
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microscale synthesis of multiblock copolymers using ultrafast RAFT polymerisation†
Joji
Tanaka
 *a,
Pratik
Gurnani
a,
Alexander B.
Cook
a,
Satu
Häkkinen
a,
Junliang
Zhang
a,
Jie
Yang
a,
Andrew
Kerr
a,
David M.
Haddleton
a,
Sébastien
Perrier
*ab and
Paul
Wilson
*ab
*a,
Pratik
Gurnani
a,
Alexander B.
Cook
a,
Satu
Häkkinen
a,
Junliang
Zhang
a,
Jie
Yang
a,
Andrew
Kerr
a,
David M.
Haddleton
a,
Sébastien
Perrier
*ab and
Paul
Wilson
*ab
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, CV4 7AL Coventry, UK. E-mail: j.tanaka@warwick.ac.uk
bMonash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), 399 Royal Parade, Parkville, Victoria 3152, Australia
First published on 7th February 2019
Abstract
We demonstrate that ultrafast RAFT in the presence of air can be scaled down to 2 μL with good control using microvolume insert vials as the polymerisation vessel. By careful cooling and mixing of the sequential monomers, well-defined pentablock copolymers were generated with a final volume of 10 μL.
High-throughput screening is becoming increasingly prevalent industrially and academically in identifying pharmaceutical targets and in optimising synthetic routes.1 Consequently, scaling down reactions is of paramount importance for exploring enormous numbers of possible permutations of parameters involved in a chemical synthesis.2 Polymerisation in standard chemistry laboratory reaction vessels becomes increasingly difficult at smaller scales. The transfer of advanced polymer synthesis techniques to smaller scales will allow for the high-throughput screening of polymer compositions for biomaterial discovery, and has been previously exploited in step-growth polymerisations.3,4 However, small scale screening using controlled polymerisation methods was only recently achieved by Boyer et al. in the investigation of the influence of polymer architecture on material properties.5,6
Polymerisations are typically carried out with reaction volumes between 50 mL and 0.5 mL.7–15 These ranges are practical for the conventional reaction vessels and deoxygenation processes necessary for typical Reversible Deactivation Radical Polymerisation (RDRP). Note that the latter condition limits scales of the reactions, because using nitrogen sparging or freeze–pump–thaw cycles to deoxygenate the reaction media is not practical at ultralow volumes, due to the inherent loss of volatile monomers and solvents. Hence, oxygen tolerant RDRP protocols are necessary to allow polymerisations to be carried out at the microscale. To this end, Boyer et al. have performed ultralow volume reactions (20 μL) in 96 well plates, using photo-catalysed redox Reversible Addition–Fragmentation Chain Transfer (RAFT) polymerisation without deoxygenation in the presence of air.16 This enabled screening of different homopolymers, diblock copolymers, star architectures and nanoparticle formulations.
RDRP protocols without deoxygenation have become an emerging topic;17,18 however, many of these protocols require external stimuli,19 additives16 or oxygen scavenging enzymes,5,20,21 resulting in the deviation from the simplicity of RDRP protocols. To address this, Gody et al. demonstrated standard RAFT polymerisation using only conventional ingredients without deoxygenation, in vessels open to air.22 This elegant and simple approach takes advantage of the fast propagation of acrylamidic monomers in water, a solvent known to increase the rate of radical polymerisation, which is further accelerated at elevated temperatures. This ultrafast RAFT polymerisation was generally demonstrated with acrylamide-based monomers with 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) as the initiator (10 h t1/2 = 44 °C) and at 100 °C. This allowed the synthesis of multiblock copolymers (MBCPs) through iterative chain extensions where full monomer conversion was achieved within 3 minutes per block, before the initiator was fully decomposed (approximately 80%).22
MBCPs are macromolecules with defined control over the block sequence that can be synthesised from just simple chemical ingredients without complex biological machineries, and are amenable for industrial scales.23–25 The synthesis of MBCPs has progressed more recently with RDRP, using copper-mediated polymerisation26–34 and RAFT polymerisation.35–41 In spite of the inevitable small number of radical termination events, 21 iterative block extensions have been reliably demonstrated with RDRP.42 Furthermore, these routes are popular as they allow the incorporation of monomers of various functional groups43–48 and do not require immaculately dry reagents and environments, necessary for ionic living polymerisation systems.49 Recently, work on sulfur-free RAFT polymerisation has offered the potential for MBCP synthesis amenable for industrial scales.42,50,51 However, the possible benefits of scaling down MBCP synthesis are often overlooked in academic settings. Industries often rely on the inexpensive small scale combinatorial reactions for optimisation before larger scale synthesis. Microscale MBCP synthesis could have applicability in high-throughput microarrays,2 thus allowing the rapid investigation of permutations of monomers and block lengths of MBCPs.
We postulate that the aforementioned ultrafast RAFT protocol could be applicable in the microliter scale due to the rapid consumption of the monomer without deoxygenation; thus demonstrating the synthesis of MBCPs at a microscale suitable for potential applications such as microarray patterning and combinatorial chemistry with only conventional ingredients used for RAFT polymerisation.
To counteract the inherent problem of the increased air/water interface when scaling down the protocol proposed by Gody et al., we used narrow micro-volume glass inserts (4.6 mm diameter, 200 μL capacity) with conical bottoms (cone volume = approximately 20 μL) that are typically fitted into standard 2 mL vials for low volume HPLC/GPC analysis.52 A master mix of the RAFT agent, monomer, solvent and initiator was made as an “all-in-one” stock solution and added into the insert using a standard micropipette (Scheme 1). This mix was made following the published protocol,22 using VA-044 as the initiator (3 × 10−3 M, [CTA]/[I]0 = 40), N-acryloylmorpholine (NAM) as a suitable acrylamidic monomer ([M]0 = 3 M) in an aqueous mixture and 2-(((butylthio)-carbonothioyl)thio)propanoic acid (PABTC) as the RAFT agent. The inserts were then heated in an oil bath at 100 °C for 3 minutes. In contrast to the previous study where the increase in reaction temperature was gradual, taking 80 seconds to reach 96 °C,22 we assume the temperature of the reaction to reach equilibrium almost immediately. Conveniently, as the polymerisations were carried out in SEC insert vials, the reaction mixture could be directly diluted with the SEC eluent within the insert, and injected directly for SEC analysis (Fig. S2†). A duplicate reaction was carried out to dilute with NMR solvent (DMSO-d6) to measure the monomer conversion by NMR.
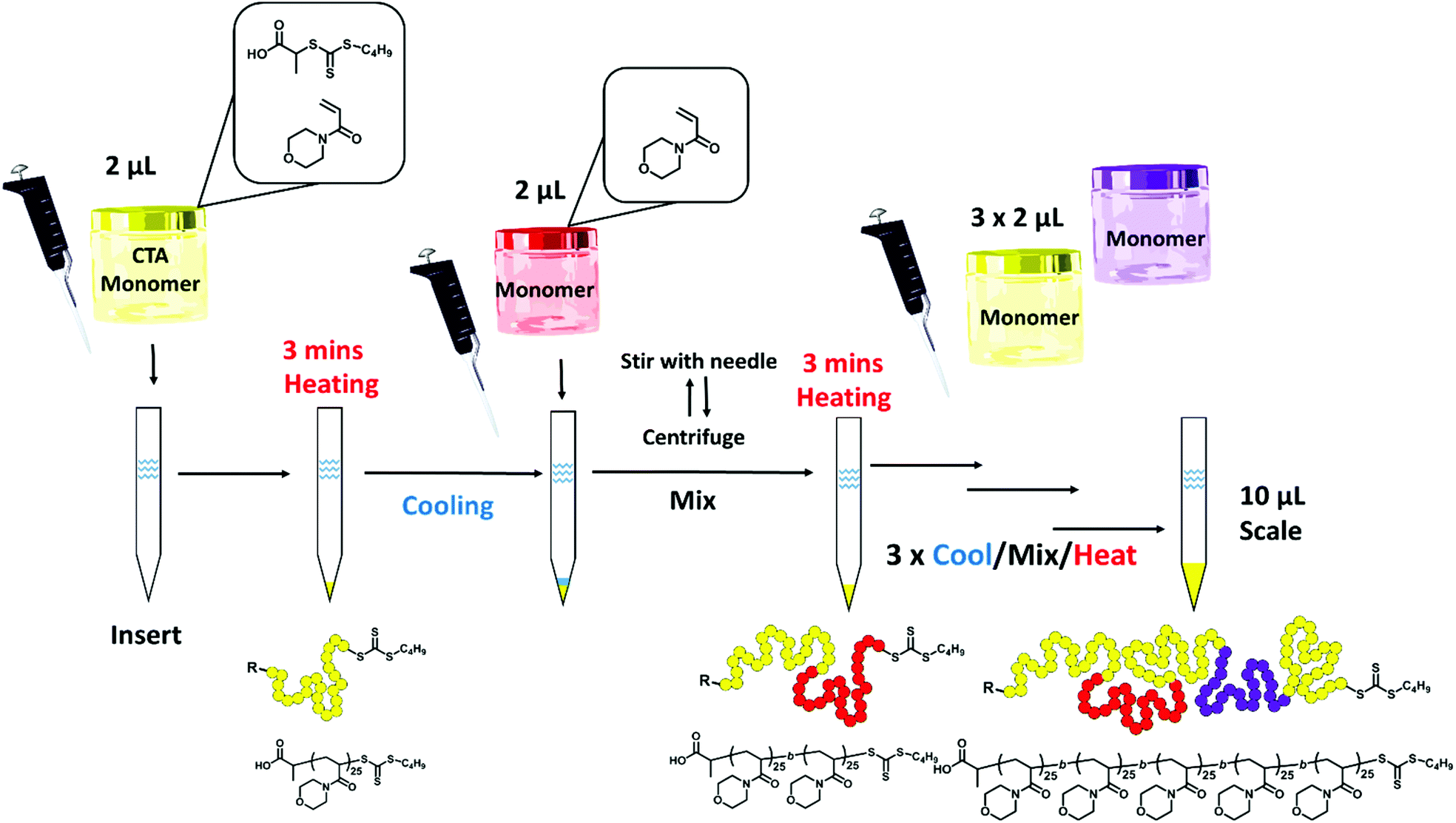 | ||
| Scheme 1 General scheme: “master mix” (with a monomer, CTA, initiator and solvent) was added into the microvolume insert using a regular air displacement micropipette. After 3 min of heating at 100 °C in an oil bath, polymerisation was complete and the insert was cooled with liquid nitrogen. For sequential chain extension, a separate monomer master mix was directly added and mixed by stirring with a needle and centrifugation, before reheating for further 3 minutes for block extension. This cycle was repeated to yield a pentablock copolymer. All the polymerisations were carried out without deoxygenation and in the presence of open air. | ||
Preliminary experiments were designed to investigate the absolute limit of scale for the polymerisation. Initially this was investigated with a targeted degree of polymerisation (DP) of 25 using 20 vol% dioxane in water to aid the solubility of the CTA. The experimental number-average molar mass (Mn,SEC) and dispersity (Đ) were measured using SEC analysis to investigate the control maintained at lower volumes. At 10 μL (Mn,SEC = 2200 g mol−1; Đ = 1.23), 5 μL (Mn,SEC = 2600 g mol−1; Đ = 1.23), and 2 μL (Mn,SEC = 2600 g mol−1; Đ = 1.29), we were able to reproducibly obtain PNAM25 as observed by SEC analysis (Fig. S1†) with only a slight increase in dispersity, nevertheless they maintained good control comparable to the polymerisation carried out in a 5.4 mL test tube (termed macroscale in this paper) (Mn,SEC = 2700 g mol−1; Đ = 1.19). At 1 μL scale polymers were obtained (Mn,SEC = 2600 g mol−1); however, the Đ increased to 1.42. The weight loss due to the evaporation of the reaction mixture was also noted (Table S1†), which seemingly increased at lower scales and may have been a contributing factor to the loss of control at the 1 μL scale. Hence we concluded that 2 μL is the lowest scale to maintain good control. Our next objective was to apply this protocol to longer polymer chain lengths of PNAMn (Fig. 1). Increasing the chain length fourfold (DP = 100) required a slight modification of the master mix (10 vol% dioxane in water; [I]0 = 1 × 10−3 M, [CTA]/[I]0 = 30). Pleasingly, polymerisation yielded PNAM100 at the 2 μL scale (Mn,SEC = 9200 g mol−1; Đ = 1.36), and in contrast to the macroscale its molecular weight distribution was relatively broader as revealed by SEC analysis (Mn,SEC = 8900 g mol−1; Đ = 1.19). Increasing the length further (DP = 200), increased the dispersity at the 2 μL scale (PNAM200, Mn,SEC = 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1; Đ = 1.43), compared to the macroscale equivalent (Mn,SEC = 15000 g mol−1; Đ = 1.23). SEC analysis in all cases revealed a slightly higher dispersity due to the appearance of low molecular tailing. Also the 1H NMR spectra revealed that more residual monomer was present (approximately 2–3% more). As the targeting DP of 25 of NAM yielded a relatively narrow dispersity at the 2 μL scale, we therefore decided to keep this a constant block length for our MBCPs. In order to generate MBCPs through iterative chain extension with the current protocol, it was important to consider the limitation of mixing sequential monomers in the polymerisation mixture, as stirring during polymerisation is unfeasible at the 2 μL scale. To maximise the mixing of each monomer aliquot the polymerisation reaction mixture was cooled prior to the addition of a new monomer and stirred before heating again at 100 °C for a successful sequential chain extension. This circumvented the need for continual stirring during the addition of sequential monomers. Thus by adopting this necessary measure of cooling and mixing before reheating (Scheme 1), we were able to successfully demonstrate successive chain extensions within the insert vials to synthesize a homopolymer in five successive chain extensions, P(NAM25)5, at 5 μL per block (final Mn,SEC = 10600 g mol−1; Đ = 1.25) and 2 μL per block (final Mn,SEC = 10000 g mol−1; Đ = 1.35) (Fig. 2). Centrifugation was necessary to collect the new monomer solution at the bottom of the insert before stirring (see the ESI† for full details). It is important to note that the monomer concentration of the chain extension stock solution was kept constant at 3 M, such that the same DP per chain extension could be targeted by sequentially adding the same volume as the original block. It is noteworthy that all the chain extension stock solutions had contained the same initiator concentration of 2.2 × 10–3 M. This was designed to give a constant overall macroCTA/initiator ratio of 40 per block, whilst assuming that 20% of the initiator is still remaining from the previous block. This gave a good balance of quantitative monomer consumption (>96%, Table 1) at each block whilst keeping the theoretical livingness of each block high (>98%), thus minimising the number of dead chains being formed (see the ESI for calculation and S6–S10† for detailed experimental conditions).
800 g mol−1; Đ = 1.43), compared to the macroscale equivalent (Mn,SEC = 15000 g mol−1; Đ = 1.23). SEC analysis in all cases revealed a slightly higher dispersity due to the appearance of low molecular tailing. Also the 1H NMR spectra revealed that more residual monomer was present (approximately 2–3% more). As the targeting DP of 25 of NAM yielded a relatively narrow dispersity at the 2 μL scale, we therefore decided to keep this a constant block length for our MBCPs. In order to generate MBCPs through iterative chain extension with the current protocol, it was important to consider the limitation of mixing sequential monomers in the polymerisation mixture, as stirring during polymerisation is unfeasible at the 2 μL scale. To maximise the mixing of each monomer aliquot the polymerisation reaction mixture was cooled prior to the addition of a new monomer and stirred before heating again at 100 °C for a successful sequential chain extension. This circumvented the need for continual stirring during the addition of sequential monomers. Thus by adopting this necessary measure of cooling and mixing before reheating (Scheme 1), we were able to successfully demonstrate successive chain extensions within the insert vials to synthesize a homopolymer in five successive chain extensions, P(NAM25)5, at 5 μL per block (final Mn,SEC = 10600 g mol−1; Đ = 1.25) and 2 μL per block (final Mn,SEC = 10000 g mol−1; Đ = 1.35) (Fig. 2). Centrifugation was necessary to collect the new monomer solution at the bottom of the insert before stirring (see the ESI† for full details). It is important to note that the monomer concentration of the chain extension stock solution was kept constant at 3 M, such that the same DP per chain extension could be targeted by sequentially adding the same volume as the original block. It is noteworthy that all the chain extension stock solutions had contained the same initiator concentration of 2.2 × 10–3 M. This was designed to give a constant overall macroCTA/initiator ratio of 40 per block, whilst assuming that 20% of the initiator is still remaining from the previous block. This gave a good balance of quantitative monomer consumption (>96%, Table 1) at each block whilst keeping the theoretical livingness of each block high (>98%), thus minimising the number of dead chains being formed (see the ESI for calculation and S6–S10† for detailed experimental conditions).
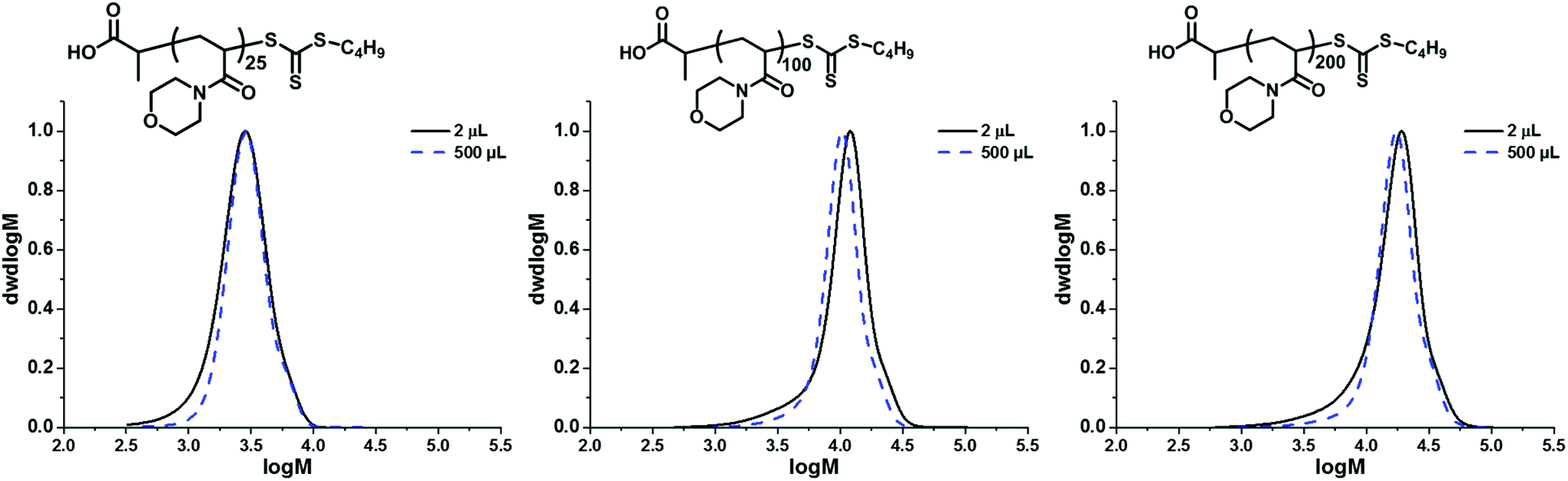 | ||
| Fig. 1 SEC analysis (dRI, THF) of PNAMn (n = 25, 100 and 200) prepared at the microscale (2 μL) in microvolume inserts and at the normal scale (500 μL) in conventional test tubes (5.4 ml). All the polymerisations were carried out in 3 minutes open to air without stirring and deoxygenation. PMMA was used as the standard for SEC analysis. | ||
 | ||
| Fig. 2 Multichain extension to generate P(NAM25)5 with ultrafast RAFT polymerisation at different scales: Top row = 2.5 ml, 0.5 ml per block; middle row = 25 μL, 5 μL per block; bottom row = 10 μL, 2 μL per block. Left column: Photograph after the reaction next to a British penny coin (20.3 mm in diameter) as a reference to the size of the scale. Middle column: SEC chromatograms for successive chain extensions. Right column: Evolution of number-average molar masses and dispersity values with the succession of chain extensions during the preparation of P(NAM25)5. The black line represents the theoretical molar mass calculated using eqn (2) (see the ESI†). The filled squares represent the experimental molar masses and empty squares represent the dispersity values, both determined by SEC in THF with PMMA standards. | ||
| Polymer | Scalea (μL) | Conv.b % |
M
n,thc (g mol−1) |
M
n,SECd (g mol−1) |
Đ |
|---|---|---|---|---|---|
| a All polymerisations were carried out in insert vials unless the scale is above or equal to 500 μL in which case they were carried out in a test tube (5.4 mL). b Determined by 1H-NMR (DMSO-d6) and calculated using eqn (1) in the ESI. c As calculated from eqn (2) in the ESI. d Determined by SEC in THF with PMMA standards, unless stated otherwise. e Determined by SEC in DMF with PMMA standards. | |||||
| PNAM25 | 500 | >97 | 3600 | 2700 | 1.18 |
| 10 | >98 | 3600 | 2200 | 1.23 | |
| 5 | >98 | 3600 | 2600 | 1.23 | |
| 2 | >98 | 3600 | 2600 | 1.29 | |
| 1 | >96 | 3600 | 2600 | 1.42 | |
| PNAM100 | 500 | >99 | 14000 |
8900 | 1.19 |
| 2 | >97 | 13600 |
9200 | 1.36 | |
| PNAM200 | 500 | >98 | 26800 |
15000 |
1.23 |
| 2 | >98 | 26800 |
13000 |
1.43 | |
| PNAM25-b-PNAM25-b-PNAM25-b-PNAM25-b-PNAM25 | 2500 | >99.9 | 17200 |
12600 |
1.23 |
| 25 | >99 | 17200 |
10600 |
1.25 | |
| 10 | >99 | 17200 |
10000 |
1.35 | |
| PNAM25-b-PDMA25-b-PNAM25-b-PHEAm25-b-PNAM25 | 2500 | >96 | 15400 |
15100e | 1.32e |
| 10 | >98 | 15500 |
14000e | 1.35e | |
The monomer consumption was followed by 1H NMR spectroscopy and the succession of the sequential chain extension was confirmed by GPC analysis of polymerisation at each block. To analyse each block extension, the same number of replicate reactions as the number of iterative blocks was prepared, whereby the representative vessel at each stage was used as a whole for each analysis (see the ESI† for the detailed procedure). In all cases, a linear increase in Mn,SEC was observed with the increasing number of iterative block extensions (Fig. 2), suggesting excellent control in polymerisation at the microscale. In comparison the molecular weight distributions of P(NAM25)5 were only slightly broader at the microscale compared to the macroscale with our protocol (final Mn,SEC = 12600 g mol−1; Đ = 1.23). At the macroscale, bimodal distributions were observed, due to backbiting induced β-scission and subsequent branching, with successive chain extensions.53 Although this is typically a characteristic of more labile methine backbone hydrogens of acrylic monomer families, this was observed here with an acrylamide monomer as a result of high temperature. This was indeed the case in previous work manifested as a high molecular skew.22 We suspect that at the microscale (25 μL and 10 μL), this feature is still present despite molecular weight distributions appearing to be unimodal, due to the broadening of the molecular weight distribution. We attribute this as a result of the increased interface between the air and solution phase when scaling down, leading to an increased viscosity and uneven reaction mixture within the reaction vessel from evaporation as well as increased oxygen related termination events being more prevalent. It is noteworthy that the relative weight loss for each chain extension was considerably greater at the microscale compared to the conventional scale (Table S2†).
To further demonstrate the robustness of this method, MBCPs were synthesized with blocks of different monomers: N,N-dimethylacrylamide (DMA) and N-hydroxyethylacrylamide (HEAm). Pentablocks of PNAM25-b-PDMA25-b-PNAM25-b-PHEAm25-b-PNAM25 were prepared in the inserts at the microliter scale (2 μL per block) following our protocol, with a well-defined molecular weight distribution (Mn,SEC = 14000 g mol−1; Đ = 1.35) which was comparable to the macroscale synthesis (Mn,SEC = 15100 g mol−1; Đ = 1.32). Note that switching the SEC eluent to DMF was necessary to aid the solubility of the HEAm incorporated pentablock. The resulting Mn,SEC showed better agreement with the theoretical number-average molar mass (Mn,th) owing to a better comparison with the PMMA calibrant in DMF (Fig. S4 and S5†).
Conclusions
To conclude, we have demonstrated the downscaling of the robust, oxygen-tolerant ultrafast RAFT polymerisation method, suitable for acrylamidic monomers in water in microvolume insert vials. Increased evaporation of the reaction mixture was observed with an increasing surface-to-volume ratio of the reaction mixture. This set a practical lower limit; however, we found that good control was still maintained at the 2 μl scale. The resulting polymer was successfully chain extended by sequential monomer additions. The reported method enables the synthesis of pentablock copolymers using only a final volume of 10 μl of the reaction mixture, thus demonstrating the high potential of ultrafast RAFT as a high-throughput screening method for MBCPs. Further studies are being carried out to investigate the robustness of the protocol in the synthesis of complex architectures and using different solvent systems, as well as its applicability to biological sciences.Conflicts of interest
The authors declare no conflict interest.Acknowledgements
The authors gratefully acknowledge financial support from the Engineering and Physical Sciences Research Council (EPSRC) under grant EP/F500378/1 through the Molecular Organisation and Assembly in Cells Doctoral Training Centre (MOAC-DTC). S. H. acknowledges Lubrizol for financial support. A. B. C. acknowledges Syngenta for funding. The authors also wish to acknowledge the facilities and personnel (S. P., P. W.) enabled by the Monash–Warwick Alliance. S. P. acknowledges a Royal Society Wolfson Merit Award (WM130055). P. W. thanks the Leverhulme Trust for the award of an Early Career Fellowship (ECF/2015-075). The authors would like to thank Dr Daniel Lester and the Polymer Characterisation Research Technology Platform for GPC facilities.Notes and references
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188 CrossRef CAS PubMed.
- N. J. Gesmundo, B. Sauvagnat, P. J. Curran, M. P. Richards, C. L. Andrews, P. J. Dandliker and T. Cernak, Nature, 2018, 557, 228–232 CrossRef CAS PubMed.
- D. G. Anderson, S. Levenberg and R. Langer, Nat. Biotechnol., 2004, 22, 863 CrossRef CAS PubMed.
- J. J. Green, R. Langer and D. G. Anderson, Acc. Chem. Res., 2008, 41, 749–759 CrossRef CAS PubMed.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Angew. Chem., Int. Ed., 2016, 55, 4500–4503 CrossRef CAS PubMed.
- G. Ng, J. Yeow, R. Chapman, N. Isahak, E. Wolvetang, J. J. Cooper-White and C. Boyer, Macromolecules, 2018, 51, 7600–7607 CrossRef CAS.
- G. Moriceau, G. Gody, M. Hartlieb, J. Winn, H. Kim, A. Mastrangelo, T. Smith and S. Perrier, Polym. Chem., 2017, 8, 4152–4161 RSC.
- J. Tanaka, S. Tani, R. Peltier, E. H. Pilkington, A. Kerr, T. P. Davis and P. Wilson, Polym. Chem., 2018, 9, 1551–1556 RSC.
- J. Tanaka, A. S. Gleinich, Q. Zhang, R. Whitfield, K. Kempe, D. M. Haddleton, T. P. Davis, S. Perrier, D. A. Mitchell and P. Wilson, Biomacromolecules, 2017, 18, 1624–1633 CrossRef CAS PubMed.
- J. Zhang, J. Tanaka, P. Gurnani, P. Wilson, M. Hartlieb and S. Perrier, Polym. Chem., 2017, 8, 4079–4087 RSC.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2018, 1800460 Search PubMed.
- A. M. Lunn and S. Perrier, Macromol. Rapid Commun., 2018, 39, 1800122 CrossRef PubMed.
- A. B. Cook, R. Peltier, M. Hartlieb, R. Whitfield, G. Moriceau, J. A. Burns, D. M. Haddleton and S. Perrier, Polym. Chem., 2018, 9, 4025–4035 RSC.
- P. Gurnani, C. Sanchez-Cano, K. Abraham, H. Xandri-Monje, A. B. Cook, M. Hartlieb, F. Lévi, R. Dallmann and S. Perrier, Macromol. Biosci., 2018, 1800213 CrossRef PubMed.
- P. Gurnani, A. M. Lunn and S. Perrier, Polymer, 2016, 106, 229–237 CrossRef CAS.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS PubMed.
- J. Tan, D. Liu, Y. Bai, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromolecules, 2017, 50, 5798–5806 CrossRef CAS.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- L. Zhifen, L. Yue and A. Zesheng, Angew. Chem., Int. Ed., 2017, 129, 14040–14044 CrossRef.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- M. Zamfir and J.-F. Lutz, Nat. Commun., 2012, 3, 1138 CrossRef.
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917 CrossRef CAS PubMed.
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. C. van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483–14487 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 5517–5525 CrossRef CAS.
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 6421–6432 CrossRef CAS.
- C. Waldron, Q. Zhang, Z. Li, V. Nikolaou, G. Nurumbetov, J. Godfrey, R. McHale, G. Yilmaz, R. K. Randev, M. Girault, K. McEwan, D. M. Haddleton, M. Droesbeke, A. J. Haddleton, P. Wilson, A. Simula, J. Collins, D. J. Lloyd, J. A. Burns, C. Summers, C. Houben, A. Anastasaki, M. Li, C. R. Becer, J. K. Kiviaho and N. Risangud, Polym. Chem., 2014, 5, 57 RSC.
- Q. Zhang, A. Anastasaki, G.-Z. Li, A. J. Haddleton, P. Wilson and D. M. Haddleton, Polym. Chem., 2014, 5, 3876–3883 RSC.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox and V. Percec, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- A. Kerr, M. Hartlieb, J. Sanchis, T. Smith and S. Perrier, Chem. Commun., 2017, 53, 11901–11904 RSC.
- J. Zhang, R. Deubler, M. Hartlieb, L. Martin, J. Tanaka, E. Patyukova, P. D. Topham, F. H. Schacher and S. Perrier, Macromolecules, 2017, 50, 7380–7387 CrossRef CAS.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS PubMed.
- J. Zhang, G. Gody, M. Hartlieb, S. Catrouillet, J. Moffat and S. Perrier, Macromolecules, 2016, 49, 8933–8942 CrossRef CAS.
- C. Bray, R. Peltier, H. Kim, A. Mastrangelo and S. Perrier, Polym. Chem., 2017, 8, 5513–5524 RSC.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2016, 9, 171 CrossRef PubMed.
- T. R. Barlow, J. C. Brendel and S. Perrier, Macromolecules, 2016, 49, 6203–6212 CrossRef CAS.
- C. Footman, P. A. de Jongh, J. Tanaka, R. Peltier, K. Kempe, T. P. Davis and P. Wilson, Chem. Commun., 2017, 53, 8447–8450 RSC.
- V. Nikolaou, A. Simula, M. Droesbeke, N. Risangud, A. Anastasaki, K. Kempe, P. Wilson and D. M. Haddleton, Polym. Chem., 2016, 7, 2452–2456 RSC.
- J. Collins, J. Tanaka, P. Wilson, K. Kempe, T. P. Davis, M. P. McIntosh, M. R. Whittaker and D. M. Haddleton, Bioconjugate Chem., 2015, 26, 633–638 CrossRef CAS PubMed.
- Q. Zhang, Z. Li, P. Wilson and D. M. Haddleton, Chem. Commun., 2013, 49, 6608–6610 RSC.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- D. R. Carroll, A. P. Constantinou, N. Stingelin and T. K. Georgiou, Polym. Chem., 2018, 9, 3450–3454 RSC.
- N. G. Engelis, A. Anastasaki, R. Whitfield, G. R. Jones, E. Liarou, V. Nikolaou, G. Nurumbetov and D. M. Haddleton, Macromolecules, 2018, 51, 336–342 CrossRef CAS.
- A. Lotierzo, R. M. Schofield and S. A. F. Bon, ACS Macro Lett., 2017, 6, 1438–1443 CrossRef CAS.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019 10.1039/C8PY01720D.
- A. Postma, T. P. Davis, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5307–5318 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01437j |
| This journal is © The Royal Society of Chemistry 2019 |