
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer synthesis in non-ionic deep eutectic solvents
Natacha
Ndizeye
,
Subramanian
Suriyanarayanan
 * and
Ian A.
Nicholls
*
* and
Ian A.
Nicholls
*
Bioorganic & Biophysical Chemistry Laboratory, Centre for Biomaterials Chemistry, Department of Chemistry & Biomedical Sciences, Linnaeus University, SE-391 82 Kalmar, Sweden. E-mail: esusu@lnu.se; ian.nicholls@lnu.se
First published on 2nd September 2019
Abstract
Herein, we report the use of the use of non-ionic deep eutectic solvents (ni-DESs) as porogens in polymer synthesis. Three ni-DES systems, acetamide-N-methylacetamide (AA-NMA), N-methylacetamide-N-methylurea (NMA-NMU) and N-methylacetamide-N,N′-dimethylurea (NMA-NN'DMU), were deployed in the synthesis of a series of cross-linked copolymer monoliths comprised of a functional monomer, methacrylic acid (MAA) or hydroxyethylmethacrylate (HEMA), and a cross-linking monomer, ethylene glycol dimethylacrylate (EGDMA) or divinylbenzene (DVB) or 1,4-bis(acryloyl)piperazine (BAP). Polymers were synthesized under thermally initiated conditions with 2,2′-azobis(2-methylpropionitrile) (AIBN) or 2,2′-azobis(2-amidinopropane) dihydrochloride (ABAH) as an initiator. The resulting polymer monoliths were ground and sieved to yield particles of 63–125 μm. Corresponding polymers prepared in conventional porogens, acetonitrile, toluene and water were synthesized to serve as controls. The influence of the respective ni-DESs on polymer morphologies was examined by Brunauer–Emmett–Teller (BET) N2-adsorption, Fourier transform infrared spectroscopy (FT-IR), elemental analysis, scanning electron microscopy (SEM) and zeta potential measurements. The materials displayed surface areas, pore volumes and pore diameters of 115–532 m2 g−1, 0.1–1.4 cm3 g−1 and 5.2–12.5 nm, generally comparable with those of polymers obtained using conventional solvents, thus presenting these ni-DESs as viable alternatives to conventional organic solvents. The post-polymerization recovery of the ni-DESs (>80%) was demonstrated, highlighting the potential for using these novel liquids as alternatives to conventional, and often more expensive, toxic, flammable or volatile solvents in polymer synthesis.
1. Introduction
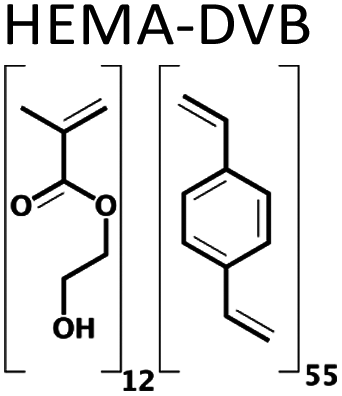
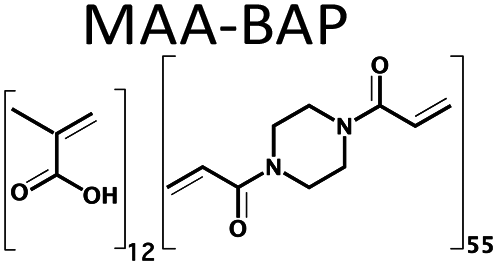
The need for new solvents for use in the production of fine chemicals and materials is driven by the requirement for more effective and more sustainable alternatives to current modalities.1 Over the past decades, the unique properties of deep eutectic solvents (DESs) have been exploited in areas as diverse as the synthesis of fine chemicals and materials and in drug delivery.2–14 They are traditionally comprised of two or more components, where at least one is ionic, accordingly. Soviet scientists aiming to develop liquid fertilizers discovered the first non-ionic deep eutectic liquid, a mixture of acetamide (67 wt%) and urea (33 wt%), with melting points of 80 °C and 133 °C, respectively, and a eutectic temperature of 56 °C. The relatively low toxicities of acetamide and urea together with their availability from renewable sources motived our search for novel non-ionic DESs (ni-DESs) for use as new alternatives to traditional organic solvents.5,15 Computational studies of the acetamide–urea (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) system indicated that higher-order complexes of the components were the basis for the deviation from Raoult's law that underlies the melting point depression.5,15 Based upon these observations, a family of amide/urea-based systems was designed where N-methyl substitutions were used to attenuate interaction between proposed flickering cluster-like species, to produce systems with deep eutectic behaviour. This series of ni-DESs included examples with sub-room temperature liquid states. This, in turn, motivated studies to explore their solvent properties in a range of application areas including their use as alternatives to conventional solvents in synthesis and, as reported here, as alternatives to conventional organic solvents in free-radical polymerization reactions. In particular, we have explored the use of three members of this family of novel solvents in the synthesis of a series of polymer monoliths including two functional monomers and three cross-linking monomers, respectively (Chart 1 and Table 1): methacrylic acid (MAA), hydroxyethylmethacrylic acid (HEMA), ethylene glycol dimethacrylate (EGDMA), divinyl benzene (DVB) and 1,4-bis(acryloyl)piperazine (BAP). The battery of twelve polymers was characterized using FT-IR, BET, SEM and zeta potential analysis. Moreover, the capacity to recycle the DES was demonstrated, using a simple water-based extraction and subsequent evaporation/heating for regeneration. The cheap, low toxicity and biodegradable nature of these amide-based DES components, together with the possibility for their recovery and reuse, makes them potentially attractive for use as a substitute for more toxic or flammable organic solvents in laboratory or industrial scale processes.16,17
:1) system indicated that higher-order complexes of the components were the basis for the deviation from Raoult's law that underlies the melting point depression.5,15 Based upon these observations, a family of amide/urea-based systems was designed where N-methyl substitutions were used to attenuate interaction between proposed flickering cluster-like species, to produce systems with deep eutectic behaviour. This series of ni-DESs included examples with sub-room temperature liquid states. This, in turn, motivated studies to explore their solvent properties in a range of application areas including their use as alternatives to conventional solvents in synthesis and, as reported here, as alternatives to conventional organic solvents in free-radical polymerization reactions. In particular, we have explored the use of three members of this family of novel solvents in the synthesis of a series of polymer monoliths including two functional monomers and three cross-linking monomers, respectively (Chart 1 and Table 1): methacrylic acid (MAA), hydroxyethylmethacrylic acid (HEMA), ethylene glycol dimethacrylate (EGDMA), divinyl benzene (DVB) and 1,4-bis(acryloyl)piperazine (BAP). The battery of twelve polymers was characterized using FT-IR, BET, SEM and zeta potential analysis. Moreover, the capacity to recycle the DES was demonstrated, using a simple water-based extraction and subsequent evaporation/heating for regeneration. The cheap, low toxicity and biodegradable nature of these amide-based DES components, together with the possibility for their recovery and reuse, makes them potentially attractive for use as a substitute for more toxic or flammable organic solvents in laboratory or industrial scale processes.16,17
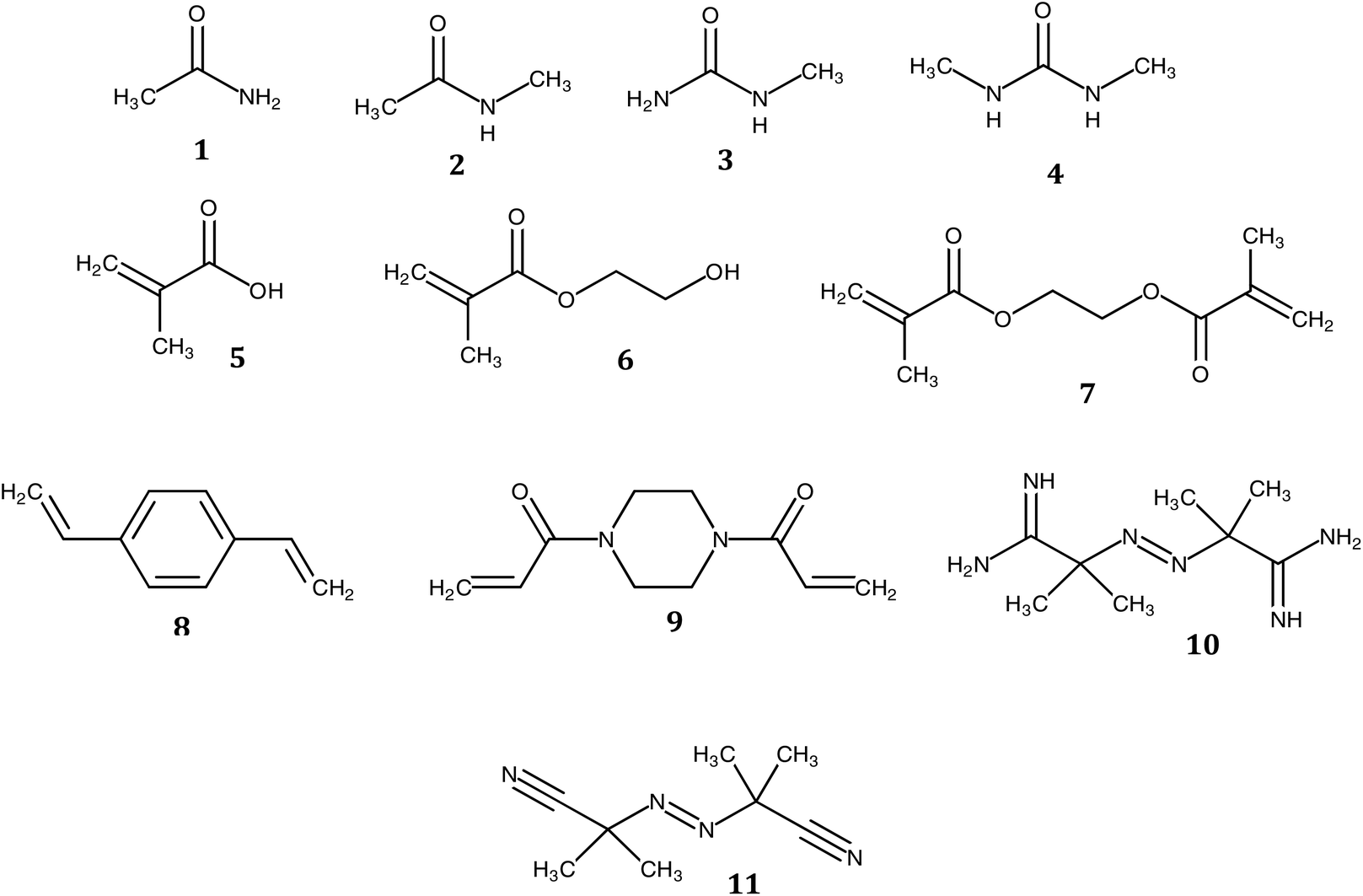 | ||
| Chart 1 Molecular structures of ni-DES components, functional monomers, crosslinker and initiators used in this study: acetamide (AA, 1), N-methylacetamide (NMA, 2), N-methylurea (NMU, 3), N,N′-dimethylurea (NN'DMU, 4), methacrylic acid (MAA, 5), hydroxyethylmethacrylate (HEMA, 6), ethylene glycol dimethylmethacrylate (EGDMA, 7), divinylbenzene (DVB, 8), 1,4 bis(acryloyl)piperazine (BAP, 9), 2,2′-azobis(2-amidinopropane) dihydrochloride (ABAH, 10) and 2,2′-azobis (2-methylpropionitrile (AIBN, 11). | ||

2. Results and discussion
A series of twelve co-polymer monoliths was synthesized by free radical polymerization using three different crosslinked polymers HEMA-DVB (P1–P4), HEMA-EGDMA (P5–P8) and MAA-BAP (P9–P12), in three different ni-DESs: N-methylacetamide-acetamide (NMA-AA), N-methylacetamide-N-methylurea (NMA-NMU) and N-methylacetamide-N,N′-dimethylurea (NMA-NN'DMU), and traditional porogens, respectively. The eutectic temperatures of NMA-AA, NMA-NMU and NMA-NN'DMU are 15, 14 and 12 °C, respectively. The polymers selected for synthesis in these ni-DESs have previously been used in the synthesis of highly crosslinked molecularly imprinted polymers using porogens of widely different character; toluene (P4),18 acetonitrile (P8)19 and water (P12),20Table 1.Transmission mode FT-IR analyses were conducted to establish which functionalities had been incorporated into the respective polymers, and to determine whether porogen had been removed, Fig. 1. In all the cases, the polymers prepared using a ni-DES revealed similar spectral features to those prepared in conventional solvents. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching from the ester/acid carbonyl groups of the functional monomers show sharp peaks around 1705–1750 cm−1. The CC stretching from the arene group of the DVB cross-linker moieties show a band located at 1500–1600 cm−1. Discernible bands corresponding to ν(CO), ν(CC), ν(C–OH), ν(C–N), and ν(CO–O) vibrational modes around 1710, 1605, 1010 and 1000–1200 cm−1, reflect the presence of the anticipated functionalities. Importantly, the absence of strong primary and secondary amide stretching modes at 1680 cm−1, in all cases, confirmed the successful extraction of the eutectic mixtures from the ground polymers.
O stretching from the ester/acid carbonyl groups of the functional monomers show sharp peaks around 1705–1750 cm−1. The CC stretching from the arene group of the DVB cross-linker moieties show a band located at 1500–1600 cm−1. Discernible bands corresponding to ν(CO), ν(CC), ν(C–OH), ν(C–N), and ν(CO–O) vibrational modes around 1710, 1605, 1010 and 1000–1200 cm−1, reflect the presence of the anticipated functionalities. Importantly, the absence of strong primary and secondary amide stretching modes at 1680 cm−1, in all cases, confirmed the successful extraction of the eutectic mixtures from the ground polymers.
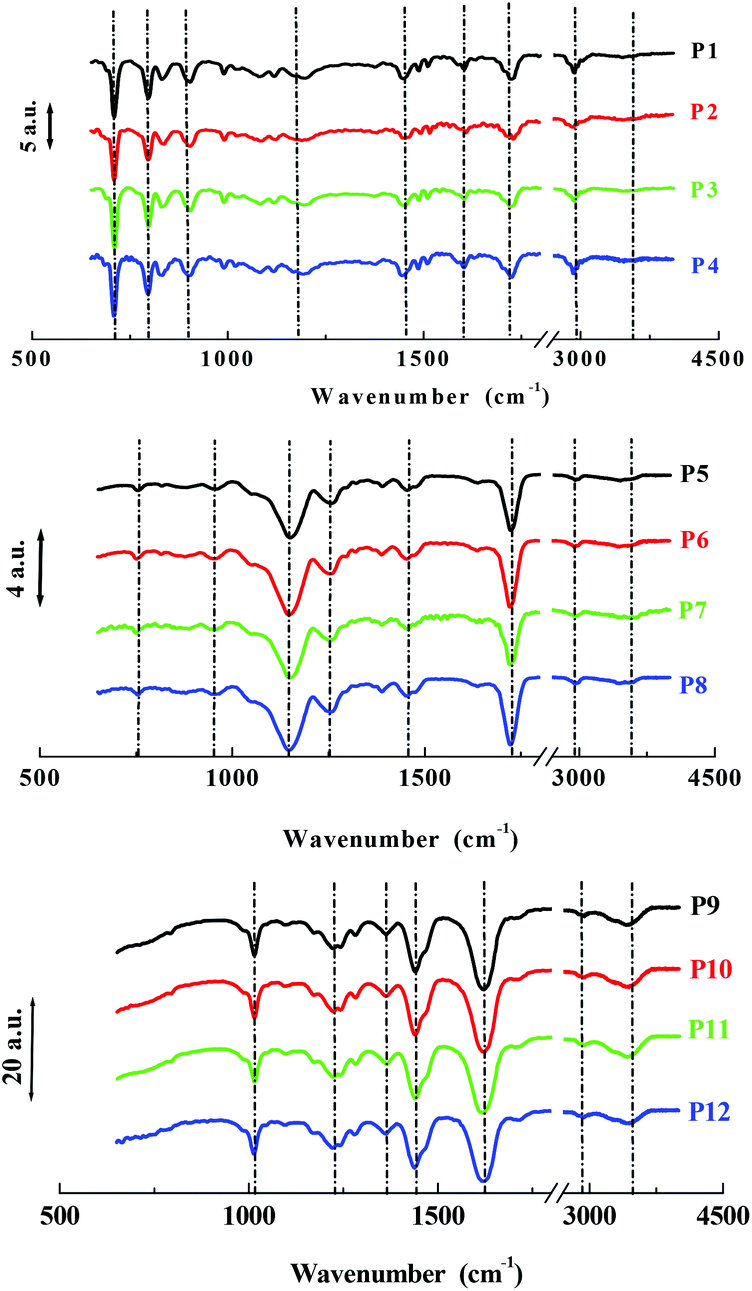 | ||
| Fig. 1 FT-IR spectra of polymers particles from P1–P4 (HEMA-DVB), P5–P8 (HEMA-EGDMA) and P9–P12 (MAA-BAP) prepared in different solvents. | ||
Elemental analysis (Table 2) of the twelve polymers revealed comparable elemental compositions for HEMA-DVB (P1–P4) and HEMA-EGDMA (P5–P8) polymers prepared in the three ni-DESs and conventional organic solvents, toluene and acetonitrile, respectively, which is indicative of a comparable incorporation of monomers. In the case of the MAA-BAP polymers (P9–P12), those prepared in the various ni-DESs (P9–P11) were comparable, though P12, prepared in water, was shown to have a marginally higher carbon content, corresponding to a lower oxygen content. In all cases a slightly lower carbon content was observed, reflecting entrapped water. Together, the nitrogen elemental analysis data and the FT-IR data clearly show that the amide-based DESs are neither entrapped in the resultant polymers nor are they incorporated into the polymer structure.
| Polymers | Carbona (%) | Hydrogena (%) | Nitrogena (%) |
|---|---|---|---|
| a Calculated values are given in parentheses. | |||
| P1 | 84.7 (85.7) | 8.2 (7.7) | <0.5 |
| P2 | 84.7 (85.7) | 8.2 (7.7) | <0.5 |
| P3 | 84.8 (85.7) | 8.3 (7.7) | <0.5 |
| P4 | 84.9 (85.7) | 8.2 (7.7) | <0.5 |
| P5 | 59.3 (60.1) | 7.9 (7.1) | <0.4 |
| P6 | 59.9 (60.1) | 7.4 (7.1) | <0.4 |
| P7 | 59.3 (60.1) | 7.4 (7.1) | <0.4 |
| P8 | 59.4 (60.1) | 7.4 (7.1) | <0.4 |
| P9 | 57.1 (60.6) | 7.9 (7.2) | 12.3 (13.1) |
| P10 | 57.5 (60.6) | 8.1 (7.2) | 12.5 (13.1) |
| P11 | 57.7 (60.6) | 8.0 (7.2) | 12.6 (13.1) |
| P12 | 59.4 (60.6) | 8.0 (7.2) | 12.6 (13.1) |
Initial studies on the recycling of the ni-DESs showed that they could be recovered (80–86%) by first washing the ground polymers with water, filtering and removal of water in vacuo. Optimization of this process and the exploring of other methods for the recycling of the ni-DESs is ongoing. BET and BJH analyses were used to evaluate morphological features; surface area, pore volume, cavity size and their distributions, Table 3. Nitrogen adsorption and desorption loops for the DVB, EGDMA and BAP cross-linked polymers are shown in Fig. 2.
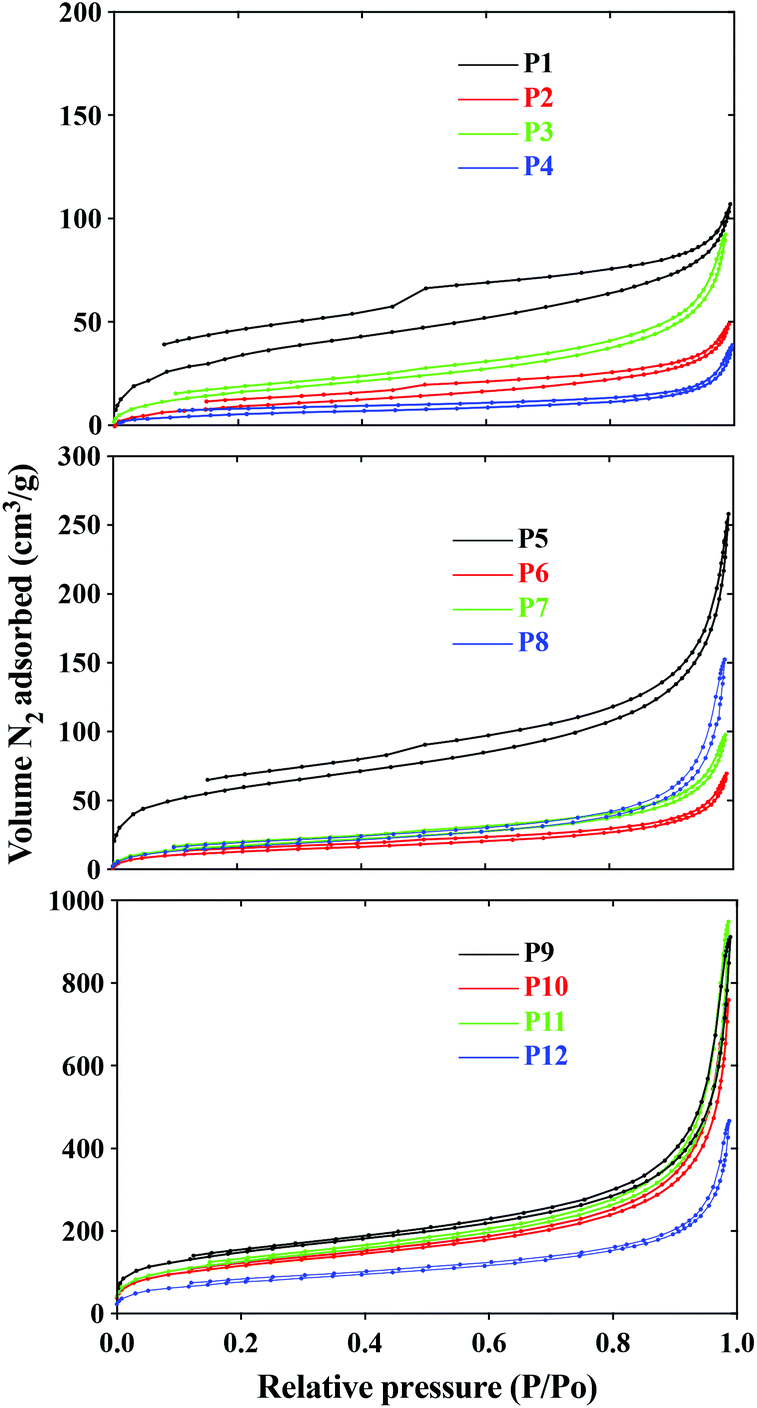 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms of polymers P1 to P12. | ||
| Polymers | Surface areaa (m2 g−1) [r] | Pore volumeb (cm3 g−1) | Pore diameterc (nm) | Zeta potential (mV) | |
|---|---|---|---|---|---|
| H2O | Methanol | ||||
| a Surface area presented as mean ± standard deviation from linear regression where r is the correlation coefficient. b Single point total pore volume. c Average pore diameter (4 V A−1). | |||||
| P1 | 127.4 ± 2.3 [0.999] | 0.1 | 5.1 | 9.08 ± 0.71 | 10.80 ± 0.57 |
| P2 | 234.4 ± 3.4 [0.999] | 0.4 | 6.9 | 12.41 ± 0.97 | 1.24 ± 0.06 |
| P3 | 365.6 ± 1.8 [0.999] | 0.7 | 8.1 | 15.80 ± 1.24 | 9.83 ± 0.52 |
| P4 | 115.0 ± 1.8 [0.999] | 0.3 | 10.6 | −11.20 ± 0.87 | −0.74 ± 0.03 |
| P5 | 212.4 ± 1.3 [0.999] | 0.3 | 7.1 | 2.13 ± 0.16 | 4.81 ± 0.25 |
| P6 | 213.7 ± 1.7 [0.999] | 0.3 | 7.1 | 6.97 ± 0.54 | 11.30 ± 0.63 |
| P7 | 269.4 ± 1.8 [0.999] | 0.5 | 7.8 | 2.84 ± 0.22 | 5.36 ± 0.28 |
| P8 | 258.6 ± 0.9 [0.999] | 0.8 | 12.5 | −7.47 ± 0.58 | −11.70 ± 0.62 |
| P9 | 532.8 ± 2.6 [0.999] | 1.4 | 10.5 | −10.80 ± 0.84 | −8.24 ± 0.43 |
| P10 | 477.7 ± 2.6 [0.999] | 1.2 | 10.0 | −9.25 ± 0.72 | −10.80 ± 0.57 |
| P11 | 516.4 ± 2.6 [0.999] | 1.4 | 11.5 | −7.55 ± 0.59 | −8.26 ± 0.44 |
| P12 | 335.0 ± 1.9 [0.999] | 0.7 | 8.9 | −8.57 ± 0.67 | −4.88 ± 0.26 |
In general, the polymers synthesized in the ni-DESs exhibited behavior comparable to Type IV isotherms, implying mesoporous polymer structures (2 nm < pore size > 50 nm).21 Some of these polymers follow a H4 hysteresis loop (associated with the secondary process of capillary condensation) indicating polymer scaffolds that are composed of mesopores that are limited by the micropores. Four polymers, P1–P3 and P5 showed hysteresis loop shapes representative of a broader pore size distribution that is close to microporous in nature. It was observed that the three ni-DESs produced similar pore characteristics in each of the three polymer systems, HEMA-DVB (P1–P3), HEMA-EGDMA (P5–P7) and MAA-BAP (P9–P11), though in all cases somewhat lower than those of the corresponding polymers synthesized in toluene, (P4), acetonitrile (P8) and water (P12), respectively. No significant correlations between surface areas and porosities of the polymers synthesized in the ni-DESs was revealed, though larger surface areas and porosities were observed for the MAA-BAP polymers prepared in the ni-DESs (P9–P11) than that prepared in water (P12). This we suggest may be due to water being so much smaller than the ni-DES components, which themselves are postulated to form larger flickering cluster-like aggregates.5
Zeta potential measurements of the polymers in H2O and methanol, Table 3, revealed that polymers prepared in the ni-DESs possessed very low zeta potentials relative to those of polymers prepared in traditional solvents. In the cases of the DVB- and EGDMA-crosslinked polymers synthesized in the ni-DESs (P1–P3 and P5–P7, respectively), positive surface charge was observed, though negative values were obtained for those synthesized in toluene (P4) and acetonitrile (P8). This is indicative of relatively limited charge distribution at the surface of the polymers prepared in the ni-DESs, as compared to those prepared in the traditional solvents, which may be explained by a greater degree of solvation of functionalities by the ni-DESs facilitating a more even distribution of these functionalities throughout the polymer particles. This postulate finds support in the reported high conductivities of these ni-DESs.5
SEM studies were used to further assess the effect of using the ni-DESs as alternative porogens on polymer structural and morphological features (Fig. 3). Again, essentially comparable morphologies were observed confirming that ni-DESs provide similar structural features to those obtained when using conventional solvents, ranging from mesoporous monolith-like structures (e.g. P9) to mosaic microspheres (P1).
 | ||
| Fig. 3 Surface topographies of polymer particles P1 to P12 imaged using scanning electron microscopy (SEM). | ||
3. Experimental section
3.1 Materials
Acetamide (AA), N-methylacetamide (NMA), N-methylurea (NMU), N,N′-dimethylurea (NN'DMU), methacrylic acid (MAA), hydroxyethylmethacrylate (HEMA), ethylene glycol dimethylmethacrylate (EGDMA), divinylbenzene (DVB), 1,4 bis(acryloyl)piperazine (BAP), 2,2′-azobis(2-amidinopropane) dihydrochloride (ABAH) and 2,2′-azobis (2-methylpropionitrile (AIBN) (Chart 1) were purchased from Sigma-Aldrich Inc., Sweden. Analytical grade sodium hydroxide, acetone, methanol, ethanol and acetic acid, procured from Sigma-Aldrich, were used without further purification. Milli-Q grade (Millipore AB, Sweden) water was used. Prior to use, HEMA was passed through a column of activated basic aluminum oxide then fractional distillation was carried out before storage at −20 °C until use. MAA was vacuum distilled and stored at −20 °C. EGDMA (100 mL) was extracted three times with NaOH (aq, 0.1 M, 75 mL) and saturated NaCl (25 mL). It was then dried over anhydrous MgSO4 for 15 min and stored at −20 °C. Acetonitrile, toluene and DVB were distilled then dried over 4 Å molecular sieves before storage at −8 °C. ABAH was recrystallized from a acetone:water mixture (1:1, v/v) and AIBN was recrystallized from methanol. The eutectic mixtures were prepared by heating a known amount of NMA at 40 °C until a clear liquid was obtained, before the appropriate quantity (w/w) of the other component, AA (30%), or NMU (20%) or NN'DMU (30%), was added to the molten NMA and the mixture was stirred until a homogenous phase was formed.
3.2 Polymer synthesis
Three different polymers were synthesized in each of the three ni-DESs using MAA or HEMA as a functional monomer and DVB, EGDMA or BAP (Table 1) as the crosslinker. In each case, the molar ratio of functional monomer: cross-linker was maintained at 12:55.19 The pre-polymerization mixture was dissolved in the eutectic mixture and saturated with nitrogen for 5 min to purge dissolved oxygen. The solution was then brought to the polymerization temperature (70 °C) before addition of the initiator; AIBN for HEMA-DVB and HEMA-EGDMA and ABAH for the MAA-BAP polymer system. The polymerization reaction mixtures were maintained at 70 °C for 6 h using a heated oil bath. Following polymerization, the resultant bulk polymer was ground with a mortar and pestle before sieving to yield polymer particles of 63–125 μm size. The polymer particles (∼1 g) were slurry packed into an HPLC column to be washed from unreacted particles and porogen following a procedure reported by Karlsson et al.22 The polymer was emptied from the column by pumping acetone in the column, it was collected, air-dried and stored in vials at ambient temperature until use. The three polymer systems were also synthesized using conventional solvents and were processed using the same procedure. The yields of 63–125 μm size polymer particles after the work-up process were all in the range 86–90%.3.3 Physical characterization of synthesized polymers
4. Conclusions
In this study we have demonstrated the use of ni-DESs as porogens in polymer synthesis, deploying them as alternatives to the use of toluene, acetonitrile and water. A series of cross-linked co-polymers, HEMA-DVB, HEMA-EGDMA and MAA-BAP, was prepared using three different ni-DESs and conventional solvents. The ni-DESs afforded polymers with comparable surface areas and pore sizes to those prepared in conventional solvents. An interesting difference between polymers was the suppression of surface charge obtained when syntheses were performed in the ni-DESs, as reflected in the zeta potential results. Initial studies on the potential for recycling of these ni-DESs have demonstrated that recoveries of >80% are possible. Finally, the wide range of chemical functionalities soluble in these ni-DESs, together with their low flammabilities, volatilities and toxicities can make them attractive alternatives to many conventional organic solvents for polymer synthesis and other applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swedish Research Council (Vetenskapsrådet, grant number 2014-4573), the Swedish Knowledge Foundation (BIO-QC, grant number 20170059) and Linnaeus University for financial support.References
- J. H. Clark, Green Chem., 2006, 8, 17 RSC.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108 RSC.
- N. Adawiyah, M. Moniruzzaman, S. Hawatulaila and M. Goto, MedChemComm, 2016, 7, 1881 RSC.
- M. Usanovich, Dokl. Akad. Nauk, 1958, 120, 1304 CAS.
- S. Suriyanarayanan, G. D. Olsson, S. Kathiravan, N. Ndizeye and I. A. Nicholls, Int. J. Mol. Sci., 2019, 20, 2857 CrossRef PubMed.
- L. S. Longo and M. V. Craveiro, J. Braz. Chem. Soc., 2018, 29, 1999 CAS.
- M. Francisco, A. v. d. Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074 CrossRef CAS PubMed.
- K. E. Plass, K. Kim and A. J. Matzger, J. Am. Chem. Soc., 2004, 126, 9042 CrossRef CAS PubMed.
- Y. Y. Zhang, X. Y. Ji and X. H. Lu, Renewable Sustainable Energy Rev., 2018, 97, 436 CrossRef CAS.
- F. del Monte, D. Carriazo, M. C. Serrano, M. C. Gutiérrez and M. L. Ferrer, ChemSusChem, 2014, 7, 999 CrossRef CAS PubMed.
- J. D. Mota-Morales, R. J. Sánchez-Leija, A. Carranza, J. A. Pojman, F. del Monte and G. Luna-Bárcenas, Prog. Polym. Sci., 2018, 78, 139 CrossRef CAS.
- D. Carriazo, M. C. Serrano, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Soc. Rev., 2012, 41, 4996 RSC.
- M. Mokhtary, Acad. J. Polym. Sci., 2019, 2, 555586 Search PubMed.
- J. Wang, J. Han, M. Y. Khan, D. He, H. Peng, D. Chen, X. Xie and Z. Xue, Polym. Chem., 2017, 8, 1616 RSC.
- S. Suriyanarayanan and I. A. Nicholls, Swedish Patent Application, SE-1850195-7, 2018 Search PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060 CrossRef CAS PubMed.
- P. Liu, J.-W. Hao, L.-P. Mo and Z.-H. Zhang, RSC Adv., 2015, 5, 48675 RSC.
- N. Ndizeye, S. Suriyanarayanan and I. A. Nicholls, Eur. Polym. J., 2018, 106, 223 CrossRef CAS.
- S. Shoravi, G. D. Olsson, B. C. G. Karlsson, F. Bexborn, Y. Abghoui, J. Hussain, J. G. Wiklander and I. A. Nicholls, Org. Biomol. Chem., 2016, 14, 4210 RSC.
- J. P. Rosengren-Holmberg, J. Andersson, J. R. Smith, C. Alexander, M. R. Alexander, G. Tovar, K. N. Ekdahl and I. A. Nicholls, Biomater. Sci., 2015, 3, 1208 RSC.
- S.-S. Chang, B. Clair, J. Ruelle, J. Beauchêne, F. Di Renzo, F. Quignard, G.-J. Zhao, H. Yamamoto and J. Gril, J. Exp. Bot., 2009, 60, 3023 CrossRef CAS PubMed.
- J. G. Karlsson, L. I. Andersson and I. A. Nicholls, Anal. Chim. Acta, 2001, 435, 57 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |