
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-reporting visible light-induced polymer chain collapse†
Janin T.
Offenloch
a,
Eva
Blasco
 a,
Simon
Bastian
a,
Christopher
Barner-Kowollik
*ab and
Hatice
Mutlu
*ac
a,
Simon
Bastian
a,
Christopher
Barner-Kowollik
*ab and
Hatice
Mutlu
*ac
aMacromolecular Architectures, Institut für Technische Chemie und Polmerchemie, Karlsruhe Institute of Technology (KIT), Engesserstraße 18, 76128 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; hatice.mutlu@kit.edu
bSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, QLD 4000, Brisbane, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
cSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Karlsruhe, Germany
First published on 15th July 2019
Abstract
We introduce a facile photoinduced self-reporting crosslinking methodology for the compaction of polymer chains in highly diluted solution. The chain compaction, which is readily monitored via the change in optical properties due to the release of the chromophore, is achieved via mild visible light irradiation (430–435 nm) of statistically distributed pyrene-substituted oxime ester derivatives. In-depth characterization via size exclusion chromatography (SEC), UV/Vis and fluorescence spectroscopy, as well as NMR measurements including diffusion ordered spectroscopy (DOSY), reveals an efficient system, which may serve as a blueprint for reading out the state of SCNPs in more complex scenarios.
Chemical transformations driven by light absorption are nature's choice for executing precision chemistry. In contemporary synthetic chemistry, the benefits of the application of light1 include its spatiotemporal controllability,2 or the utilization of the sun as infinite light source.3,4 Particularly, reactions which are stimulated within the visible light regime (400–800 nm) exhibit enhanced penetration depths into soft matter materials at low energies. Therefore, such reactions are applicable in the presence of sensitive structural motifs such as those contained in biomolecules. Specifically, pyrene has proven to be a valuable chromophore for modular light-induced reactions without the need of external additives or laser systems.3 On the one hand, in polymer chemistry highly efficient ligation techniques triggered by light are advantageous for the design of complex macromolecular architectures including block copolymers5,6 and star-shaped polymeric species.7 On the other hand, photoinitiators (e.g., benzoin derivatives)8,9 play a vital role in lithography10,11 and coatings as important industrial application.12 Particularly, O-acyl oximes (or oxime esters) present versatile chemical motifs since they allow N-heterocycle formation via metal catalysis,13 or light-induced homolytic scission of the N–O bond followed by CO2 release (refer to Scheme 1). Their initiation characteristics were in-depth investigated,14–16 and can be readily tuned by suitable substitution or coinitiators.17,18 For instance, poly(methyl methacrylate) species with pendant O-methacryloyl oxime units either undergo chain scission without suitable reaction partners, or initiate graft polymerization in the presence of monomers as a photochemical macroinitiator.19 In contrast, the irradiation of the same photoreactive moiety incorporated into a polystyrene backbone predominantly affords unsaturated alkene groups along the polymer chain.20 However, radicals derived from O-acryloyl oxime species incline to couple forming imine units, which are prone to hydrolysis.21,22
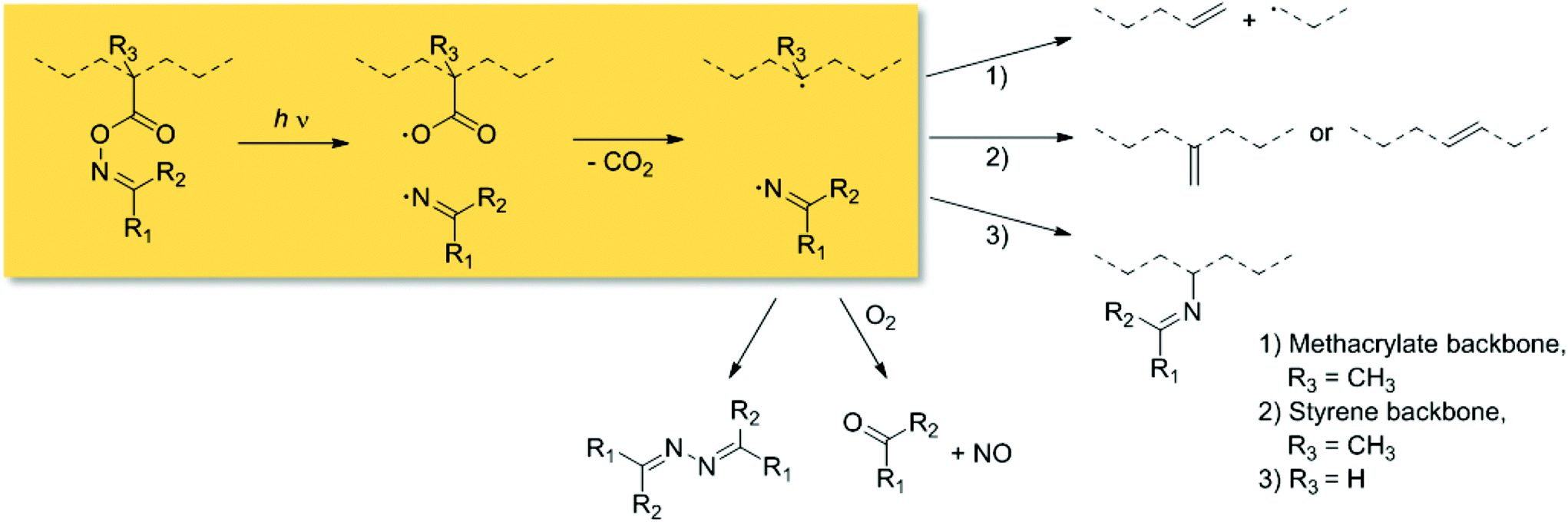 | ||
| Scheme 1 Mechanism of the light-induced fragmentation of O-acyl oxime derivatives tethered to polymeric species (with R1,R2 = hydrogen, aliphatic or aromatic substituents). | ||
In a similar fashion, the field of so-called single-chain nanoparticles (SCNPs) is a fast-growing contemporary topic in polymer chemistry and materials science,23–25 due to their potential application as protein mimic,26,27 drug carrier,28 or catalyst.29 Depending on the synthesis strategy, SCNP generation is classified into different approaches, i.e. the repeating unit approach,30–32 or the synthetically more demanding selective point folding method.33 Furthermore, SCNPs are categorized into different classes depending on the nature of the crosslinking motif. Radically induced chain collapse affords covalent bonds. Typical procedures include radical-mediated crosslinking of pendant olefinic moieties,34,35 metal-catalyzed atom transfer radical coupling of halogen species,36 and light-induced fragmentation of a bifunctional crosslinker followed by alkoxamine formation via reaction with pendant nitroxide units.36 Other photochemical pathways for chain compaction include dimerization,37,38 or the utilization of light-induced generation of highly reactive species for crosslinking.39,40
Critically, to the best of our knowledge, there exists no example of SCNPs that exploit the combination of visible light-induced chemistry and oxime ester derivatives to induce the formation of single-chain folded structures. Thus, we herein designed a photoreactive styrene derivative, which is tethered to a pyrene-substituted oxime ester (PyOHSty). Upon copolymerization with various amounts of styrene, three distinct copolymer species were obtained (refer to Table S1 in the ESI†). The irradiation of the polymer samples in highly diluted solution (c = 0.02 mg mL−1) with an LED light source (3 × 3 W, 430–435 nm, for the emission spectrum refer to Fig. S6 in the ESI†), followed by purification via precipitation in ice-cold methanol afforded the collapsed polymeric species (refer to Fig. 1), which were characterized by careful nuclear magnetic resonance (NMR) spectroscopy including diffusion ordered spectroscopy (DOSY) and size exclusion chromatography (SEC) in addition to optical spectroscopic investigations.
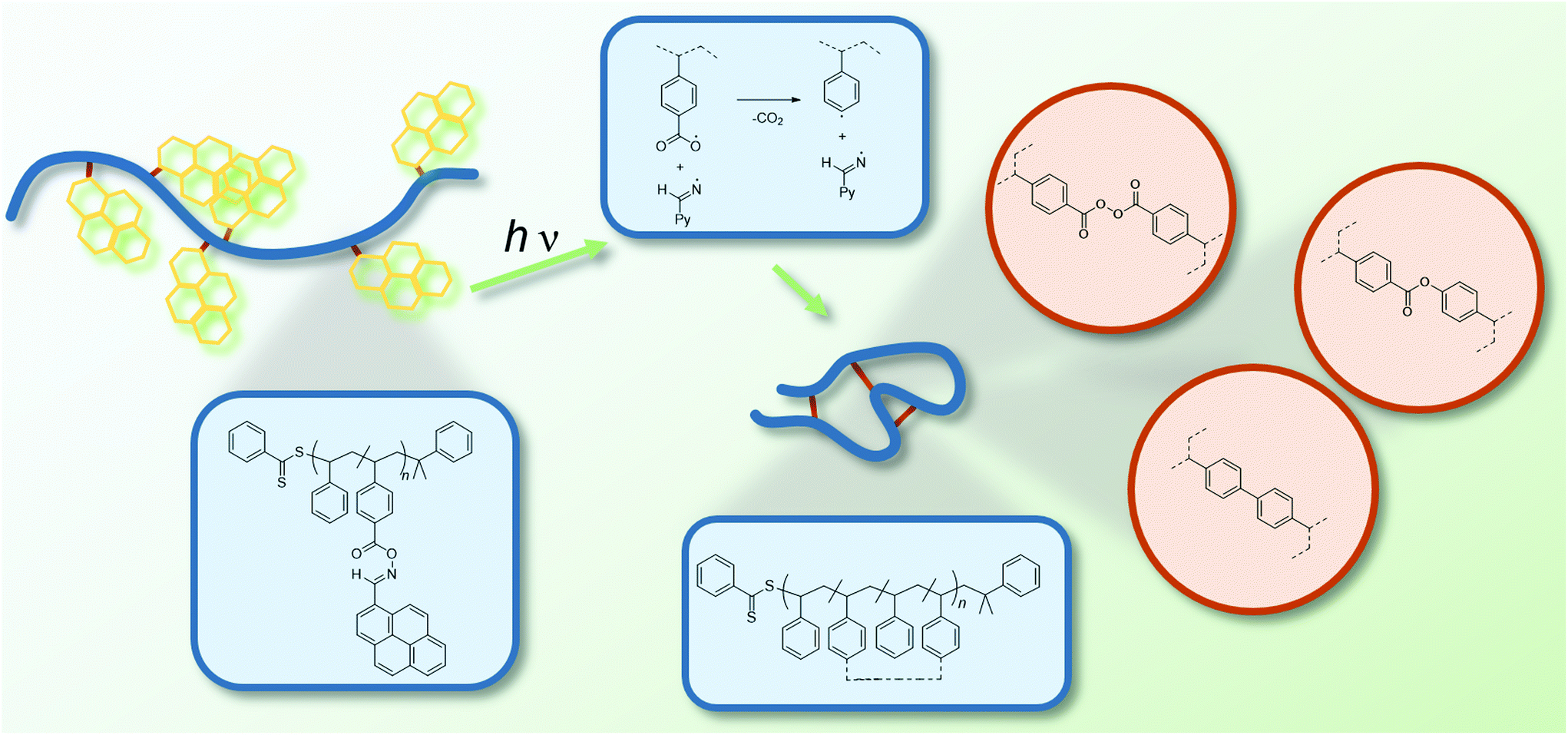 | ||
| Fig. 1 Light-induced fragmentation of pyrene-substituted oxime ester moieties along a polymer chain providing reactive radicals for chain compaction via radical coupling. The proposed crosslinked structures are envisaged in the red circles (with Py = pyrene). | ||
The photoreactive monomer was synthesized in a consecutive two-step synthesis. According to literature,41 1-pyrenecarboxaldehyde was converted into the corresponding oxime derivative followed by straightforward esterification with 4-vinylbenzoic acid. After purification via column chromatography, the targeted styrene derivative PyOHSty was obtained in very good yields (95%). The 1H NMR spectrum of PyOHSty in Fig. S1† indicates magnetic resonances of the olefin protons at 6.81, 5.93 and 5.45 ppm. Concomitantly, the aldehyde proton is visible as a singlet at 9.55 ppm. The magnetic resonances at 8.76 and 8.62 ppm, and in the region between 8.29 and 8.05 ppm are associated with the pyrenyl protons, while the phenyl protons are represented by doublets at 8.19 and 7.56 ppm. Important features of the 13C NMR spectrum in Fig. S2† are the magnetic resonances at 164.06, along at 136.13 and 117.07 ppm, which are associated with the carbon atom of the oxime unit and the carbon atoms of the double bond, respectively. In addition, high resolution electrospray ionization mass spectrometry (ESI-MS) measurements confirm the successful synthesis of PyOHSty. The absorption spectrum of PyOHSty in Fig. S3A† indicates a broad absorbance between 320 and 420 nm with two maxima at 365 and 395 nm, while fluorescence is detected in the range of 400 and 520 nm with a maximum at 409 nm in the fluorescence spectrum in Fig. S3B† (λexc. = 344 nm).
Subsequently, the light-sensitive monomer species was copolymerized with styrene in order to assess the behavior of the oxime ester units upon light exposure. Intramolecular crosslinking reactions of polymer chains cause changes in the hydrodynamic diameter as evidenced by SEC and DOSY experiments. Therefore, well-defined polymer species were prepared via RAFT copolymerization employing cumyl dithiobenzoate (CDB) as chain transfer agent (CTA). Table S1† presents the conditions and results of the copolymerization process employing variable equivalents of the pyrene monomer derivative in the presence of azobisisobutyronitrile (AIBN) as initiator at 60 °C.
Usually, the ratio of CTA to initiator should be kept low. However, higher amounts of initiator are necessary for the polymerizations since pyrene acts as a radical scavenger during the process. Nevertheless, the evaluation of the molar masses Mn,SEC(THF) based on a polystyrene calibration affords values in the range of 17 000 to 12 000 g mol−1, which are suitable to monitor changes in hydrodynamic diameter via SEC or DOSY NMR, and the polydispersity Đ lies between 1.15 and 1.22 indicating good control over the polymerization process. In addition, the SEC traces of CDB PS PyOHSty 1–3 depicted in Fig. 3 show a monomodal distribution of molar masses. The concomitant analysis of the 1H NMR spectra in Fig. 2 reveals characteristic magnetic resonances in the range of 8.80 and 7.50 ppm, stemming from the pyrene unit. Furthermore, the magnetic resonance associated with the polymer-bound aldehyde proton is visible between 9.60 and 9.20 ppm. The polystyrene backbone is detected as broad magnetic resonances between 7.40 and 6.20 ppm, and 2.30 and 1.20 ppm, which are assigned to the phenyl moieties including the aliphatic backbone, respectively.
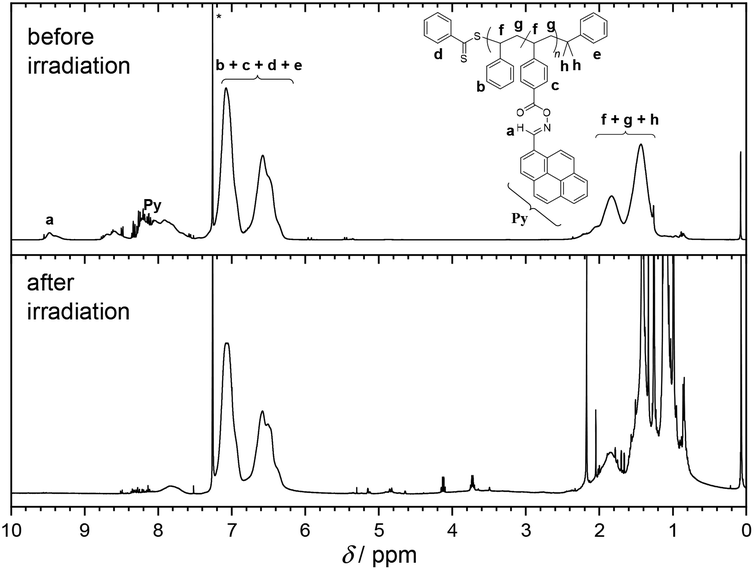 | ||
| Fig. 2 1H NMR spectrum (400 MHz, CDCl3, 298 K) of CDB PS PyOHSty 1 before and after LED irradiation (430–435 nm). The magnetic resonance at 4.12 and 3.73 ppm originate by the fragmentation of the RAFT moiety upon light exposure. The magnetic resonance marked with an asterisk is assigned to CHCl3. | ||
 | ||
| Fig. 3 SEC (THF, RI, 35 °C) traces and results of CDB PS PyOHSty 1 to 3 before and after exposure to LED irradiation (430–435 nm). Mn,SEC(THF) was calculated by employing a polystyrene calibration. The number of photoreactive units was obtained via1H NMR analysis of the block copolymers prior to light exposure. | ||
With the successful synthesis of the copolymer species, the properties of CDB PS PyOHSty 1–3 after the light irradiation were investigated. Based on the UV/Vis spectrum of the pyrene-substituted monomer in Fig. S3A† and recent investigations on the reactivity of photosensitive moieties,15,42 an LED setup (430–435 nm, 3 × 3 W, refer to Fig. S6† for the emission spectrum) was selected as light source. The copolymer species were dissolved in DCM under highly diluted conditions (c = 0.02 mg mL−1), and the solution was deoxygenated via purging with argon. After the irradiation of the dissolved polymeric species for 2.5 h while continuous stirring the solution, the solvent was removed under reduced pressure, and CDB PS PyOHSty 1–3 were purified via precipitation in ice-cold methanol.
The analysis of the 1H NMR spectra in Fig. 2 and S4† of the irradiated polymeric species indicates the disappearance of the magnetic resonances associated with the pyrene chromophore. In contrast, the magnetic resonances associated with the polystyrene backbone are still detectable between 7.40 and 6.20 ppm, and 2.30 and 1.20 ppm. As a result, upon cleavage of the oxime moiety, no recombination between the iminyl radical and the carboxyl or phenyl species is evident. Concomitantly, no magnetic resonances associated with unsaturated groups along the polymer backbone are detected. Therefore, NMR analysis suggests no H-abstraction and subsequent olefin formation as main process after the irradiation of the oxime ester units.
The SEC analysis of CDB PS PyOHSty 1 and 2 in Fig. 3 display a shift to higher retention, and therefore lower apparent molar mass compared to their pristine polymer species. Mn,SEC(THF) is reduced by 9000 and 5000 g mol−1 for CDB PS PyOHSty 1 and 2, which corresponds to a reduction of 50 and 40%, respectively. Higher retention times in SEC analysis are translated into lower hydrodynamic radii, which are caused by lower molecular weights (due to the loss of pyrene units) or intramolecular crosslinking of polymer chains. Consequently, the irradiation of CDB PS PyOHSty 1 and 2 either results in chain scission or intramolecular coupling of phenyl or carboxyl radicals. In contrast, the SEC trace of CDB PS PyOHSty 3 is virtually unaltered compared to the SEC trace of the parent polymer. The presence of only approximately two photolabile units per chain explains the low probability of CDB PS PyOHSty 3 to form a collapsed chain upon light-induced radical formation. Ring formation is still possible, yet not observed. Since the SEC trace of CDB PS PyOHSty 3 does not display polymeric species at higher retention times, no chain scission of the copolymers takes place caused by radicals. As a result, chain scission can be excluded as main process for CDB PS PyOHSty 1 and 2. However, carboxyl units, which are most probably formed via H-abstraction as side reaction, interact with the material of the SEC columns resulting in broadening of the SEC traces. Further confirmation of the chain collapse is provided by DOSY NMR measurements. Applying the Stokes–Einstein equation (refer to section B.6. in the ESI†), a hydrodynamic diameter of 4.3 nm for CDB PS PyOHSty 2 in CDCl3 is determined. After irradiation, the value decreases by 30% to 2.9 nm. The DOSY data is supported by the DOSY NMR fit data in Fig. S7 and S8,† and by the good agreement of the diffusion coefficients with each other (refer to Table S2†).
The proposed structural units formed during chain collapse are displayed in Fig. 1. According to the fragmentation of the oxime esters illustrated in Scheme 1, the coupling of two phenyl and carboxyl radicals affords a biphenyl and a peroxyanhydride motif as crosslinking unit. The reaction between the aforehand mentioned radicals generates a phenyl ester bond.
Importantly, the herein established radical formation process is self-reporting via luminescence, i.e., the folded state is quenched, while the luminescence of the exited states in the folded polymers does luminesce in the open chain. The UV/Vis and fluorescence spectra are collated in Fig. 4A and C. The non-folded initial polymer species CDB PS PyOHSty 1 and 2 display an absorbance between 320 and 420 nm with two maxima at 365 and 395 nm stemming from the pyrene unit, in a similar manner as for the corresponding pyrene monomer. Therefore, the intensity of the absorbance is lower for CDB PS PyOHSty 2 due to a lower content of the chromophore. However, after irradiation and purification, the optical properties caused by the presence of pyrene are not detectable anymore. Instead, merely a weak absorbance for the phenyl groups of the polystyrene backbone is visible between 250 and 300 nm. Further fluorescence spectra in Fig. 4B and D indicate characteristic monomer (between 375 and 460 nm) and excimer fluorescence (between 460 and 675 nm) of pyrene units upon irradiation with λexc. = 344 nm. Due to a higher loading of the chromophore, the excimer fluorescence is more pronounced for CDB PS PyOHSty 1. After the chain compaction induced by light irradiation, the intensity of the fluorescence is significantly reduced (refer to dashed line in Fig. 4B and D). The combined results of the UV/Vis and fluorescence spectroscopy support the light-induced fragmentation of the oxime ester units releasing pyrene, and exclude the recombination of iminyl radicals and the polymer backbone.
 | ||
| Fig. 4 UV Vis (A and C) and fluorescence spectra (B and D, λexc. = 344 nm) of CDB PS PyOHSty 1 and 2 before and after exposure to LED irradiation (430–435 nm) showing the presence of pyrene monomers and excimers before light-induced cleavage. The fluorescence of pyrene can also be detected via exposure of the NMR samples to a simple hand-held UV lamp (λexc. = 366 nm, part D, left: parent polymer, right: after LED light-induced cleavage). | ||
In summary, we introduce a mild light induced methodology for single chain collapse based on radical species by employing visible light activation within the range of 430 nm. Specifically, a novel photoreactive monomer with a pyrene-substituted oxime ester moiety was designed and copolymerized with different amounts of the photoreactive unit. Subsequently, this polymer scaffold, which critically expands the field of visible light-sensing self-reporting systems, was collapsed exclusively to SCNPs in highly mildest and efficient way leading to a substantial reduction in size with regard to the decrease in hydrodynamic volume and required reactive unit. The formation of the SCNPs was carefully confirmed using spectroscopic techniques such as 1H and DOSY-NMR, UV/Vis and fluorescence spectroscopy as well as SEC. Importantly, we submit that due to their fluorescent features fused with the use of visible light, the photoreactive pyrene-substituted oxime ester moiety presents a valuable novel visible light reactive compound, which further has the potential to be examined, for example, as polymerization initiator, radical catalyst or handle for network formation.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
C. B.-K. acknowledges continued support from the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz BioInterfaces in Technology and Medicine (BIFTM) and Science and Technology of Nanosystems (STN) programs as well as from the Queensland University of Technology (QUT) and the Australian Research Council (ARC) in the form of a Laureate Fellowship enabling his photo-chemical research program. J. T. O.'s PhD studies are funded by the Fonds der Chemischen Industrie (FCI) as well as the Karlsruhe Institute of Technology (KIT).References
- A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1–6 RSC.
- G. Delaittre, A. S. Goldmann, J. O. Mueller and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2015, 54, 11388–11403 CrossRef CAS PubMed.
- J. T. Offenloch, M. Gernhardt, J. P. Blinco, H. Frisch, H. Mutlu and C. Barner-Kowollik, Chem. – Eur. J., 2019, 25, 3700–3709 CrossRef CAS PubMed.
- J. O. Mueller, N. K. Guimard, K. K. Oehlenschlaeger, F. G. Schmidt and C. Barner-Kowollik, Polym. Chem., 2014, 5, 1447–1456 RSC.
- J. T. Offenloch, S. Norsic, H. Mutlu, M. Taam, O. Boyron, C. Boisson, F. D'Agosto and C. Barner-Kowollik, Polym. Chem., 2018, 9, 3633–3637 RSC.
- Y. Yagci and W. Schnabel, Prog. Polym. Sci., 1990, 15, 551–601 CrossRef CAS.
- K. Hiltebrandt, M. Kaupp, E. Molle, J. P. Menzel, J. P. Blinco and C. Barner-Kowollik, Chem. Commun., 2016, 52, 9426–9429 RSC.
- C. Barner-Kowollik, F. Günzler and T. Junkers, Macromolecules, 2008, 41, 8971–8973 CrossRef CAS.
- D. S. Esen, N. Arsu, J. P. Da Silva, S. Jockusch and N. J. Turro, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1865–1871 CrossRef CAS.
- E. C. Hagberg, M. Malkoch, Y. Ling, C. J. Hawker and K. R. Carter, Nano Lett., 2007, 7, 233–237 CrossRef CAS PubMed.
- E. Blasco, M. Wegener and C. Barner-Kowollik, Adv. Mater., 2017, 29, 1604005 CrossRef PubMed.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- H. Huang, J. Cai and G. J. Deng, Org. Biomol. Chem., 2016, 14, 1519–1530 RSC.
- J. Xu, G. Ma, K. Wang, J. Gu, S. Jiang and J. Nie, J. Appl. Polym. Sci., 2011, 123, 725–731 CrossRef.
- D. E. Fast, A. Lauer, J. P. Menzel, A. M. Kelterer, G. Gescheidt and C. Barner-Kowollik, Macromolecules, 2017, 50, 1815–1823 CrossRef CAS.
- T. Imamoglu, A. Onen and Y. Yagci, Angew. Makromol. Chem., 1995, 224, 145–151 CrossRef CAS.
- C. Dworak and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5865–5871 CrossRef CAS.
- M. Makino, K. Uenishi and T. Tsuchimura, J. Photopolym. Sci. Technol., 2018, 31, 37–44 CrossRef CAS.
- G. A. Delzenne, U. Laridon and H. Peeters, Eur. Polym. J., 1970, 6, 933–943 CrossRef CAS.
- K. Suyama and M. Tsunooka, Polym. Degrad. Stab., 1994, 45, 409–413 CrossRef CAS.
- K. Song, M. Tsunooka and M. Tanaka, Makromol. Chem., Rapid Commun., 1988, 524, 519–524 CrossRef.
- K. H. Song, A. Urano, M. Tsunooka and M. Tanaka, J. Polym. Sci., Part C: Polym. Lett., 1987, 25, 417–421 CrossRef CAS.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef CAS PubMed.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef CAS.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- J. P. Cole, A. M. Hanlon, K. J. Rodriguez and E. B. Berda, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 191–206 CrossRef CAS.
- A. Sanchez-Sanchez, S. Akbari, A. Etxeberria, A. Arbe, U. Gasser, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 491–495 CrossRef CAS.
- A. P. P. Kröger and J. M. J. Paulusse, J. Controlled Release, 2018, 286, 326–347 CrossRef PubMed.
- H. Rothfuss, N. D. Knöfel, P. W. Roesky and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 5875–5881 CrossRef CAS.
- E. Harth, B. Van Horn, V. Y. Lee, D. S. Germack, C. P. Gonzales, R. D. Miller and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 8653–8660 CrossRef CAS.
- D. E. Whitaker, C. S. Mahon and D. A. Fulton, Angew. Chem., Int. Ed., 2013, 52, 956–959 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, I. Asenjo-Sanz, L. Buruaga and J. A. Pomposo, Macromol. Rapid Commun., 2012, 33, 1262–1267 CrossRef CAS.
- O. Altintas, E. Lejeune, P. Gerstel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 640–651 RSC.
- J. Jiang and S. Thayumanavan, Macromolecules, 2005, 38, 5886–5891 CrossRef CAS.
- D. Mecerreyes, V. Lee, C. J. Hawker, J. L. Hedrick, A. Wursch, W. Volksen, T. Magbitang, E. Huang and R. D. Miller, Adv. Mater., 2001, 13, 204–208 CrossRef CAS.
- R. Chen, J. G. Dickinson, K. J. Rodriguez, A. M. Hanlon, E. B. Berda, C. Willis and M. Cashman, Macromolecules, 2017, 50, 2996–3003 CrossRef.
- H. Frisch, J. P. Menzel, F. R. Bloesser, D. E. Marschner, K. Mundsinger and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 9551–9557 CrossRef CAS PubMed.
- P. G. Frank, B. T. Tuten, A. Prasher, D. Chao and E. B. Berda, Macromol. Rapid Commun., 2014, 35, 249–253 CrossRef CAS PubMed.
- J. T. Offenloch, J. Willenbacher, P. Tzvetkova, C. Heiler, H. Mutlu and C. Barner-Kowollik, Chem. Commun., 2017, 53, 775–778 RSC.
- C. Heiler, S. Bastian, P. Lederhose, J. P. Blinco, E. Blasco and C. Barner-Kowollik, Chem. Commun., 2018, 54, 3476–3479 RSC.
- J. T. Offenloch, S. Bastian, H. Mutlu and C. Barner-Kowollik, ChemPhotoChem, 2019, 3, 66–70 CAS.
- J. P. Menzel, B. B. Noble, A. Lauer, M. L. Coote, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2017, 139, 15812–15820 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py00834a |
| This journal is © The Royal Society of Chemistry 2019 |