
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-induced copper-RDRP in continuous flow without external deoxygenation†
Arkadios
Marathianos
 ,
Evelina
Liarou
,
Athina
Anastasaki
,
Richard
Whitfield
,
Matthew
Laurel
,
Alan M.
Wemyss
and
David M.
Haddleton
*
,
Evelina
Liarou
,
Athina
Anastasaki
,
Richard
Whitfield
,
Matthew
Laurel
,
Alan M.
Wemyss
and
David M.
Haddleton
*
Department of Chemistry, University of Warwick Library Road, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk
First published on 19th July 2019
Abstract
Photo-induced Cu-RDRP of acrylates in a continuous flow reactor without the need for deoxygenation or externally added reagents. Optimization of the catalyst concentration and the flow rate/residence time leads to well-defined polyacrylates with controlled molecular weights, excellent initiator efficiency, high end-group fidelity polymers and product uniformity. A multifunctional initiator was also used to demonstrate the versatility of the system.
Controlled radical polymerization (CRP) techniques such as atom-transfer radical polymerization (ATRP),1–3 single electron transfer-living radical polymerization (SET-LRP),4,5 reversible addition–fragmentation chain-transfer polymerization (RAFT),6–9 and nitroxide-mediated polymerization (NMP),10 have expanded the capability of polymer synthesis, allowing access to a plethora of new materials.11,12 Among the benefits, the ability to externally regulate these techniques with various stimuli has further expanded the scope of their applications. The use of light as a stimulus allows for excellent spatial and temporal control thus expanding their applications.13–17
Nevertheless, CRP techniques are often not available to undergraduate laboratories or those lacking specialist equipment for efficient deoxygenation. Oxygen is a radical inhibitor, scavenging both primary and propagating radicals leading to the formation of peroxy radicals and hydroperoxides having an overall detrimental effect.18 However, the various traditional deoxygenation techniques applied prior to polymerization (freeze–pump–thaw, N2/Ar sparging, glove box equipment, etc.) can be disadvantageous due to their high cost and implementation time. In order to circumvent this, different approaches have been made so as to replace conventional deoxygenation in Cu-mediated reversible deactivation radical polymerization19–24 and photo-induced electron transfer (PET) RAFT.25–27 PET-RAFT, has employed various reducing agents (i.e. ascorbic acid,28,29 photo-redox catalysts30,31) which have been successfully used for the efficient removal of oxygen in both batch reactions and in continuous flow processes.32
Polymerizations in continuous flow (CF) reactors are of interest since they have been proved to be efficient alternatives to batch reactions33–36 and by providing the ability to produce large volumes in short times,37 have introduced an industrialized way of materials production.38,39 Continuous flow RAFT has been developed and exploited, for example by CSIRO where the ingress of oxygen through the tubing was problematic and avoided by using steel tubing to prevent quenching of the radical process by oxygen,40 and more recently making use of the light penetration of millimetre-size fluoropolymer tubing giving multigrams/kgs of RAFT polymer per day.41 CF processes have been fully exploited when combined with light as an external stimulus.34,42–45 Junkers and colleagues has recently reported on the CF synthesis of core crosslinked star polymers via a photo induced copper mediated system. This system required prior nitrogen sparging and showed an elegant route to an interesting tool to continuously produce star polymers without intermediate purification46 Efficiency of light penetration is increased due to the high surface area-to-volume ratio leading to more uniform irradiation and resulting in better control over the polymerization.35 As a result, significant amounts of polymers are obtained through a user-friendly approach, with the ability to easily regulate the reaction parameters (flow rate, residence time, light intensity, etc.). On account of this, the scope of CF polymerizations has been expanded with the replacement of traditional deoxygenation in PET-RAFT polymerization.30,47 However, to the best of our knowledge, there is no example of a photoinduced copper mediated process that does not require deoxygenation in CF reactors.
Herein, we introduce the photo-induced Cu-RDRP of acrylates in a continuous flow reactor without the requirement of applying any type of deoxygenation or using externally added reagents. Optimization of the flow rate/residence time as well as the [EBiB]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Cu(II)Br2]:[Me6Tren] ratio leads to the synthesis of well-defined poly(acrylates). The versatility of this approach is further confirmed with the synthesis of different molar mass polymers (Mn,SEC ∼ 2300–26000 g mol−1) and the high end-group fidelity maintained is demonstrated through nucleophilic thio-bromine substitution with thioglycerol.
:[Cu(II)Br2]:[Me6Tren] ratio leads to the synthesis of well-defined poly(acrylates). The versatility of this approach is further confirmed with the synthesis of different molar mass polymers (Mn,SEC ∼ 2300–26000 g mol−1) and the high end-group fidelity maintained is demonstrated through nucleophilic thio-bromine substitution with thioglycerol.
In order to explore the ability of this system to perform without deoxygenation in a continuous flow reactor, methyl acrylate (MA) was used as monomer, ethyl α-bromoisobutyrate (EBiB) as initiator, tris(2-(dimethylamino)ethyl)-amine (Me6Tren) as ligand, Cu(II)Br2 as the copper source, and DMSO as solvent. In this chemistry the copper(II) is reduced following photoexcitation of the ligand.48 The copper(I) is prone to both oxidation and disproportionation.49 Disproportionation leads to copper(0) which is also prone to rapid oxidation.50 One of the primary aims was to design a user-friendly process, able to provide uniform irradiation for the continuous flow. Consequently, the set-up included a dual syringe pump, a mixing tee, 3-meter PFA tubing located in the chamber of a UVP crosslinker with λmax = 365 nm, ending outside of the UVP chamber and connected with the foil-wrapped collection vessel (Scheme 1, Fig. S1†). Initially, based on our findings in the batch system (near-quantitative conversion after ∼2.5 hours) (Fig. S2†), the polymerization of MA with targeted DPn = 200 was conducted using the ratio [MA]:[I]:[Cu(II)Br2]:[Me6Tren] = [200]:[1]:[0.02]:[0.12] with a flow rate of 8 μL min−1 (residence time = 2.5 hours). In contrast to the equivalent batch process, no polymerization took place. The presence of oxygen in this system is significant since it can be found dissolved in the reaction solution or localized in the tubing acting as “headspace”. Based on our previous work using non-deoxygenated systems, where an increase of the copper complex concentration contributed to fast oxygen consumption,20 we doubled both the concentration of Cu(II)Br2 and Me6Tren. However, no polymerization was seen under these conditions. The lack of polymerization (Table S1,† entries 1 & 2) was attributed to the low amounts of copper complex which proved insufficient to both participate in oxygen consumption and generate active species for the polymerization. Thus, higher amounts of the copper complex were used with [Cu(II)Br2]:[Me6Tren] = [0.08]:[0.48], yielding PMA with 46% monomer conversion (Table S1, entry 3, Fig. S3†). It is noted that copper(II) salts are classified as Generally Regarded As Safe (GRAS) compounds by the FDA. The deviations between theoretical and experimental Mn values for the latter might be attributed to the oxygen consumption, taking place at this stage of the polymerization, leading to a reduction of initiator efficiency. As low monomer conversion and initiator efficiency were obtained, we envisaged that further increase of the copper complex concentration was needed in order to achieve sufficient oxygen consumption and higher monomer conversions. Consequently, [Cu(II)Br2]:[Me6Tren] = [0.16]:[0.96] was used, resulting in 77% monomer conversion, good agreement between theoretical and experimental molecular weights (Mn,SEC = 13600, Mn,th. = 13500) and low dispersity (Đ = 1.17) (Table S1, entry 4, Fig. S3 & S4†). Interestingly, the continuous flow process provided high initiator efficiency when compared with a batch process, where deviations between experimental and theoretical Mn values were present (Fig. S2†).20 It is noteworthy that when higher amounts of Cu(II)Br2 and Me6Tren were used (0.32 eq. and 1.92 eq., respectively), a slightly higher dispersity was observed (Đ = 1.23) and no further increase in the monomer conversion was obtained (Fig. S3, Table S1,† entry 5). Hence, the ratio [EBiB]:[Cu(II)Br2]:[Me6Tren] = [1]:[0.16]:[0.96] was selected for further investigation of this non-deoxygenated system in continuous flow process (Table S1, Fig. S3†).
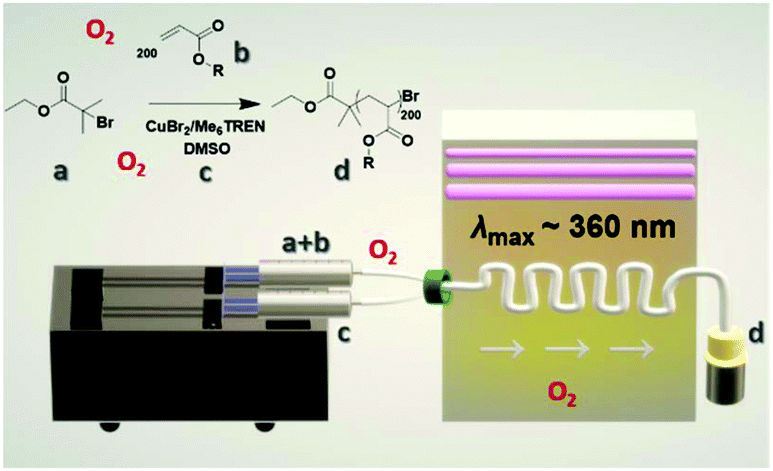 | ||
| Scheme 1 Reaction scheme and setup for the photo-induced Cu-RDRP in continuous flow with a: EBiB, b: MA, c: CuBr2/Me6Tren/DMSO and d: targeted PMA200. | ||
Consequently, identical samples were prepared and polymerized with different flow rates leading to different residence times (Table 1). MA with targeted DPn = 200 was polymerized with [Cu(II)Br2]:[Me6Tren] = [0.16]:[0.96] (Fig. 1a and b & c). With a flow rate of 80 μL min−1, the monomer conversion was as low as 4%, indicating that longer residence times are needed for the polymerization to proceed to acceptable conversions. From a flow rate of 40 μL min−1 (Table 1, entry 2) the conversion increased steadily (Fig. 1b & c) and continued for all the samples with residence times up to 200 minutes (Table 1, entries 2–9) where good control over the polymerization was achieved, with low dispersities, high conversions and good agreement between Mn,th and Mn,SEC suggesting good initiator efficiency. In order to achieve even higher conversions, lower flow rates (longer residence times) were used. When a flow rate of 5 μL min−1 was used, the results were similar to 6 μL min−1 (Table 1, entry 9). With 4 μL min−1 and 2 μL min−1, the monomer conversion remained constant at 85%, but higher MWts were obtained (Table 1, entry 11 & 12). This might be attributed to the prolonged residence times in the reactor and the extended exposure to oxygen, which can induce termination events. It is noteworthy that although the ratio [Cu(II)Br2]:[Me6Tren] = [0.04]:[0.24] resulted in zero monomer conversion with 8 μL min−1 (Table S1,† entry 2), it resulted in 46% monomer conversion when 6 μL min−1 was applied. Furthermore, the molecular characteristics of the sample run at different times in the reactor were the same for the whole volume of the polymer, corroborating the uniformity of the system (Fig. 1d, Table S2†).
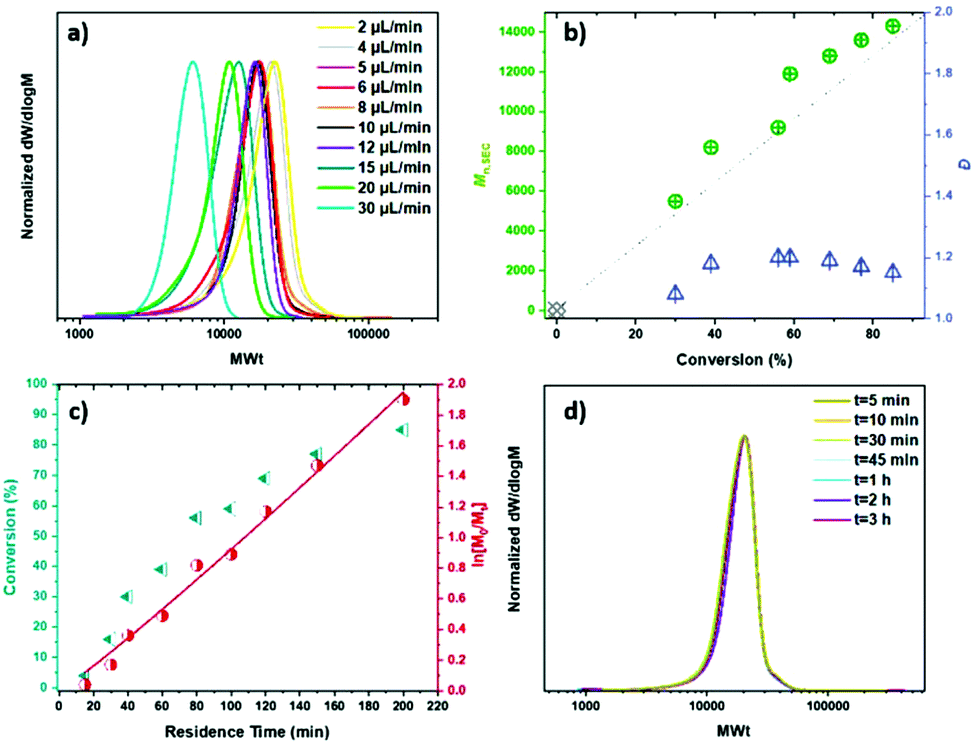 | ||
| Fig. 1 (a) THF-SEC derived molecular weight distributions showing the evolution of MWts, (b) plots of Mnversus conversion (left, green) and dispersity (Đ) versus conversion (right, blue), (c) kinetic plots of ln[M0/Mt] (right, red) and conversion (left, dark cyan) versus residence time showing linear trend, (d) THF-SEC derived molecular weight distributions for targeted PMA200 passed through the tubing reactor at different times. Conditions: [MA]:[I]:[Cu(II)Br2]:[Me6Tren] = [200]:[1]:[0.16]:[0.96]. | ||
a
| Entry | Flow rate (μL min−1) | Res. Time (min) | Mon. Conv.b (%) | M n,th. (g mol−1) |
M
n,SECc |
Đ |
|---|---|---|---|---|---|---|
|
a In all polymerizations, the volume ratio of monomer to solvent was maintained 1:1.
b Conversion was calculated via1H NMR in CDCl3.
c Determined by THF SEC analysis and expressed as molecular weight equivalents to PMMA narrow molecular weight standards.
|
||||||
| 1 | 80 | 15 | 4 | — | — | — |
| 2 | 40 | 30 | 16 | — | — | — |
| 3 | 30 | 40 | 30 | 5400 | 5500 | 1.08 |
| 4 | 20 | 60 | 39 | 6900 | 8200 | 1.18 |
| 5 | 15 | 80 | 56 | 9800 | 9200 | 1.20 |
| 6 | 12 | 100 | 59 | 10400 |
11900 |
1.20 |
| 7 | 10 | 120 | 69 | 12100 |
12800 |
1.19 |
| 8 | 8 | 150 | 77 | 13500 |
13600 |
1.17 |
| 9 | 6 | 200 | 85 | 14800 |
14300 |
1.15 |
| 10 | 5 | 240 | 84 | 14700 |
14300 |
1.15 |
| 11 | 4 | 300 | 85 | 14800 |
15400 |
1.21 |
| 12 | 2 | 600 | 84 | 14700 |
16400 |
1.23 |
A further requirement for a controlled radical polymerization is the retention of the chain end, which enables the functionalization of the obtained polymers.51 In order to explore the ω-Br functionality in this system, matrix assisted laser desorption–ionization time-of-flight mass spectrometry (MALDI-ToF-MS) was employed for PMA25,52 revealing a predominant single peak distribution corresponding to the bromine-capped polymer chains (Fig. 2b and c). A small second distribution observed was attributed to a small degree of fragmentation during the MALDI-ToF-MS process. Since this suggested that the active end-groups were preserved, thioglycerol was used for the thio-bromine substitution of the well-defined PMA25 so as to introduce a different functionality for the non-deoxygenated polymer (Fig. 2a). After the thio-bromine substitution, MALDI-ToF-MS showed full shift of the –Br terminated chains and revealed the thioglycerol-functionalized PMA25 (Fig. 2c and d, Fig. S5†). The single peak distribution observed for the substituted PMA, corroborated our hypothesis that the small distribution observed in the –Br capped sample corresponded to the MALDI-ToF-MS process.
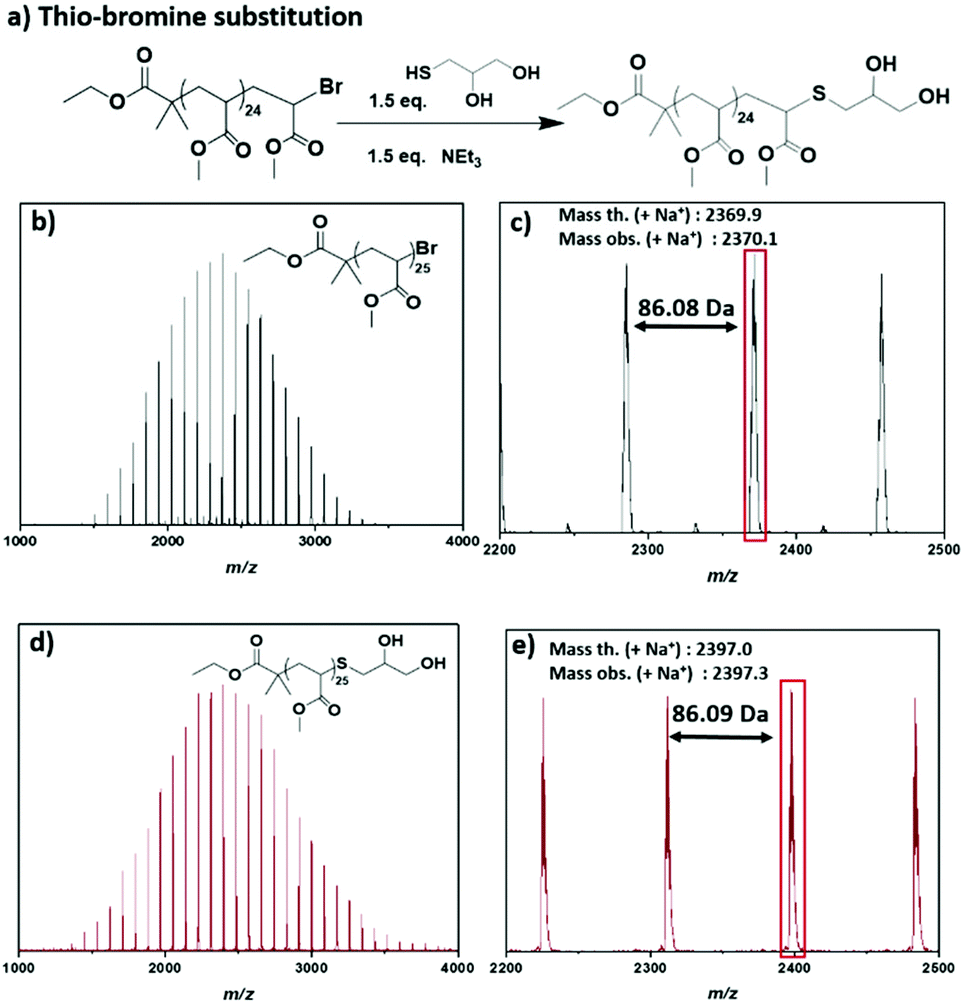 | ||
| Fig. 2 (a) Reaction scheme for the thio-bromine substitution of PMA25 with thioglycerol and MALDI-ToF spectra for (b, c) –Br substituted PMA25 and (d, e) –thioglycerol substituted PMA25. | ||
In order to examine the ability to produce different molar masses, various DPs of PMA (25–400) were targeted with [Cu(II)Br2]:[Me6Tren] = [0.16]:[0.96]. Since the production of different molar masses requires different polymerization times, different flow rates (residence times) were applied for this purpose (Table S3†). As a result, molecular weights from 2300 to 26400 g mol−1 were achieved (Fig. S6†). Apart from various molar masses, the non-deoxygenated CF polymerization of hydrophobic and hydrophilic monomers was examined with both n-butyl acrylate (n-BA) (Fig. S7†) and poly(ethylene glycol) methyl ether acrylate (PEGA480) (Fig. S8†). For both, the optimum conditions of [Cu(II)Br2]:[Me6Tren] = [0.16]:[0.96] with a flow rate of 6 μL min−1 were used, leading to good agreement between theoretical and experimental Mn values, low dispersities (Đ = 1.14–1.16) and high conversions (Fig. S9†). In addition to the synthesis of linear polymers, we were interested in different architectures, since their properties have gained a lot of academic and industrial interest.53,54 For this purpose, an 8-arm initiator (octa-O-isobutyryl bromide lactose initiator) was used for the synthesis of a PMA star homopolymer with targeted DPn = 200. Following [Cu(II)Br2]:[Me6Tren] = [0.16]:[0.96] and 6 μL min−1 flow rate, a well-defined PMA star was obtained (Fig. S10†).
In summary, a photo-induced Cu-RDRP of acrylates in continuous flow without the requirement for deoxygenation is reported. The photo reduction of copper(II) and disproportion of copper(I) to copper(0) provide a regenerating process for the rapid consumption of oxygen. Low dispersities, control over the molecular weights and high conversions were obtained, after optimization of the copper catalyst loadings and residence times. Without external deoxygenation, good initiator efficiency was evident and high end-group fidelity was maintained, allowing for post polymerization modification. The robustness of the system is further corroborated with the synthesis of sophisticated architectures, as well as hydrophobic and hydrophilic polymers through a user-friendly setup.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the University of Warwick (A. M., E. L., A. M. W.), and Syngenta (A. M.) is gratefully acknowledged. A. A. acknowledges the Global Marie Curie Fellowship (BINAMA 705041). R. W. acknowledges the Institute of Advanced Study (UoW) for an Early Career Innovation Fellowship. We are also grateful for the Polymer Characterization RTP, Dr D. Lester and D. Coursari for providing use of SEC equipment.Notes and references
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Ng, J. Yeow, R. Chapman, N. Isahak, E. Wolvetang, J. J. Cooper-White and C. Boyer, Macromolecules, 2018, 51, 7600–7607 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- G. R. Jones, R. Whitfield, A. Anastasaki, N. Risangud, A. Simula, D. J. Keddie and D. M. Haddleton, Polym. Chem., 2018, 9, 2382–2388 RSC.
- R. Whitfield, A. Anastasaki, G. R. Jones and D. M. Haddleton, Polym. Chem., 2018, 9, 4395–4403 RSC.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963–971 RSC.
- Y. Wang, L. Fu and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 1317–1321 CrossRef CAS.
- A. E. Enciso, L. Fu, S. Lathwal, M. Olszewski, Z. Wang, S. R. Das, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 16157–16161 CrossRef CAS.
- Q. Yang, J. Lalevée and J. Poly, Macromolecules, 2016, 49, 7653–7666 CrossRef CAS.
- S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1190–1196 CrossRef CAS.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Angew. Chem., Int. Ed., 2016, 55, 4500–4503 CrossRef CAS.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- J. Tanaka, P. Gurnani, A. B. Cook, S. Häkkinen, J. Zhang, J. Yang, A. Kerr, D. M. Haddleton, S. Perrier and P. Wilson, Polym. Chem., 2019, 10, 1186–1191 RSC.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 RSC.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS.
- J. Yeow, S. Joshi, R. Chapman and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 10102–10106 CrossRef CAS.
- N. Corrigan, D. Rosli, J. W. J. Jones, J. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef CAS.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
- T. Junkers and B. Wenn, React. Chem. Eng., 2016, 1, 60–64 RSC.
- B. L. Buss and G. M. Miyake, Chem. Mater., 2018, 30, 3931–3942 CrossRef CAS.
- M. H. Reis, C. L. G. Davidson and F. A. Leibfarth, Polym. Chem., 2018, 9, 1728–1734 RSC.
- C. Diehl, P. Laurino, N. Azzouz and P. H. Seeberger, Macromolecules, 2010, 43, 10311–10314 CrossRef CAS.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- J. L. Steinbacher and D. T. McQuade, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6505–6533 CrossRef CAS.
- C. H. Hornung, C. Guerrero-Sanchez, M. Brasholz, S. Saubern, J. Chiefari, G. Moad, E. Rizzardo and S. H. Thang, Org. Process Res. Dev., 2011, 15, 593–601 CrossRef CAS.
- J. Gardiner, C. H. Hornung, J. Tsanaktsidis and D. Guthrie, Eur. Polym. J., 2016, 80, 200–207 CrossRef CAS.
- B. L. Ramsey, R. M. Pearson, L. R. Beck and G. M. Miyake, Macromolecules, 2017, 50, 2668–2674 CrossRef CAS.
- B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Polym. Chem., 2014, 5, 3053–3060 RSC.
- M. Chen and J. A. Johnson, Chem. Commun., 2015, 51, 6742–6745 RSC.
- A. Melker, B. P. Fors, C. J. Hawker and J. E. Poelma, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2693–2698 CrossRef CAS.
- J. H. Vrijsen, C. Osiro Medeiros, J. Gruber and T. Junkers, Polym. Chem., 2019, 10, 1591–1598 RSC.
- J. Peng, Q. Xu, Y. Ni, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2019, 10, 2064–2072 RSC.
- E. Frick, A. Anastasaki, D. M. Haddleton and C. Barner-Kowollik, J. Am. Chem. Soc., 2015, 137, 6889–6896 CrossRef CAS PubMed.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6880–6895 CrossRef CAS.
- A. Anastasaki, J. Willenbacher, C. Fleischmann, W. R. Gutekunst and C. J. Hawker, Polym. Chem., 2017, 8, 689–697 RSC.
- J. S. Town, G. R. Jones and D. M. Haddleton, Polym. Chem., 2018, 9, 4631–4641 RSC.
- R. Whitfield, A. Anastasaki, N. P. Truong, P. Wilson, K. Kempe, J. A. Burns, T. P. Davis and D. M. Haddleton, Macromolecules, 2016, 49, 8914–8924 CrossRef CAS.
- B. L. Buss, L. R. Beck and G. M. Miyake, Polym. Chem., 2018, 9, 1658–1665 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00945k |
| This journal is © The Royal Society of Chemistry 2019 |