
Metal-dependent electronic and photochromic behaviors of dimethylbenzotriazolium iodometallate hybrids†
Pengfei
Hao
 ,
Weipin
Wang
,
Lifang
Zhang
,
Junju
Shen
and
Yunlong
Fu
*
,
Weipin
Wang
,
Lifang
Zhang
,
Junju
Shen
and
Yunlong
Fu
*
Key Laboratory of Magnetic Molecules, Magnetic Information Materials Ministry of Education, School of Chemical and Material Science, Shanxi Normal University, Linfen 041004, China. E-mail: yunlongfu@sxnu.edu.cn; Fax: (+86) 357 2053716; Tel: +86 (0) 357 2053716
First published on 29th November 2018
Abstract
Two isostructural organic–inorganic hybrids, [DMBTz][Ag3I4] (1) and [DMBTz][Cu3I4] (2) (DMBTz+ = dimethylbenzotriazolium), have been constructed from [Ag3I4]−/[Cu3I4]− inorganic chains and [DMBTz]+ cations, which exhibit metal-dependent electronic behaviors and adjustable photoresponsive properties. In contrast to obvious intermolecular charge transfer (CT) and optically inert response of an iodocuprate hybrid, an iodoargentate hybrid exhibits photoinduced electron transfer (ET) photochromism and phase-transition enhanced photosensitivity, suggesting a delicate modulating effect of the composition and packing modes on the intermolecular CT and ET as well as consequently photoresponsive behaviors.
Introduction
Organic–inorganic hybrid materials composed of electron donor–acceptor (D–A) units have received considerable attention due to their flexible composition–function advantages, unique responses to multiple external stimuli on the basis of intermolecular charge transfer/electron transfer (CT/ET), and primary importance in functional materials, such as protection, decoration, displays, memory, switches, photography, and so on.1–3 In order to rationally construct D–A pairs and facilitate the correlatively functional requirement, the use of strong electron acceptors is prevalent, such as viologens,4,5 1,4,5,8-naphthalenediimides (NDIs),6,7 and tris(4-pyridiniumyl)-1,3,5-triazine (TPT),8–10 which are usually matched with strong Lewis bases such as chlorometallates, carboxylates, zeolites, and phosphates. Also, the effects on the detailed variation of substituted groups of the organic acceptors and the composition of inorganic moieties as well as organic–inorganic interfaces are extensively investigated.4–10Iodometallates are well-known inorganic semiconductors, which exhibit structural diversity and flexibility,11–14 and tunable energy gaps as well as fascinating properties for applications in photovoltaic cells and catalysis.15–17 The introduction of unsaturated organic countercations into iodometallates usually leads to obvious intermolecular CT, which is not beneficial for the occurrence of photoinduced ET due to the so-called heavy-atom effect18,19 and polarization effect.20,21 Noteworthily, as recently reported by our group, effectively photoinduced ET and chromic behaviors can be realized in a few iodometallates by introducing moderately electron-deficient monocyclic pyridine derivatives such as methylnicotinohydrazide, pyrazinium and carbomethoxypyridinium,22–26 which implies the critical roles of suitable electron acceptors in the intermolecular CT and/or ET processes. The factors related to the photoresponsive behaviors in the iodometallate hybrid system, such as the electron-donating ability of the iodoargentates22–25 and the electron-accepting ability of organic cations26 have been investigated in detail. However, the subtle balance between intermolecular CT and ET as well as photoresponsive behaviors modulated by the composition of iodometallates and/or the change of packing modes has not been documented so far.
Structurally different from monocyclic pyridine derivatives, benzotriazolium derivatives possess moderately electron-accepting abilities and consequently stable radical forms, which have been widely applied in photovoltaic27,28 and electrochromic29–31 polymer systems but seldom in photochromic crystalline states.32 Therefore, the combination of electron-deficient benzotriazolium and electron-rich iodometallates should be an effective route to rationally modulate intermolecular CT and ET and consequently improve photochromic behaviors.
On the basis of the above consideration, by using dimethylbenzotriazolium as a moderate electron acceptor and semiconductive iodoargentate/iodocuprate as an electron donor, we herein reported a pair of isostructural organic–inorganic hybrid iodometallates, [DMBTz][Ag3I4] (1) and [DMBTz][Cu3I4] (2) (DMBTz+ = dimethylbenzotriazolium). 1 shows ET photochromism and phase-transition enhanced photochromic behavior, while 2 is optically inert, which reveals the delicate modulating effect of the flexibility of semiconductive iodoargentate/iodocuprate on the intermolecular CT and ET accompanying consequently photoresponsive behaviors.
Experimental
Materials and methods
All chemicals except for [DMBTz]I (dimethylbenzotriazolium iodate) were commercially available and used as received without further purification. IR spectra were recorded with a Nicolet 5DX spectrometer as KBr disks (4000–500 cm−1) (Fig. S1†). Elemental analyses for C, H and N were performed using a PerkinElmer 240 elemental analyzer. Thermogravimetric analysis-differential scanning calorimetry (TG-DSC) data were collected under a nitrogen atmosphere (flow rate 100 cm3 min−1, thermal ramp 10 °C min−1, and temperature range 35–800 °C) using a HCT-2 thermogravimetric analyzer. UV-vis absorption spectra were recorded at room temperature with a Varian Cary 5000 UV-vis spectrophotometer. Powder X-ray diffraction (PXRD) patterns were obtained with a Rigaku UItima IV-185 diffractometer at room temperature, and BaSO4 plates were used as the reference. Electron paramagnetic resonance (EPR) spectra were recorded on a Bruker A300-10/12 electron paramagnetic resonance spectrometer at room temperature. A 300 W mercury lamp (∼365 nm) system with temperature control equipped with an IR filter was used to prepare colored samples for EPR, and UV-Vis spectrophotometer studies, and the distances between these samples and the mercury lamp were around 25 cm. A visible light source is generated from a 500 W xenon lamp (>420 nm) with temperature control equipped with a UV filter.Synthesis of [DMBTz][Ag3I4] (1)
A mixture of benzotriazole (0.077 g, 0.65 mmol), AgI (0.306 g, 1.3 mmol), NaI·2H2O (0.186 g, 1.0 mmol), HI (0.1 mL), CH3OH (3.0 mL) and acetone (4.0 mL) was stirred for 30 min at room temperature, then sealed in a 25 mL Teflon-lined stainless steel autoclave and then kept under autogenous pressure at 110 °C for 3 days. Colorless stick crystals of 1 were obtained (yield: 39.8% based on Ag). Anal. calcd for C8H10N3Ag3I4: C 9.81, H 1.03, N 4.29%. Found: C 9.84, H 1.01, N 4.32%. IR (KBr, cm−1): 3063(w), 2993(w), 2941(w), 2366(w), 2324(w), 1607(m), 1501(w), 1477(w), 1435(m), 1392(w), 1347(m), 1316(m), 1258(m), 1143(m), 1102(w), 1051(w), 1017(w), 946(w), 780(s), 746(s), 636(w), 564(w).Synthesis of [DMBTz][Cu3I4] (2)
A mixture of [DMBTz]I (0.179 g, 0.65 mmol), CuI (0.437 g, 2.3 mmol), HI (0.1 mL), and acetonitrile (7.0 mL) was stirred for 30 min at room temperature. Brown plate crystals of 2 were obtained (yield: 45.3% based on Cu). Anal. calcd for C8H10N3Cu3I4: C, 11.35; H, 1.19; N, 4.96%. Found: C, 11.43; H, 1.13; N, 4.87%. IR (KBr, cm−1): 3090(w), 3048(m), 2922(w), 2849(w), 2362(s), 2333(m), 1627(s), 1603(s), 1421(w), 1388(w), 1338(w), 1312(w), 1262(w), 1123(m), 1043(m), 765(s), 668(w), 630(w), 563(w).X-Ray crystallography
Single crystals of 1, 1P and 2 were mounted with glue on a glass fiber and crystal data were collected at 293 K on the Oxford Gemini diffractometer using graphite monochromated Mo-Kα (λ = 0.71073 Å). Data reduction was accomplished using the CrysAlisPro (Oxford Diffraction Ltd, version 1.171.33.55) program. An empirical absorption correction was done using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm.33 All the structures were solved by direct methods and refined on F2 by full-matrix least-squares techniques with the SHELXTL-97 program.34,35 All non-hydrogen atoms were refined anisotropically. A summary of the crystal data is shown in Table S1.† The selected bond lengths and bond angles are listed in Table S2 of the ESI.† CCDC 1487190, 1487191 and 1867441† contain the supplementary crystallographic data for 1, 1P and 2.Results and discussion
Crystal structures
Compounds [DMBTz][Ag3I4] (1) and [DMBTz][Cu3I4] (2) are isostructural and crystallize in the monoclinic space group P21/c. Both compounds possess 1-D anionic chains ([Ag3I4]− in 1 and [Cu3I4]− in 2) and charge compensating agents of [DMBTz]+ cations. Their asymmetric unit contains three crystallographically independent metal ions (Ag+ in 1 and Cu+ in 2), and four I− ions together with one [DMBTz]+ cation (Fig. S2 and S3†). All Ag+ ions in 1 (Cu+ ions in 2) are surrounded by four I− ions in slightly distorted tetrahedral coordination environments. Ag–I bond distances 2.7784(9)–3.0449(11) Å and I–Ag–I angles in 1 are in the range of 88.93(3)–119.41(3)°, respectively, while Cu–I bond distances and I–Cu–I angles in 2 vary from 2.6020(9) to 2.7730(11) Å and 91.80(3) to 116.05(4)° respectively. The Ag–Ag distances of 3.0470(19)–3.209(2) Å in 1 are much shorter than the sum of the van der Waals radii of silver (3.44 Å), while the Cu–Cu distances of 2.8462(12)–2.9852(12) Å in 2 are comparable to the sum of the van der Waals radii of copper (2.80 Å), indicating the existence of weak metal⋯metal interactions.36–42 AgI4/CuI4 tetrahedra are joined to each other via the edge-sharing mode to form [Ag3I4]−/[Cu3I4]− chains43,44 (Fig. 1a) with discrete DMBTz+ as counterions (Fig. 1b). The short contact between I(3) and the centroid of the triazole ring is 3.567 Å in 1 and 3.512 Å in 2, and the angle of the I⋯centroid axis to the plane of the triazole ring is 83.17° in 1 and 76.70° in 2, indicating strong anion–π interactions.45,46 | ||
| Fig. 1 (a) View of [Ag3I4]n−/[Cu3I4]n− anionic chains. (b) View of the packing diagram of 1 and 2. All H atoms are omitted for clarity. | ||
TG-DSC studies and phase transition
TG-DSC studies were performed for 1 and 2 under an argon atmosphere (Fig. 2 and Fig. S4†). The TGA curves show that 1 and 2 are stable up to about 272 °C and 252 °C, respectively. The weight losses of 30.59% (233–332 °C in 1) and 31.6% (206–321 °C in 2) are attributed to the loss of [DMBTz]I (calc. 31.91% for 1 and 31.35% for 2) followed by the decomposition of inorganic frameworks.47 Interestingly, there are two strong endothermic peaks at 137 °C and 163 °C in 1 (Fig. 2, blue line), which correspond to the possible phase transition and melting point of the iodoargentate hybrid. In contrast, only one strong endothermic peak at 172 °C is observed in 2 and it corresponds to the melting point of the iodocuprate hybrid (Fig. S4†), which is due to the difference of the metal ions. Furthermore, after heating 1 at 137 °C for 30 min in air, the colorless crystal can transform into a surprisingly yellow crystal. Silent electron paramagnetic resonance (EPR) signals of 1T rule out the thermo-induced ET and formation of colored [DMBTz]˙ radicals. Although the structure of 1T cannot be obtained because of the loss of the single crystal character, the powder X-ray diffraction (PXRD) pattern of 1T (Fig. 3) is recorded and carefully analyzed. The diffraction intensity of 1T with respect to that of 1 (Fig. 3, blue line vs. red line) becomes weak and the position of some peaks is slightly changed. In particular, the [001] peak at 9.64° shifts to a lower angle of 8.25° and the [020] peak at 7.14° shifts to a higher angle of 7.46° suggesting lattice expansion along the a-axis and lattice contraction along the b-axis, respectively, which are likely consistent with the modulation of the packing modes between iodoargentate and [DMBTz]+, aggregation of [DMBTz]+ cations and the consequent enhancement of intermolecular CT.32 The thermal-induced phase transition is not reversible, which is proved by the retained color and PXRD (Fig. S5†) as well as the disappearance of the strong endothermic peaks at 137 °C in the DSC curve of 1T (Fig. S6†). This phenomenon exhibits the thermodynamic stability of 1T with respect to 1, which reveals the structural flexibility of semiconductive iodoargentate responding to the external stimuli of temperature. | ||
| Fig. 2 Thermo-gravimetric (TG) and differential scanning calorimetry (DSC) curves of 1. | ||
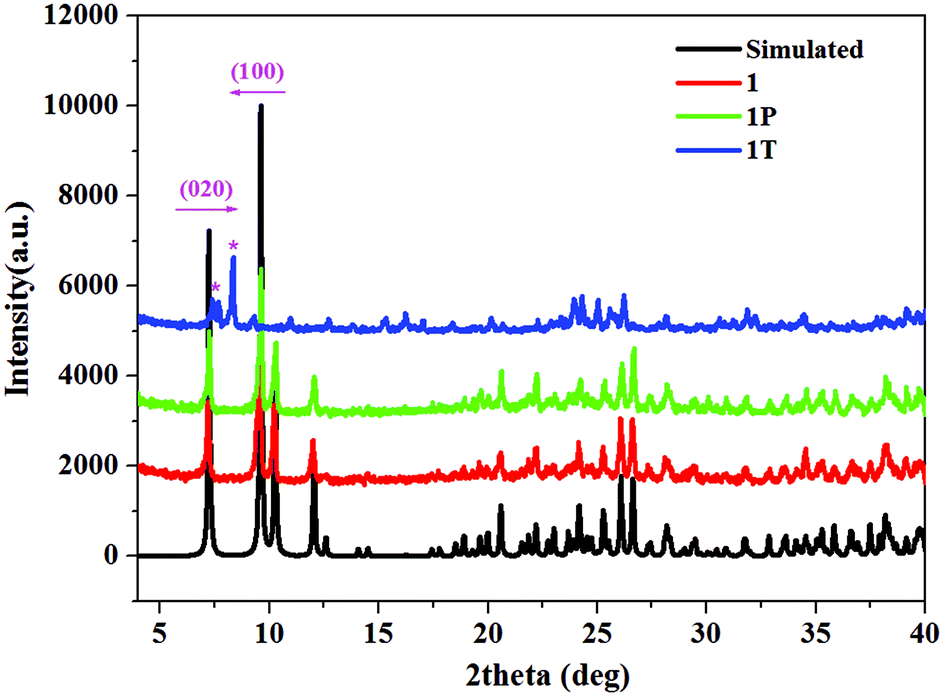 | ||
| Fig. 3 Powder X-ray diffraction (PXRD) of 1, 1P and 1T at room temperature. | ||
Electronic and photochromic behaviors: intermolecular charge transfer and electron transfer
The initial color of 1 is nearly colorless, corresponding to the obvious absorption bands of 370–450 nm, while 2 presents as a brown color, featuring a strong absorption band between 370 and 550 nm and a weak absorption band between 550 and 800 nm, which are ascribed to intermolecular CT from iodoargentate/iodocuprate anions to [DMBTz]+ cations. The obviously red-shifted absorption edge of 2 implies the stronger electron-donating ability of [Cu3I4]− than [Ag3I4]− due to the difference of metal ions and consequent binding abilities with I− ions.Upon irradiation by the mercury (Hg) lamp (300 W, ∼365 nm) at room temperature in air, compound 1 changes from colorless to dark red (denoted as 1P) within 3 min and tends to be saturated for 40 min (Fig. 4). Although it is difficult to obtain the crystal structure after photo-stimuli due to the loss of the single crystal character, the crystal structure of 1P is fortunately obtained and exhibits the same crystal structure besides the subtle differences of bonding lengths in their organic moieties (Table S3†), which are further confirmed by identical PXRD (Fig. 3), ruling out possible photolysis and/or photoinduced isomerization.23 As depicted in Fig. 5, the UV-vis absorption spectrum of 1P displays a new broad absorption band in the range of 450–750 nm, which is consistent with the characteristic absorption of colored organic radicals [DMBTz]˙![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48,49 and supported by theoretical value (450–700 nm) (Fig. S7†), implying a possible photoinduced ET process. The EPR signal at 2.0049 for 1P (Fig. 6, green line) further confirms that the photochromism of 1 is due to the photoinduced ET from [Ag3I4]− anions to organic cations and consequent generation of free radicals in [DMBTz]+ cations. Meanwhile, 1P can completely return to its initial color after being kept in the dark for about three months or heating at 100 °C for 24 h, accompanying the disappearance of the new absorption band (Fig. S8†) and the EPR signal. Therefore, the photoinduced coloration–decoloration process is reversible with no noticeable loss of photochromic properties after more than 8 cycles (Fig. S9†). In addition, compound 1P can directly transform into 1T after heating at 137 °C for about 12 h in the dark, and undergoes two processes, the disappearance of the colored radicals [DMBTz]˙ and the thermal-induced phase transition. However, 1T cannot transform into 1P due to the irreversible process of the thermal-induced phase transition at 137 °C.
48,49 and supported by theoretical value (450–700 nm) (Fig. S7†), implying a possible photoinduced ET process. The EPR signal at 2.0049 for 1P (Fig. 6, green line) further confirms that the photochromism of 1 is due to the photoinduced ET from [Ag3I4]− anions to organic cations and consequent generation of free radicals in [DMBTz]+ cations. Meanwhile, 1P can completely return to its initial color after being kept in the dark for about three months or heating at 100 °C for 24 h, accompanying the disappearance of the new absorption band (Fig. S8†) and the EPR signal. Therefore, the photoinduced coloration–decoloration process is reversible with no noticeable loss of photochromic properties after more than 8 cycles (Fig. S9†). In addition, compound 1P can directly transform into 1T after heating at 137 °C for about 12 h in the dark, and undergoes two processes, the disappearance of the colored radicals [DMBTz]˙ and the thermal-induced phase transition. However, 1T cannot transform into 1P due to the irreversible process of the thermal-induced phase transition at 137 °C.
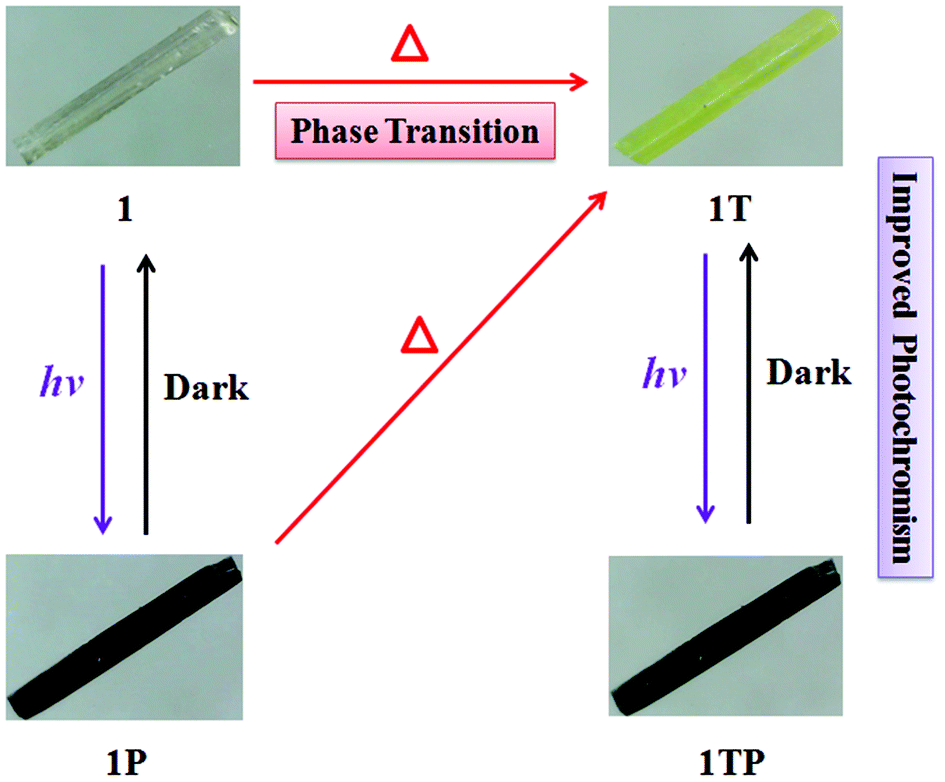 | ||
| Fig. 4 Photographs showing the photochromic behaviors of 1 and 1T. | ||
 | ||
| Fig. 5 UV-vis absorption spectra of 1, 1T, 1P and 1TP. | ||
 | ||
| Fig. 6 EPR spectra of 1, 1T, 1P and 1TP in the solid state at room temperature. | ||
Noteworthily, compound 1T is more photosensitive than 1 and turns from yellow to the black form (denoted as 1TP) for only 8 s and tends to be saturated in 10 min (Fig. 4), which is proved by intense absorption in the range of 450–750 nm (Fig. 5, blue line) and enhanced EPR signals (Fig. 6, blue line). As proved by the UV-vis absorption spectra (Fig. S8†) and EPR analysis, 1TP can also completely transform into 1T after placing it in the dark for about three months or heating at 137 °C for 12 h, showing that this process is also reversible and can be repeated at least for 8 cycles (Fig. S10†). Interestingly, the [DMBTz]˙ radicals are stable in dark air for about three months at room temperature implying an ultra-long-lived charge-separated state, which could be potentially applied to construct molecular electrodes.50 Compared with the well-studied ET photochromic hybrids of chlorometallates, carboxylates, zeolites, and phosphates matched with the strong electron acceptors of viologens, naphthenediimides (NDIs) and tris(4-pyridiniumyl)-1,3,5-triazine (TPT),4–10 such photochromic performance of the iodoargentate hybrid is unique, in which the photoresponsive rate and color range could be enhanced by the thermal-induced phase transition.
Comparatively, as proved by the unchanged UV-vis absorption spectra (Fig. S11†), consistent PXRD patterns (Fig. S12†) and silent EPR signals (Fig. S13†), compound 2 remains silent even after exposure to Hg/Xe light for 72 h. This phenomenon is totally different from that of the photochromic iodocuprate hybrid {[MNH][Cu2I3]2}n (MNH2+ = methylnicotinohydrazide dication),23,24 which largely originates from the stronger electron-accepting ability of [MNH]2+ compared to [DMBTz]+ as evidenced by theoretical calculations (Table S4†).
Intermolecular CT and ET are two important internal electron behaviors in the electron donating–accepting system, typically in the family of electron-rich iodometallate hybrids, which are largely dependent on the electron-donating/accepting ability of the electron donor/acceptor and the packing modes of two components. Although intensive CT is usually considered as an unfavorable factor for photoinduced ET,18–20 rational modulation of the composition and/or packing modes can realize a delicate balance between intermolecular CT and ET. As discussed above, matched with the same electron-deficient [DMBTz]+ cations, iodoargentate hybrids show suitable matching performance and consequently photochromic properties,22–26 while iodocuprate hybrids prefer the formation of strong intermolecular CT and show no response to light irradiation. Similar to the larger band gap of cuprous iodide compared to silver iodide, the band gap of the inorganic moiety of iodocuprate is also larger than that of iodoargentate, which is proved by DFT calculations.51,52 In other words, there exists a stronger restriction for electrons in [Cu3I4]− than that for [Ag3I4]− due to the smaller sum of the van der Waals radii of Cu with respect to Ag. Therefore, it is the key factor that the electrons (e−) and holes (h+) could not be generated from the inorganic moiety of iodocuprate under irradiation by ultraviolet light, which is not beneficial for the photoinduced ET process from iodocuprate anions to [DMBTz]+ cations (Fig. 7) and lead to optical inertness for iodocuprate hybrids. Meanwhile, compared with relatively weaker intermolecular CT in iodoargentate hybrids, stronger intermolecular CT in iodocuprate hybrids largely reduces the electron-accepting ability of [DMBTz]+ cations, which should be another important factor for the suppression of the net photoinduced ET as well as the optically inert iodocuprate hybrid because of rapid charge recombination. Noteworthily, although the phase transition in iodoargentate hybrids leads to slightly stronger intermolecular CT and reduces the electron-accepting ability of [DMBTz]+ cations to some extent, the changes of packing modes are more beneficial for the photoinduced ET process and the improvement of photochromic properties.
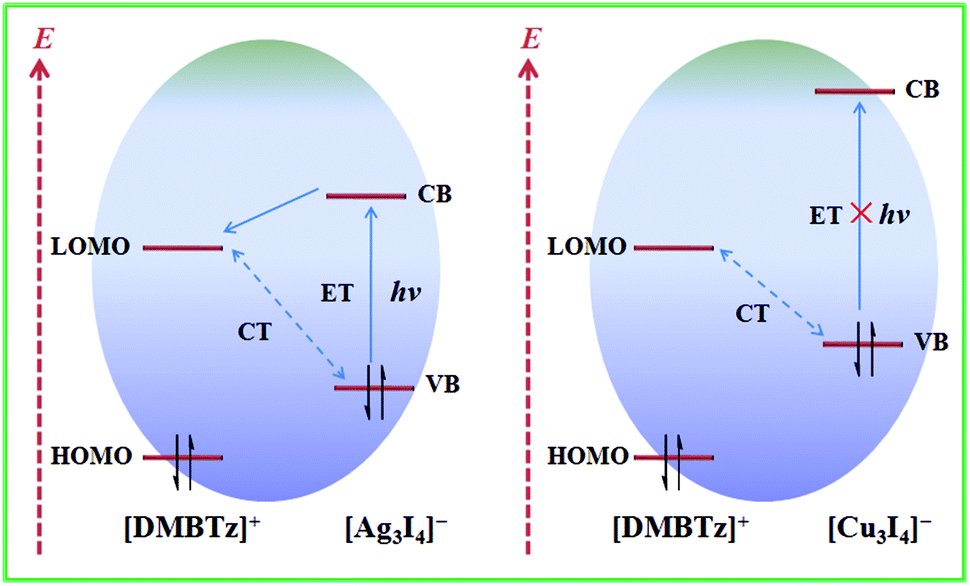 | ||
| Fig. 7 Energy diagrams showing the intermolecular CT and ET process in the iodometallate hybrids and relative positions of the HOMO and LUMO levels of [DMBTz]+ with respect to the CB and VB of anions. | ||
Conclusions
In summary, a pair of isostructural organic–inorganic hybrid iodometallates, [DMBTz][Ag3I4] (1) and [DMBTz][Cu3I4] (2) (DMBTz+ = dimethylbenzotriazolium) is constructed from [Ag3I4]−/[Cu3I4]− inorganic chains and discrete [DMBTz]+ cations. The combination of electron-rich iodoargentate/iodocuprate and electron-deficient benzotriazolium realizes an effective route to rationally modulate intermolecular CT and ET as well as adjust photoresponsive behaviors. The present study provides a simple and effective method for the construction of photochromic iodometallate hybrid materials with tunable responsive properties. Further studies are still ongoing in our laboratory.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21171110), the Research Fund for the Doctoral Program of Higher Education of China (20131404110001), the Natural Science Foundation of Science and Technology Agency of Shanxi Province (201601D202025, 201701D121022), and the 1331 Project of Shanxi Province.Notes and references
- Y. Tan, H. J. Chen, J. Zhang, S. J. Liao, J. C. Dai and Z. Y. Fu, CrystEngComm, 2012, 14, 5137–5139 RSC.
- C. Y. Tao, J. B. Wu, Y. Yan, C. Shi and J. Y. Li, Inorg. Chem. Front., 2016, 3, 541–546 RSC.
- G. Xu, G. C. Guo, M. S. Wang, Z. J. Zhang, W. T. Chen and J. S. Huang, Angew. Chem., Int. Ed., 2007, 46, 3249–3251 CrossRef CAS PubMed.
- S. Guha, F. S. Goodson, S. Roy, L. J. Corson, C. A. Gravenmier and S. Saha, J. Am. Chem. Soc., 2011, 133, 15256–15259 CrossRef CAS PubMed.
- L. Han, L. Qin, L. P. Xu, Y. Zhou, J. L. Sun and X. D. Zou, Chem. Commun., 2013, 49, 406–408 RSC.
- A. H. Sun, S. D. Han, J. Pan, J. H. Li, G. M. Wang and Z. H. Wang, Cryst. Growth Des., 2017, 17, 3588–3591 CrossRef CAS.
- Z. Y. Fu, J. Zhang, Y. Zeng, Y. Tan, S. J. Liao, H. J. Chen and J. C. Dai, CrystEngComm, 2012, 14, 786–788 RSC.
- B. Yoon, J. Lee, I. S. Park, S. Jeon, J. Lee and J.-M. Kim, J. Mater. Chem. C, 2013, 1, 2388–2403 RSC.
- D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed.
- S. Nishikiori, H. Yoshikawa, Y. Sano and T. Iwamoto, Acc. Chem. Res., 2005, 38, 227–234 CrossRef CAS PubMed.
- L. M. Wu, X. T. Wu and L. Chen, Coord. Chem. Rev., 2009, 253, 2787–2804 CrossRef CAS.
- R. Peng, M. Li and D. Li, Coord. Chem. Rev., 2010, 254, 1–18 CrossRef CAS.
- S. A. Adonin, M. N. Sokolov and V. P. Fedin, Coord. Chem. Rev., 2016, 312, 1–21 CrossRef CAS.
- A. H. Sun, J. Pan, S. D. Han, X. Y. Xue, Q. Wei, J. H. Li and G. M. Wang, Inorg. Chem., 2017, 56, 13785–13793 CrossRef CAS PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- D. H. Wang, L. M. Zhao, X. Y. Lin, Y. K. Wang, W. T. Zhang, K. Y. Song, H. H. Li and Z. R. Chen, Inorg. Chem. Front., 2018, 5, 1162–1173 RSC.
- X. W. Lei, C. Y. Yue, J. Q. Zhao, Y. F. Han, J. T. Yang, R. R. Meng, C. S. Gao, H. Ding, C. Y. Wang and W. D. Chen, Cryst. Growth Des., 2015, 15, 5416–5426 CrossRef CAS.
- J. K. Sun, P. Wang, Q. X. Yao, Y. J. Chen, Z. H. Li, Y. F. Zhang, L. M. Wu and J. Zhang, J. Mater. Chem., 2012, 22, 12212–12219 RSC.
- X. Y. Lv, M. S. Wang, C. Yang, G. E. Wang, S. H. Wang, R. G. Lin and G. C. Guo, Inorg. Chem., 2012, 51, 4015–4019 CrossRef CAS PubMed.
- S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674–17677 CrossRef CAS PubMed.
- H. T. Chifotides, B. L. Schottel and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 7202–7207 CrossRef CAS.
- P. F. Hao, H. H. Li, T. L. Yu and Y. L. Fu, Dyes Pigm., 2017, 136, 825–829 CrossRef CAS.
- J. J. Shen, F. Wang, X. X. Li, T. L. Yu, P. F. Hao and Y. L. Fu, RSC Adv., 2016, 6, 98916–98920 RSC.
- J. J. Shen, X. X. Li, T. L. Yu, F. Wang, P. F. Hao and Y. L. Fu, Inorg. Chem., 2016, 55, 8271–8273 CrossRef CAS.
- T. L. Yu, P. F. Hao, J. J. Shen and Y. L. Fu, Dalton Trans., 2016, 45, 16505–16510 RSC.
- T. L. Yu, G. X. Wu, M. Xue, Z. H. Wang and Y. L. Fu, Dalton Trans., 2018, 47, 12172–12180 RSC.
- D. Baran, A. Balan, S. Celebi, B. M. Esteban, H. Neugebauer, N. S. Sariciftci and L. Toppare, Chem. Mater., 2010, 22, 2978–2987 CrossRef CAS.
- D. Barana, A. Balan, T. Stubhana, T. Ameri, L. Toppare and C. J. Brabec, Synth. Met., 2012, 162, 2047–2051 CrossRef.
- A. Balan, G. Gunbas, A. Durmus and L. Toppare, Chem. Mater., 2008, 20, 7510–7513 CrossRef CAS.
- A. Balan, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt and L. Toppare, Chem. Commun., 2009, 6768–6770 RSC.
- D. G. Patel, F. Feng, Y. Y. Ohnishi, K. A. Abboud, S. Hirata, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 2599–2612 CrossRef CAS PubMed.
- P. F. Hao, L. F. Zhang, J. J. Shen and Y. L. Fu, Dyes Pigm., 2018, 153, 284–290 CrossRef CAS.
- CrysAlisPro, version 1.171.33.56, Oxford Diffraction Ltd., Oxfordshire, UK, 2010 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97, A program for X-ray crystal structure solution, and SHELXL-97, A program for X-ray structure refinement, Göttingen University, Germany, 1997 Search PubMed.
- J. W. Liu, L. Feng, H. F. Su, Z. Wang, Q. Q. Zhao, X. P. Wang, C. H. Tung, D. Sun and L. S. Zheng, J. Am. Chem. Soc., 2018, 140, 1600–1603 CrossRef CAS.
- Z. Wang, H. F. Su, M. Kurmoo, C. H. Tung, D. Sun and L. S. Zheng, Nat. Commun., 2018, 9, 2094 CrossRef PubMed.
- Z. Wang, H. F. Su, X. P. Wang, Q. Q. Zhao, C. H. Tung, D. Sun and L. S. Zheng, Chem. – Eur. J., 2018, 24, 1640–1650 CrossRef CAS PubMed.
- Y. M. Su, W. Liu, Z. Wang, S. A. Wang, Y. A. Li, F. Yu, Q. Q. Zhao, X. P. Wang, C. H. Tung and D. Sun, Chem. – Eur. J., 2018, 24, 4967–4972 CrossRef CAS PubMed.
- Z. Wang, H. F. Su, Y. Z. Tan, S. Schein, S. C. Lin, W. Liu, S. A. Wang, W. G. Wang, C. H. Tung, D. Sun and L. S. Zheng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12132–12137 CrossRef CAS PubMed.
- R. W. Huang, Y. S. Wei, X. Y. Dong, X.-H. Wu, C. X. Du, S. Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS.
- Z. H. Yan, X. Y. Li, L. W. Liu, S. Q. Yu, X. P. Wang and D. Sun, Inorg. Chem., 2016, 55, 1096–1101 CrossRef CAS PubMed.
- C. J. Gilmore, P. A. Tucker and P. Woodward, Inorg. Phys. Theor., 1971, 1337–1341 RSC.
- G. N. Liu, X. Zhang, H. M. Wang, H. Xu, Z. H. Wang, X. L. Meng, Y. N. Dong, R. Y. Zhao and C. Li, Dalton Trans., 2017, 46, 12474–12486 RSC.
- A. G. Saiz, I. D. Pedro, P. Migowski, O. Vallcorba, J. Junquera, J. A. Blanco, O. Fabelo, D. Sheptyakov, J. C. Waerenborgh, M. T. Fernandez-Díaz, J. Rius, J. Dupont, J. A. Gonzaleź and J. R. Fernandez, Inorg. Chem., 2014, 53, 8384–8396 CrossRef PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS.
- P. F. Hao, Y. R. Qiao, T. L. Yu, J. J. Shen, F. Liu and Y. L. Fu, RSC Adv., 2016, 6, 53566–53572 RSC.
- M. Asay, C. E. Kefalidis, J. Estrada, D. S. Weinberger, J. Wright, C. E. Moore, A. L. Rheingold, L. Maron and V. Lavallo, Angew. Chem., Int. Ed., 2013, 52, 11560–11563 CrossRef CAS PubMed.
- Y. Jiang and J. B. Adams, Surf. Sci., 2003, 529, 428–442 CrossRef CAS.
- A. J. Cowana and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC.
- W. X. Chai, L. M. Wu, J. Q. Li and L. Chen, Inorg. Chem., 2007, 46, 1042–1044 CrossRef CAS.
- W. X. Chai, L. M. Wu, J. Q. Li and L. Chen, Inorg. Chem., 2007, 46, 8698–8704 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra, asymmetric units, TG-DSC curves, PXRD patterns, UV-vis absorption spectra, EPR spectra and the coloration–decoloration process with repeated UV irradiation/heating of compounds. CCDC 1487190, 1487191 and 1867441. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qi01067f |
| This journal is © the Partner Organisations 2019 |