
Heavy-atom-free BODIPY photosensitizers with intersystem crossing mediated by intramolecular photoinduced electron transfer
Mikhail A.
Filatov

School of Chemical and Pharmaceutical Sciences, Technological University Dublin, City Campus, Kevin Street, Dublin 8, Ireland. E-mail: mikhail.filatov@tudublin.ie
First published on 12th November 2019
Abstract
Organic photosensitizers possessing efficient intersystem crossing (ISC) and forming long-living triplet excited states, play a crucial role in a number of applications. A common approach in the design of such dyes relies on the introduction of heavy atoms (e.g. transition metals or halogens) into the structure, which promote ISC via spin–orbit coupling interaction. In recent years, alternative methods to enhance ISC have been actively studied. Among those, the generation of triplet excited states through photoinduced electron transfer (PET) in heavy-atom-free molecules has attracted particular attention because it allows for the development of photosensitizers with programmed triplet state and fluorescence quantum yields. Due to their synthetic accessibility and tunability of optical properties, boron dipyrromethenes (BODIPYs) are so far the most perspective class of photosensitizers operating via this mechanism. This article reviews recently reported heavy-atom-free BODIPY donor–acceptor dyads and dimers which produce long-living triplet excited states and generate singlet oxygen. Structural factors which affect PET and concomitant triplet state formation in these molecules are discussed and the reported data on triplet state yields and singlet oxygen generation quantum yields in various solvents are summarized. Finally, examples of recent applications of these systems are highlighted.
 Mikhail A. Filatov | Mikhail Filatov received his PhD degree in the field of organic chemistry from Moscow State University in 2008. He has conducted postdoctoral research in the University of Burgundy and Max Planck Institute for Polymer Research. In 2015 he was awarded Marie Curie Individual Fellowship to perform a two-year research project at Trinity College Dublin. He joined TU Dublin as a faculty member in 2017. His current research spans the areas of multistep organic synthesis, photochemistry, spectroscopy and the development of light-harvesting materials for energy applications and healthcare. |
1. Introduction
The development of innovative photonic technologies critically depends on the availability of photoactive materials with strong absorption across the visible spectrum and tunable excited state properties. In this context, organic dyes have an important advantage compared to common inorganic photocatalysts: their excited state energies and lifetimes can be finely tuned by rational design of molecular structures to match the desired range.Normally, excitation of a chromophore, leads to the lowest singlet excited state S1, which possesses rather short lifetimes (nanoseconds or less) and rapidly relaxes back to the ground state.1 Alternatively, lower-lying triplet excited states Tn can be populated from S1 state via a spin-forbidden intersystem crossing (ISC) process. Due to their long lifetimes (up to seconds), triplet excited states can efficiently transfer energy to other molecules and mediate chemical transformations. Dyes possessing efficient ISC, referred to as triplet sensitizers, are used to harvest light energy and found applications in various fields of technology, e.g. in solar fuel generation,2 photovoltaics,3 photoredox catalysis for organic synthesis,4 photooxidation of organic pollutants,5 photoinitiated polymerization,6 triplet–triplet annihilation upconversion (TTA-UC)7 and photodynamic therapy (PDT).8
Triplet photosensitizers are commonly obtained through complexation of organic chromophores with transition metals (e.g. Ru, Pd or Pt) or introduction of halogens (Br or I) into the structure.9 ISC in such derivatives is usually efficient due to spin–orbital interaction – a relativistic effect pronounced in atoms with large nuclei (heavy atoms). This mechanism is known as a spin–orbit coupling intersystem crossing (SO-ISC). The effect of heavy atoms on photophysical properties is illustrated in Fig. 1 on an example of boron dipyrromethene (BODIPY)101 and its 2,6-diiodo derivative 2. Compound 1 possesses intense fluorescence, while its ISC is inefficient due to a weak spin–orbit coupling, giving a triplet state yield (ΦT) of less than 1%. On the other hand, enhanced spin–orbit coupling in BODIPY 2 results in a triplet excited state yield of >80%, making it suitable for use as a triplet sensitizer.11
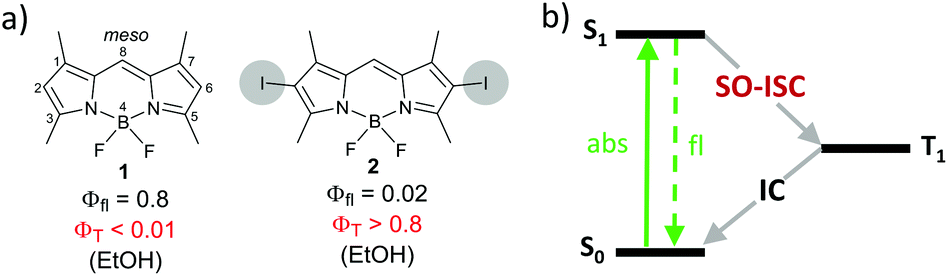 | ||
| Fig. 1 (a) Structures of BODIPYs 1 and 2. IUPAC numbering system is shown in the structure of 1. Φfl – fluorescence quantum yield, ΦT – triplet state yield. (b) Jablonski diagram illustrating excited state transitions in 2. S0 – ground state, S1 – lowest singlet excited state, T1 – lowest triplet excited state. SO-ISC – spin–orbit coupling intersystem crossing, IC – internal conversion. Solid arrow: most likely process; dashed arrow: less likely process. | ||
Although this approach for enhancing triplet state yields in organic molecules seems convenient, the introduction of heavy atoms often results in issues such as tedious synthesis, increased cost, low solubility and other unwanted side effects. For instance, in photoredox catalysis much effort is currently focused on replacing costly transition metal-based photosensitizers with heavy-atom-free organic dyes,12 because on an industrial scale their application is expected to be more economical and will reduce environmental impact.13 For this reasons, alternative methods to promote ISC, e.g. using a spin converter,14 introduction of carbonyl groups,15 radical-enhanced ISC16 and twist-induced ISC17 have been actively studied in recent years. However, it is still difficult to design heavy-atom-free sensitizers due to the lack of established relationships between ISC and molecular structure.
The formation of triplet excited states by way of intramolecular photoinduced electron transfer (PET) was studied for the first time by Okada and co-workers on a series of aminopyrenes.18 Recently, unexpectedly efficient ISC has been reported for various heavy-atom-free BODIPYs19 and other difluoroboron complexes,20 metal dipyrrins,21 phenoxazines,22 biphenyls,23 naphthalene and perylene imides.24 For many of these systems, very high triplet state yields (>90%) and long triplet lifetimes (up to a few hundreds of microseconds) have been observed. Notably, triplet state and fluorescence quantum yields in these systems strongly depend on molecular geometry and polarity of the media, providing outstanding possibilities for “programming” excited state behavior via rational design of the structures.
Polarity-controlled triplet states generation is particularly advantageous for applications involving reactive oxygen species (ROS). In PDT, interaction of the sensitizer triplet states with molecular oxygen (3O2), results in the formation of highly reactive singlet oxygen (1O2) which causes oxidative stress and ultimately cell death.25 Formation of 1O2 in selected sites of the cell via polarity-controlled PET in diiodo-substituted BODIPY derivatives was demonstrated for deactivation of specific proteins by the Nagano group.26 Activatable photosensitizers based on transition metal complexes were reported in a number of works.27 However, the use of this methodology in photomedicine is still limited, because molecules containing heavy atoms often possess rather high dark cytotoxicity,28i.e. can be harmful to the tissue in the absence of light. On the other hand, the scope of available heavy-atom-free photosensitizers which selectively generate singlet oxygen in polar/non-polar environments or in response to activation stimuli is still quite narrow and principles for their design are not sufficiently elaborated.
In this review, the progress in the development of heavy-atom-free BODIPY photosensitizers achieved over the past several years is discussed. The paper is structured as follows. Background information on photoinduced electron transfer and triplet state formation from charge transfer states (CT) is presented in section 2. In sections 3 and 4, data on electron transfer, triplet state and singlet oxygen quantum yields for the reported BODIPY donor–acceptor dyads and dimers are summarized. Correlations between molecular structures and the observed photophysical properties in different solvents are discussed. On the basis of this information, criteria for the design of efficient photosensitizers operating via PET are highlighted in section 5. Examples of recent applications of such photosensitizers in photon upconversion and PDT are presented in section 6.
2. Photoinduced electron transfer and triplet states formation from charge transfer states
Photoinduced electron transfer in donor–acceptor dyads, i.e. molecules in which electron donor (D) and acceptor (A) subunits are chemically connected, is a very general and well-studied phenomenon.29 A schematic frontier molecular orbital diagram for the PET process, outlining the requirements towards HOMO and LUMO energy levels of the subunits, is shown in Fig. 2a. Upon light absorption, electron transfer within the dyad results in the formation of a highly polar excited state, usually called a charge-transfer state (CT), or a charge-separated state (CSS).30 This state can be described as a radical ion-pair, in which a radical cation is localized on the donor subunit (D˙+) and a radical anion is localized on the acceptor subunit (A˙−).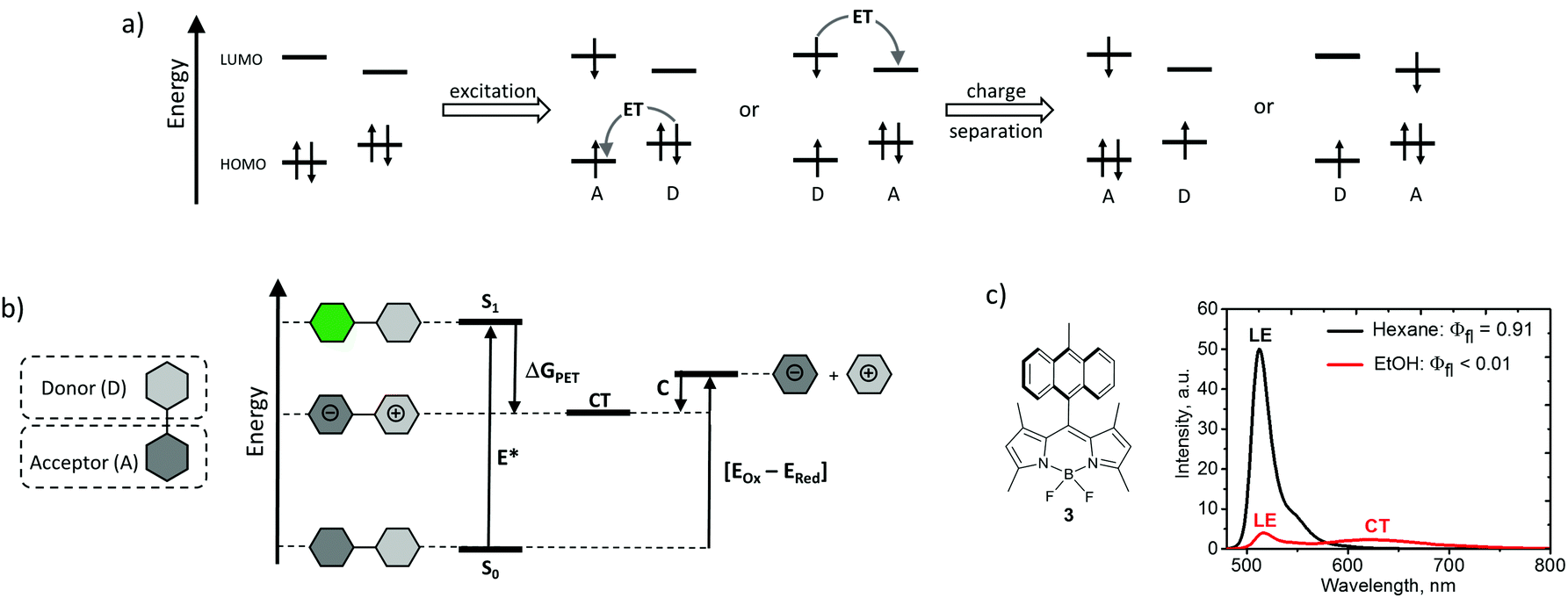 | ||
| Fig. 2 (a) Schematic frontier molecular orbital diagram for the PET process in electron donor–acceptor dyads. (b) Energy level diagram of PET in polar solvent. (c) Structure of BODIPY-anthracene dyad 3 and its fluorescence spectra in non-polar (hexane) and polar (ethanol) solvents. | ||
The thermodynamic feasibility of PET in dyad molecules can be estimated from spectroscopic and electrochemical data by calculating the free energy change using the Rehm-Weller eqn (1):31
| ΔGPET = e[EOx(D) − ERed(A)] − E* − C | (1) |
where EOx(D) and ERed(A) are one-electron oxidation and reduction potentials of the donor and acceptor, respectively, E* is the energy of the excited state (S1) and C represents the coulombic interactions between two ions produced at a distance rDA in a solvent with a dielectric constant εr (Fig. 2b).
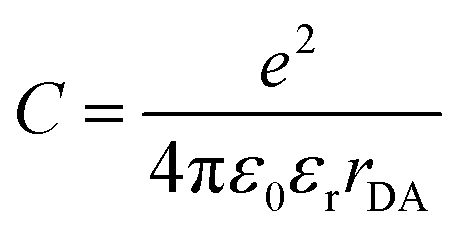
Efficient PET in donor–acceptor dyads is usually manifested by the profound effect of solvent on the emission properties. A progressive red-shift in the emission maxima, accompanied by a concomitant broadening and decrease in emission quantum yields, is observed for such compounds with increasing solvent polarity.32 This effect is illustrated in Fig. 2c for dyad 3, composed of a tetramethyl-substituted BODIPY (electron acceptor) and 9-methylanthracene (electron donor) subunits. The intense emission observed in hexane (εr = 4.81) corresponds to the fluorescence from a local excited (LE) state of the BODIPY subunit. It is strongly quenched in ethanol (εr = 24.5) due to the PET process leading to a poorly emissive CT state.19a
Solvent dependence in dyad emission can be rationalized by taking into account the dipolar nature of the CT state being formed. While the energy of the LE state is virtually unchanged in various solvents, the CT state energy level is strongly dependent on the possibility of dipole–dipole interactions with solvent molecules.33 In non-polar solvents, such as hexane, the CT state does not get stabilized, resulting in a situation where it resides in a higher energy state than the LE state. In this case the electron transfer process is thermodynamically unfavorable (ΔGPET > 0) and the dyad exhibits intense LE emission. More polar solvents render the energy level of the CT state lower than LE state, making the electron transfer process thermodynamically allowed (ΔGPET < 0).
Charge-transfer states undergo a non-radiative charge recombination (CR), also known as a back electron transfer (BET), to restore the ground state of the dyad.30a The free energy change associated with the recombination process can have rather large negative values due to a large energy gap between the CT state and the ground state (e.g. >1.5 eV). Under these circumstances, thermodynamics of the process falls into the so-called Marcus “inverted” region, where the activation energy is substantially increased, consequently decreasing the CR rate. Because of slow charge recombination, CT states can have rather long lifetimes, often reaching the microsecond range.34
Over the past decades, a number of efforts have been devoted to the design of electron donor–acceptor systems which efficiently produce long-living CT states.35 For many of these systems an alternative charge recombination process is observed, namely the recombination into local triplet excited states.36 This process is recognized as one of the most serious bottlenecks in the design of artificial photosynthetic systems.37
Two general pathways for the formation of triplets from CT states have been investigated and are schematically presented in Fig. 3a. The mechanism involving the formation of an intermediate triplet charge-transfer state (3CT) is known as a radical-pair intersystem crossing (RP-ISC).38 This process was found to occur in natural photosynthetic reaction centers39 and various electron donor–acceptor dyads which exhibit a weak electronic coupling between the donor and acceptor subunits due to long separation distances (e.g. >15 Å).40 ISC in the initially formed singlet charge-transfer state (1CT) happens via hyperfine interaction (HFI) – an interaction between an electron spin and a nuclear spin. ISC is followed by a fast charge recombination populating the lowest triplet excited state of either donor or acceptor subunit. The rate of RP-ISC is very sensitive to external magnetic fields and can be studied with several spectroscopic methods, e.g. time-resolved EPR and chemically induced dynamic nuclear polarization (CIDNP).41
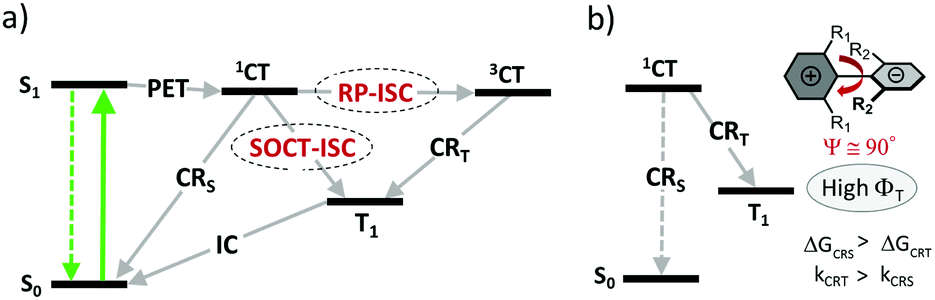 | ||
| Fig. 3 (a) Jablonski diagram showing possible mechanisms of triplet state formation in electron donor–acceptor dyads. PET – photoinduced electron transfer, 1CT – singlet charge-transfer state, 3CT – triplet charge-transfer state. RP-ISC – radical pair intersystem crossing, SOCT-ISC – spin–orbit charge transfer intersystem crossing. (b) Schematic representation of conditions required for efficient SOCT-ISC in a closely-spaced donor–acceptor dyad. CRT and CRs – charge recombination into the local triplet state and into the ground state, respectively. | ||
For donor–acceptor systems with stronger electronic couplings, ISC via HFI is less probable, since the energy splitting between 1CT and 3CT states becomes larger than the HFI energy. An alternative ISC pathway which can take place in such molecules is a direct conversion of 1CT into T1 state, involving a back electron transfer and a spin inversion. This process is referred to as a spin–orbit charge transfer intersystem crossing (SOCT-ISC). It is enhanced if the subunits are in a near perpendicular orientation, which allows to compensate electron spin angular momentum changes during ISC by molecular orbit angular momentum changes.42 This mechanism is similar to the ISC in aromatic carbonyl compounds, where the S1(n,π*) → T2(n,π*) transition can be regarded as a transfer of an electron from the lone pair of the oxygen atom to the π* orbital located on the carbon atom.43
SOCT-ISC was recognized as a major mechanism responsible for the formation of triplets in closely-spaced dyads, i.e. those in which the donor and the acceptor are directly linked through a single C–C bond. Steric hindrance between the subunits in such dyads leads to their orthogonal arrangement, which induces a large variation of the orbital magnetic momentum during electron transfer. This compensates the change of spin magnetic momentum, essential for the occurrence of ISC. The probability of SOCT-ISC is substantially reduced for dyads with dihedral angles between the subunits of less than 70°, leading to reduced triplet state yields. Nevertheless, triplet states formation in non-orthogonal BODIPY dyads was noted in several works, which are discussed in sections 3.2 and 3.3.
As was demonstrated for various dyads and dimers, the triplet state yield from 1CT state depends on the rates of two competitive relaxation pathways: charge recombination into the local triplet state (CRT) and recombination into the ground singlet state (CRS), kCRT and kCRS, respectively (Fig. 3b).44 High triplet state yields can be achieved if kCRS is substantially lower than kCRT. This condition is met, for example, if the driving force of the CRS process (ΔGCRS) has large negative values and falls within the Marcus inverted region. In this case, charge recombination into the lowest triplet excited state can be considerably faster because the corresponding Gibbs free energy change (ΔGCRT) is smaller due to a smaller 1CT–T1 energy gap.
BODIPYs have been employed both as electron donors and acceptors in a number of dyads undergoing PET.45 Surprisingly, the development of triplet sensitizers operating via SOCT-ISC has attracted attention only recently. The formation of triplets upon CT state recombination in the absence of heavy atoms was studied for the first time in BODIPYs covalently attached to buckminsterfullerene, C60.46 Applications of these systems as triplet sensitizers in photocatalysis47 and photon upconversion,48 have been demonstrated and are discussed in a recent review by Zhao.14 However, preparation of such compounds costs a considerable synthetic effort, limiting the opportunities for their practical use. For this reason, compact dyad molecules capable of triplet state formation, discussed in the following section, are particularly interesting.
3. BODIPY donor–acceptor dyads
3.1 Meso-phenyl, naphthyl- and pyridyl BODIPYs
The presence of aryl substituents in the BODIPY core is known to have a strong influence on its excited state dynamics and luminescent properties.49 As reported by the groups of Daub50 and Nagano,51 various BODIPYs bearing an electron donating meso-aryl group undergo PET and form charge-transfer excited states. Zhang and co-workers systematically studied singlet oxygen generation for a series of molecules 5–12, in which the aryl group plays the role of electron donor (Fig. 4a). Compared to the reference meso-phenyl BODIPY 4 possessing intense fluorescence and low singlet oxygen quantum yields (ΦΔ) in all solvents, dyad 5 bearing a 2-methoxyphenyl group exhibited a progressive quenching of the fluorescence and singlet oxygen quantum yield values, which increased with solvent polarity (Table 1). By changing the number and the position of methoxy substituents in meso-phenyl group (compounds 5–9), singlet oxygen generation was optimized to reach 46% yield.52,54a Characteristic charge-transfer emission bands were observed for compounds 6 and 9, having substituents in ortho positions of the aryl group, which hinder its rotation and secure orthogonal arrangement with respect to the BODIPY subunit. Formation of BODIPY triplets upon CT state recombination was confirmed by transient absorption (TA) experiments for 9, with a lifetime estimated to be 6.4 μs.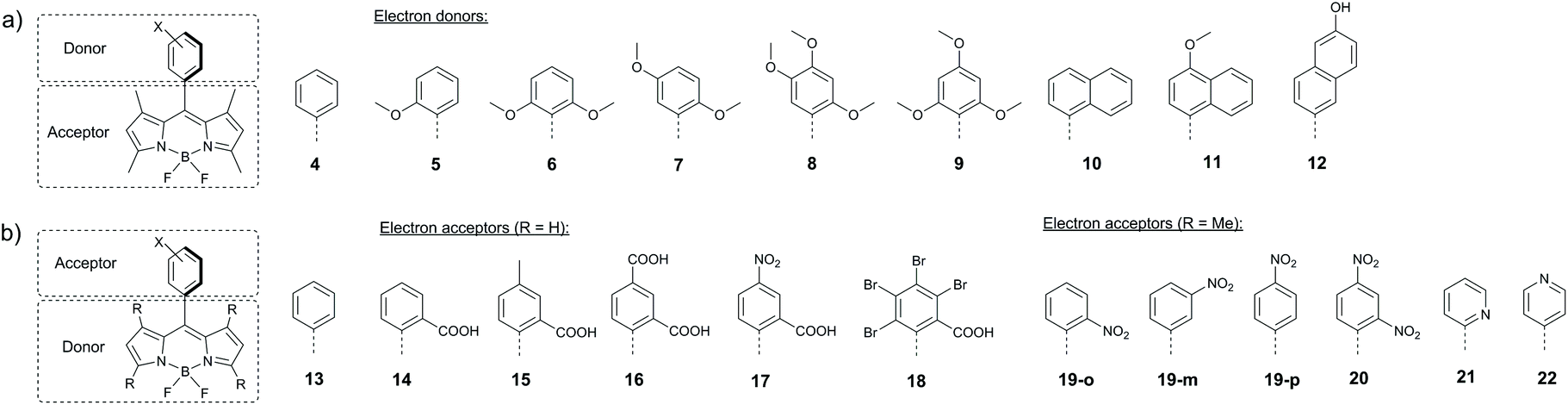 | ||
| Fig. 4 Structures of the BODIPYs incorporating electron donating (a) and electron accepting (b) meso-aryl groups and reference compounds (4 and 13). | ||
| Compound | Solvent (εr)a |
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (nm) b (nm) |
λ fl (nm) | Φ fl |
Φ
Δc |
Ref. |
|---|---|---|---|---|---|---|
| a εr – dielectric constant of the solvent. b Low energy band corresponding to the BODIPY chromophore. c Determined using singlet oxygen trapping with diphenylisobenzofuran (DPIBF). d Not reported. e Not soluble. | ||||||
| 4 | Hexane (1.89) | 501 | 511 | 0.56 | 0.05 | 53 |
| CH3CN (37.5) | 497 | 508 | 0.52 | 0.017 | 53 | |
| 5 | Hexane (1.89) | 504 | 516 | 0.98 | 0.029 | 52 |
| THF (7.58) | 503 | 516 | 0.87 | 0.061 | 52 | |
| CH3CN (37.5) | 499 | 512 | 0.57 | 0.18 | 52 | |
| 6 | Hexane (1.89) | 507 | 519 | 0.71 | 0.04 | 52 |
| THF (7.58) | 506 | 520 | 0.64 | 0.051 | 52 | |
| CH3CN (37.5) | 500 | 513 | 0.54 | 0.18 | 52 | |
| 7 | Hexane (1.89) | 504 | 517 | 0.971 | 0.026 | 54a |
| THF (7.58) | 503 | 514 | 0.457 | 0.462 | 54a | |
| CH3CN (37.5) | 499 | 510 | 0.01 | 0.125 | 54a | |
| 8 | Hexane (1.89) | 504 | 518 | 0.863 | 0.11 | 54a |
| THF (7.58) | 503 | 514 | 0.004 | 0.357 | 54a | |
| CH3CN (37.5) | 500 | 510 | 0.001 | 0.033 | 54a | |
| 9 | Hexane (1.89) | 505 | 520 | 0.95 | 0.02 | 52 |
| THF (7.58) | 506 | 517 | 0.78 | 0.06 | 52 | |
| CH3CN (37.5) | 501 | 513 | 0.55 | 0.31 | 52 | |
| 10 | Hexane (1.89) | 503 | 513 | 0.87 | 0.05 | 53 |
| THF (7.58) | 505 | 514 | 0.85 | 0.13 | 53 | |
| CH3CN (37.5) | 500 | 510 | 0.83 | 0.057 | 53 | |
| 11 | Hexane (1.89) | 503 | 515 | 0.906 | 0.011 | 54a |
| THF (7.58) | 503 | 516 | 0.438 | 0.232 | 54a | |
| CH3CN (37.5) | 499 | 512 | 0.123 | 0.872 | 54a | |
| 12 | Hexane (1.89) | 500 | 511 | 0.637 | 0.047 | 54a |
| THF (7.58) | 500 | 513 | 0.581 | 0.442 | 54a | |
| CH3CN (37.5) | 498 | 508 | 0.118 | 0.081 | 54a | |
| 13 | Hexane (1.89) | 498 | 513 | 0.031 | 55 | |
| EtOH (24.5) | 498 | 514 | 0.025 | 0.03 | 55 | |
| 14 | Hexane (1.89) | 500 | 519 | 0.61 | 55 | |
| EtOH (24.5) | 496 | 513 | 0.66 | 0.04 | 55 | |
| 15 | Hexane (1.89) | 499 | 516 | 0.69 | 55 | |
| EtOH (24.5) | 496 | 513 | 0.66 | 0.06 | 55 | |
| 16 | Hexane (1.89) | 55 | ||||
| EtOH (24.5) | 500 | 523 | 0.47 | 0.07 | 55 | |
| 17 | Hexane (1.89) | 505 | 527 | 0.35 | 55 | |
| EtOH (24.5) | 502 | 522 | 0.08 | 0.16 | 55 | |
| 18 | Hexane (1.89) | 516 | 526 | 0.49 | 55 | |
| EtOH (24.5) | 514 | 528 | 0.75 | 0.45 | 55 | |
| 19-o | Hexane (1.89) | 505 | 521 | 0.027 | 0.018 | 56 |
| MeOH (32.7) | 501 | 512 | 0.023 | 0.0083 | 56 | |
| 19-m | Hexane (1.89) | 505 | 521 | 0.42 | 0.0062 | 56 |
| MeOH (32.7) | 501 | 512 | 0.14 | 0.01 | 56 | |
| 19-p | Hexane (1.89) | 505 | 521 | 0.19 | 0.0067 | 56 |
| MeOH (32.7) | 501 | 512 | 0.03 | 0.0036 | 56 | |
| 20 | Hexane (1.89) | 514 | 510 | 0.033 | 0.021 | 56 |
| MeOH (32.7) | 510 | 508 | 0.045 | 0.0055 | 56 | |
| 21 | Hexane (1.89) | 505 | 515 | 0.059 | 0.0052 | 56 |
| MeOH (32.7) | 503 | 513 | 0.071 | 0.013 | 56 | |
| 22 | Hexane (1.89) | 503 | 517 | 0.25 | 0.0091 | 56 |
| MeOH (32.7) | 501 | 517 | 0.12 | 0.012 | 56 | |
Introduction of electron donating substituents was found to activate PET and singlet oxygen generation in meso-naphthyl BODIPYs.53,54 For dyad 10, electron transfer is thermodynamically unfavourable (ΔGPET > 0.2 eV) and it exhibits strong fluorescence even in polar solvents. On the other hand, dyads 11 and 12 with naphthyl subunits having lower oxidation potentials (EOx(D)) undergo PET and efficiently form triplet excited states, as indicated by high ΦΔ values (Table 1).
Notably, while the fluorescence quenching for compounds 5–12 becomes more profound in strongly polar solvents, singlet oxygen quantum yields do not correlate with solvent polarity. As is evident from the data presented in Table 1, for some representatives of this series ΦΔ values are much higher in the moderately polar tetrahydrofuran (εr = 7.58) than in acetonitrile (εr = 37.5). This is consistent with the solvent polarity effect on the rates of CRS and CRT processes and has been observed for many other BODIPY dyads (vide infra).
The BODIPY moiety can behave as an electron donating subunit, when combined with an appropriate electron acceptor. Particularly, dyads 14–18 were found to undergo electron transfer from the BODIPY to the meso-aryl group (Fig. 4b).55 ΔGPET value for compound 14 (−0.01 eV) indicates feasibility of electron transfer from the S1 state of the BODIPY to the meso-2-carboxyphenyl group in polar solvents. Introduction of extra carboxy or nitro substituents into the meso-aryl group increases its reduction potential (ERed(A)) and consequently the driving force for electron transfer. For instance, ΔGPET value of −0.84 eV was computed for meso-2,4-dicarboxyphenyl BODIPY 16. Sensitization experiments with 14–17 in ethanol showed up to five-fold enhancement of singlet oxygen generation with respect to the reference compound 13 (Table 1). At the same time, dyad 18 bearing four bromine atoms exhibited a much higher ΦΔ value of 0.45, due to the heavy atom-promoted ISC.
However, as was further demonstrated by Zhang and co-workers on systems 19–22, efficient PET from the BODIPY subunit to the electron-accepting aryl group does not always result in triplet state formation.56 ΔGPET for nitrophenyl-substituted BODIPYs 19 (ortho-, metha- and para-isomers) and 20 were found to be −0.34 and −0.73 eV, respectively, and the fluorescence parameters evidence the occurrence of PET even in non-polar hexane (Table 1). However, as was confirmed by TA data, charge recombination in these molecules leads only to the ground state. Similar behaviour was observed for pyridyl-substituted BODIPYs 21 and 22.
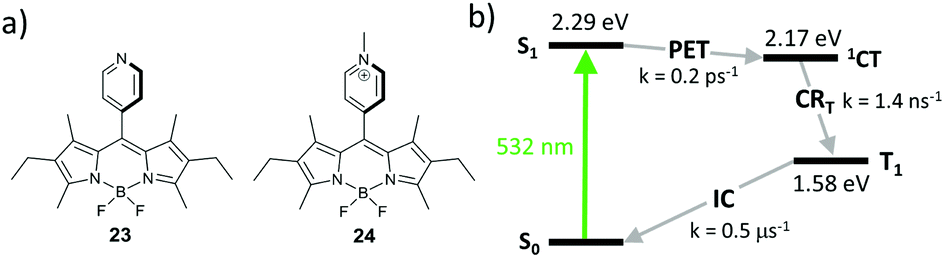
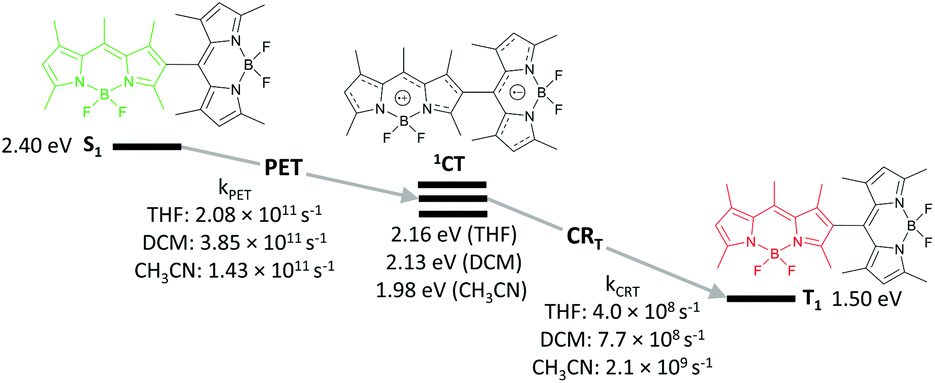
Harriman and co-workers reported hexaalkyl-substituted BODIPYs 23 and 24, bearing pyridyl and N-methylpyridinium groups in the meso-position, respectively (Fig. 5a).57 In contrast to the highly fluorescent 23 (Φfl = 0.78), in dyad 24 showed a rapid CT state formation in acetonitrile (kPET = 0.2 ps−1). This behaviour was attributed to pyridyl group methylation, which transforms it into a more easily reducible species, making the electron transfer from the BODIPY thermodynamically favourable (ΔGPET = −0.12 eV). The CT state in 24 recombines into the BODIPY triplet with a rate of 1.4 ns−1 (Fig. 5b), resulting in a triplet state yield of up to 75% in acetonitrile.
 | ||
| Fig. 5 (a) Structures of BODIPYs 23 and 24. (b) Jablonski diagram showing the excited state processes in 24 and corresponding kinetic parameters measured in acetonitrile. | ||
3.2 BODIPY dyads containing anthracene, pyrene and perylene groups
In 2010 Benniston and co-workers reported a solvent-dependent emission for meso-anthryl BODIPY 25 (Fig. 6), which indicate excited state intramolecular electron transfer between the subunits.58 The decay of CT state in 25 was proposed to lead to the BODIPY triplet, however it was not experimentally proved. Later, Filatov and co-workers demonstrated that a wide range of directly-linked BODIPY-anthracene dyads undergo PET from the anthracene to the BODIPY subunit, evidenced by the observation of radical-anion (BDP˙−) and radical-cation (Ant˙+) species in TA spectra.19a,59 Notably, charge recombination in these systems was found to produce long-living BODIPY triplet states in high yields. Singlet oxygen generation was investigated for a series of dyads based on BODIPY scaffolds with a different number (0 to 6) of alkyl substituents in the pyrrole rings, for instance, dyads 3 and 26–30 (Fig. 6). It was shown that the substituents in positions 1 and 7 (β-positions of pyrrole rings) control the molecular geometry and absence of alkyl groups in these positions opens the possibility of mutual rotation of the subunits, whereas introduction of methyl or ethyl groups results in a near orthogonal arrangement. Besides that, electron donating alkyl groups affect the reduction potentials of the BODIPY core,60 thus affecting the driving force of PET. The fluorescence parameters and ΦΔ values of these dyads in non-polar and polar solvents are compared in Table 2. The most efficient singlet oxygen generation (ΦΔ up to 0.67) was observed in ethanol for dyads 3, 29 and 30, based on a tetramethyl-substituted BODIPY scaffold, which are rigidly constrained in the orthogonal geometry. Alternatively, in hexane electron transfer is thermodynamically unfavourable and the dyads display low ΦΔ values and intense fluorescence emission.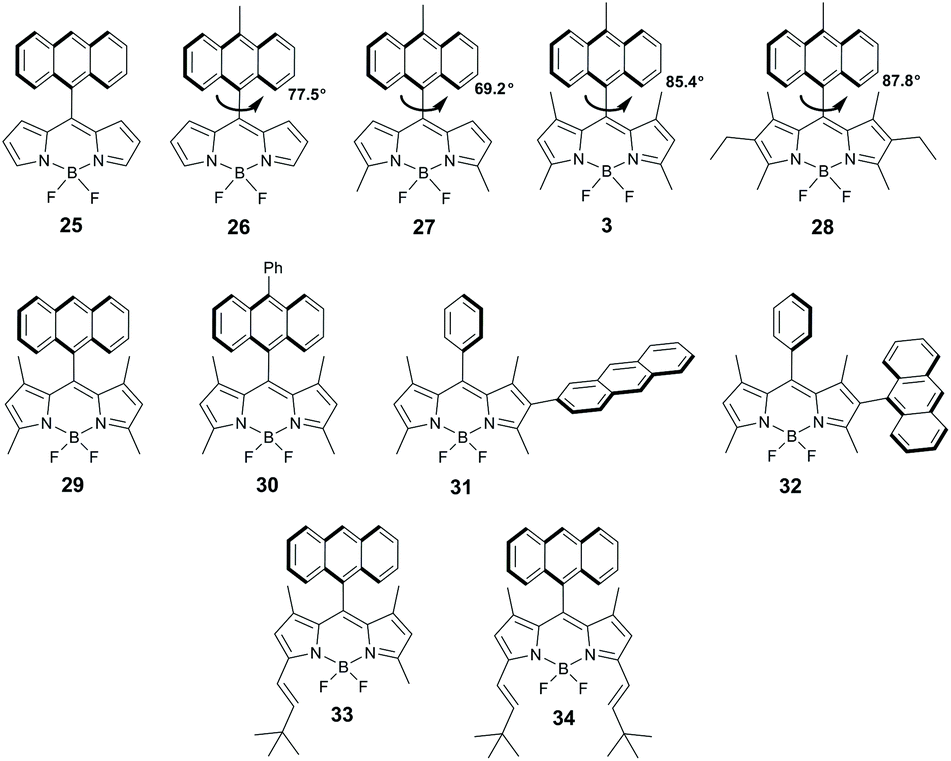 | ||
| Fig. 6 (a) Structures of BODIPY-anthracene dyads 25–34. The values of dihedral angles between the subunits (determined from single crystal X-ray data) are shown for dyads 3 and 26–28 to demonstrate the effect of alkyl substituents on the molecular geometry. | ||
| Compound | Solvent (εr)a |
λ
absb (nm) |
λ fl (nm) | Φ fl |
Φ
Δc |
τ
Td, μs |
Ref. |
|---|---|---|---|---|---|---|---|
| a εr – dielectric constant of the solvent. b Low energy band corresponding to the BODIPY chromophore. c Determined using singlet oxygen trapping with diphenylisobenzofuran (DPIBF). d BODIPY triplet state lifetime determined from TA spectroscopy data. e Not reported. | |||||||
| 3 | Hexane (1.89) | 505 | 512 | 0.91 | 0.04 | 59 | |
| DCM (8.93) | 506 | 519, 643 | 0.01 | 246 | 93 | ||
| EtOH (24.5) | 504 | 516, 627 | <0.01 | 0.67 | 59 | ||
| DMF (36.7) | 506 | 519, 612 | <0.01 | 41 | 19a | ||
| 25 | Hexane (1.89) | 505 | 521 | 0.135 | 0.39 | 59 | |
| EtOH (24.5) | 503 | 520, 716 | <0.01 | 0.11 | 59 | ||
| 26 | Hexane (1.89) | 504 | 566 | 0.04 | 0.38 | 59 | |
| EtOH (24.5) | 503 | 528 | <0.01 | 0.05 | 59 | ||
| 27 | Hexane (1.89) | 515 | 525 | 0.37 | 0.17 | 59 | |
| EtOH (24.5) | 515 | 533, 670 | <0.01 | 0.38 | 59 | ||
| 28 | Hexane (1.89) | 529 | 540 | 0.88 | 0.03 | 59 | |
| EtOH (24.5) | 528 | 538 | 0.31 | 0.32 | 59 | ||
| 29 | Hexane (1.89) | 504 | 515 | 0.99 | 0.01 | 59 | |
| Toluene (2.38) | 508 | 521 | 0.84 | 0.04 | 345 | 19c | |
| DCM (8.93) | 506 | 518 | 0.14 | 0.82 | 82 | 19c | |
| EtOH (24.5) | 505 | 513 | 0.04 | 0.53 | 59 | ||
| CH3CN (37.5) | 502 | 510 | 0.01 | 0.86 | 78 | 19c | |
| 30 | Hexane (1.89) | 505 | 514 | 0.9 | 0.04 | 59 | |
| Toluene (2.38) | 508 | 521 | 0.81 | 0.10 | 317 | 19c | |
| DCM (8.93) | 506 | 518, 595 | 0.01 | 0.95 | 85 | 19c | |
| EtOH (24.5) | 504 | 512, 606 | 0.02 | 0.59 | 59 | ||
| CH3CN (37.5) | 503 | 512 | <0.01 | 0.84 | 68 | 19c | |
| 31 | Toluene (2.38) | 520 | 568 | 0.42 | 0.11 | 102 | 19c |
| DCM (8.93) | 517 | 575 | 0.20 | 0.13 | 116 | 19c | |
| CH3CN (37.5) | 513 | 587 | 0.04 | 0.05 | 125 | 19c | |
| 32 | Toluene (2.38) | 516 | 556 | 0.39 | 0.20 | 127 | 19c |
| DCM (8.93) | 513 | 584 | 0.10 | 0.24 | 118 | 19c | |
| CH3CN (37.5) | 510 | 524, 627 | 0.01 | 0.11 | 137 | 19c | |
| 33 | Chloroform (4.81) | 549 | 556 | 0.32 | 62 | ||
| 34 | Chloroform (4.81) | 582 | 589 | 0.58 | 62 | ||
| 35 | Hexane (1.89) | 503 | 520 | 0.16 | 0.02 | 64 | |
| EtOH (24.5) | 503 | 660 | <0.01 | 0.75 | 64 | ||
| 36 | Hexane (1.89) | 514 | 526 | 0.75 | 0.01 | 64 | |
| EtOH (24.5) | 513 | 524 | 0.06 | 0.25 | 64 | ||
| 37 | Hexane (1.89) | 503 | 513 | 0.97 | 0.01 | 64 | |
| EtOH (24.5) | 502 | 511 | 0.65 | 0.34 | 64 | ||
| 38 | Hexane (1.89) | 538 | 542 | 0.755 | 0.01 | 64 | |
| EtOH (24.5) | 534 | 542 | 0.7 | 0.04 | 64 | ||
| 39 | Toluene (2.38) | 500, 560 | 690 | 0.067 | 0.31 | 196 | 65 |
| 40 | DCM (8.93) | 506 | 522, 677 | 0.037 | 0.42 | 216 | 65 |
| 41 | Hexane (1.89) | 529 | 540 | 0.92 | 0.01 | 64 | |
| EtOH (24.5) | 529 | 536 | 0.07 | 0.13 | 64 | ||
| 43 | Toluene (2.38) | 0.673 | 116 | 66 | |||
| DCM (8.93) | 502 | 510 | 0.027 | 0.013 | 66 | ||
| 44 | Toluene (2.38) | 0.246 | 13 | 66 | |||
| DCM (8.93) | 502 | 510 | 0.072 | <0.001 | 66 | ||
| 54 | Hexane (1.89) | 512 | 526 | 0.20 | 0.01 | 59 | |
| EtOH (24.5) | 511 | 526 | 0.04 | 0.01 | 59 | ||
| 55 | Hexane (1.89) | 511 | 526 | 0.23 | 0.01 | 64 | |
| EtOH (24.5) | 510 | 522 | 0.14 | 0.01 | 64 | ||
The mechanism of the BODIPY triplet state formation in several BODIPY-anthracene dyads, including 3, was later investigated by Mani and co-workers.44 The absence of RP-ISC mechanism contribution to the formation of triplets was confirmed by conducting TA in the presence of an external magnetic field of up to 3000 Gauss, which showed no effect on ΦT values. The lack of a magnetic field effect (MFE) proves that the triplets are formed via SOCT-ISC mechanism and RP-ISC is not involved. In addition, the absence of the anthracene triplet state was noted, indicating that the charge recombination in these systems leads exclusively to the BODIPY triplet state.
Comparison of kinetic parameters for two competing CT state recombination pathways – into the ground state (CRS) and into the triplet state (CRT, Fig. 7) has shown that high triplet state yield (ΦT = 0.9) in 3 is associated with a large difference in the rates of these processes. Due to the large negative Gibbs free energy change for the ground state recombination (ΔGCRS = −2.42 eV), the process exhibits Marcus inverted region behavior. On the other hand, the free energy change associated with the CRT process is significantly smaller (ΔGCRT = −0.81 eV), resulting in kCRT value of an order of magnitude higher, compared to kCRS. Importantly, similar behavior was observed for several types of dyads, e.g. having different substitution patterns of the BODIPY core or containing other electron donors (vide infra), proving the versatility of the inverted region effect.
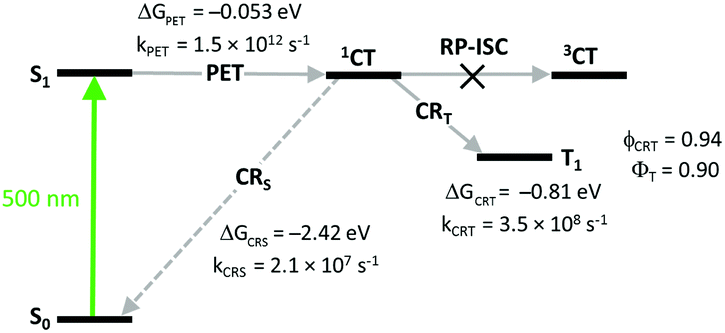 | ||
| Fig. 7 Jablonski diagram showing excited state transitions in dyad 3 and corresponding kinetic parameters measured in acetonitrile. | ||
Zhao and co-workers described dyads 31 and 32, in which the anthracene subunit is attached to the 2-position of the BODIPY core (Fig. 6). Both compounds showed efficient PET in acetonitrile, but modest triplet state yields (0.06 and 0.16 for 31 and 32, respectively) in contrast to meso-anthracenyl dyads 29 and 30 (0.96 and 0.92, respectively).19c In the case of 31 this was attributed to the effect of twisted geometry, the dihedral angle between the BODIPY and anthracene subunits (52°) does not satisfy the requirements for SOCT-ISC. Low triplet state yield in orthogonal dyad 32 was proposed to be associated with an unfavourable mutual orientation of the transition dipole moments of the subunits, reducing the efficiency of the CRT process.
In a follow-up report of Dick and co-workers, the formation of triplet states in dyads 29–32 was studied with time-resolved electron paramagnetic resonance (TREPR) spectroscopy.61 For dyad 30, three different triplet states were detected: the triplet state localized on the anthracene (3Ant) subunit, the BODIPY triplet state (3BDP) and the triplet charge-transfer state (3CT). Although the pathway for the 3CT state generation was not unequivocally revealed, based on the electron spin polarization (ESP) data for these three triplet states, it was concluded that the 1CT → 3CT transition is not the dominant pathway and the role of SOCT-ISC as the main intersystem crossing mechanism was confirmed.
Styryl-substituted BODIPY-anthracene dyads 33 and 34 were prepared by Yang and co-workers from the corresponding tetramethyl-substituted precursor 29.62 Both dyads exhibited significant bathochromic shifts in absorption and fluorescence spectra, with respect to 29 (Table 2); however, PET between the BODIPY and anthracene subunits was not observed.
Energy and electron transfer processes in BODIPY-pyrene dyads have been reported in several works,63 but the formation of triplets via SOCT-ISC in these systems was only recognized very recently. Compounds 35–38 (Fig. 8) were shown to generate singlet oxygen in polar solvents (Table 2) with ΦΔ values depending on the number of alkyl substituents in the BODIPY subunit (Table 2).64 All dyads of this series have close to orthogonal geometries (dihedral angles 81–89°). Highly efficient sensitization was observed for alkyl-unsubstituted 35 (ΦΔ = 0.75 in ethanol), which exhibited an ultrafast electron transfer (k−1 = 0.49 ps) from the pyrene to the BODIPY subunit. Dyads 36 and 37, based on dimethyl and tetramethyl-substituted BODIPY scaffolds, respectively, display lower ΦΔ values in ethanol (0.25–0.34) and negligible sensitization ability in hexane. Dyad 38 containing six alkyl groups showed much lower ΦΔ values in both solvents. Mani and co-workers were able to demonstrate that the presence of methyl groups in the BODIPY core makes electron transfer from the pyrene subunit in 37 thermodynamically unfavourable even in acetonitrile (ΔGPET = 0.083 eV).44 The presence of two additional ethyl groups in 38 further reduces the driving force of the process (ΔGPET = 0.27 eV). As a result, the S1 state of the BODIPY in 38 decays predominantly via fluorescence emission, resulting in a rather low triplet state yield (ΦT = 0.35).
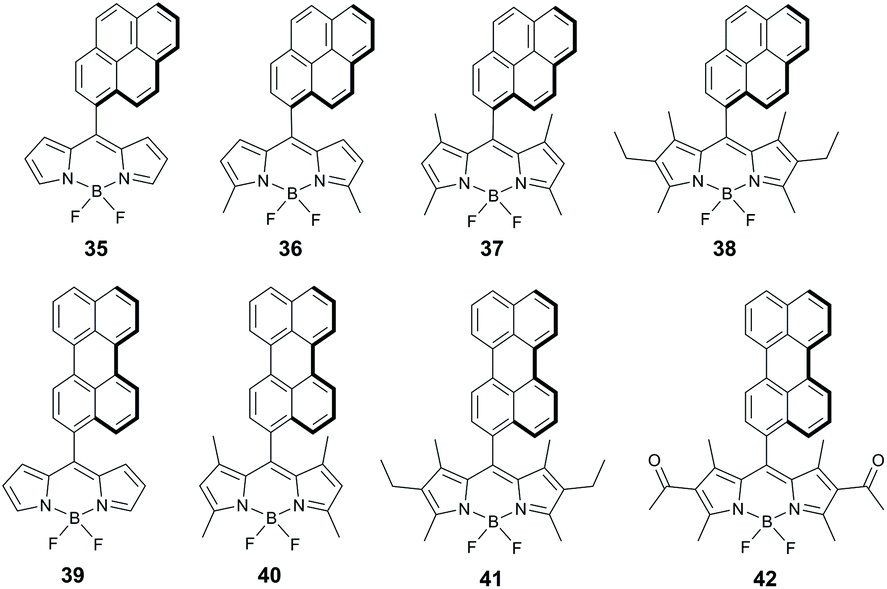 | ||
| Fig. 8 Structures of dyads 35–41 containing pyrene and perylene groups as electron donors. | ||
Zhao and co-workers studied dyads 39 and 40 (Fig. 8), employing unsubstituted and tetramethyl-substituted BODIPY scaffolds as electron accepting subunits, respectively.65 Both dyads were found to generate long-living triplet states (196 μs and 216 μs for 39 and 40, respectively) from the corresponding CT states. Noteworthy, dyad 39 showed a high triplet state yield although the molecule is twisted (dihedral angle 66°) due to the absence of methyl groups in positions 1 and 7 of the BODIPY. A remarkable feature of 39, which was not observed for 40, is a moderate, broad absorption in the range of 535–635 nm (Fig. 9a). On the basis of DFT computations this band was assigned to a charge-transfer absorption, i.e. a direct S0 → 1CT transition. The appearance of this band was explained by an enhanced electronic coupling between the BODIPY and the pyrene chromophores in this dyad. The triplet state yield, upon excitation into the CT band at 589 nm (Fig. 9b), was determined to be 0.43, comparable with excitation into the BODIPY absorption band at 500 nm (ΦT = 0.66). Excitation of 39 into the CT absorption band was demonstrated for anti-Stokes shift enhancement in photon upconversion.
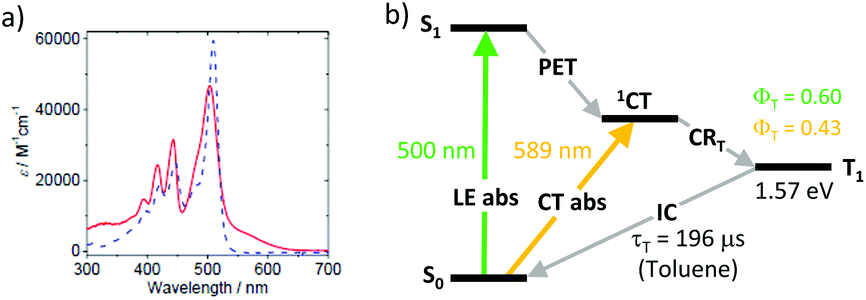 | ||
| Fig. 9 (a) Absorption spectra of dyads 39 (red line) and 40 (blue dashed line) in toluene. Adapted from ref. 65. (b) Jablonski diagram showing two pathways for the BODIPY triplet state formation in 39. | ||
BODIPY-perylene dyad 41 was reported to have rather low sensitization efficiency in ethanol (ΦΔ = 0.13).64 Mani and co-workers showed that the electron transfer from perylene to the BODIPY subunit in 41 is thermodynamically favourable in polar solvents (e.g. ΔGPET = −0.031 eV in acetonitrile).44 However, the rate of ground state recombination was found to be substantially higher than for recombination into triplet state (3.1 × 108 s−1 and 2.0 × 108 s−1, respectively). This accounts for the modest triplet state yield (ΦT = 0.33) in this dyad.
Introduction of electron accepting acyl groups was demonstrated to promote PET from perylene to the BODIPY subunit in dyad 42 (ΔGPET = −0.69 eV in acetonitrile). Negligible triplet state yield (ΦT = 0.005) observed for this dyad in acetonitrile was attributed to a strong stabilization of CT state in highly polar media and a reduced energy gap between 1CT and S0 states, which promotes ground state recombination.44 The rate of CRS was found to be two orders of magnitude higher than for CRT (1.9 × 1010 s−1 and 0.97 × 108 s−1, respectively) in this solvent. The triplet state yield in 42 greatly increases in less polar toluene (ΦT = 0.2), due to the increased CT state energy level and 1CT–S0 energy gap, unfavourable for the CRS process.
3.3 Directly linked dyads vs. spacer-separated BODIPY dyads
Excited state dynamics of directly-linked and phenylene-separated BODIPY-phenothiazine dyads 43 and 44 (Fig. 10a) was compared by Di Donato and co-workers.66 The geometry of 43 is almost orthogonal (dihedral angle 87°) due to the steric hindrance caused by methyl groups in positions 1 and 7 of the BODIPY. Introduction of the phenylene group in 44 allows mutual rotation of the subunits and leads to a twisted geometry (dihedral angle ∼40°). The electron transfer from phenothiazine to the BODIPY subunit is thermodynamically favourable for both dyads in acetonitrile (ΔGPET < −0.8 eV) and, in the case of 43, even in toluene (ΔGPET = −0.03 eV). An increased distance between the BODIPY and phenothiazine results in almost an order of magnitude slower PET, the rates in acetonitrile were measured to be 2 × 1012 s−1 and 2.2 × 1011 s−1 for 43 and 44, respectively. Importantly, the rate of CRT process is also substantially reduced for 44, compared to 43 (4.5 × 1010 s−1 and 1.5 × 1011 s−1, respectively). Very high triplet state yield was found for 43 in toluene (ΦT = 0.975). Dyad 44 showed much lower triplet state yield in this solvent (ΦT = 0.134) due to inefficient charge separation step. In more polar solvents triplet state formation is reduced for both dyads. As can be seen from Fig. 10a, this correlates with the energies of CT states. In toluene the energy gap between the 1CT and S0 states is larger than in acetonitrile, which makes the combination of ΔGCRT and ΔGCRS values favorable for the triplet state recombination.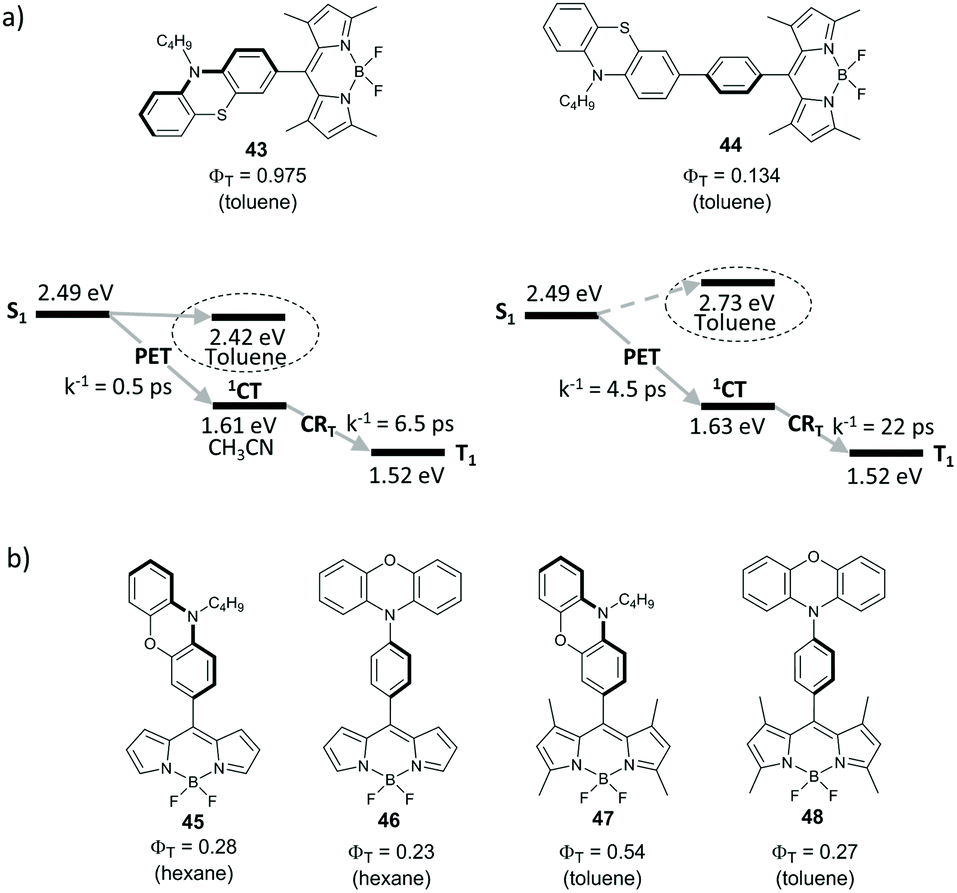 | ||
| Fig. 10 (a) Structures of phenothiazine-BODIPY dyads 43 and 44 and energy level diagrams comparing the parameters of excited state processes in these dyads in acetonitrile and toluene. (b) Structures of phenoxazine-BODIPY dyads 45–48. | ||
Voronkova and co-workers investigated triplet state formation via SOCT-ISC in BODIPY dyads 45–48 (Fig. 10b), employing a phenoxazine group as an electron donor.67 Efficient electron transfer was observed for all of these dyads in both non-polar and polar solvents. This was explained by higher stability of charge-transfer states formed in 45–48, compared to corresponding phenothiazine dyads. For instance, the CT state energy level in 47 was estimated to be 2.04 eV in toluene, i.e. by 0.38 eV lower than for dyad 43 in this solvent. Dyad 45, based on alkyl-unsubstituted BODIPY, possesses twisted geometry (dihedral angle between the subunits 49.6°) and in the spacer-separated analogue 46 the subunits are in an almost coplanar orientation. Despite non-orthogonal geometry, dyads 45 and 46 showed appreciable triplet state yields of 28% and 25% respectively, in hexane. On the other hand, for orthogonal dyad 47 based on tetramethyl-substituted BODIPY scaffold, the triplet state yield reaches 54% in toluene. Separation of the subunits by the phenylene spacer in 48 results in a reduced triplet state yield (ΦT = 0.27) in the same solvent.
Benniston and co-workers studied dyads 49–52, with a benzoquinone group anchored either directly or through a phenylene spacer at the meso- and 2-positions of the BODIPY (Fig. 11).68 In toluene and more polar solvents electron transfer from the BODIPY to the quinone group occurs for all members of this series on the sub-picosecond timescale, leading to corresponding CT states. Decay times of CT states were found to be 11.5–24 ps and the decay pathway depends on the structure of the dyad and solvent polarity. Only ground state recombination processes were observed for 49–51 in all solvents. Alternatively, formation of the BODIPY triplet state with up to 21% yield was found for dyad 52 in solvents of intermediate polarity (ethyl acetate, diethyl ether, dichlorobenzene, toluene).
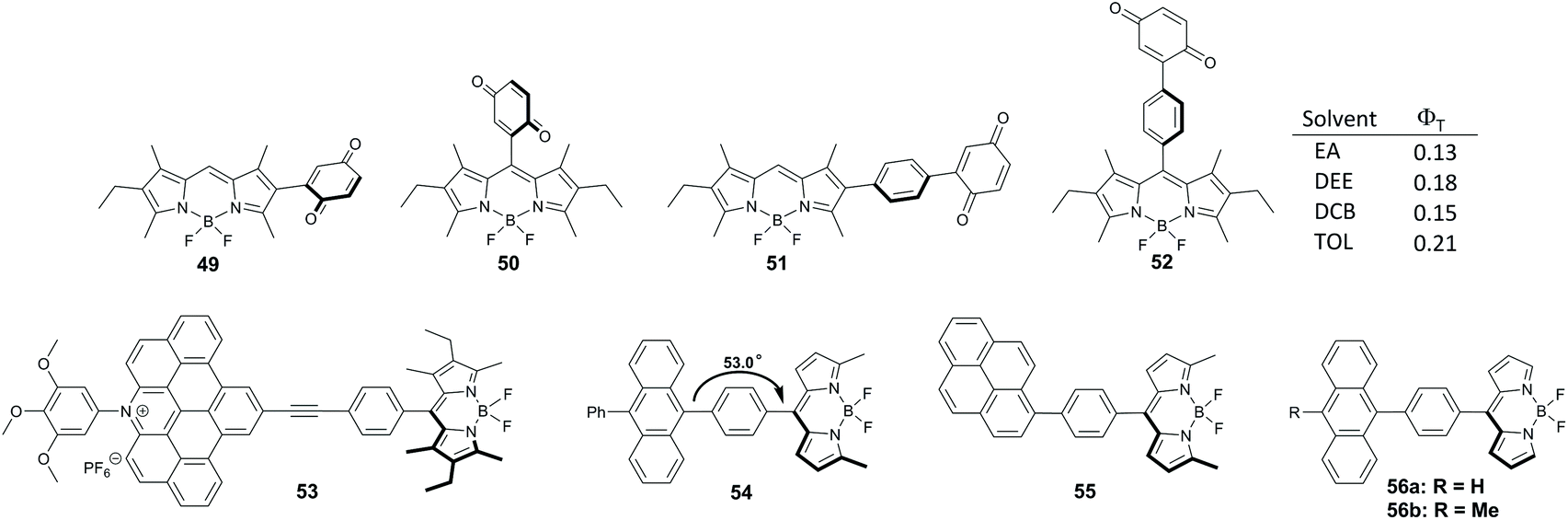 | ||
| Fig. 11 (a) Structures of dyads 49–56. The values of triplet state yields upon photoexcitation of 52 in different solvents (EA – ethylacetate, DEE – diethyl ether, DCB – dichlorobenzene, TOL – toluene) are shown. The value of dihedral angle between the subunits is shown for dyad 54. | ||
Tkachenko and co-workers described dyad 53 incorporating hexalkyl-substituted BODIPY and an expanded acridinium cation as electron donor and acceptor subunits, respectively, separated by a phenylethynyl spacer (Fig. 11).69 Excitation of 53 in acetonitrile leads to a CT state in which a positive charge is localized on the BODIPY and a negative on the acridinium subunit. Although this state recombines on the picosecond timescale predominantly back to the ground state, formation of the BODIPY triplet state with 15% yield was noted.
Phenylene-separated BODIPY-anthracene and pyrene dyads 54 and 55 were reported to undergo PET in polar solvents, however both exhibit very low singlet oxygen quantum yields (ΦΔ ≈ 0.01), evidencing low efficiency of the triplet state formation.59,64 The dihedral angle between the subunits in 54 is approximately 53°, suggesting that SOCT-ISC is not operative in this system.
Majima and co-workers described structurally similar BODIPY-anthracene dyads 56a–b, which do not exhibit PET in dimethyl sulfoxide and methanol.19d Interestingly, an intermolecular electron transfer between the anthracene and BODIPY moieties was found to be triggered by aggregation of these hydrophobic molecules in aqueous media. Singlet oxygen generation by the aggregates was confirmed by near-IR phosphorescence, although ΦΔ values were not reported. Based on the X-ray crystallographic data obtained for 56a–b, it was proposed that the lattice-like molecular packing between dyad molecules in the aggregates facilitates the electron transfer and triplet state formation through SOCT-ISC. Although the efficiency of the process has not been studied in detail, these results indicate that SOCT-ISC can be regulated by self-assembly and is likely to be feasible in the solid state.
4. BODIPY dimers
4.1 Symmetrical dimers
BODIPY dimers and oligomers have attracted attention due to their interesting properties linked to charge delocalization and exciton coupling. Depending on the nature of a bridge between the BODIPY subunits and its position, very different photophysical properties have been observed.70 Among these systems, directly linked symmetrical BODIPY dimers exhibit particularly interesting behaviour and were the first type of BODIPY compounds in which, a symmetry-breaking charge separation (SBCS) process was observed.SBCS or photoinduced electron transfer between two identical subunits in a single molecule was known to occur in several types of biaryl compounds, e.g. biphenyls, bianthryls and diperylenyls.71 For 9,9′-bianthryl derivatives, solvent-dependent fluorescence, showing characteristic broad and red-shifted emission bands along with a decrease in Φfl values is observed in polar solvents.72 In these molecules SBCS results in a radical pair species in which one anthracene subunit has a radical-anion (Ant˙−) character and the second is a radical-cation (Ant˙+). This process is not observed in a non-polar environment due to insufficient stabilization of the radical pair.
Occurrence of SBCS in BODIPY derivatives was shown for the first time by Thompson and co-workers on symmetrical dimer 57 (Fig. 12).73 The dimer possesses strong fluorescence in cyclohexane and toluene, while in polar acetonitrile excitation into the S1 state leads to an ultrafast charge separation (k−1 < 170 fs), which populates the CT state as evidenced by the appearance of a BODIPY radical-anion (BDP˙−) signal in TA spectra. Thus formed CT state was found to be rather long-living (kr−1 = 0.65 ns), proposed to be an effect of steric congestion hindering the rotation of the subunits and inhibiting charge recombination. The possibility of triplet state formation upon CT state recombination in 57 could be anticipated, however it has not been studied in detail.
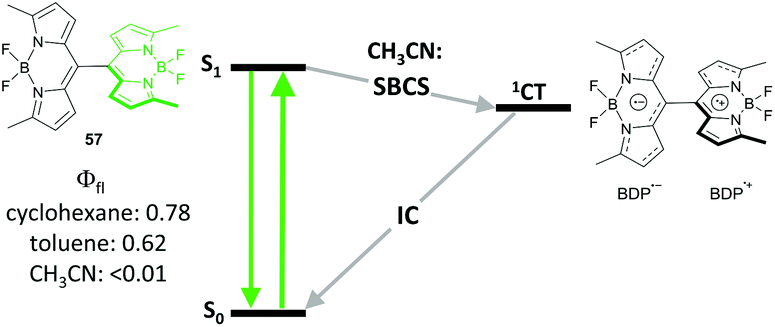 | ||
| Fig. 12 Jablonski diagram illustrating symmetry-breaking charge separation (SBCS) in dimer 57 in polar solvent. | ||
A series of symmetrical dimers 58–61 (Fig. 13) with orthogonal arrangement of the BODIPY subunits was studied by Bröring and co-workers and revealed enhanced ISC compared to the corresponding monomer 62.74 While the fluorescence characteristics of 62 are almost unchanged in different solvents (Table 3), the dimers displayed a notable dependence of the emission on solvent polarity.75 For compound 58 the fluorescence quantum yield decreases from toluene (0.71) to dichloromethane (0.56), and to a greater extent in acetonitrile, where the emission is strongly quenched (0.036). TA experiments proved that a decrease in Φfl values is due to the BODIPY triplet state formation. Triplet state lifetimes of 122 and 118 μs were measured for dimers 58 and 59, respectively, in toluene. The effect of solvent on the triplet state yield in 58–61 is evident from singlet oxygen quantum yield values, collected in Table 3. For all dimers of this series, ΦΔ is ca. 0.4 in toluene and 0.5 in dichloromethane, indicating high triplet state yields in these solvents. At the same time, triplet state formation was found to be substantially less efficient in acetonitrile (ΦΔ < 0.1).
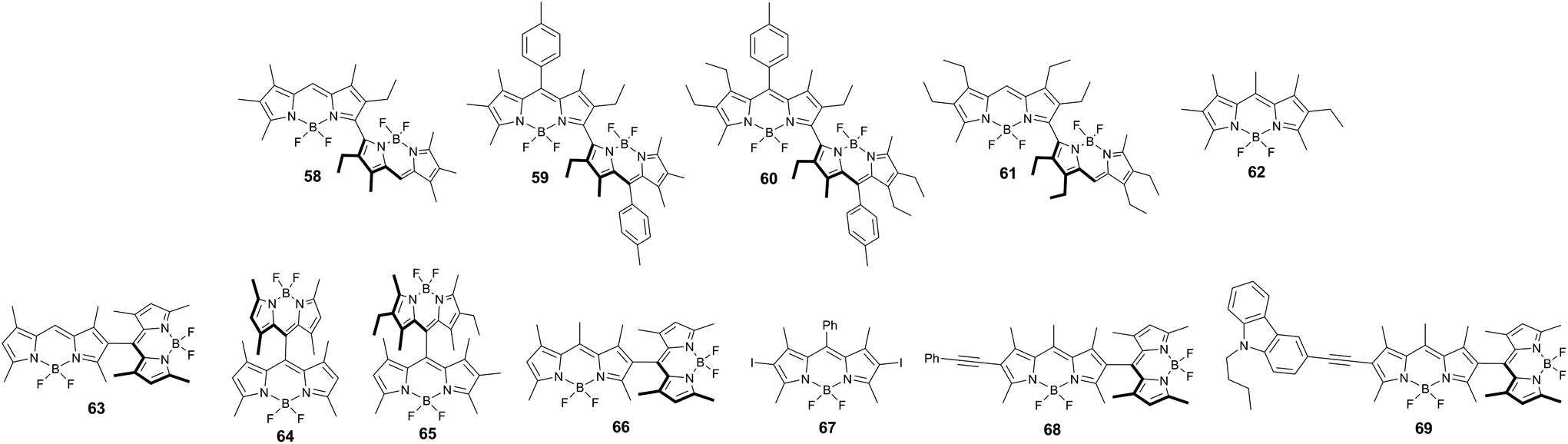 | ||
| Fig. 13 Structures of BODIPY dimers and reference compounds (62 and 67). | ||
| Compound | Solvent (εr)a | λ abs (nm) | λ fl (nm) | Φ fl | Φ Δ |
τ
Td (μs) |
Ref. |
|---|---|---|---|---|---|---|---|
| a εr – dielectric constant of the solvent. b Determined from singlet oxygen phosphorescence data. c Determined using singlet oxygen trapping with diphenylisobenzofuran (DPIBF). d Determined from TA spectroscopy data. e Not reported. | |||||||
| 58 | Toluene (2.38) | 462, 565 | 648 | 0.71 | 0.4b | 122 | 75 |
| DCM (8.93) | 489, 559 | 651 | 0.56 | 0.5b | 75 | ||
| CH3CN (37.5) | 484, 553 | 648 | 0.036 | <0.1b | 100 | 75 | |
| 59 | Toluene (2.38) | 489, 558 | 638 | 0.67 | 0.4b | 118 | 75 |
| DCM (8.93) | 487, 555 | 650 | 0.60 | 0.5b | 75 | ||
| CH3CN (37.5) | 483, 550 | 646 | 0.15 | <0.1b | 72 | 75 | |
| 60 | Toluene (2.38) | 490, 559 | 638 | 0.69 | 0.3b | 75 | |
| DCM (8.93) | 488, 557 | 650 | 0.62 | 0.5b | 75 | ||
| CH3CN (37.5) | 485, 553 | 650 | 0.24 | <0.1b | 75 | ||
| 61 | Toluene (2.38) | 494, 567 | 650 | 0.76 | 0.3b | 75 | |
| DCM (8.93) | 491, 562 | 652 | 0.63 | 0.5b | 75 | ||
| CH3CN (37.5) | 486, 557 | 652 | 0.086 | <0.1b | 75 | ||
| 62 | Toluene (2.38) | 534 | 540 | 1.0 | <0.1b | 75 | |
| DCM (8.93) | 531 | 537 | 1.0 | 75 | |||
| CH3CN (37.5) | 525 | 532 | 0.98 | <0.1b | 75 | ||
| 63 | Hexane (1.89) | 509 | 527 | 0.87 | 0.13c | 78 | |
| Chloroform (4.81) | 514 | 527 | 0.03 | 0.51c | 76 | ||
| DCM (8.93) | 511 | 528 | 0.0029 | 0.45c | 36 | 78 | |
| EtOH (24.5) | 506 | 521 | 0.0016 | 0.10c | 78 | ||
| 64 | Chloroform (4.81) | 515 | 588 | 0.31 | 0.46c | 76 | |
| 65 | Chloroform (4.81) | 542 | 605 | 0.49 | 0.21c | 76 | |
| 66 | Cyclohexane (2.0) | 509 | 524 | 0.92 | <0.01c | 19b | |
| Toluene (2.38) | 509 | 529 | 0.75 | 0.24c | 232 | 79, 19b | |
| Chloroform (4.81) | 505 | 527 | 0.22 | 0.75c | 19b | ||
| THF (7.58) | 505 | 525 | 0.17 | 0.86c | 21 | 79, 82, 19b | |
| DCM (8.93) | 506 | 527 | 0.022 | 0.64c | 115 | 79, 82 | |
| Acetone (20.7) | 503 | 532, 660 | 0.01 | 0.50c | 19b | ||
| CH3CN (37.5) | 501 | 530, 715 | <0.01 | 0.25c | 86 | 79, 19b | |
| 67 | DCM (8.93) | 529 | 552 | 0.03 | 0.92b | 57 | 82 |
| 68 | DCM (8.93) | 509, 541 | 578 | 0.176 | 0.42c | 141 | 82 |
| 69 | Toluene (2.38) | 510, 544 | 522, 615 | 0.083 | <0.01c | 79 | |
| DCM (8.93) | 508, 542 | 519, 644 | 0.023 | <0.01c | 82 | ||
Later Akkaya and co-workers reported symmetrical dimers 63 and 64 (Fig. 13) which generate singlet oxygen in chloroform with quantum yields of 0.51 and 0.46, respectively, and demonstrated their suitability for PDT.76 The non-symmetrical dimer 65, studied in this work, displayed much lower photosensitization efficiency (ΦΔ = 0.21). Based on theoretical calculations,77 it was argued that high ISC in these dimers is due to the formation of degenerate HOMOs and LUMOs provided by orthogonal molecular geometry, in which π-mixing is avoided, allowing the subunits to retain undisturbed monomeric orbital energies.
4.2 Non-symmetrical dimers
Ortiz and co-workers investigated singlet oxygen generation by the non-symmetrical orthogonal dimer 66 in different solvents and proposed that intramolecular electron transfer and the subsequent SOCT-ISC process are responsible for BODIPY triplet state formation in this molecule.19b Consistent with this model, the fluorescence quantum yield of 66, which is very high in non-polar solvents (e.g. Φfl = 0.92 in cyclohexane), greatly decreases as the polarity of the solvent increases, accompanied by the appearance of broad red-shifted bands, characteristic for CT states. ΦΔ values of 66 in solvents of intermediate polarity are comparable with those of the reference iodinated compound 67 (Table 3), while in acetonitrile the photosensitization efficiency is substantially reduced.Han and co-workers confirmed that the formation of triplet states in 66 takes place upon the CT state recombination.79 PET between the BODIPY subunits was monitored by the appearance of bands corresponding to the radical-cation (BDP˙+)80 and radical-anion (BDP˙−)81 species in TA spectra. In non-polar toluene, these species were not observed, consistent with the calculated Gibbs free energy change for the electron transfer process (ΔGPET = 0.24 eV). In THF, DCM and acetonitrile, the electron transfer is thermodynamically allowed (ΔGPET < −0.2 eV) and the CT state is populated on the picosecond timescale with rate constants correlating with solvent polarity (Fig. 14). Its recombination was found to produce local triplet state of the BODIPY and larger recombination rates were found in DCM and THF, than in acetonitrile, in agreement with the reported singlet oxygen quantum yield values in these solvents (Table 3).
 | ||
| Fig. 14 Energy level diagram showing excited state processes in dimer 66 and corresponding kinetic parameters in different solvents. The values of TD-DFT calculated excited states energies and rate constants of PET and CRT processes, determined by TA spectroscopy, are presented. | ||
Dimers 68 and 69, bearing phenylethynyl and carbazole groups appended to one of the BODIPY subunits, respectively, were studied by Zhao and co-workers. Compound 68 exhibited long-living triplet excited state (τT = 140.9 μs) formation and a high singlet oxygen generation quantum yield (ΦΔ = 0.42) in dichloromethane.82 Alternatively, dimer 69 shows negligible triplet state formation, although efficient PET between the subunits was observed in this molecule.
Jiao and co-workers reported a series of meso,β-linked dimers 70a–d (Fig. 15a) having different substitution patterns at the meso-aryl and/or pyrrolic positions of the BODIPY subunits.83 The ability of these dimers to generate triplet excited states in toluene was demonstrated by singlet oxygen trapping experiments, showing the highest efficiency for dimers 70b and 70d having orthogonal geometry. Dimers 70a and 70c in which the dihedral angles between the BODIPYs are 34° and 39°, respectively, exhibited substantially lower rates of DPIBF oxidation (ΦΔ values not reported). Triplet state and singlet oxygen quantum yields for dimer 70d were later measured by Zhang in different solvents.84 Photosensitization experiments with 70d reveal that it generates singlet oxygen only in hexane, cyclohexane and toluene, but not in polar solvents (Fig. 15b).
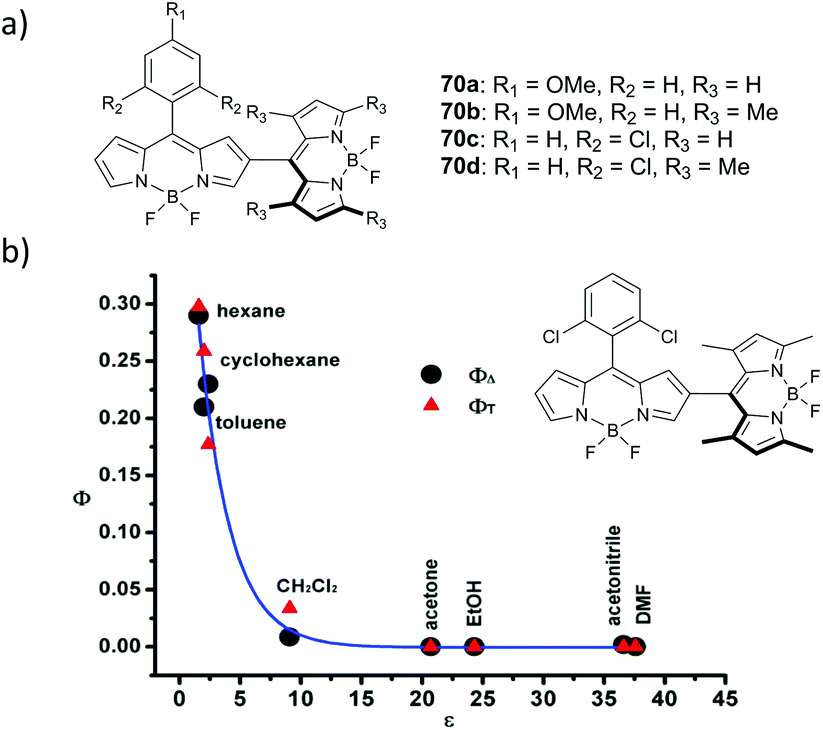 | ||
| Fig. 15 (a) Structures of dimers 70a–d. (b) Triplet state yield and singlet oxygen quantum yield values for 70d as a function of solvent polarity. Adapted with permission from ref. 84. Copyright (2013) American Chemical Society. | ||
Ortiz and co-worker examined the effects of electron-donating and accepting substituents on oxygen sensitization by asymmetrical dimers 71 and 72 (Fig. 16). Dimer 71, containing a 4-aminophenyl group showed higher singlet oxygen generation quantum yield (0.85) in chloroform compared to 66 (0.75).19b Alternatively, a 4-nitrophenyl-substituted dimer 72 showed reduced sensitization efficiency (ΦΔ = 0.43 in chloroform). This was attributed to the additional stabilization of the CT state by the electron-withdrawing effect of the nitrophenyl group.19b Han and co-workers estimated the CT state energy level in 72 to be 2.23 eV in toluene, making electron transfer thermodynamically allowed in this solvent (ΔGPET = −0.15 eV).79 A rather high singlet oxygen quantum yield was found for 72 in toluene (ΦΔ = 0.44), while in more polar solvents lower values were obtained, e.g. ΦΔ = 0.2 in dichloromethane.
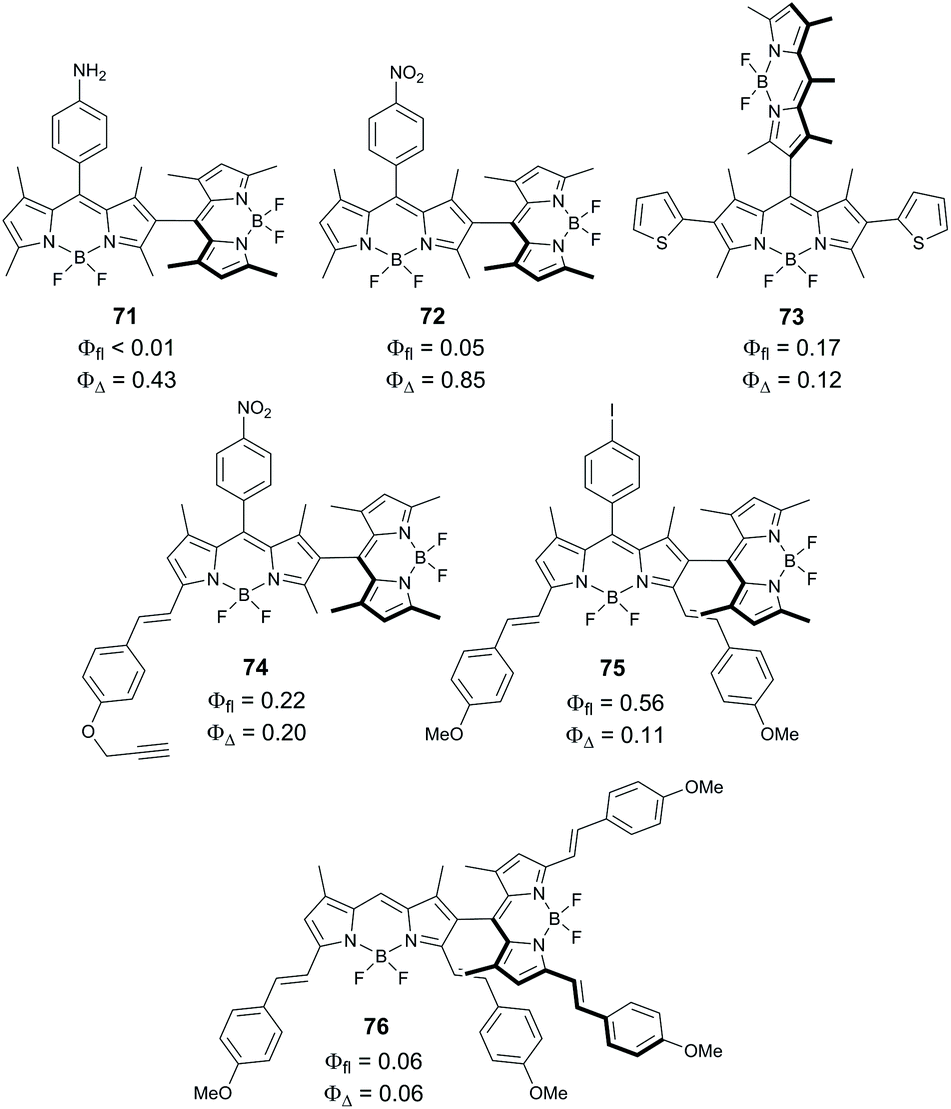 | ||
| Fig. 16 Structures of non-symmetrical BODIPY dimers 71–76 and photophysical parameters measured in chloroform.19b,77 | ||
Reports on triplet state formation via SOCT-ISC in dimers with extended π-conjugation are still very scarce. As shown by Ortiz and co-workers, the introduction of thienyl moieties into one of the BODIPY subunits in dimer 73 results in a red shift of the absorption (λmax = 525 nm) and emission (λmax = 627 nm) bands, however a modest singlet oxygen quantum yield was observed (ΦΔ = 0.12 in chloroform).19b In the case of the styryl-substituted dyad 74, selective excitation of the tetramethyl-substituted BODIPY subunit at 510 nm resulted in ΦΔ of 0.2. The yield dropped to 0.11 when the dimer was selectively excited at the absorption band corresponding to the styryl-BODIPY subunit (λmax = 589 nm). For the distyryl-substituted dimer 75, no singlet oxygen formation was detected under red light excitation. It was proposed that the extension of the BODIPY π-system enhances radiative processes in the CT state, thus hindering SOCT-ISC and generation of singlet oxygen. A similar result was reported by Akkaya and co-workers for the tetrastyryl-substituted dimer 76. Despite having orthogonal geometry, it shows a rather low singlet oxygen quantum yield of 0.06.77
4.3 Dimers with BODIPY subunits separated through a spacer
BODIPY dimers, in which the subunits are separated by a spacer have been reported in several works, with no appreciable triplet state formation observed. Zhao and co-workers studied the excited state dynamics of dimers 77 and 78 (Fig. 17) in which the BODIPY subunits are linked via phenylene group.79 Electron transfer is thermodynamically allowed for these systems in acetonitrile, ΔGPET were calculated to be −0.42 and −0.39 eV for 77 and 78, respectively. Transient absorption data proved that the formation of CT states does occur for both compounds. However, only charge recombination into the ground state was observed for these dimers in all solvents studied. Solid-state structure of 77 showed that the two BODIPY subunits are almost parallel to each other (dihedral angle of 2°), and it was concluded that the SOCT-ISC process does not occur in these systems.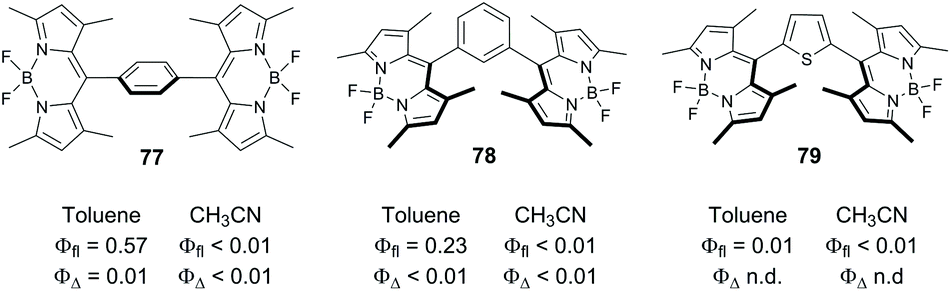 | ||
| Fig. 17 Structures of spacer-separated BODIPY dimers 77–79 and photophysical parameters measured in toluene and acetonitrile (n.d. = not determined). | ||
Recently Kuang and co-workers claimed that a thiophene-bridged dimer, 79, efficiently generates singlet oxygen in various solvents. However the mechanism of triplet state formation in this system has not been studied in detail.85
5. Design criteria for photosensitizers operating via SOCT-ISC
Photophysical and structural data for the BODIPY dyads and dimers demonstrate that the key requirements for achieving high triplet state yields via SOCT-ISC are: (1) orthogonal mutual orientation of the donor and acceptor subunits, (2) favorable Gibbs free energy change for the PET process and (3) suppressed charge recombination of CT states into the ground state.In order to secure orthogonal geometry, the subunits in a molecule must be directly coupled through a single C–C bond. As is evident from the data reported for spacer-separated dyads and dimers, the introduction of a spacer inevitably leads to twisted geometry, diminishing triplet state yields due to inefficient SOCT-ISC. However, even for closely-spaced systems containing rather bulky subunits, e.g. anthracenyl or perylenyl groups, significant deviation from orthogonality is possible (e.g. in dyads 27 and 39). To block the mutual rotation of the subunits, substituents in positions 1 and 7 of the BODIPY must be present. Other potential approaches towards orthogonal systems come to mind, e.g. using rigid polycyclic hydrocarbon scaffolds as spacers.
Rational choice of substituents in the pyrrole rings of the BODIPY also allows control of the direction and driving force of the PET process, since the oxidation and reduction potentials of the BODIPY core are strongly affected by its substitution pattern. Introduction of a single methyl group in the BODIPY core can decrease the reduction potential by up to 100–150 mV, depending on the substitution position.86 Increasing the number of alkyl substituents in pyrrolic rings results in more facile oxidation of the BODIPY. This can be employed for promoting electron transfer from the BODIPY subunit to the electron-accepting aryls, as seen from the results obtained with dyads 14–24.
If the donor and acceptor subunits possess low reduction and high oxidation potentials, respectively, large negative values of ΔGPET can be expected. This implies that the CT state energy level is well below the S1 state and is advantageous for rapid electron transfer, ensuring a high yield of the CT state. For instance, the presence of electron-withdrawing groups in the acceptor subunit of dimers 70d and 72 allows the PET process to take place even in non-polar solvents. Increasing the solvent polarity further promotes PET due to the additional stabilization of the CT state.
However, strong stabilization of the CT state due to structural factors or high polarity of the media leads to a reduced energy gap between the CT and S0 states. As a result, the charge recombination into the ground state shifts from the inverted Marcus region to the normal region, leading to increased rates of the CRS process with respect to CRT. This effect was observed for many of the discussed systems possessing reduced ΦT and ΦΔ values in highly polar solvents, e.g. dimers 58–61 and 66 in acetonitrile.
To secure high triplet state yields, the CT state must be close in energy to S1 state. This maximizes the inverted region effect and suppresses recombination into the ground state. One way to achieve this relies on the introduction of electron-donating groups into the BODIPY core, as shown for dimer 71. Another possibility is using less polar solvents, in which the CT state energy level and the CT–S0 energy gap are increased. In this situation, CRT is expected to be the predominant process, given that the dyad possesses orthogonal geometry essential for SOCT-ISC. Thus by tuning the electronic effects in the BODIPY subunits or aryl substituents, triplet state formation can be optimized for a desired range of polarities. Moreover, applying these principles allows to achieve high triplet state yields not only in heavy-atom-free BODIPYs but in other electron donor–acceptor systems, as was recently demonstrated for perylene derivatives.44
6. Applications
6.1 Triplet–triplet annihilation upconversion
TTA-UC is a process wherein lower-energy photons are converted into photons of higher energy.7 The mechanism of TTA-UC involves a series of energy transfers in a two-component system containing a sensitizer and an emitter (annihilator). Light absorbed by the sensitizer leads to the triplet excited state via ISC, which then transfers energy to the emitter molecule (triplet–triplet energy transfer, TTET). Triplet states of two emitter molecules can undergo triplet–triplet annihilation (TTA), in which one molecule returns back to its singlet ground state and the other gains the energy of both triplet states. Thus formed singlet excited states decay radiatively to the ground state and the resulting fluorescence is strongly blue-shifted with respect to the excitation light. TTA-UC has been experimentally demonstrated to operate with sunlight87 and improve the efficiencies of solar cell devices.88 Moreover, during the past decade this phenomenon has found unique applications in material science,89 solar fuels,90 and bio-imaging.91Fluorescent BODIPYs are widely used in TTA-UC as emitters in combination with sensitizers having appropriate triplet energy levels, such as Pd(II)/Pt(II) porphyrins.92 Alternatively, BODIPYs containing heavy atoms and giving high triplet state yields have been thoroughly investigated as sensitizers for TTA-UC systems.14
TTA-UC sensitization by heavy-atom-free BODIPYs, relying on a SOCT-ISC mechanism is currently a subject of active research.19c,65,66 The ability of BODIPY donor–acceptor dyads to switch between strong fluorescence and efficient triplet state generation, depending on the media polarity, provides an opportunity for their use either as sensitizer or emitter components. A unique dual performance of dyad 3 was demonstrated by Turshatov and co-workers in the following TTA-UC systems: (1) based on dyad 3 and perylene in dichloromethane (Fig. 18a) and (2) based on dyad 3 and Pd(II) tetrabenzoporphyrin (Fig. 18b) in toluene.93 In dichloromethane, where intramolecular PET in 3 is thermodynamically allowed, excitation of system (1) with 525 nm light (absorption band of 3) leads to the BODIPY triplet state generation, followed by TTET to perylene and results in an anti-Stokes emission at 445 nm (perylene fluorescence) via TTA. Alternatively, in toluene 3 behaves as a typical fluorophore, exhibiting negligible ISC and high fluorescence quantum yield (0.95). Excitation of system (2) with 638 nm light (absorption band of tetrabenzoporphyrin) leads to emission at 529 nm originating from the BODIPY LE state. The use of BODIPY donor–acceptor dyads with such dual performance promises to provide new advances in the TTA-UC field in the near future. The photophysical performance of these compounds make them suitable for simultaneous use in several types of TTA-UC devices (e.g. green-to-blue, red-to-orange light converters), which ultimately could reduce the cost of this technology.
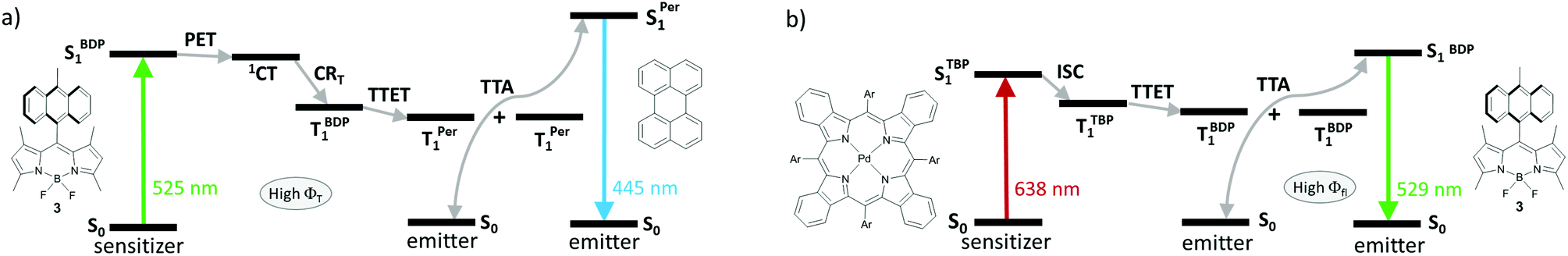 | ||
| Fig. 18 Jablonski diagrams showing the TTA-UC process in (a) a system employing dyad 3 as a triplet sensitizer and perylene as an emitter in dichloromethane; (b) a system based on Pd(II) tetrabenzoporphyrin as a sensitizer and 3 as an emitter in toluene. | ||
6.2 Photodynamic therapy
Photodynamic therapy is used for the destruction of malignant cells and tissues via photosensitized reactive oxygen species formation. Currently, most of the clinically-approved PDT photosensitizers are cyclic tetrapyrroles (porphyrins, chlorins, and bacteriochlorins). There is a growing interest in non-porphyrin photosensitizers, based on small photoactive molecules, such as BODIPYs.94One of the key requirements for potential PDT agents is minimal toxicity to the target tissue in the absence of light. Introduction of heavy atoms into BODIPYs has been shown to result in significant dark cytotoxicity of these compounds, limiting the utility in PDT.8,28 Several recent works demonstrated that heavy-atom-free BODIPY dimers and donor–acceptor dyads induce strong cytotoxic effect in cells under light illumination due to the triplet state formation via SOCT-ISC and the subsequent generation of singlet oxygen.76,19a–d,95 However, the potential of this class of photosensitizers is still underscored. Apart from solvent polarity, the PET process can be turned on–off in response to complexation with metal ions and biomolecules, changes in pH, hydrogen bond formation and other stimuli,96 thus allowing to control generation of triplet states and singlet oxygen.97 In particular, photosensitizers with 1O2 generation, depending on the applied light dose and localization within the cell, can prevent unintended damage of healthy tissue by singlet oxygen during light treatment. An example of such an advanced photodynamic treatment of cancer cells was recently demonstrated by Akkaya and co-workers using dyad 80.98 The Zn(II)-terpyridyl fragment in this system plays the role of electron acceptor, promoting PET from the BODIPY subunit (Fig. 19a). The dyad generates singlet oxygen (ΦΔ = 0.1) and triggers apoptosis in cancer cells under light irradiation. Subsequently, the PET process in the dyad terminates, resulting in a strong fluorescence emission from the BODIPY LE state. Such behavior was explained by interaction of the Zn(II) ion in 80 with phosphatidylserine – a lipid normally facing the inner side of the cell membrane. During apoptosis, phosphatidylserine molecules can exchange between the two sides of the membrane and become exposed on the cell surface. Complexation between the Zn(II) ion and phosphate group of phosphatidylserine terminates electron transfer within the dyad due to increased ΔGPET (Fig. 19b).
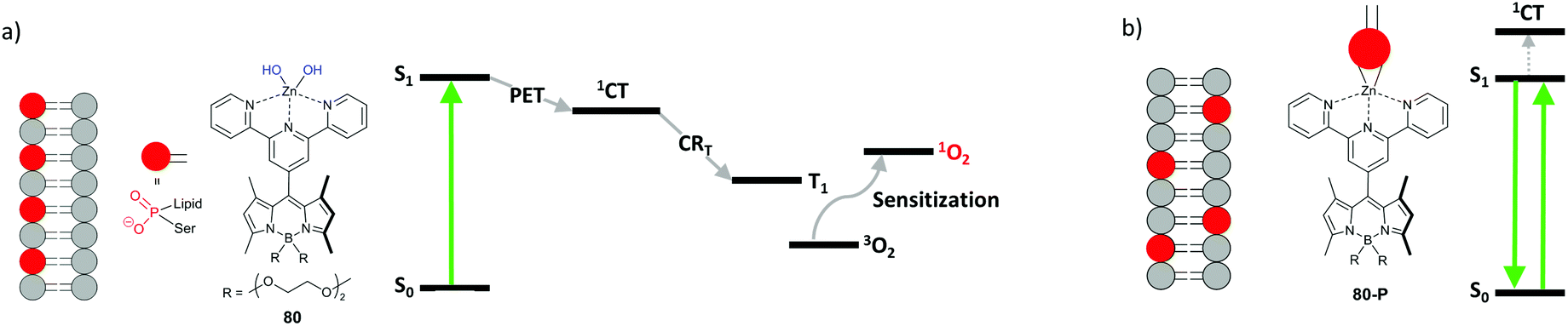 | ||
| Fig. 19 (a) Triplet state generation from the CT state in dyad 80 induces apoptosis. (b) Loss of membrane asymmetry accompanying the apoptosis results in the complexation of Zn(II) with the phosphate group which terminates the electron transfer and results in a fluorescent complex 80-P. Red polar heads represent phosphotidylserine lipids. | ||
7. Conclusions
Chromophores with tunable ISC, showing either strong fluorescence or efficient triplet state formation in different environments would be beneficial for a range of applications. The design of such systems is still challenging due to the lack of established relationships between molecular structure and intersystem crossing. The formation of triplets upon recombination of CT states in BODIPY dimers and donor acceptor dyads via SOCT-ISC represents is an appealing alternative to the heavy atom-promoted ISC, enabling access to photosensitizers with programmed triplet states and fluorescence quantum yields.As shown in the works discussed herein, the photophysical behavior of such systems depends on structural factors, such as the substitution pattern of the BODIPY core, which determine dihedral angles between the subunit and consequently the efficiency of SOCT-ISC. Furthermore, the PET process between the subunits in dyads and dimers can be controlled by the media polarity and external stimuli (e.g. complexation with metal ions or biomolecules). Thus triplet state formation, mediated by PET, can be selectively switched on–off in response to these stimuli.
Importantly, BODIPY donor–acceptor dyads are easy to synthesize compared to conventional photosensitizers, such as porphyrins and transition metal complexes. A number of advanced applications for these multifunctional dyes can be expected over the coming years. However, despite the breadth of scope of the reported systems, there are still unsolved challenges with these molecules, or areas in which the SOCT-ISC mechanism remains underexplored, e.g. generation of triplets in near infra-red absorbing BODIPYs. So far, rather low triplet state yields have been obtained for dyads and dimers with extended π-systems. Hence, new design strategies are needed in this area and SOCT-ISC is likely to remain a rich research topic in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support from the European Commission (CONSORT, Grant No. 655142) is acknowledged.Notes and references
- B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag, GmbH, 2001 Search PubMed.
- O. Inganäs and V. Sundström, Ambio, 2016, 45(Suppl. 1), S15 CrossRef PubMed.
- F. Etzold, I. A. Howard, N. Forler, A. Melnyk, D. Andrienko, M. R. Hansen and F. Laquai, Energy Environ. Sci., 2015, 8, 1511 RSC.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- M. L. Mari, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710 CrossRef.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245 CrossRef CAS.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS; (b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162 CrossRef CAS.
- (a) S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286 CrossRef CAS; (b) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353 CrossRef CAS; (c) N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096 CrossRef CAS; (d) S. M. Sartor, B. G. McCarthy, R. M. Pearson, G. M. Miyake and N. H. Damrauer, J. Am. Chem. Soc., 2018, 140, 4778 CrossRef CAS.
- (a) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS; (b) C. Li, Y. Xu, W. Tu, G. Chen and R. Xu, Green Chem., 2017, 19, 882 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904 RSC.
- S. K. Rajagopal, A. R. Mallia and M. Hariharan, Phys. Chem. Chem. Phys., 2017, 19, 28225 RSC.
- (a) Z. Wang, J. Zhao, A. Barbon, A. Toffoletti, Y. Liu, Y. An, L. Xu, A. Karatay, H. G. Yaglioglu, E. A. Yildiz and M. Hayvali, J. Am. Chem. Soc., 2017, 139, 7831 CrossRef CAS; (b) Z. Wang, Y. Gao, M. Hussain, S. Kundu, V. Rane, M. Hayvali, E. A. Yildiz, J. Zhao, H. G. Yaglioglu, R. Das, L. Luo and J. Li, Chem. – Eur. J., 2018, 24, 18663 CrossRef CAS.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776 RSC.
- T. Okada, I. Karaki, E. Matsuzawa, N. Mataga, Y. Sakata and S. Misumi, J. Phys. Chem., 1981, 85, 3957 CrossRef CAS.
- (a) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282 CrossRef CAS PubMed; (b) N. Epelde-Elezcano, E. Palao, H. Manzano, A. Prieto-CastaCeda, A. R. Agarrabeitia, A. Tabero, A. Villanueva, S. de la Moya, C. Ljpez-Arbeloa, V. Martinez-Martinez and M. J. Ortiz, Chem. – Eur. J., 2017, 23, 4837 CrossRef CAS; (c) Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492 CrossRef CAS; (d) S. Kim, Y. Zhou, N. Tohnai, H. Nakatsuji, M. Matsusaki, M. Fujitsuka, M. Miyata and T. Majima, Chem. – Eur. J., 2018, 24, 636 CrossRef CAS PubMed.
- (a) J. H. Golden, L. Estergreen, T. Porter, A. C. Tadle, D. M. R. Sylvinson, J. W. Facendola, C. P. Kubiak, S. E. Bradforth and M. E. Thompson, ACS Appl. Energy Mater., 2018, 1, 1083 CrossRef CAS; (b) W. Huang, X. Zhang, B. Chen, H. Miao, C. O. Trindle, Y. Wang, Y. Luo and G. Zhang, Chem. Commun., 2019, 55, 67 RSC; (c) J.-X. Wang, L.-Y. Niu, P.-Z. Chen, Y.-Z. Chen, Q.-Z. Yang and R. Boulatov, Chem. Commun., 2019, 55, 7017 RSC.
- (a) C. Trinh, K. O. Kirlikovali, S. Das, M. E. Ener, H. B. Gray, P. I. Djurovich, S. Bradforth and M. E. Thompson, J. Phys. Chem. C, 2014, 118, 21834 CrossRef CAS; (b) S. Das, W. G. Thornbury, A. N. Bartynski, M. E. Thompson and S. E. Bradforth, J. Phys. Chem. Lett., 2018, 9, 3264 CrossRef CAS.
- S. M. Sartor, B. G. McCarthy, R. M. Pearson, G. M. Miyake and N. H. Damrauer, J. Am. Chem. Soc., 2018, 140, 4778 CrossRef CAS.
- Y. Tsuga, M. Katou, S. Kuwabara, T. Kanamori, S. Ogura, S. Okazaki, H. Ohtani and H. Yuasa, Chem. – Asian J., 2019, 14, 2067 CrossRef CAS.
- (a) Y. Zhao, X. Li, Z. Wang, W. Yang, K. Chen, J. Zhao and G. G. Gurzadyan, J. Phys. Chem. C, 2018, 122, 3756 CrossRef CAS; (b) Y. Zhao, X. Li, Z. Wang, W. Yang, K. Chen, J. Zhao and G. G. Gurzadyan, J. Phys. Chem. C, 2018, 122, 3756 CrossRef CAS; (c) Y. Zhao, R. Duan, J. Zhao and C. Li, Chem. Commun., 2018, 54, 12329 RSC.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181 RSC.
- T. Yogo, Y. Urano, A. Mizushima, H. Sunahara, T. Inoue, K. Hirose, M. Iino, K. Kikuchi and T. Nagano, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 28 CrossRef CAS.
- (a) J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839 CrossRef CAS; (b) Y. You, Org. Biomol. Chem., 2016, 14, 7131 RSC.
- C. S. Kue, S. Y. Ng, S. H. Voon, A. Kamkaew, L. Y. Chung, L. V. Kiew and H. B. Lee, Photochem. Photobiol. Sci., 2018, 17, 1691 RSC.
- D. Escudero, Acc. Chem. Res., 2016, 49, 1816 CrossRef CAS PubMed.
- (a) Z. R. Grabowski and K. Rotkiewicz, Chem. Rev., 2003, 103, 3899 CrossRef PubMed; (b) S. Sasaki, G. P. C. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2731 RSC.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CrossRef CAS.
- M. Maus and W. Rettig, J. Phys. Chem. A, 1999, 103, 3388 CrossRef CAS.
- H. Heitele, P. Finckh, S. Weeren, F. Poellinger and M. E. Michel-Beyerle, J. Phys. Chem., 1989, 93, 5173 CrossRef CAS.
- J. W. Verhoeven, H. J. van Ramesdonk, M. M. Groeneveld, A. C. Benniston and A. Harriman, ChemPhysChem, 2005, 6, 2251 CrossRef CAS.
- (a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed; (b) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS PubMed; (c) S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575 RSC; (d) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS; (e) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS.
- (a) J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40 CrossRef CAS.
- A. C. Benniston, A. Harriman, P. Y. Li, J. P. Rostron, H. J. van Ramesdonk, M. M. Groeneveld, H. Zhang and J. W. Verhoeven, J. Am. Chem. Soc., 2005, 127, 16054 CrossRef CAS.
- H. van Willigen, G. Jones and M. S. Farahat, J. Phys. Chem., 1996, 100, 3312 CrossRef CAS.
- (a) M. C. Thurnauer, J. J. Katz and J. R. Norris, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3270 CrossRef CAS; (b) H. Levanon and J. R. Norris, Chem. Rev., 1978, 78, 185 CrossRef CAS.
- (a) M. R. Wasielewski, D. G. Johnson, W. A. Svec, K. M. Kersey and D. W. Minsek, J. Am. Chem. Soc., 1988, 110, 7219 CrossRef CAS; (b) G. P. Wiederrecht, W. A. Svec, M. R. Wasielewski, T. Galili and H. Levanon, J. Am. Chem. Soc., 2000, 122, 9715 CrossRef CAS; (c) P. L. Dutton, J. S. Leigh and M. Seibert, Biochem. Biophys. Res. Commun., 1972, 46, 406 CrossRef CAS; (d) K. Hasharoni, H. Levanon, S. R. Greenfield, D. J. Gosztola, W. A. Svec and M. R. Wasielewski, J. Am. Chem. Soc., 1995, 117, 8055 CrossRef CAS; (e) D. Carbonera, N. DiValentin, C. Corvaja, G. Agostini, G. Giacometti, P. A. Liddell, D. Kuciauskas, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1998, 120, 4398 CrossRef CAS.
- M. Goez, Concepts Magn. Reson., 1995, 7, 69 CrossRef CAS.
- (a) Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194 CrossRef CAS; (b) Z. E. X. Dance, Q. Mi, D. W. McCamant, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. B, 2006, 110, 25163 CrossRef CAS PubMed.
- I. V. Khudyakov, Y. A. Serebrennikov and N. J. Turro, Chem. Rev., 1993, 93, 537 CrossRef CAS.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2018, 5, 1 Search PubMed.
- R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611 RSC.
- R. Ziessel, B. D. Allen, D. B. Rewinska and A. Harriman, Chem. – Eur. J., 2009, 15, 7382 CrossRef CAS.
- (a) L. Huang, X. Yu, W. Wu and J. Zhao, Org. Lett., 2012, 14, 2594 CrossRef CAS; (b) L. Huang and J. Zhao, Chem. Commun., 2013, 49, 3751 RSC.
- (a) W. Wu, J. Zhao, J. Sun and S. Guo, J. Org. Chem., 2012, 77, 5305 CrossRef CAS PubMed; (b) D. Huang, J. Zhao, W. Wu, X. Yi, P. Yang and J. Ma, Asian J. Org. Chem., 2012, 1, 264 CrossRef CAS.
- H. L. Kee, C. Kirmaier, L. Yu, P. Thamyonkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433 CrossRef CAS.
- (a) M. Kollmannsberger, K. Rurack, U. Resch-Genger and J. Daub, J. Phys. Chem. A, 1998, 102, 10211 CrossRef CAS; (b) M. Kollmannsberger, K. Rurack, U. Resch-Genger, W. Rettig and J. Daub, J. Chem. Phys. Lett., 2000, 329, 363 CrossRef CAS; (c) K. Rurack, M. Kollmannsberger, U. Resch-Genger and J. Daub, J. Am. Chem. Soc., 2000, 122, 968 CrossRef CAS.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597 CrossRef CAS.
- W. Hu, Y. Lin, X.-F. Zhang, M. Feng, S. Zhao and J. Zhang, Dyes Pigm., 2019, 164, 139 CrossRef CAS.
- X.-F. Zhang and N. Feng, Chem. – Asian. J., 2017, 12, 2447 CrossRef CAS.
- (a) W. Hua, X.-F. Zhang, X. Lu, S. Lan, D. Tian, T. Li, L. Wang, S. Zhao, M. Feng and J. Zhang, J. Lumin., 2018, 194, 185 CrossRef; (b) X.-F. Zhang and N. Feng, Spectrochim. Acta, Part A, 2018, 189, 13 CrossRef CAS PubMed.
- X.-F. Zhang, Y. Zhang and B. Xu, J. Photochem. Photobiol., A, 2017, 349, 197 CrossRef CAS.
- W. Hu, M. Liu, X.-F. Zhang, Y. Wang, Y. Wang, H. Lan and H. Zhao, J. Phys. Chem. C, 2019, 123, 15944 CrossRef CAS.
- A. Harriman, L. J. Mallon, G. Ulrich and R. Ziessel, ChemPhysChem, 2007, 8, 1207 CrossRef CAS.
- A. C. Benniston, A. Harriman, V. L. Whittle, M. Zelzer, R. W. Harrington and W. Clegg, Photochem. Photobiol. Sci., 2010, 9, 1009 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Phys. Chem. Chem. Phys., 2018, 20, 8016 RSC.
- A. B. Nepomnyashchii and A. J. Bards, Acc. Chem. Res., 2012, 45, 1844 CrossRef CAS PubMed.
- Z. Wang, A. A. Sukhanov, A. Toffoletti, F. Sadiq, J. Zhao, A. Barbon, V. K. Voronkova and B. Dick, J. Phys. Chem. C, 2019, 123, 265 CrossRef CAS.
- L. Yang, Y. Liu, W. Liu, C. Ma, C. Zhang and Y. Li, Bioorg. Med. Chem. Lett., 2015, 25, 5716 CrossRef CAS.
- (a) C. Caltagirone, M. Arca, A. M. Falchi, V. Lippolis, V. Meli, M. Monduzzi, T. Nylander, A. Rosa, J. Schmidt, Y. Talmon and S. Murgia, RSC Adv., 2015, 5, 23443 RSC; (b) Y. Yang, L. Zhang, C. Gao, L. Xu, S. Bai and X. Liu, RSC Adv., 2014, 4, 38119 RSC; (c) F. Bizet, M. Ipuy, Y. Bernhard, V. Lioret, P. Winckler, C. Goze, J.-M. Perrier-Cornet and R. A. Decreau, Bioorg. Med. Chem., 2018, 26, 413 CrossRef CAS PubMed; (d) J. P. Rostron, G. Ulrich, P. Retailleau, A. Harriman and R. Ziessel, New J. Chem., 2005, 29, 1241 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606 CrossRef CAS.
- Z. Wang, J. Zhao, M. Di Donato and G. Mazzone, Chem. Commun., 2019, 55, 1510 RSC.
- K. Chen, W. Yang, Z. Wang, A. Iagatti, L. Bussotti, P. Foggi, W. Ji, J. Zhao and M. Di Donato, J. Phys. Chem. A, 2017, 121, 7550 CrossRef CAS.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, J. Phys. Chem. C, 2019, 123, 22793 CrossRef CAS.
- A. C. Benniston, G. Copley, H. Lemmetyinen and N. V. Tkachenko, ChemPhysChem, 2010, 11, 1685 CrossRef CAS.
- X. He, A. C. Benniston, H. Lemmetyinen and N. V. Tkachenko, ChemPhotoChem, 2018, 2, 277 CrossRef CAS.
- (a) S. Rihn, M. Erdem, A. De Nicola, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 1916 CrossRef CAS; (b) Y. Hayashi, S. Yamaguchi, W. Y. Cha, D. Kim and H. Shinokubo, Org. Lett., 2011, 13, 2992 CrossRef CAS; (c) R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611 RSC; (d) A. Poirel, A. D. Nicola, P. Retailleau and R. Ziessel, J. Org. Chem., 2012, 11, 7512 CrossRef; (e) L. Gai, B. Lu, B. Zou, G. Lai, Z. Shen and Z. Li, RSC Adv., 2012, 2, 8840 RSC; (f) S. Kusaka, R. Sakamoto, Y. Kitagawa, M. Okumura and H. Nishihara, Chem. – Asian. J., 2013, 8, 723 CrossRef CAS; (g) V. Yang, L. Li, B. Zhang, L. Zhanga and X. Liu, RSC Adv., 2013, 3, 16933 Search PubMed; (h) H. Qi, J. J. Teesdale, R. C. Pupillo, J. Rosenthal and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 13558 CrossRef CAS; (i) A. V. Benniston, G. Copley, A. Harriman, D. Howgego, R. S. Harrington and W. Clegg, J. Org. Chem., 2010, 75, 2018 CrossRef CAS; (j) N. Saki, T. Dinc and E. U. Akkaya, Tetrahedron, 2006, 62, 2721 CrossRef CAS; (k) P. E. Kesavan, S. Das, M. Y. Lone, P. C. Jha, S. Moric and I. Gupta, Dalton Trans., 2015, 44, 17209 RSC.
- E. Vauthey, ChemPhysChem, 2012, 13, 2001 CrossRef CAS.
- (a) F. Schneider and E. Lippert, Ber. Bunsen-Ges. Phys. Chem., 1968, 72, 1155 CrossRef CAS; (b) F. Schneider and E. Lippert, Ber. Bunsen-Ges. Phys. Chem., 1970, 74, 624 CrossRef CAS; (c) F. C. Grozema, M. Swart, R. W. J. Zijlstra, J. J. Piet, L. D. A. Siebbeles and P. T. van Duijnen, J. Am. Chem. Soc., 2005, 127, 11019 CrossRef CAS PubMed.
- M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Chem. Commun., 2012, 48, 284 RSC.
- M. Bröring, R. Krüger, S. Link, C. Kleeberg, S. Köhler, X. Xie, B. Ventura and L. Flamigni, Chem. – Eur. J., 2008, 14, 2976 CrossRef PubMed.
- B. Ventura, G. Marconi, M. Bröring, R. Krüger and L. Flamigni, New J. Chem., 2009, 33, 428 RSC.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937 CrossRef CAS.
- S. Duman, Y. Cakmak, S. Kolemen, E. U. Akkaya and Y. Dede, J. Org. Chem., 2012, 77, 4516 CrossRef CAS.
- X.-F. Zhang, J. Photochem. Photobiol., A, 2018, 355, 431 CrossRef CAS.
- L. Ya, J. Zhao, A. Iagatti, L. Bussotti, P. Foggi, E. Castellucci, M. Di Donato and K. Han, J. Phys. Chem. C, 2018, 122, 2502 CrossRef.
- (a) G. Duvanel, N. Banerji and E. Vauthey, J. Phys. Chem. A, 2007, 111, 5361 CrossRef CAS; (b) A. Iagatti, L. Cupellini, G. Biagiotti, S. Caprasecca, S. Fedeli, A. Lapini, E. Ussano, S. Cicchi, P. Foggi, M. Marcaccio, B. Mennucci and M. Di Donato, J. Phys. Chem. C, 2016, 120, 16526 CrossRef CAS.
- (a) S. Hattori, K. Ohkubo, Y. Urano, H. Sunahara, T. Nagano, Y. Wada, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, J. Phys. Chem. B, 2005, 109, 15368 CrossRef CAS; (b) A. M. Lifschitz, R. M. Young, J. Mendez-Arroyo, V. V. Roznyatovskiy, C. M. McGuirk, M. R. Wasielewski and C. A. Mirkin, Chem. Commun., 2014, 50, 6850 RSC.
- W. Wu, X. Cui and J. Zhao, Chem. Commun., 2013, 49, 9009 RSC.
- W. Pang, X.-F. Zhang, J. Zhou, C. Yu, E. Hao and L. Jiao, Chem. Commun., 2012, 48, 5437 RSC.
- X.-F. Zhang and X. Yang, J. Phys. Chem. B, 2013, 117, 9050 CrossRef CAS PubMed.
- D.-G. Wang, L.-N. Zhang, Q. Li, Y. Yang, Y. Wu, X. Fan, M. Song and G.-C. Kuang, Tetrahedron, 2017, 73, 6894 CrossRef CAS.
- B. L. Thompson and Z. Heiden, Redox Chemistry of BODIPY Dyes, IntechOpen, 2018, DOI:10.5772/intechopen.79704.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, S. Chernov, S. Aleshchenkov, A. Cheprakov, A. Yasuda and G. Wegner, Appl. Phys. Lett., 2007, 90, 181103 CrossRef.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103 RSC.
- S. Ji, W. Wu, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626 CrossRef CAS.
- K. Börjesson, D. Dzebo, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. A, 2013, 1, 8521 RSC.
- C. Wohnhaas, V. Mailänder, M. Dröge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422 CrossRef CAS.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54 CrossRef CAS.
- N. Kiseleva, M. A. Filatov, M. Oldenburg, D. Busko, M. Jakoby, I. A. Howard, B. S. Richards, M. O. Senge, S. M. Borisov and A. Turshatov, Chem. Commun., 2018, 54, 1607 RSC.
- S. Callaghan and M. O. Senge, Photochem. Photobiol. Sci., 2018, 17, 1490 RSC.
- S. Callaghan, M. A. Filatov, H. Savoie, R. W. Boyle and M. O. Senge, Photochem. Photobiol. Sci., 2019, 18, 495 RSC.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS.
- (a) B. Turfan and E. U. Akkaya, Org. Lett., 2002, 4, 2857 CrossRef CAS; (b) S. Kolemen, M. Isık, G. Mi Kim, D. Kim, H. Geng, M. Buyuktemiz, T. Karatas, X.-F. Zhang, Y. Dede, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2015, 54, 5340 CrossRef CAS.
- I. S. Turan, G. Gunaydin, S. Ayan and E. U. Akkaya, Nat. Commun., 2018, 9, 805 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |