stability of the catalyst. This work provides a feasible strategy for the rational design of full-spectrum-activated photoactive materials for effectively harvesting solar energy, which are potential candidates as photocatalysts for environmental applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 61172027), the Guangdong Natural Science Foundation (No. 2014A030311049) and the Science and Technology Planning Project of Guangdong Province (No. 2017A010103035).
References
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 8, 6904–6920 RSC.
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed.
- J. Di, J. Xia, M. Ji, B. Wang, S. Yin, Q. Zhang, Z. Chen and H. Li, ACS Appl. Mater. Interfaces, 2015, 7, 20111 CrossRef CAS PubMed.
- E. Lhuillier, S. Pedetti, S. Ithurria, B. Nadal, H. Heuclin and B. Dubertret, Acc. Chem. Res., 2015, 48, 22–30 CrossRef CAS PubMed.
- M. Chatti, V. N. Adusumalli, S. Ganguli and V. Mahalingam, Dalton Trans., 2016, 45, 12384–12392 RSC.
- Q. Wang, W. Wang, L. Zhong, D. Liu, X. Cao and F. Cui, Appl. Catal., B, 2018, 220, 290–302 CrossRef CAS.
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2016, 50, 112–121 CrossRef PubMed.
- L. Ye, Y. Su, X. Jin, H. Xie and C. Zhang, Environ. Sci.: Nano, 2014, 1, 90–112 RSC.
- J. Li, Y. Yu and L. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC.
- Y. Huo, J. Zhang, M. Miao and Y. Jin, Appl. Catal., B, 2012, 111, 334–341 CrossRef.
- X. C. Song, Y. F. Zheng, H. Y. Yin, J. N. Liu and D. R. Xue, New J. Chem., 2016, 40, 130–135 RSC.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- H. Huang, X. Han, X. Li, S. Wang, P. K. Chu and Y. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 482–492 CrossRef CAS PubMed.
-
(a) C. Ma, X. Xu, F. Wang, Z. Zhou, S. Wen, D. Liu, J. Fang, C. I. Lang and D. Jin, J. Phys. Chem. Lett., 2016, 7, 3252–3258 CrossRef CAS PubMed;
(b) C. Ma, X. Xu, F. Wang, Z. Zhou, D. Liu, J. Zhao, M. Guan, C. I. Lang and D. Jin, Nano Lett., 2017, 17, 2858 CrossRef CAS PubMed.
-
(a) S. Huang, L. Gu, C. Miao, Z. Lou, N. Zhu, H. Yuan and A. Shan, J. Mater. Chem. A, 2013, 1, 7874–7879 RSC;
(b) Q. Tian, W. Yao, Z. Wu, J. Liu, L. Liu, W. Wu and C. Jiang, J. Mater. Chem. A, 2017, 5, 23566–23576 RSC.
- W. Wang, W. Huang, Y. Ni, C. Lu and Z. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 340–348 CrossRef CAS PubMed.
- Q. Zhang, J. Deng, Z. Xu, M. Chaker and D. Ma, ACS Catal., 2017, 7, 6225–6234 CrossRef CAS.
- Z. Xu, M. Quintanilla, F. Vetrone, A. O. Govorov, M. Chaker and D. Ma, Adv. Funct. Mater., 2015, 25, 2950–2960 CrossRef CAS.
- Q. Tian, W. Yao, W. Wu, J. Liu, Z. Wu, L. Liu, Z. Dai and C. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 10889–10899 CrossRef CAS.
- S. Niu, R. Zhang, X. Zhou, X. Zhao, H. Suo, Y. Jiao, H. Yao and C. Guo, Dyes Pigm., 2018, 149, 462–469 CrossRef CAS.
- W. Wu, D. Chen, Y. Zhou, Z. Wan and Z. Ji, J. Alloys Compd., 2016, 682, 275–283 CrossRef CAS.
- L. Xu, F. He, C. Wang, S. Gai, A. Gulzar, D. Yang, C. Zhong and P. Yang, J. Mater. Chem. B, 2017, 5, 7939–7948 RSC.
- Y. Li, Z. Song, Z. Yin, Q. Kuang, R. Wan, Y. Zhou, Q. Liu, J. Qiu and Z. Yang, Spectrochim. Acta, Part A, 2015, 150, 135–141 CrossRef CAS PubMed.
- S. Liang, M. He, J. Guo, J. Yue, X. Pu, B. Ge and W. Li, Sep. Purif. Technol., 2018, 206, 69–79 CrossRef CAS.
- P. Ramasamy, P. Chandra, S. W. Rhee and J. Kim, Nanoscale, 2013, 5, 8711–8717 RSC.
- H. Khan and I. K. Swati, Ind. Eng. Chem. Res., 2016, 55, 6619–6633 CrossRef CAS.
- S. Chala, K. Wetchakun, S. Phanichphant, B. Inceesungvorn and N. Wetchakun, J. Alloys Compd., 2014, 597, 129–135 CrossRef CAS.
- Y. Mi, L. Wen, Z. Wang, D. Cao, R. Xu, Y. Fang, Y. Zhou and Y. Lei, Nano Energy, 2016, 30, 109–117 CrossRef CAS.
- M. Yuan, F. Tian, G. Li, H. Zhao, Y. Liu and R. Chen, Ind. Eng. Chem. Res., 2017, 56, 5935–5943 CrossRef CAS.
- Y. Liu, G. Zhu, J. Gao, R. Zhu, M. Hojamberdiev, C. Wang, X. Wei and P. Liu, Appl. Catal., B, 2017, 205, 421–432 CrossRef CAS.
- J. Tang, L. Chen, J. Li, Z. Wang, J. Zhang, L. Zhang, Y. Luo and X. Wang, Nanoscale, 2015, 7, 14752–14759 RSC.
- Y. Zhang, Y. Shen, M. Liu, Y. Han, X. Mo, R. Jiang, Z. Lei, Z. H. Liu, F. Shi and W. Qin, CrystEngComm, 2017, 19, 1304–1310 RSC.
- G. Jiang, X. Wang, Z. Wei, X. Li, X. Xi, R. Hu, B. Tang, R. Wang, S. Wang and T. Wang, J. Mater. Chem. A, 2013, 1, 2406–2410 RSC.
- C. Liu, X. Dong, Y. Hao, X. Wang, H. Ma and X. Zhang, RSC Adv., 2017, 7, 22415–22423 RSC.
- Y. Guo, Z. Zhang, G. Zhu, W. Zhang and W. Yang, RSC Adv., 2017, 7, 2629–2636 RSC.
- J. Li, K. Zhao, Y. Yu and L. Zhang, Adv. Funct. Mater., 2015, 25, 2189–2201 CrossRef CAS.
- C. Y. Wang, Y. J. Zhang, W. K. Wang, D. N. Pei, G. X. Huang, J. J. Chen, X. Zhang and H. Q. Yu, Appl. Catal., B, 2017, 221, 320–328 CrossRef.
- Q. Wang, Z. Liu, D. Liu, G. Liu, M. Yang, F. Cui and W. Wang, Appl. Catal., B, 2018, 236, 222–232 CrossRef CAS.
-
(a) W. L. Huang, J. Comput. Chem., 2010, 30, 1882–1891 CrossRef PubMed;
(b) J. Guo, X. Liao, M. Lee, G. Hyett, C. Huang, D. W. Hewak, S. Mailis, W. Zhou and Z. Jiang, Appl. Catal., B, 2019, 243, 502–512 CrossRef CAS.
- S. Liang, T. Zhang, D. Zhang, X. Pu, X. Shao, W. Li and D. Jianmin, J. Am. Ceram. Soc., 2018, 101, 3424–3436 CrossRef CAS.
- P. Xiao, J. Lou, H. Zhang, W. Song, X. Wu, H. Lin, J. Chen, S. Liu and X. Wang, Catal. Sci. Technol., 2018, 8, 201–209 RSC.
- H. Suo, X. Zhao, Z. Zhang, R. Shi, Y. Wu, J. Xiang and C. Guo, Nanoscale, 2018, 10, 9245–9251 RSC.
- D. Tu, Y. Liu, H. Zhu, R. Li, L. Liu and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 1128–1133 CrossRef CAS PubMed.
- H. Jia, Z. Chen, P. Chen, C. Xu, C. Liu, Z. Zhao, X. Liu and J. Qiu, J. Am. Ceram. Soc., 2017, 100, 697–704 CrossRef CAS.
- H. Li, H. Hao, S. Jin, W. Guo, X. Hu, H. Hou, G. Zhang, S. Yan, W. Gao and G. Liu, Catal. Commun., 2017, 97, 60–64 CrossRef CAS.
- S. Ganguli, C. Hazra, M. Chatti, T. Samanta and V. Mahalingam, Langmuir, 2015, 32, 247–253 CrossRef PubMed.
- S. Huang, N. Zhu, Z. Lou, L. Gu, C. Miao, H. Yuan and A. Shan, Nanoscale, 2014, 6, 1362–1368 RSC.
- C. Regmi, Y. K. Kshetri, S. K. Ray, R. P. Pandey and S. W. Lee, Appl. Surf. Sci., 2017, 392, 61–70 CrossRef CAS.
- S. Ding, X. Xiong, X. Liu, Y. Shi, Q. Jiang and J. Hu, Catal. Sci. Technol., 2017, 7, 3791–3801 RSC.
- M. Wang, J. Gao, G. Zhu, N. Li, R. Zhu, X. Wei, P. Liu and Q. Guo, RSC Adv., 2016, 6, 85962–85969 RSC.
- C. Huang, J. Hu, S. Cong, Z. Zhao and X. Qiu, Appl. Catal., B, 2015, 174–175, 105–112 CrossRef CAS.
- J. Yu, Q. Xiang and M. Zhou, Appl. Catal., B, 2009, 90, 595–602 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and Fig. S1–S11. See DOI: 10.1039/c8qi01098f |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Lu
Yao
a,
Dekang
Xu
b,
Yulin
Hu
a,
Shenghong
Yang
a and
Yueli
Zhang
*a
a,
Lu
Yao
a,
Dekang
Xu
b,
Yulin
Hu
a,
Shenghong
Yang
a and
Yueli
Zhang
*a