
Tuning PBT vitrimer properties by controlling the dynamics of the adaptable network†
Yanwu
Zhou
 a,
Ramon
Groote
b,
Johannes G. P.
Goossens
*b,
Rint P.
Sijbesma
a and
Johan P. A.
Heuts
*a
a,
Ramon
Groote
b,
Johannes G. P.
Goossens
*b,
Rint P.
Sijbesma
a and
Johan P. A.
Heuts
*a
aSupramolecular Polymer Chemistry group, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, the Netherlands. E-mail: j.p.a.heuts@tue.nl
bSABIC T&I, Plasticslaan 1, 4612 PX Bergen op Zoom, the Netherlands. E-mail: Han.Goossens@sabic.com
First published on 15th November 2018
Abstract
Vitrimers, which form a bridge between thermosets and thermoplastics, are a class of materials with promising opportunities for modern material innovations. Poly(butylene terephthalate) (PBT) vitrimers that combine the properties of neat PBT and those of adaptable networks are expected to greatly extend the potential applications of this industrially important engineering plastic. The current study aims at building up a tailor-made semi-crystalline vitrimer through understanding the effect of the dynamics and density of the adaptable network on the physical properties of PBT vitrimers. We show that the rubber plateau modulus of PBT vitrimers is almost exclusively governed by the cross-linker (glycerol) content, whereas the ratio of the glycerol to the Zn(II) catalyst content strongly influences both the elastic and stress relaxation properties. This enables independent tuning of the tensile storage modulus (E′) and rubber plateau modulus. The PBT vitrimer exhibits a better creep resistance than neat PBT at service temperature while a similar crystallinity is maintained.
Introduction
Vitrimers, pioneered by Leibler and co-workers,1–3 are a new type of covalent adaptable networks based on an associative exchange mechanism.4–7 These dynamically cross-linked networks undergo associative exchange reactions at elevated temperature permitting a change of the network topology while keeping the number of bonds and cross-links constant. The network rearrangement and the concomitant stress relaxation rates for vitrimers are determined by the rate of the chemical exchange reactions.1–3 Although catalyst-free vitrimers have been reported,8–16 catalysis offers an efficient way to control vitrimer properties (e.g., stress relaxation time, weldability and flowability), and a wide range of catalysts, such as organic bases or inorganic salts, have been reported to control transesterification reactions.17 Recently, a new type of semi-crystalline vitrimer was developed by us based on poly(butylene terephthalate) (PBT) and glycerol and prepared via solid-state (co-)polymerization (SSP, Scheme 1A).18 The glycerol content of these vitrimers ranged from 2 to 18 mol%, while the Zn(II) catalyst was kept fixed at 2 mol% with respect to the PBT repeat unit. These vitrimers exhibited macroscopic flow that could be tuned from liquid-like to solid-like by increasing the glycerol content, while maintaining the ratio of catalyst-to-PBT repeat units constant.18 Since the network rearrangement (Scheme 1B), responsible for flow and stress relaxation, is mediated by the Zn(II) catalyst, it is expected that the stress relaxation will be faster when the number of Zn(II) catalytic centers per cross-link increases. The number of catalytic centers per cross-link, here denoted by r, is given by eqn (1):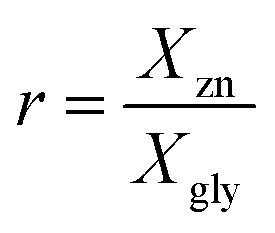 | (1) |
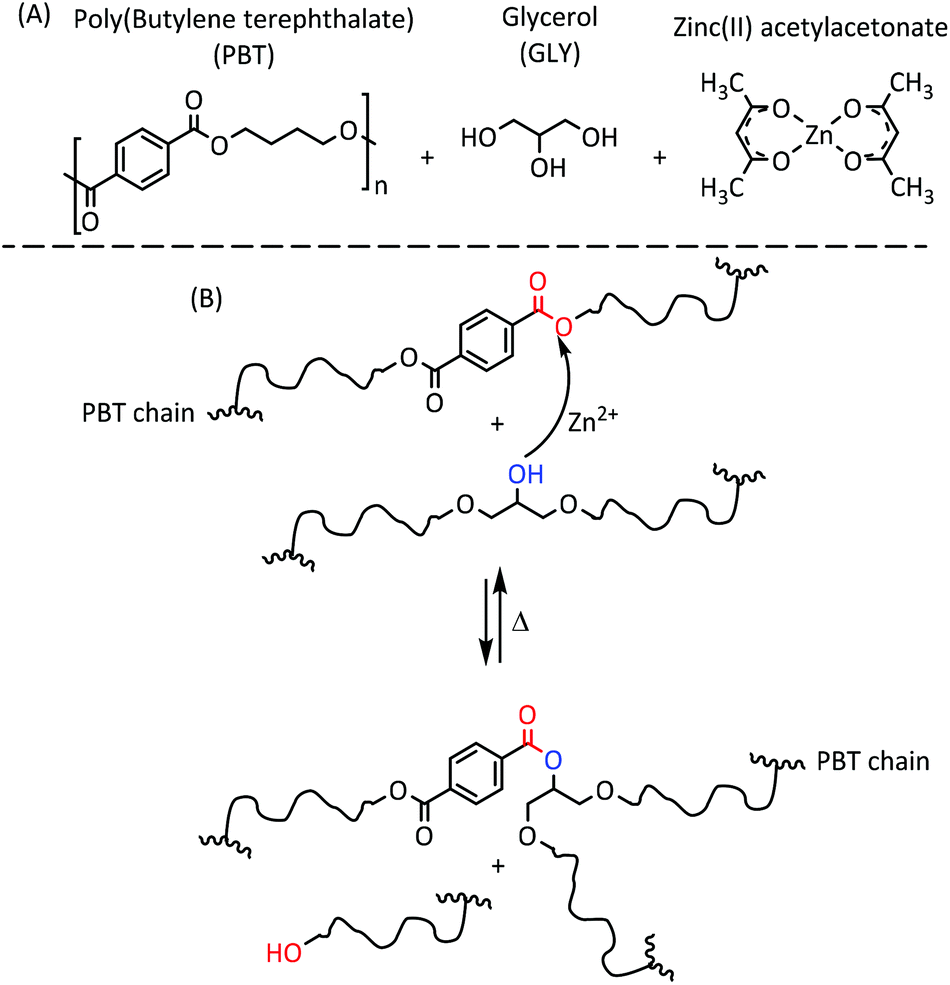 | ||
| Scheme 1 (A) The molecular structure of the compounds used for synthesizing the PBT/glycerol-based vitrimer. (B) Illustration of the network undergoing a bond exchange reaction between hydroxyl and ester group in the presence of Zn(II) catalyst. | ||
In the current study we correlate (and control) the stress relaxation, rubber plateau modulus, thermo-mechanical properties and creep resistance of these semi-crystalline vitrimers to Xgly and r.
Experimental section
Materials
Special grade poly(butylene terephthalate) pellets (Mn = 21.2 kg mol−1, Mw = 46.6 kg mol−1 against poly(methyl methacrylate) (PMMA) standards in 1,1,1,3,3,3-hexafluoroisopropanol) were provided by SABIC (Bergen op Zoom, the Netherlands) and used as received. Glycerol (≥99.5%), zinc(II) acetylacetonate hydrate (Zn(acac)2) (powder), potassium nitrate (BioXtra, ≥99.0%), sodium nitrate (≥99.0%), and sodium nitrite (≥97.0%) were all obtained from Sigma-Aldrich. Hydrochloric acid, anhydrous dichloromethane, methanol, 1,1,1,3,3,3-hexafluoroisopropanol (HFiP, 99%) and MilliQ water (LC-MS grade) were obtained from Biosolve. Deuterated chloroform (CDCl3, 99.8 atom% D) and deuterated trifluoroacetic acid (TFA-d, 99 atom% D) were obtained from Cambridge Isotope Laboratories. All chemicals were used as received unless denoted otherwise.Solution preparation of a physical PBT/glycerol mixture
Physical mixtures of PBT and glycerol were prepared from solution using a common solvent approach.18 As a representative example, for the physical mixture containing 14.1 mol% of glycerol and 2 mol% Zn(acac)2, the dried PBT powder (9.14 g, 41.55 mmol), glycerol (0.64 g, 6.92 mmol) and Zn(acac)2 (0.22 g, 0.83 mmol) as the transesterification catalyst were dissolved in 20 mL of HFiP at 55 °C. After complete dissolution of all compounds, the HFiP was distilled off. As soon as the material started to precipitate, a vacuum (p = 10−2 mbar) was applied for complete removal of HFiP. Finally, the obtained lump residue was dried under vacuum for 24 h at 30 °C, vitrified in liquid nitrogen, and subsequently ground into powder using an IKA A11 Basic Analytical mill. This powder was subsequently dried under vacuum for a period of 24 h at 30 °C. The molecular weight of the polymer in the mixture was then checked to ascertain that no undesirable reactions, such as transesterification or degradation, occurred during the preparation procedure.Preparation of PBT/glycerol-copolyesters
PBT/glycerol-copolyesters were synthesized by a two-step solid-state polymerization (SSP) method as described previously.18 Typically, in the first step, 5 g of the PBT/glycerol powder containing a predetermined amount of Zn(acac)2 was placed in a high-pressure reactor (Fig. 1A) and pressurized with argon (p < 3 bar) to avoid the evaporation of glycerol. After a predetermined amount of time at 160 °C, the mixture was transferred to the SSP reactor, which was a glass tube (inner diameter = 2.4 cm) with a sintered glass plate at the bottom. A heat exchange glass coil (inner diameter = 0.5 mm) surrounded the reactor and entered the inner glass tube at the bottom just below the glass plate. The nitrogen gas was heated by passing through this coil prior to entering the reactor and its flow was controlled by a flow meter. The powder bed was fixed by glass pearls (diameter = 2 mm) placed on top of the powder, and the reactor was purged with a nitrogen flow of 0.5 L min−1 for 30 min. After flushing, the reactor was placed in a heated salt bath (T = 180 °C). The reaction time (tssp) was counted from the time that the temperature inside the reactor reached 180 °C, and the composition and molecular weight were followed until the gel point. After reaction, the product was cooled down to room temperature under a continuous nitrogen flow, discharged from the reactor, and the obtained polymer dried under vacuum at 120 °C for 6 h. The prepared PBT/glycerol-based copolyesters will be abbreviated as Cxy, x = mol% of glycerol and y = mol% of Zn(acac)2 with respect to the number of PBT repeat units, using the compositions as determined by 1H-NMR spectroscopy. | ||
| Fig. 1 (A) Molecular weight distributions of sample set I, 2, 0.5, and 0.05 mol% Zn2+ after prepolymerization at 160 °C and 180 °C for 0 mol% Zn2+ (reaction times see text). (B) Molecular weight distributions of sample set II (r ≈ 0.015) after prepolymerization for 48 h (sample C10.02 at 180 °C, all other samples at 160 °C). | ||
Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v CDCl3:d-TFA mixture. Chemical shifts are reported in ppm relative to the residual solvent peak of CDCl3 (δ = 77.0 ppm).
:20 v/v CDCl3:d-TFA mixture. Chemical shifts are reported in ppm relative to the residual solvent peak of CDCl3 (δ = 77.0 ppm).
Results and discussion
Two sets of materials were prepared via a two-step solid-state (co)polymerization of PBT and glycerol in the presence of Zn(II) catalyst. The procedure consisted of a prepolymerization step at 160 °C in a closed vial followed by a polymerization step at 180 °C under a nitrogen flow, as reported in our previous work.18 In material set I, Xgly was kept constant (∼13.2 mol%) while varying XZn from 0 to 2 mol%, corresponding to r ranging from 0 to 0.15 (= 2/13.2). In material set II, XZn and Xgly were varied simultaneously while the molar ratio between glycerol and Zn(II) catalyst was kept constant (r ≈ 0.015). Here we use the following sample notation: Cxy where x = mol% of glycerol and y = mol% of Zn(acac)2 with respect to the number of PBT repeat units.Synthesis and characterization of the PBT vitrimers
In the current study, the prepolymerization step was performed at 160 °C in a high-pressure reactor (∼80 g scale), which was pressurized with inert gas to avoid the evaporation of glycerol. The polymerization conditions were slightly adjusted to the changing XZn because of its strong influence on the polymerization kinetics. For instance, to obtain materials with the same molecular weight after prepolymerization step at 160 °C, the following reaction times were required for set I: 24 h for C132 (2 mol% Zn2+), 48 h for C130.2 (0.2 mol% Zn2+) and 96 h for C130.05 (0.05 mol% Zn2+). Only sample C130 (0 mol% Zn2+, only containing a residual amount of catalyst used in the PBT production) was prepolymerized at 180 °C for 48 h. After the total incorporation of the glycerol, the mixture was transferred to an SSP reactor (5–40 g scale), and the reaction was continued at 180 °C with a N2 flow of 0.5 L min−1. The molecular weights after prepolymerization (see Fig. 1) and the compositions before “gelation” of the resulting copolyesters are summarized in Table 1.| Samplea | r |
M
nc (kg mol−1) |
M w (kg mol−1) | Thermal properties | ||||
|---|---|---|---|---|---|---|---|---|
|
T
gd (°C) |
T
ce (°C) |
T
me (°C) |
χ
ce (%) |
|||||
a Sample notation: Cxy, x = mol% of glycerol and y = mol% of Zn(acac)2 with respect to the number of PBT repeat units.
b
r = XZn/Xgly, where the mol fraction of glycerol Xgly was determined by 1H NMR spectroscopy in the samples just before gelation.
c Number- (Mn) and weight-average (Mw) molecular weights of the PBT/glycerol copolyesters before gelation determined by SEC in HFiP relative to PMMA standards.
d The glass transition temperature (Tg) was determined from the peak maximum of the loss modulus in DMTA.
e The melting (Tm) and crystallization (Tc) temperatures were the peak values of the melting endotherms and crystallization exotherms (in DSC), respectively. The degree of crystallinity (χc) was determined by dividing the melting enthalpy (ΔHmelting) (obtained via DSC measurements) by the melting enthalpy for 100% crystalline PBT  .19 Linear integration errors of melting peak <2%. .19 Linear integration errors of melting peak <2%.
|
||||||||
| Neat PBT | PBT | — | 21.2 | 46.6 | 55 | 194 | 222 | 38 |
| I. Varying r: Xgly constant, varying XZn | C 13 2 | 0.15 (2/13.0) | 3.1 | 9.3 | 80 | 167 | 201 | 32 |
| C 13 0.2 | 0.015 (0.2/13.0) | 3.8 | 8.1 | 66 | 181 | 214 | 34 | |
| C 13 0.05 | 0.0038 (0.05/13.0) | 3.2 | 9.6 | 69 | 182 | 215 | 35 | |
| C 13 0 | 0 (0/13.0) | 6.5 | 15.0 | 62 | 189 | 217 | 35 | |
| II. Constant r: varying Xgly and XZn | C 13 0.2 | 0.015 (0.2/13.0) | 3.8 | 8.1 | 66 | 181 | 214 | 34 |
| C 6 0.1 | 0.016 (0.1/6.2) | 6.8 | 13.6 | 64 | 192 | 218 | 35 | |
| C 3 0.05 | 0.016 (0.05/3.0) | 10.8 | 21.7 | 59 | 193 | 222 | 38 | |
| C 1 0.02 | 0.016 (0.02/1.2) | 17.0 | 31.9 | 59 | 194 | 222 | 38 | |
It should be noted here that gelation is defined as the point where the materials are not fully soluble in HFiP anymore and that the characterization of gel fraction will always be more qualitative than in the case of permanently cross-linked materials because of the dynamic nature of the vitrimer network.1,2 This was also demonstrated by Williams et al. who showed that the transesterification reactions in their vitrimer systems increase the cross-link density and simultaneously generate pendant chains and a sol fraction.20
Influence of r on the network dynamics
In our previous study18 we reported on the effect of a varying glycerol content (Xgly) while maintaining the catalyst content (XZn) constant (hence, a varying r with constant XZn) and found a clear effect on both the rubber plateau modulus and the stress relaxation: both increased with an increasing glycerol content. In the current study the effects of catalyst (XZn) and glycerol (Xgly) contents were investigated in more detail.The melt rheology of PBT/glycerol vitrimers was investigated with frequency sweeps in oscillatory shear experiments at sufficiently low strain amplitudes (1%) to probe the linear viscoelastic properties. A 25 mm plate-plate geometry at various temperatures over an angular frequency (ω) range of 100–0.01 rad s−1 was used. Sample C130.2 (XZn ≈ 0.2 mol% and Xgly ≈ 13 mol%) with r ≈ 0.015 from set I is used as a representative example to demonstrate the time-temperature dependent dynamic mechanical properties in Fig. 2.
 | ||
| Fig. 2 Frequency dependence of the storage modulus G′ of C130.2 (r ≈ 0.015) at different temperatures. Please note that for clarity reasons, only the loss modulus at 280 °C is displayed. | ||
This sample displays an almost frequency-independent rubber plateau modulus G′ ≈ 0.46 MPa for ω < 10−1 rad s−1 at temperatures below 240 °C. Above this temperature, the rubber plateau does not exist anymore and a drop in modulus of the material at lower frequencies is observed. The transition point where the modulus drops shifts to higher frequencies with increasing temperature. This behaviour is consistent with the dynamically cross-linked nature of a polyester-based vitrimer,5 in which the changes in temperature primarily affect the transesterification kinetics but do not change the cross-link density. Thus, the stress relaxation time (τ) is temperature dependent since it is determined by the exchange rate, while the plateau modulus is not (or, at least to a much lower extent).
The effect of varying XZn for a constant Xgly on the time- and temperature-dependent stress relaxation dynamics of the vitrimers was further studied by stress relaxation experiments. Before performing these experiments, the compression-molded materials (for a description of the compression-molding process see ESI, Fig. S1†) were subjected to an oscillatory time sweep at constant temperature until a steady-state plateau was reached (C130.2 was shown as an example in Fig. S2†) to guarantee complete equilibration of the system and a constant network density. Similar to what was observed in our previous work,18 the “virgin” compression-molded material [C132] showed a significant increase in storage modulus with increasing temperature above the melting temperature. Normalized stress relaxation plots for material set I at 270 °C are shown in Fig. 3A, and it is immediately clear from this plot that the stress relaxation is faster at higher catalyst contents. For this set of materials, one dominant relaxation time is observed; the data can be fitted with the Maxwell model using a single characteristic relaxation time, τ.1 Interestingly, C130 (the material without added Zn(acac)2) shows the same characteristic stress relaxation behavior as the other materials in material set I. This unexpected dynamic behavior is most likely caused by the presence of a residual amount of catalyst, used in the production of PBT. Above the melting temperature of the PBT vitrimer, the stress relaxation times display an Arrhenius-type temperature dependence as shown in Fig. 3B. Linear fitting of the data gives activation energies in the range of 150–170 kJ mol−1, with only C130.05 as the exception with a slightly higher Ea (200 kJ mol−1). Fitted activation energies (Ea) and pre-exponential factor (A) for PBT/glycerol-based vitrimers are summarized in Table S1.†
 | ||
| Fig. 3 Dynamic properties of material set I (constant Xgly, changing r). (A) Normalized stress relaxation curves of at 270 °C (a version with linear axes is shown in the ESI, Fig. S3†). (B) Arrhenius plot of the stress relaxation times. | ||
The results of tests in which the XZn and Xgly were simultaneously varied in order to maintain a constant r ≈ 0.015 (material set II) are presented in Fig. 4. The frequency sweep experiments shown in Fig. 4 illustrate that all the materials show solid-like network characteristics at 250 °C (above Tm), with G′ > G′′ in the experimental angular frequency range of 102–10−2 rad s−1. The storage modulus at the frequency where G′′ displays a minimum, increases with increasing Xgly and the data of C10.02 show much faster exchange dynamics than the other materials. No terminal flow regime was observed at 250 °C for any of the studied compositions.
 | ||
| Fig. 4 Frequency dependence of the storage modulus G′ (open symbols) and loss modulus G′′ (filled symbols) of material set II at 250 °C. | ||
The stress relaxation behavior in material set II at 250 °C is shown in Fig. 5A. It is clear from this figure that despite the large variation in cross-linker content (but maintaining the number of Zn(II) centers per cross-link constant), the relaxation times are all similar, with the notable exception of C10.02. This latter material, containing only 1 mol% of cross-linker and 0.02 mol% of catalyst, displays a stress relaxation that is much faster than in the other materials. Additionally, the stress relaxation in C10.02 cannot be described using a single characteristic relaxation time, but this needs to be described by at least two, but probably even a spectrum of relaxation times; because of the very low cross-link density, it is likely that mechanisms involving (temporarily) unconnected chains also play a role in addition to and combination with the ester exchange reactions.21 It is important to note that even in C10.02 the relaxation at 250 °C is much slower than in neat PBT (which fully relaxes its stress within a second, see Fig. S4, ESI†). The Arrhenius plots of relaxation times are shown for C30.05, C60.1 and C130.2 in Fig. 5B, and in agreement with the results from set I and our previous work,17p,18 the activation energies, Ea, lie in the narrow range of 140–170 kJ mol−1 (see also Table S1, ESI†). The effects of catalyst and cross-linker content can be summarized as follows: the storage modulus (G′) of PBT/glycerol-based vitrimers in the melt is mainly controlled by the glycerol content, while above a glycerol content of 1 mol%, the dynamics of the exchangeable networks are controlled by the catalyst-to-cross-linker ratio r.
 | ||
| Fig. 5 Dynamic properties of the material set II. (A) Normalized stress relaxation curves at 250 °C (a version with linear axes is shown in the ESI, Fig. S3†). (B) Arrhenius plots of the stress relaxation times. | ||
Influence of r on thermomechanical properties
The thermomechanical properties of the PBT vitrimers were characterized by dynamic mechanical thermal analysis (DMTA). Materials were annealed at 200 °C under vacuum for 6 h before the tests. In Fig. 6, the results of these analyses are shown and are similar to what we reported previously for PBT vitrimers:18 the behavior of the PBT vitrimers is similar to that of PBT below Tm, but above Tm a rubber plateau is observed, originating from the dynamically cross-linked network. The results for material set I are shown in Fig. 6A. The materials exhibit a single melting transition, with the exception of C132, which shows two melting transitions above Tg. We think that the melting transition after the glass transition temperature is caused by a network reorganization that leads to a partitioning of the initial crystalline part of PBT at 200 °C. As shown in Table 1, this annealing temperature is close to the peak melting temperature of C132. Therefore, the low melting transition observed in the DMTA curve is propable due to the thinner lamellar crystals, while the high melting temperature transition similar to the other vitrimer samples of material set I can be ascribed to the thicker lamellar crystals. For material set I (similar network densities, νc) the moduli in the rubber region are only weakly dependent on the temperature because of an entropic temperature factor, and all moduli fall within the narrow range of 3–5 MPa at 270 °C. However, in material set II, where XZn and Xgly are simultaneously varied, the rubber plateau modulus decreases with decreasing Xgly (Fig. 6B). | ||
| Fig. 6 DMTA curves of material set I (A) and material set II (B). Heating rate = 3 °C min−1 and ω = 1 Hz. The dotted line indicates the temperature used for creep experiments (see below). | ||
Let us now consider the tensile storage modulus (E′) of PBT vitrimers in the temperature region between Tg and Tm. From Fig. 6A, it is clear that E′ at 180 °C increases with decreasing r (decreasing XZn while keeping Xgly constant). Since the modulus of semi-crystalline materials in this temperature range is controlled by the degree of crystallinity (χc),22 we compared χc for these materials (Table 1 and Fig. S5, ESI†). In general, the χc of vitrimers are lower than the χc of neat PBT, which is consistent with the lower E′ values for the vitrimers. What cannot be explained by a simple consideration of χc are the differences in E′ for the various vitrimers in set I, as all display the same χc. In principle, a lower χc leads to a decrease in E′ and the cross-linking is expected to lead to an increase. What is observed in Fig. 6A is that the more catalyst in the system (while keeping the cross-link density constant), the lower the E′. Although this suggests that a more dynamic network decreases the E′ in this temperature region, we do not think this explanation is conceivable, as above Tm we do not observe this dependence of E′ on catalyst loading. If the dynamics were to affect the plateau modulus directly, we would expect this effect to be more prominent at higher temperatures. We believe that the explanation should be sought in possible different morphologies of the materials. During compression molding, preceding the DMTA measurements, the samples have been in a molten state and it is conceivable that annealing and subsequent recrystallization at 200 °C results in different morphologies (in terms of lamellar size distribution and the rigid versus mobile amorphous fraction)23,24 depending on the dynamics of the network; the overall crystallinities may therefore be very similar, but the morphologies could be quite different. This line of reasoning is supported by another study in which we showed that different morphologies were obtained for different thermal treatments of the material.25
The DMTA curves for material set II are presented in Fig. 6B. It is evident that the materials in set II, except for C130.2, have a similar E′ as PBT and a similar χc (Table 1). As can be seen from Table 1, C130.2 has a much lower χc and is similar to all materials from set I (all the materials contain approximately 13 mol% glycerol). These results are consistent with the dependence of E′ on χc and that high glycerol concentrations lead to lower χc, which is in good agreement with our previous work.18
Hence we can conclude that it is possible to control the stiffness (∼E′) between Tg and Tm by varying χc, through the cross-linker content (Xgly), and the dynamics of the network, through r. To summarize this section, we can conclude that we can produce PBT vitrimers with a tunable stiffness and rubber plateau.
Influence of r on creep resistance
Creep is one of the most important viscoelastic properties of a polymer material.26 A potential advantage of PBT vitrimers is that they may show a better creep resistance at service temperature (e.g., T < Tm), while maintaining a high rate of the exchange reactions at processing temperatures above Tm. In creep experiments on the PBT vitrimers, a rectangular-shape material (the same as for the DMTA test) was first heated to 125 °C, kept isothermal for 30 min, and subsequently a 2 MPa step stress was applied for a period of 60 min. After 60 min, the stress was removed, and the material was allowed to recover for another 80 min. The results of these experiments are shown in Fig. 7. | ||
| Fig. 7 Creep-recovery experiments at 125 °C on PBT vitrimers: (A) with different r (set I) and (B) with the same r (set II). | ||
The final strain reflects the combined elastic and viscoelastic response (creep deformation) that occurred during application of the step stress, which was removed after 60 min. The strain-descending portion of the curve at >60 min represents the creep recovery provided by polymer chains which rearranged their conformations via changing the internal bond lengths and angles as a response to the step stress. The irrecoverable strain represents the permanent change in the material caused by the flow of polymer chains. A summary of the creep properties for the PBT vitrimers in this study is presented in Table 2.
| Material sets | Name | E′ at 125 °C [GPa] | Final creep strain [%] | Creep rate [% per hour] |
|---|---|---|---|---|
| a The creep data of PBT at different temperatures are presented in Fig. S6. Linear integration errors of the secondary steady-state creep region <5%. | ||||
| Reference | PBT | 0.61 | 1.08 | 0.05 |
| I. Varying r: Xgly constant, varying XZn | C 13 2 | 0.16 | 2.23 | 0.15 |
| C 13 0.2 | 0.20 | 1.63 | 0.01 | |
| C 13 0.05 | 0.43 | 0.93 | 0.02 | |
| C 13 0 | 0.43 | 0.83 | 0.02 | |
| II. Constant r: Varying Xgly and XZn | C 13 0.2 | 0.20 | 1.63 | 0.01 |
| C 6 0.1 | 0.42 | 1.09 | 0.02 | |
| C 3 0.05 | 0.49 | 1.08 | 0.02 | |
| C 1 0.02 | 0.60 | 0.60 | 0.01 | |
In general, addition of cross-linker and catalyst reduced the tensile modulus (E′) at 125 °C (E′ of C10.02 is identical to that of neat PBT). Nevertheless, the vitrimer materials with a catalyst content at or below 0.1 showed a better creep resistance than neat PBT at 125 °C, regardless of cross-linker content. They have lower final creep strain, lower creep rate, and a lower irrecoverable strain. In these samples, the creep resistance at elevated temperature has improved despite the presence of exchangeable cross-links.
Conclusions
The effects of the molar ratio between cross-linker (glycerol) and catalyst (Zn(II)) on the viscoelastic properties of PBT vitrimers have been characterized and it has been demonstrated that the (thermo)mechanical and rheological properties of PBT vitrimers can be tuned by controlling the exchange dynamics and cross-link density. The rubber plateau modulus of PBT vitrimers depends on/increases with Xgly, indicating that the cross-link density primarily controls the static elasticity of PBT vitrimers, while the stress relaxation time is governed by XZn or XZn/Xgly. For materials with the same thermo-mechanical history, the modulus below Tg is slightly higher than that of neat PBT, while the modulus between Tg and Tm is controlled by r. A higher stiffness can be obtained in two ways: (1) for a range of materials with constant Xgly by decreasing r; (2) for materials with constant r by lowering the Xgly and XZn simultaneously. The creep experiments demonstrate that the creep resistance at service temperature (Tg < T < Tm), i.e., both the creep strain and the steady-state creep rate can be controlled by the cross-link density and the dynamics of the exchangeable network. While the PBT vitrimers maintained a similar temperature-dependent crystallinity behavior as neat PBT, up to a catalyst content of 0.1, they exhibited equal or superior creep resistance over neat PBT. The ability to tune both the E′ and dynamics of semi-crystalline vitrimers will ultimately open up new opportunities to design new types of vitrimers with specifically desired properties for relevant industrial application.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support by SABIC for this work is gratefully acknowledged.References
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2452 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- M. Horiz, A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, N. Renaud and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS.
- (a) K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 48682–48690 RSC; (b) K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 10108–10117 RSC; (c) Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS; (d) Y. Yang, Z. Pei, X. Zhang, L. Tao, Y. Wei and Y. Ji, Chem. Sci., 2014, 5, 3486–3492 RSC; (e) J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS; (f) F. Sordo, S.-J. Mougnier, N. Loureiro, F. Tournilhac, V. Michaud, S.-J. Mougnier and F. Tournilhac, Macromolecules, 2015, 48, 4394–4402 CrossRef CAS; (g) K. Yu, Q. Shi, M. L. Dunn, T. Wang and H. J. Qi, Adv. Funct. Mater., 2016, 26, 6098–6106 CrossRef CAS; (h) E. Chabert, J. Vial, J.-P. Cauchois, M. Mihaluta and F. Tournilhac, Soft Matter, 2016, 12, 4838–4845 RSC; (i) A. Demongeot, S.-J. Mougnier, S. Okada, C. Soulié-Ziakovic and F. Tournilhac, Polym. Chem., 2016, 7, 4486–4493 RSC; (j) Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, J. Am. Chem. Soc., 2016, 138, 2118–2121 CrossRef CAS; (k) Y. Liu, Z. Tang, Y. Chen, C. Zhang and B. Guo, ACS Appl. Mater. Interfaces, 2017, 10, 2992–3001 CrossRef; (l) H. Zhang, C. Cai, W. Liu, D. Li, J. Zhang, N. Zhao and J. Xu, Sci. Rep., 2017, 7, 1–9 CrossRef; (m) Z. Tang, Y. Liu, B. Guo and L. Zhang, Macromolecules, 2017, 50, 7584–7592 CrossRef CAS; (n) Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. J. Qi, Mater. Horiz., 2017, 4, 598–607 RSC; (o) W. Liu, D. F. Schmidt and E. Reynaud, Ind. Eng. Chem. Res., 2017, 56, 2667–2672 CrossRef CAS; (p) A. Demongeot, R. Groote, J. G. P. Goossens, T. Hoeks, F. Tournilhac and L. Leibler, Macromolecules, 2017, 50, 6117–6127 CrossRef CAS; (q) Y. Zhou, J. G. P. Goossens, S. van den Bergen, R. P. Sijbesma and J. P. A. Heuts, Macromol. Rapid Commun., 2018, 39, 1800356 CrossRef.
- Y. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2017, 50, 6742–6751 CrossRef CAS.
- T. Stukenbroeker, W. Wang, J. M. Winne, F. E. Du Prez, R. Nicolaÿ and L. Leibler, Polym. Chem., 2017, 8, 6590–6593 RSC.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Polymers, 2018, 10, 43–59 CrossRef.
- M. Pyda, E. Nowak-Pyda, J. Mays and B. Wunderlich, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 4401–4411 CrossRef CAS.
- J. M. G. Cowie and V. Arrighi, Polymers: chemistry and physics of modern materials, Blackie academic and professional, and imprint of Chapman and Hallbishopbriggs, Glasgow, 3rd edn, 1991 Search PubMed.
- M. Pyda, E. Nowak-Pyda, J. Heeg, H. Huth, A. A. Minakov, M. L. Lorenzo, C. Schick and B. Wunderlich, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1364–1377 CrossRef CAS.
- T. Konishi, W. Sakatsuji, K. Fukao and Y. Miyamoto, Macromolecules, 2016, 49, 2272–2280 CrossRef CAS.
- (a) Y. Zhou, High-performance poly(butylene terephthalate) vitrimers, Ph.D. thesis, Technische Universiteit Eindhoven, The Netherlands, 2017 Search PubMed; (b) Y. Zhou, R. Groote, J. G. P. Goossens, J. A. M. Lugger, R. P. Sijbesma and J. P. A. Heuts, Influence of morphology on the physical and mechanical properties of PBT vitrimers, to be submitted.
- J. D. Ferry, Viscoelastic properties of polymers, John Wiley & Sons, Chichester, 1980 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01156g |
| This journal is © The Royal Society of Chemistry 2019 |