
Synthesis and characterization of manganese(II) complexes supported by cyclopentadienyl-phosphine ligands†
Gao-Xiang
Wang‡
,
Jianhao
Yin‡
,
Jiapeng
Li‡
,
Zhu-Bao
Yin
,
Wen-Xiong
Zhang
 * and
Zhenfeng
Xi
*
* and
Zhenfeng
Xi
*
Beijing National Laboratory for Molecular Sciences, MOE Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wx_zhang@pku.edu.cn; zfxi@pku.edu.cn
First published on 8th December 2018
Abstract
A series of manganese(II) complexes supported by cyclopentadienyl-phosphine ligands have been synthesized and characterized. A reaction of potassium salts of a cyclopentadienyl-phosphine ligand (LK) with MnX2 provided dimeric complexes [LMn(μ-X)]2 (X = Cl, 1a; X = Br, 1b) in high yields. The monomeric complex [K(18-crown-6)][LMnCl2] (2) was obtained with the addition of 18-crown-6. [LMn(μ-Cl)]2 (1a) served as a precursor to dimeric amido and methyl manganese(II) complexes including [LMn(μ-NH2)]2 (3) and [LMn(μ-Me)]2 (4). Treatment of [LMn(μ-Cl)]2 (1a) with EtMgCl, PhMgBr or other reductants resulted in the formation of sandwich manganese(II) complex L2Mn (5). Complexes 1a,b, 2–5 were characterized by a single-crystal X-ray structural analysis.
Introduction
Since the first organomanganese(II) complexes (PhMnI and Ph2Mn) were reported in 1937,1 synthesis and reactivity of organomanganese halides (RMnX), manganese diorganyls (R2Mn) and organomanganates (R3Mn− and R4Mn2−) have been investigated for more than half a century.2 In particular, the manganese alkyl,3 aryl4 and amide5 derivatives have played an important role in the development of organomanganese chemistry.As usual, ligands play a very important role in the structure and reaction of organomanganese compounds, which are often stabilized by a variety of ligands, such as the cyclopentadienyl group6 and phosphines.7 The cyclopentadienyl-phosphine ligand (Fig. 1), in which a multi-substituted cyclopentadienyl moiety and a phosphine moiety are linked by an alkyl or aryl group, is a potential chelating ligand.8 Its cooperative coordination mode with transition metals forming so-called constrained geometry complexes (CGC) may result in resonance interactions between the lone pair on the phosphorus center and the cyclopentadienyl π system.9,10
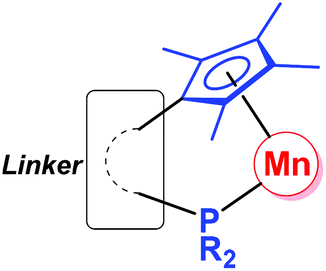 | ||
| Fig. 1 Cooperative coordination mode of organomanganese complexes bearing a cyclopentadienyl-phosphine ligand. | ||
We have developed a convenient method for the synthesis of multi-functional phosphine ligands.11 With our continued interest in their application as a chelating and stabilizing ligand for novel transition-metal complexes,12 we have now investigated their coordination behaviour with manganese. Herein, we report the synthesis and structural characterization of a series of well-defined manganese(II) complexes supported by cyclopentadienyl-phosphine ligands. The results demonstrated that the chelating and stabilizing effects of cyclopentadienyl-phosphine ligands enabled different structures and reactivities of manganese(II) complexes.
Results and discussion
Synthesis and characterization of [LMn(μ-X)]2 (X = Cl, 1a; X = Br, 1b) and [K(18-crown-6)][LMnCl2] (2)
Based on our previous work,12 we successfully synthesized the potassium salt of a cyclopentadienyl-phosphine ligand (LK) as a yellow powder (Scheme 1). The reaction of LK and anhydrous MnCl2/MnBr2 in THF at room temperature proceeded smoothly with the elimination of KX (X = Cl, Br). The dimeric complexes [LMn(μ-Cl)]2 (1a) and [LMn(μ-Br)]2 (1b) were obtained in 87% and 80% yields, respectively. 1a and 1b are sensitive to air and moisture and can be recrystallized from hexane/THF.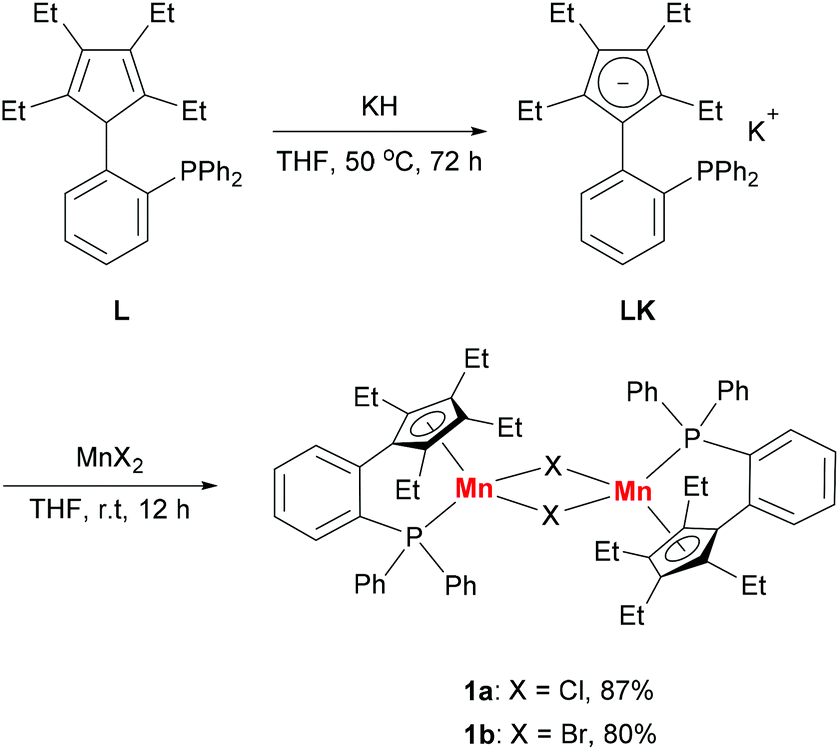 | ||
| Scheme 1 Synthesis of dimeric manganese(II) halide complexes 1a and 1b. | ||
Solution magnetic measurements of 1a and 1b in C6D6 at room temperature gave effective magnetic moments of 6.7(1)μB per dimer and 6.8(2)μB per dimer, respectively, which are consistent with weakly antiferromagnetically coupled high-spin manganese(II) centers. The molecular structures of 1a and 1b were confirmed by X-ray crystallography. Both 1a and 1b are halogen-bridged dimers in the solid state. The ORTEP drawing of 1a is shown in Fig. 2. In these two compounds, each manganese center is coordinated by the cyclopentadienyl ring, the phosphorus atom and the two bridging halogens. The structural parameters of 1a are similar to those of 1b, and only the bromine bridges in 1b cause a major lengthening of the distances of the Mn2X2 ring. The distances between two manganese atoms are 3.287 Å and 3.580 Å for 1a and 1b, respectively, indicating that there are no Mn–Mn bonds (2.170–3.291 Å) in 1a and 1b.13
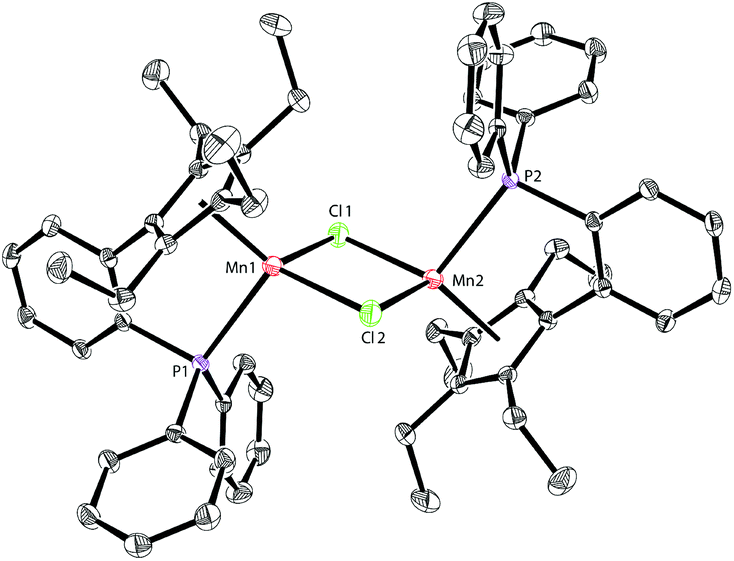 | ||
| Fig. 2 Molecular structure of 1a with thermal ellipsoids at 30% probability. H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Mn(1)–Cl(1): 2.3909(7), Mn(1)–Cl(2): 2.4603(7), Mn(1)–P(1): 2.6066(6), and Cl(1)–Mn(1)–Cl(2): 94.71(3). | ||
The addition of 18-crown-6 to a mixture of LK and MnCl2 afforded the monomeric complex 2 in 96% yield (Scheme 2). Complex 2 has a solution magnetic moment of 6.5(3)μB in d8-THF that showed a monomeric high-spin manganese(II) center. The molecular structure of complex 2 was confirmed by X-ray crystallography and the ORTEP drawing is shown in Fig. 3. The manganese centers of both 1a and 2 are in the same coordination environment. In contrast to the dimeric complex 1a, the Mn–P bond distance in complex 2 (2.726 Å) is longer than the that of dimer 1a (2.607 Å) and slightly longer than the reported Mn–P bond distances.14 The potassium atom in 2 is connected by two bridging chlorides and coordinated with six oxygen atoms in 18-crown-6.
 | ||
| Scheme 2 Synthesis of monomeric manganese(II) halide complex 2. | ||
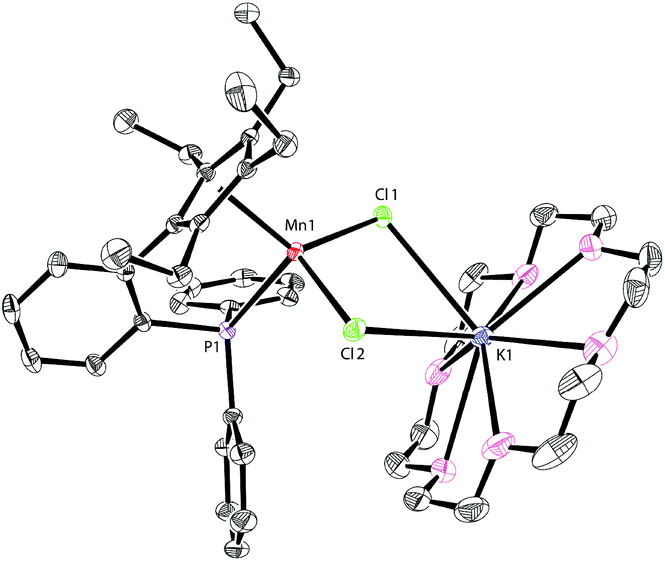 | ||
| Fig. 3 Molecular structure of 2 with thermal ellipsoids at 30% probability. H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Mn(1)–Cl(1): 2.3997(6), Mn(1)–Cl(2): 2.3844(7), Mn(1)–P(1): 2.7264(6), K(1)–Cl(1): 3.1928(8), K(1)–Cl(2): 3.2865(8), Cl(1)–Mn(1)–Cl(2): 99.83(2), and Cl(1)–K(1)–Cl(2): 65.879(16). | ||
Synthesis and characterization of organomanganese(II) derivatives: [LMn(μ-NH2)]2 (3), [LMn(μ-Me)]2 (4) and L2Mn (5)
The substitution reactions of 1a with nucleophiles were investigated to prepare organomanganese(II) derivatives (Scheme 3). Treatment of 1a with excess NaNH2 in THF at room temperature afforded the amido-bridged manganese(II) complex [LMn(μ-NH2)]2 (3) in 86% yield. As far as we know, complex 3 is a rare example of manganese complexes with parent amido (–NH2) ligands.5b The manganese(II) methyl complex [LMn(μ-Me)]2 (4) was also prepared from the addition of methyl Grignard reagent MeMgCl to 1a in THF at room temperature. By recrystallization from hexane/Et2O several times, the methyl-bridged complex was isolated as a red solid in 84% yield. Magnetic measurements showed that 3 and 4 have effective magnetic moments of 6.2(1)μB per dimer and 5.9(3)μB per dimer at room temperature in C6D6, due to the antiferromagnetic interactions between the two manganese ions.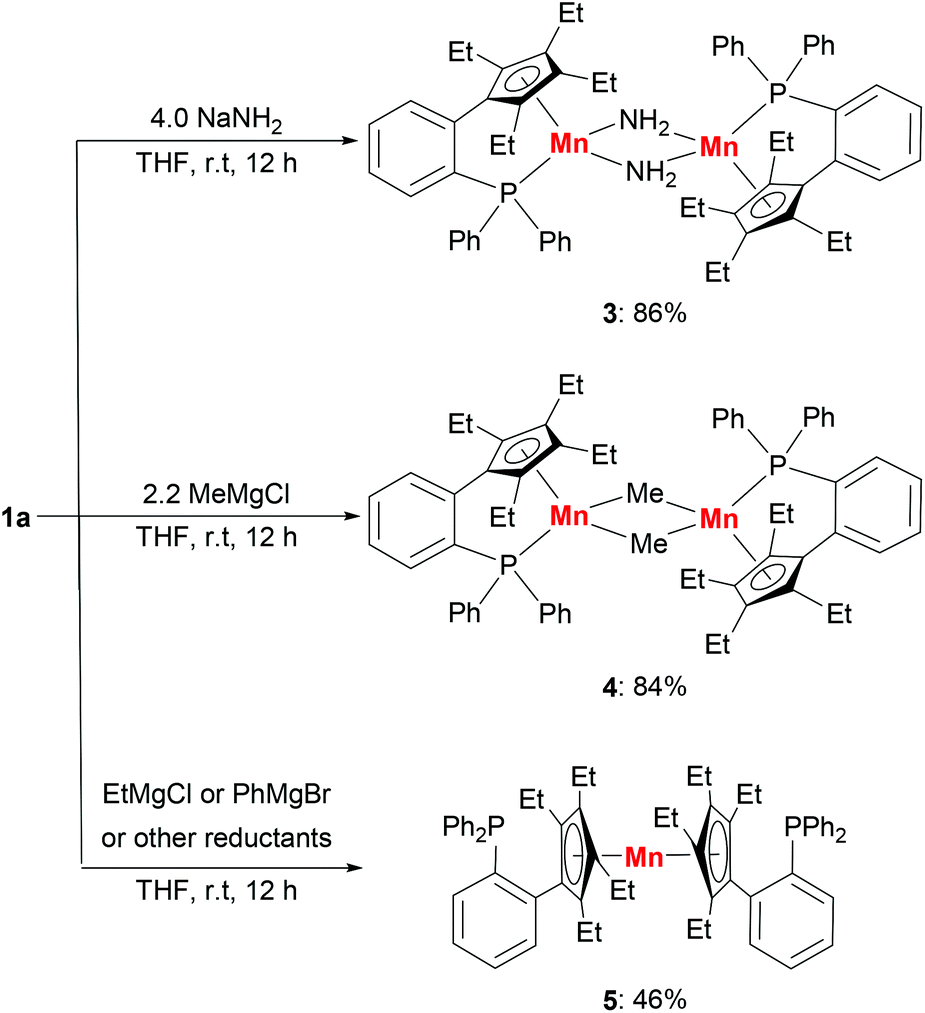 | ||
| Scheme 3 Synthesis of organomanganese(II) derivatives: amido-bridged complex 3, methyl-bridged complex 4 and sandwich complex 5. | ||
The reaction of 1a with EtMgCl or PhMgBr in THF at room temperature produced a sandwich complex 5 in 46% yield rather than the expected ethyl or phenyl complexes.15 However, the reactions between complex 1a and other reductants, such as KHBEt3, nBuLi and KC8, did not yield the expected hydride complex or the other reduction product. Only complex 5 and a black precipitate were afforded. The sandwich complex 5 can also be prepared in 79% yield by the reaction of anhydrous MnCl2 with two equivalents of LK in THF. The appearance of the sandwich complex 5 shows that the manganese center has a stronger coordination with the cyclopentadienyl moiety compared with the phosphine moiety. The solution magnetic susceptibility of complex 5 shows a magnetic moment of 5.4(2)μB at room temperature in d8-THF.
The molecular structure of complex 3 was unambiguously confirmed by X-ray crystallography. The structure of 3 (Fig. 4) shows that it is dimerized through bridging two manganese centers by two amido groups. The Mn–P bond distance in complex 3 is 2.653 Å, which is longer than 2.607 Å in 1a. The Mn2N2 core is almost a square with Mn–N bond distances of 2.123 Å and 2.096 Å and an N–Mn–N angle of 88.791°. The Mn–Mn distance in 3 is 3.019 Å, which is much shorter than the distances observed in 1a and 1b.
 | ||
| Fig. 4 Molecular structure of 3 with thermal ellipsoids at 30% probability. Hydrogen atoms, except that on nitrogen atoms N1 and N2, are omitted for clarity. Selected bond lengths [Å] and angles [°]: Mn(1)–N(1): 2.1299(19), Mn(1)–N(2): 2.096(2), Mn(1)–P(1): 2.6525(6), and N(1)–Mn(1)–N(2): 88.79(8). | ||
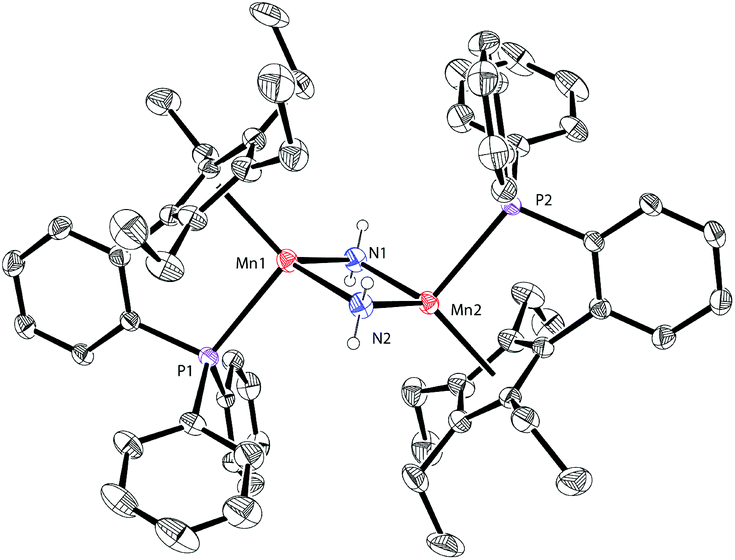
The solid-state structure of 4 is depicted in Fig. 5. The Mn–P bond distance in complex 4 (2.706 Å) is also longer than that in 1a (2.607 Å). The Mn–Mn distance of 4 (2.820 Å) is ca. 0.2 Å shorter than that of 3 (3.019 Å). This short Mn–Mn distance may indicate some interactions between the two metal atoms.3f,g The planar Mn2C2 core features the Mn–C bond distances (2.2249 Å and 2.308 Å) and the C–Mn–C angle (104.24°). These parameters are similar to those in other reported methyl-bridged dinuclear manganese(II) complexes.3c,e–g
 | ||
| Fig. 5 Molecular structure of 4 with thermal ellipsoids at 30% probability. Hydrogen atoms, except that on carbon atoms C1 and C2, are omitted for clarity. Selected bond lengths [Å] and angles [°]: Mn(1)–C(1): 2.2249(18), Mn(1)–C(2): 2.308(2), Mn(1)–P(1): 2.7056(6), and C(1)–Mn(1)–C(2): 104.24(7). | ||
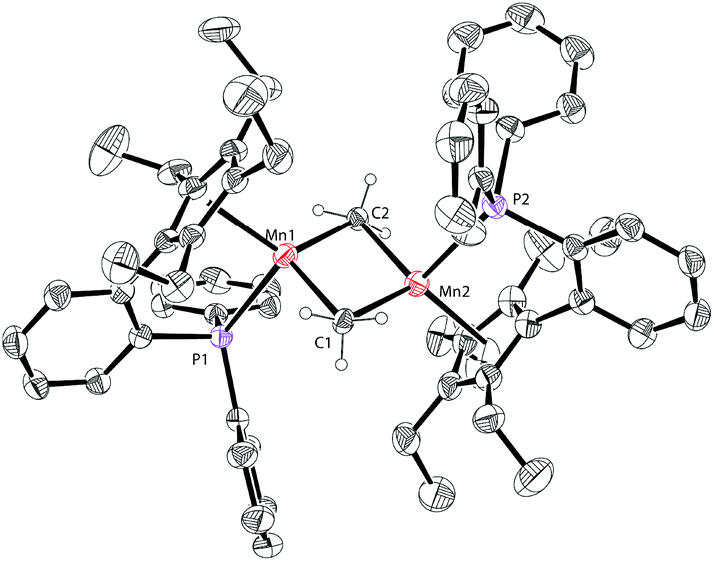
The molecular structure of complex 5 was also identified by X-ray crystallography. As shown in Fig. 6, complex 5 shows a sandwich configuration. The manganese center is coordinated with the two penta-substituted cyclopentadienyl rings in two ligand molecules, leaving the phosphines uncoordinated.
 | ||
| Fig. 6 Molecular structure of 5 with thermal ellipsoids at 30% probability. H atoms are omitted for clarity. | ||
Conclusions
In summary, we have synthesized a series of organomanganese complexes bearing the chelating and stabilizing cyclopentadienyl-phosphine ligand. Monomeric and dimeric manganese(II) halide complexes (1a, 1b and 2) were obtained by the reaction of a potassium salt of the cyclopentadienyl-phosphine ligand (LK) with MnX2 (X = Cl, Br). The reaction of 1a with NaNH2 and MeMgCl afforded amido-bridged complex 3 and methyl-bridged complex 4. In addition, we also investigated the formation of the sandwich complex 5. The reaction chemistry of these complexes is under investigation. We hope that the present results in this paper can provide some useful information for the design of phosphines and cyclopentadienyl ligands in organomanganese chemistry.Experimental
General methods
Unless otherwise mentioned, the synthesis of air and/or moisture sensitive compounds was carried out under an atmosphere of nitrogen using standard Schlenk techniques or in a nitrogen filled glovebox. Solvents were collected from an Mbraun SPS-800 Solvent Purification System. KH, nBuLi, KHBEt3, KC8, Grignard reagents, 18-crown-6, 3-hexyne, 2-bromobenzaldehyde, MnCl2, and MnBr2 were obtained from Strem, Aldrich, TCI, Alfa Aesar, Acros, J&K and others. 1H and 31P NMR spectra were recorded on a Bruker ARX500 spectrometer (FT, 400 MHz for 1H; 202 MHz for 31P) at room temperature. Ligand L can be synthesized based on the reported procedures.12a Elemental analysis was performed on a Vario EL elemental analyzer at the Analytical Center of Peking University. Infrared spectroscopy was performed on a Bruker Tensor 27 using a KBr cell. Solution magnetic susceptibility measurements were performed by the Evans method and experimental solutions contain a known amount of a TMSOTMS standard.16X-ray crystallography
Data collection for 1a,b, 2–5 was performed at 180 K on a Rigaku diffractometer, using monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were solved using the SHELXTL or Olex program.17 Refinement was performed on F2 anisotropically for all the non-hydrogen atoms by the full-matrix least-squares method. The hydrogen atoms were placed at the calculated positions and were included in the structure calculation without further refinement of the parameters. Crystal data, data collection and processing parameters for compounds 1a,b, 2–5 are summarized in the ESI.† Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1870180 (1a), 1870203 (1b), 1870204 (2), 1870205 (3), 1870206 (4) and 1870207 (5).†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21690061 and 21725201).Notes and references
- (a) H. Gilman and R. Kirby, J. Am. Chem. Soc., 1941, 63, 2046–2048 CrossRef CAS; (b) H. Gilman and J. C. Bailie, J. Org. Chem., 1937, 2, 84–94 CrossRef CAS.
- (a) G. Cahiez, An. Quim., 1995, 91, 561–578 CAS; (b) K. Oshima, J. Organomet. Chem., 1999, 575, 1–20 CrossRef CAS; (c) R. A. Layfield, Chem. Soc. Rev., 2008, 37, 1098–1107 RSC; (d) G. Cahiez, C. Duplais and J. Buendia, Chem. Rev., 2009, 109, 1434–1476 CrossRef CAS PubMed; (e) M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344–4362 CrossRef CAS; (f) B. Maji and M. K. Barman, Synthesis, 2017, 49, 3377–3393 CrossRef CAS; (g) Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS PubMed; (h) X. Yang and C. Wang, Chem. – Asian J., 2018, 13, 2307–2315 CrossRef CAS PubMed.
- (a) D. Reardon, G. Aharonian, S. Gambarotta and G. P. Yap, Organometallics, 2002, 21, 786–788 CrossRef CAS; (b) J. Chai, H. Zhu, H. W. Roesky, Z. Yang, V. Jancik, R. Herbst-Irmer, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2004, 23, 5003–5006 CrossRef CAS; (c) M. Niemeyer and P. P. Power, Chem. Commun., 1996, 1573–1574 RSC; (d) C. C. Lu and J. C. Peters, Inorg. Chem., 2006, 45, 8597–8607 CrossRef CAS PubMed; (e) C. Ni and P. P. Power, Organometallics, 2009, 28, 6541–6545 CrossRef CAS; (f) M. H. Al-Afyouni, V. M. Krishnan, H. D. Arman and Z. J. Tonzetich, Organometallics, 2015, 34, 5088–5094 CrossRef CAS; (g) T. Hashimoto, Y. Kawato, Y. Nakajima, Y. Ohki, K. Tatsumi, W. Ando, K. Sato and S. Shimada, J. Organomet. Chem., 2016, 820, 14–19 CrossRef CAS; (h) M. M. Stalzer, J. Telser, J. Krzystek, A. Motta, M. Delferro and T. J. Marks, Organometallics, 2016, 35, 2683–2688 CrossRef CAS.
- (a) J. Chai, H. Zhu, H. Fan, H. W. Roesky and J. Magull, Organometallics, 2004, 23, 1177–1179 CrossRef CAS; (b) J. Chai, H. Zhu, H. W. Roesky, C. He, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2004, 23, 3284–3289 CrossRef CAS.
- (a) C. Ni, G. J. Long, F. Grandjean and P. P. Power, Inorg. Chem., 2009, 48, 11594–11600 CrossRef CAS PubMed; (b) C. Ni, H. Lei and P. P. Power, Organometallics, 2010, 29, 1988–1991 CrossRef CAS.
- (a) G. Wilkinson, F. Cotton and J. Birmingham, J. Inorg. Nucl. Chem., 1956, 2, 95–113 CrossRef CAS; (b) M. S. Kralik, L. Stahl, A. M. Arif, C. E. Strouse and R. D. Ernst, Organometallics, 1992, 11, 3617–3621 CrossRef CAS; (c) S. Kheradmandan, H. W. Schmalle, H. Jacobsen, O. Blacque, T. Fox, H. Berke, M. Gross and S. Decurtins, Chem. – Eur. J., 2002, 8, 2526–2533 CrossRef CAS; (d) M. Maekawa, M. Römelt, C. G. Daniliuc, P. G. Jones, P. S. White, F. Neese and M. D. Walter, Chem. Sci., 2012, 3, 2972–2979 RSC; (e) F. A. Stokes, R. J. Less, J. Haywood, R. L. Melen, R. I. Thompson, A. E. H. Wheatley and D. S. Wright, Organometallics, 2012, 31, 23–26 CrossRef CAS.
- (a) F. H. Köhler, N. Hebendanz, U. Thewalt, B. Kanellakopulos and R. Klenze, Angew. Chem., Int. Ed., Engl., 1984, 23, 721–722 CrossRef; (b) F. H. Köhler, N. Hebendanz, G. Müller, U. Thewalt, B. Kanellakopulos and R. Klenze, Organometallics, 1987, 6, 115–125 CrossRef; (c) D. Unseld, V. V. Krivykh, K. Heinze, F. Wild, G. Artus, H. Schmalle and H. Berke, Organometallics, 1999, 18, 1525–1541 CrossRef CAS.
- (a) J. Foerstner, R. Kettenbach, R. Goddard and H. Butenschön, Chem. Ber., 1996, 129, 319–325 CrossRef CAS; (b) X. D. He, A. Maisonnat, F. Dahan and R. Poilblanc, Organometallics, 1989, 8, 2618–2626 CrossRef CAS; (c) B. M. Trost, B. Vidal and M. Thommen, Chem. – Eur. J., 1999, 5, 1055–1069 CrossRef CAS; (d) J. Foerstner, A. Kakoschke, R. Wartchow and H. Butenschön, Organometallics, 2000, 19, 2108–2113 CrossRef CAS; (e) A. Döhring, V. R. Jensen, P. W. Jolly, W. Thiel and J. C. Weber, Organometallics, 2001, 20, 2234–2245 CrossRef; (f) R. M. Bellabarba, M. Nieuwenhuyzen and G. C. Saunders, J. Chem. Soc., Dalton Trans., 2001, 512–514 RSC; (g) K. Onitsuka, Y. Ajioka, Y. Matsushima and S. Takahashi, Organometallics, 2001, 20, 3274–3282 CrossRef CAS; (h) K. Kunz, G. Erker, S. Döring, R. Fröhlich and G. Kehr, J. Am. Chem. Soc., 2001, 123, 6181–6182 CrossRef CAS PubMed; (i) S. Ciruelos, A. Doppiu, U. Englert and A. Salzer, J. Organomet. Chem., 2002, 663, 183–191 CrossRef CAS; (j) S. R. Klei, T. D. Tilley and R. G. Bergman, Organometallics, 2002, 21, 4905–4911 CrossRef CAS; (k) D. P. Krut'ko, M. V. Borzov, E. N. Veksler, A. V. Churakov and K. Mach, Polyhedron, 2003, 22, 2885–2894 CrossRef; (l) E. Becker, K. Mereiter, M. Puchberger, R. Schmid and K. Kirchner, Organometallics, 2003, 22, 3164–3170 CrossRef CAS; (m) L. Yong, E. Hofer, R. Wartchow and H. Butenschön, Organometallics, 2003, 22, 5463–5467 CrossRef CAS; (n) A. C. McConnell, P. J. Pogorzelec, A. M. Z. Slawin, G. L. Williams, P. I. P. Elliott, A. Haynes, A. C. Marr and D. J. Cole-Hamilton, Dalton Trans., 2006, 91–107 RSC; (o) M. Hussain, D. Albert, R. Wartchow and H. Butenschön, Chem. – Asian J., 2007, 2, 782–793 CrossRef CAS PubMed; (p) I. Werner and H. Butenschön, Eur. J. Inorg. Chem., 2014, 2014, 6051–6060 CrossRef CAS; (q) K. Kamikawa, Y.-Y. Tseng, J.-H. Jian, T. Takahashi and M. Ogasawara, J. Am. Chem. Soc., 2017, 139, 1545–1553 CrossRef CAS PubMed; (r) S. Kohser and H. Butenschön, Eur. J. Inorg. Chem., 2018, 2018, 31–45 CrossRef CAS.
- H. Butenschön, Chem. Rev., 2000, 100, 1527–1564 CrossRef.
- (a) C. Charrier and F. Mathey, Tetrahedron Lett., 1978, 19, 2407–2410 CrossRef; (b) C. Charrier and F. Mathey, J. Organomet. Chem., 1979, 170, C41–C43 CrossRef CAS; (c) B. Antelmann, U. Winterhalter, G. Huttner, B. C. Janssen and J. Vogelgesang, J. Organomet. Chem., 1997, 545, 407–420 CrossRef.
- (a) Z. Xi and P. Li, Angew. Chem., Int. Ed., 2000, 39, 2950–2952 CrossRef CAS; (b) Z. Xi, Q. Song, J. Chen, H. Guan and P. Li, Angew. Chem., Int. Ed., 2001, 40, 1913–1916 CrossRef CAS.
- (a) W. Geng, W.-X. Zhang, W. Hao and Z. Xi, J. Am. Chem. Soc., 2012, 134, 20230–20233 CrossRef CAS PubMed; (b) W. Hao, H. Wang, Q. Ye, W.-X. Zhang and Z. Xi, Org. Lett., 2015, 17, 5674–5677 CrossRef CAS PubMed; (c) J. Yin, Q. Ye, W. Hao, S. Du, Y. Gu, W.-X. Zhang and Z. Xi, Org. Lett., 2017, 19, 138–141 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1–S83 RSC.
- G. S. Girolami, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, J. Am. Chem. Soc., 1983, 105, 6752–6753 CrossRef CAS.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC; (b) S. K. Sur, J. Magn. Reson., 1989, 82, 169–173 CAS.
- (a) G. M. Sheldrick, SHELXTL 5.10 for Windows NT: Structure Determination Software Programs, Bruker Analytical X-ray Systems, Inc., Madison, WI, 1997 Search PubMed; (b) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Infrared spectroscopy for 1a,b, 2–5. CCDC 1870180, 1870203, 1870204, 1870205, 1870206 and 1870207. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qi01054d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |