
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Haloperoxidases as catalysts in organic synthesis
Georg T.
Höfler
,
Andrada
But
and
Frank
Hollmann
 *
*
Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: f.hollmann@tudelft.nl
First published on 3rd October 2019
Abstract
Haloperoxidases are very active catalysts for the in situ generation of electrophilic halide species for oxidative halogenation reactions. In the synthetic community, these catalysts, however, are not widely used. The aim of this mini-review is to critically summarise the current state-of-the-art of haloperoxidase catalysis for organic synthesis. We hope that the excellent catalytic performance of these catalysts will trigger more chemists to consider them in their synthesis planning.
Introduction
Hypohalites (XO−) and elementary halogens (X2) are common oxidants in organic synthesis.1 Their uses range from oxidation of alcohols, halogenation of activated arenes to well-known name reactions such as the Hunsdieker- or Haloform reaction or the Hofmann rearrangement.Their application as reagents, however, can be challenging due to their instability and reactivity (causing safety issues) as well as the stoichiometric formation of salt waste (causing environmental issues). Furthermore the addition of high amounts of reactive hypohalites may cause undesired side-reactions, which are frequently circumvented by using organic halogen precursor such as N-bromosuccinimide (NBS).
A promising alternative is the in situ generation of hypohalites starting from the corresponding halides, an oxidant e.g. H2O2 and a catalyst. For this, especially vanadates are established catalysts.2 In recent years also (vanadium) haloperoxidases are attracting increasing attention as biocatalytic alternative.
The aim of this contribution is to critically summarise the current knowledge about haloperoxidases from a synthetic chemical point-of-view.
Biological halogenation catalysts
Halogenated compounds are rather common in nature and a range of enzymes are known to insert halogens into (non)activated starting materials. Halohydrin dehalogenases (Hhe), for example, catalyse the reversible interconversion of epoxides into halohydrins (Scheme 1).3 Also a range of halogenating monooxygenases (halogenases, MO) are known (Scheme 1).4–10 Finally, halogenating peroxidases (haloperoxidases, HPO) are enzymes which oxidatively activate halides to the corresponding hypohalites at the expense of peroxides. Further, the hypohalites react chemically with activated starting materials such as alkenes or phenols (Scheme 1).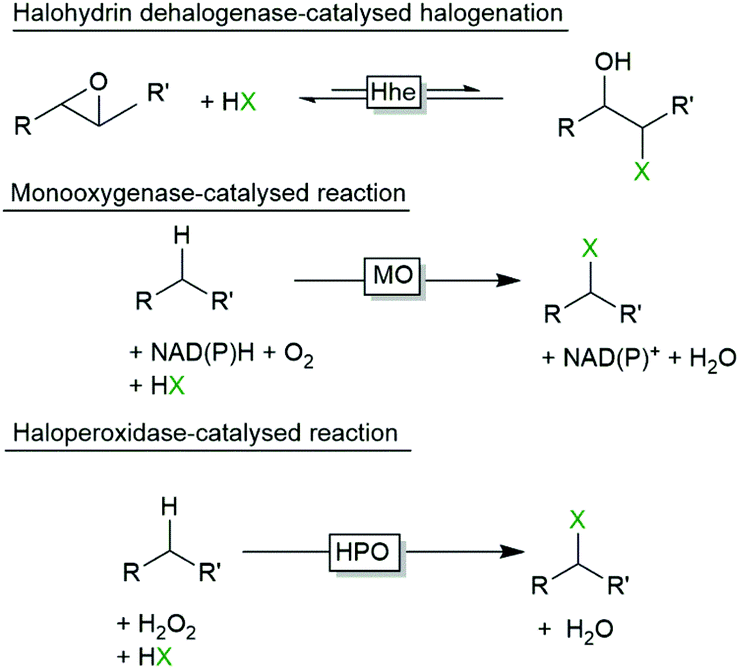 | ||
| Scheme 1 Biocatalytic halogenation reactions. Halohydrin dehalogenases (Hhe) catalyse the reversible ring opening of epoxides whereas some halogenating monooxygenases (MO) activate halides for electrophilic incorporation into C–H-bonds at the expense of NAD(P)H and O2. Haloperoxidases (HPO) utilise H2O2 to activate halides, which then spontaneously halogenate activated starting materials. | ||
From a synthetic point of view both Hhe and MO exhibit some issues that impair their preparative applicability. Halohydrin dehalogenases for example rely on epoxides as starting materials. Furthermore, the thermodynamic equilibrium of the ring opening reaction is rather unfavourable.3
Halogenating monooxygenases are more versatile converting activated arenes and even non-activated C–H-bonds.4 Most interestingly, halogenating monooxygenases are usually highly regio- and stereospecific. However, these enzymes are too slow to be of real practical value. As a consequence, catalyst loadings significantly exceed the economically reasonable range.11 Hopefully, the current activity, stability and uncoupling issues will be solved to fully exploit their synthetic potential.
In contrary, haloperoxidases excel in terms of robustness and catalytic activity making them potentially very efficient catalysts for preparative halogenation chemistry. This enormous potential motivated us to critically summarise the current state-of-art of haloperoxidases in organic synthesis, to outline the synthetic possibilities but also the their limitations.
Haloperoxidases – classification, occurrence and catalytic mechanism
Haloperoxidases are classified by the most electronegative halide ion they can oxidise. A chloroperoxidase can, for example, oxidise Cl−, Br− and I−, while a bromoperoxidase can oxidise Br− and I− but not Cl−. A fluoroperoxidase has not been identified yet (and is rather unlikely ever to be found due to the high electronegativity and oxidation potential of F−).Today, two kinds of haloperoxidases are known: the Heme-dependent haloperoxidases12 and the vanadium-dependent haloperoxidases.6,13–16 As suggested by their names, they differ with respect to the prosthetic group and consequently in their catalytic mechanisms. The Heme-dependent haloperoxidases utilise an FeIV-porphyrin+ (compound I) species formed from the heme resting state and H2O2 to oxidise halides whereas the vanadium-dependent haloperoxidases use a peroxo-vanadate species (formed by the reaction with H2O2) for the same transformation (Scheme 2).
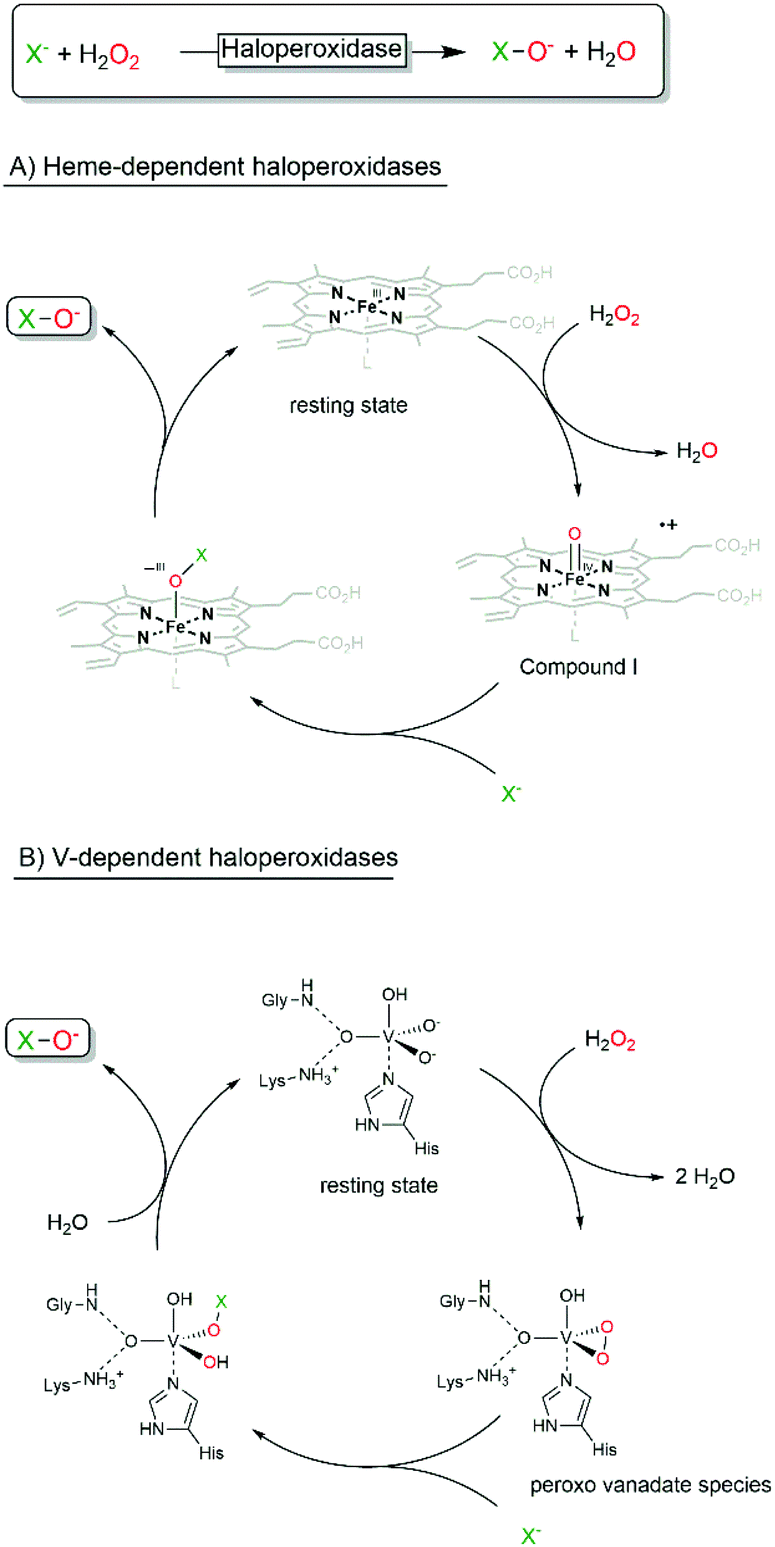 | ||
| Scheme 2 Simplified halide oxidation mechanisms of haloperoxidases. | ||
In both classes of haloperoxidases, the hypohalites after formation diffuse out of the enzyme active site;17 hence any oxidative transformation taking place is not supported by the (chiral) enzyme environment and the selectivity of the transformation is controlled by the reactivity of the starting material rather than by the enzyme (vide infra). Some exceptions (haloperoxidases of bacterial origin) to this rule have been reported.18–22 Indeed, selective halogenations would be of great interest for synthetic organic synthesis, especially if performed by highly active and stable enzymes (vide infra). As today, however, the molecular basis for the assumed selectivity remains unclear and the number of examples is yet too little. Hopefully, future engineered haloperoxidases will indeed combine the best of all: H2O2-dependent reactions, high robustness, catalytic activity and selectivity.
The archetypical Heme-dependent haloperoxidase is the one from Leptoxyphium fumago (LfCPO, formerly Caldariomyces fumago, CfCPO) first reported as early as the 1960s.23 It has been widely applied for a broad range of oxyfunctionalisation reactions, which have been summarised and discussed elsewhere.24–26 In addition, a rich literature exists dealing with LfCPO (vide infra). More recently, also a haloperoxidase from Agrocybe aegerita (AaeUPO) has been reported.27,28AaeUPO, however, is receiving more attention as selective oxyfunctionalisation catalyst.29,30 Heme-dependent haloperoxidases excel by their high catalytic activity (in the range of several dozen to hundreds per second) but are hampered by their poor robustness toward H2O2. Though this issue in principle can be overcome by slow dosing of H2O2 or in situ generation of H2O2,25 such measures usually complicate the reaction schemes.
In contrast, to the limited number of Heme-dependent haloperoxidases, a rich variety of vanadium-dependent haloperoxidases are mainly available from marine organisms31–39 but also from other sources such as lichens20,21,39,40 and terrestrial fungi.41 The rich microbiology and biochemistry of haloperoxidases has been reviewed by Wever and coworkers.13–16,42
One of the most striking differences of vanadium-dependent haloperoxidases to their heme-counterparts is the robustness against H2O2. Especially the chloroperoxidase from Curvularia inaequalis (CiVCPO) excels in this respect as the enzyme can be stored in the presence of at least 100 mM H2O2 for days without noticeable loss in catalytic activity.43 The same is true for the general stability of CiVCPO at elevated temperatures (up to 70 °C) and in the presence of organic cosolvents (ethanol, ethyl acetate, acetone).
From a practical point of view, it is, however, advisable to control the H2O2 concentration in the reaction mixture due to the spontaneous reaction between hypohalites and H2O2 yielding singlet oxygen (1O2) and halides (Scheme 3), which is well-documented for various haloperoxidases.23,31,44–47
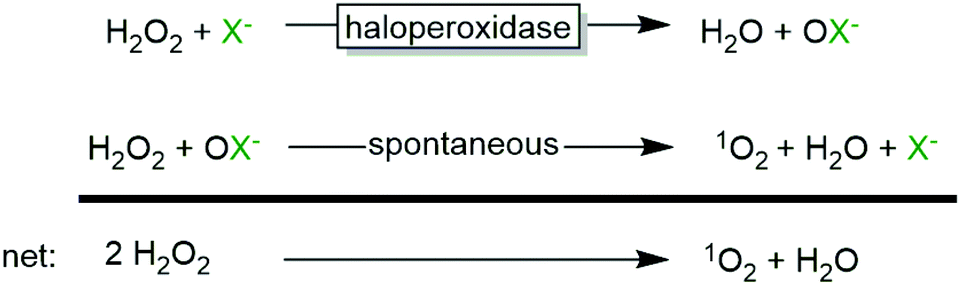 | ||
| Scheme 3 Hypohalite-dependent disproportionation of H2O2. | ||
To control the H2O2 concentration, a range of in situ H2O2 generation methods are available.25 Some of them have been evaluated in combination with haloperoxidases (Table 1). They all have their specific advantages and disadvantages. Photochemical and electrochemical methods, for example, principally bear the promise of environmentally benign reactions. However, in terms of haloperoxidase turnover numbers and ease of application, they still fall back behind the enzymatic cascades.
| Catalyst | Cosubstrate/coproduct | TNHaloperoxidase |
|---|---|---|
| AoFOx: formate oxidase from Aspergillus oryzae; HpAOx: alcohol oxidase from Hansenula polymorpha; EDTA: ethylenediamine tetraacetate; EDTriA: ethylenediamine triacetate. | ||
| AoFOx48 | HCO2H/CO2 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 500000 |
| HpAOx49 | Ethanol/acetaldehyde | n.d. |
| Au-TiO2/hν50 | H2O/O2 | 20000 |
| FMN/hν51,52 | EDTA/EDTriA + CO2 + H2CO | 2000 |
| Cathode53 | — | 164000 |
Though the above-mentioned 1O2 formation does not significantly impair the robustness of the enzyme, it lowers the yield in H2O2 and therefore is less attractive from an economical point-of-view. On the other hand, the primarily formed 1O2 is very reactive and this ‘dark reaction’ for its in situ generation may find some preparative applications such as the transformation of β-Citronellol to Rose oxide.54
Wever and coworkers45 also pointed out the efficiency of the enzymatic 1O2 synthesis reaction compared to chemical catalysts such as Na2MoO455 or La(NO3)3/NaOH½.56 While the latter exhibit turnover frequencies in the range of several dozen per hour, the enzyme shows turnover frequencies of dozens to thousands per second.
Reactions mediated by haloperoxidases
Haloperoxidases have been applied to a broad range of different oxidation and halogenation reactions. Amongst them halogenation of electron-rich aromatic compounds, halohydroxylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-double bonds, heteroatom oxidation, oxidative decarboxylation and more. In the following sections, these reactions will be discussed in more detail and critically compared to their ‘chemical’ counterparts.
C-double bonds, heteroatom oxidation, oxidative decarboxylation and more. In the following sections, these reactions will be discussed in more detail and critically compared to their ‘chemical’ counterparts.
Halogenation of activated arenes
One of the first synthetic applications of haloperoxidases was the electrophilic halogenation of electron-rich arenes. Due to the occurrence of diffusible hypohalites in the reaction media, the selectivity of the reaction is dictated by the chemical reactivity (degree of delocalisation and stability of the intermediate σ-complex as shown in Scheme 4) of the starting material rather than being influenced by the active site of the biocatalyst.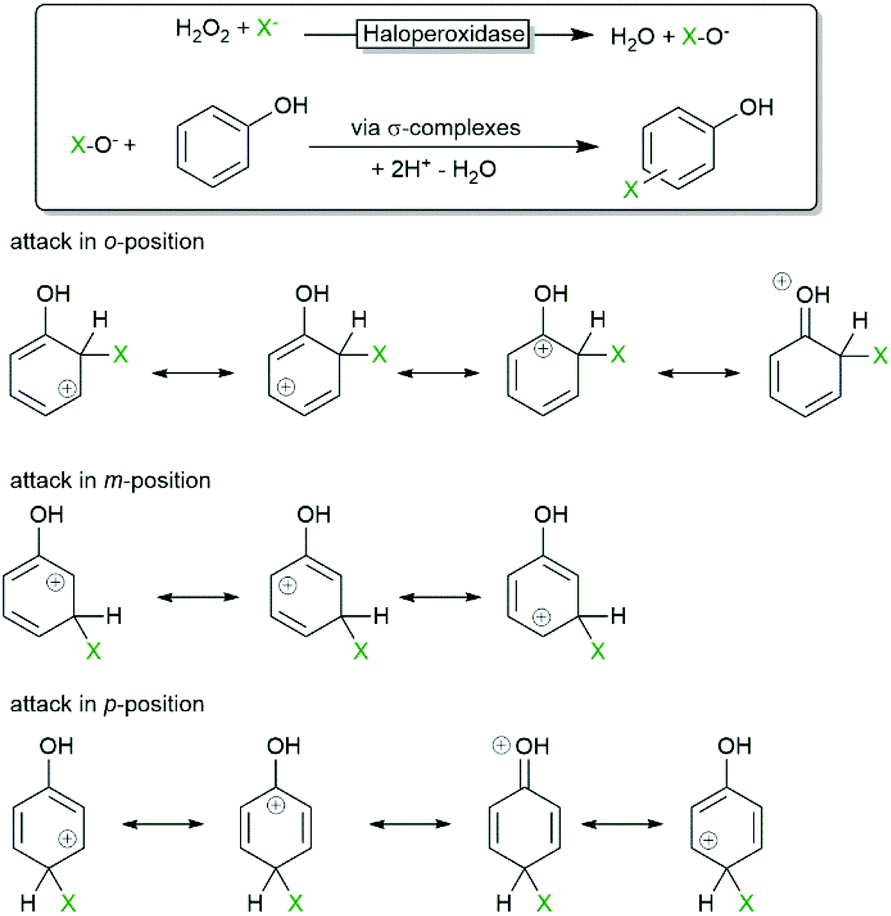 | ||
| Scheme 4 σ-Complexes involved in the haloperoxidase-initiated, electrophilic substitution of phenols. In case of o- and p-attack 4 mesomeric structures exist for the σ-complexes whereas in case of m-attack only 3 mesomeric structures can be drawn. As a consequence the σ-complexes of o- and p-attack are more stabilised and thereby favoured. | ||
An early contribution by Itoh et al. qualitatively explored the substrate scope and selectivity of the VBPO from Corallina piltllifera57 establishing that it was essentially identical to the scope and selectivity of ‘chemical methods’.
Hartung and coworkers investigated, for example, the bromination of various phenols using the V-dependent bromoperoxidase from Ascophyllum nodosum.58 Significant substitution effects on the rate of the halogenation reaction were observed. Notably the selectivity of the haloperoxidase-catalysed bromination of phenol towards the monobrominated product (90% conversion, o:p = 9:91) compared to the selectivity when using Br2 (mainly tribrominated product) under comparable conditions. The authors suspected a ‘selectivity effect’ of the biocatalyst.
Holtmann and coworkers used the heme-dependent LfCPO for the halogenation of the terpene thymol to increase its antimicrobial activity.53 Since LfCPO (as a heme-dependent haloperoxidase) suffers from severe inactivation by H2O2, electrochemical O2 reduction was used to provide the enzyme with suitable amounts of H2O2 to drive the reaction and minimise oxidative inactivation (Table 1). This way, very satisfactory turnover numbers of more than 160000 for LfCPO were achieved even though the maximal substrate loading did not exceed 2.5 mM. The regioselectivity of the halogenation reaction was (expectedly) dictated by the chemical reactivity of the starting material.
Later, using the CiVCPO, it was shown that the selectivity of the bromination of thymol is kinetically controlled.43 The monobrominated products represent the primary products, which upon reaching comparable concentrations to the starting material were further converted to the dibrominated products (Fig. 1).
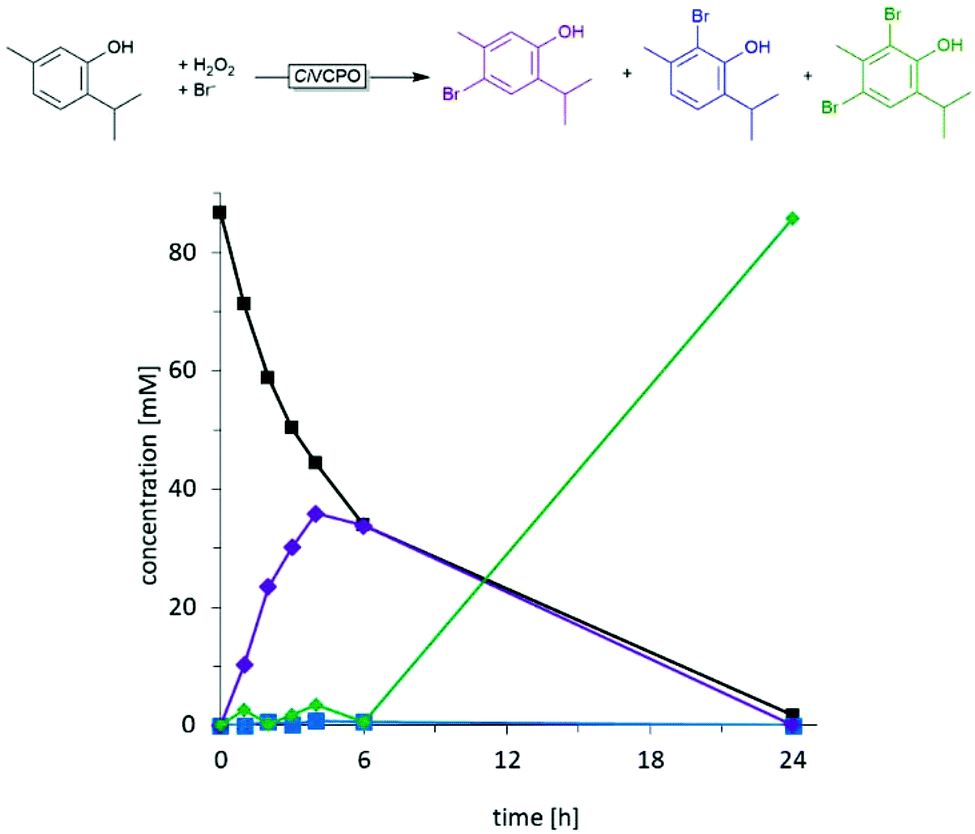 | ||
| Fig. 1 Time course of the chemoenzymatic bromination of thymol using CiVCPO as hypobromite generation catalyst. Figure reproduced from ref. 43. | ||
Over the years, a broad range of arene starting materials have been established for the chemoenzymatic halogenation (Fig. 2).
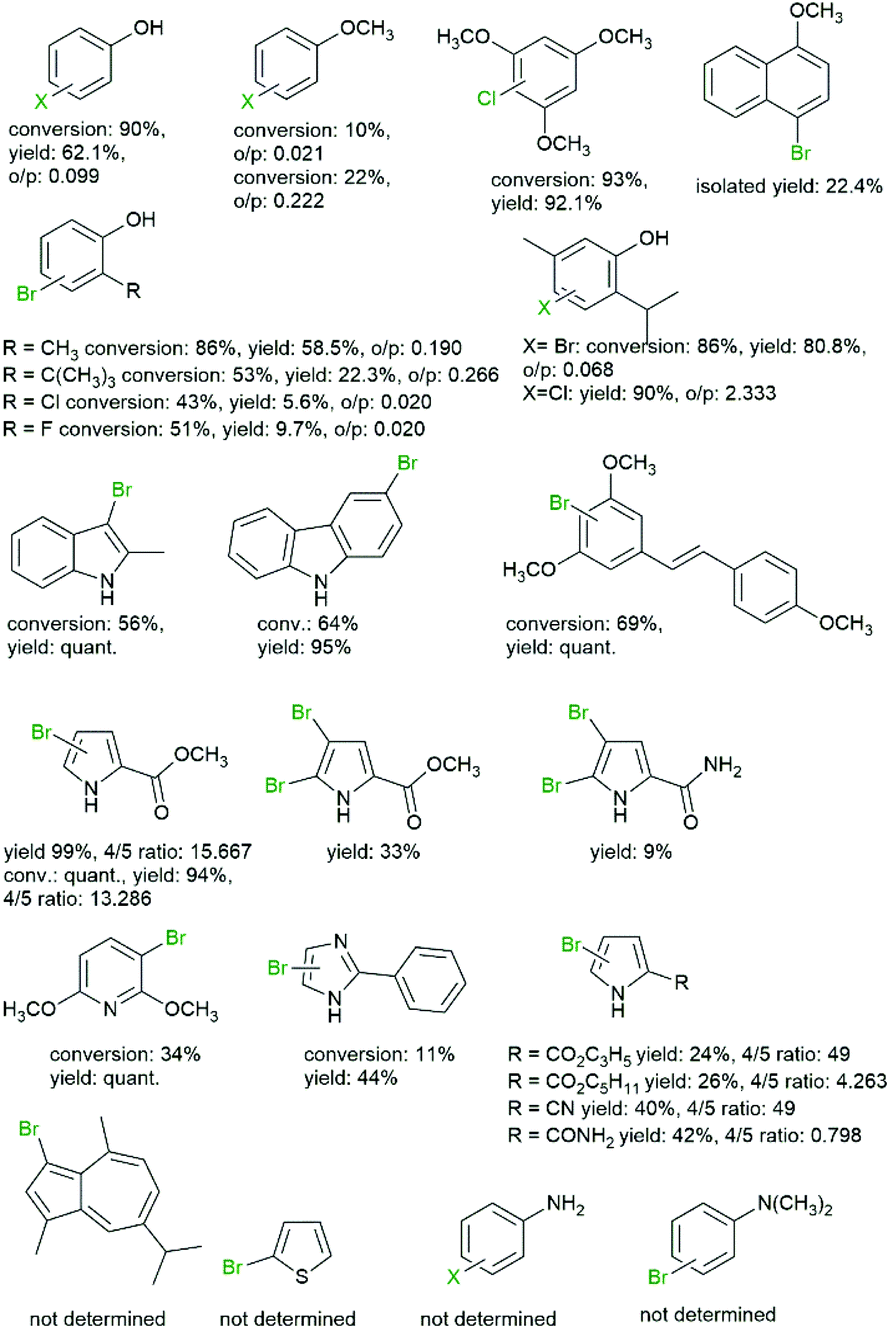 | ||
| Fig. 2 Selection of halogenated arenes obtained via chemoenzymatic halogenation.39,43,51,53,57–61 | ||
As observed for the halogenation of thymol (Table 2) the enzymatic reaction presents higher turnover numbers and frequency than the non-enzymatic reaction but the final product concentrations are still in the lower millimolar range, limiting their application for preparative scale.
| Catalyst | “X+” source | TN [mol mol−1] | TF [s−1] | Product [mM] |
|---|---|---|---|---|
|
||||
| None70 | NBS | n.a. | n.a. | 61 |
| NH4VO371 |
H2O2/Br− | 0.5 | n.a. | 50 |
| NH4VO372 |
H2O2/Br− | 0.9 | 0.00006 | 9 |
| CiVCPO65 | H2O2/Br− | 360000 |
15 | 36 |
|
||||
| [VVO(OMe)(MeOH)]73 | H2O2/Br− | 7117 | 1 | 396 |
| CiVCPO43 | H2O2/Br− | 198000 |
55 | 20 |
|
||||
| WO42− supported on [Ni, Al]-LDH74,75 | H2O2/Br− | 28 | 0.00051 | 125 |
| CiVCPO67 | H2O2/Br− | 277778 |
15 | 5 |
Halohydroxylation reactions
Another reaction that has been realised with haloperoxidases early on is the addition of (haloperoxidase-generated) hypohalites to CC-double bonds yielding in general halohydrins (Scheme 5).
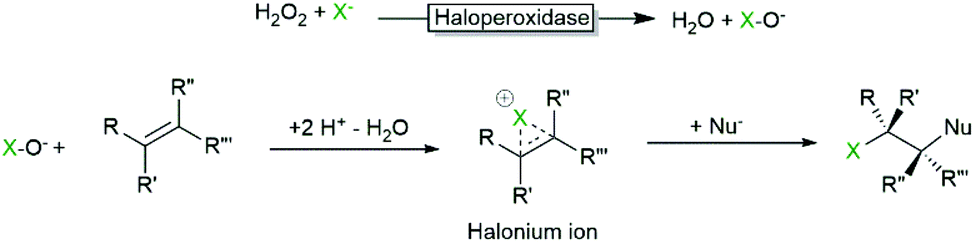 | ||
| Scheme 5 Haloperoxidase-initiated halohydroxylation of alkenes. | ||
As early as the 1980s Itoh et al. investigated the halohydroxylation of various alkenes (Fig. 3) using LfCPO62 and later CpVBPO (from Corallina pilulifera).36,57
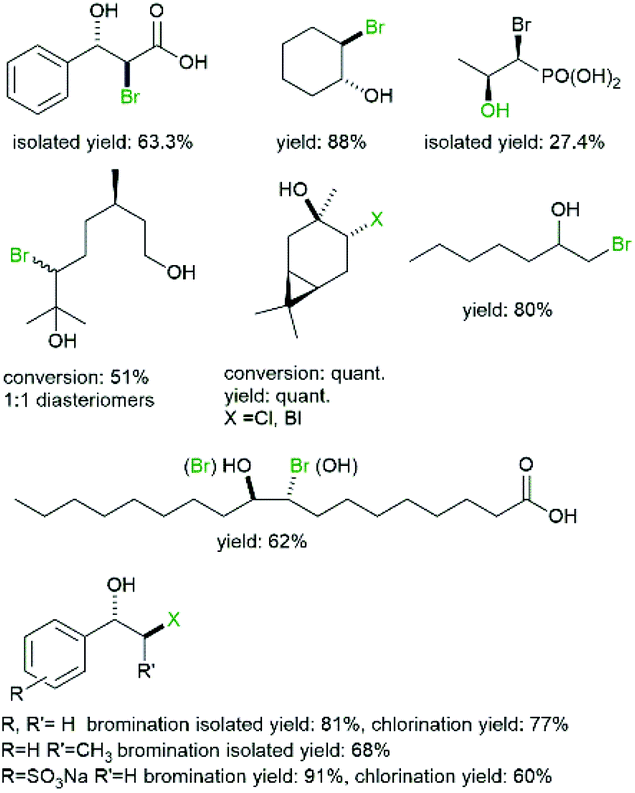 | ||
| Fig. 3 Selection of halohydrins accessible via haloperoxidase-initiated conversion of alkenes.57,62–65 | ||
Performing those reactions in aqueous media, the obvious nucleophile present in abundance is OH− resulting in the formation of halohydrins. As to be expected from a chemical reaction, the selectivity followed Markovnikov's rule. In some cases side-products such as diols or vinyl halides were observed which can be rationalised by intermediate epoxide formation (and subsequent hydrolysis) and by decarboxylation of the intermediate bromonium ion.62
More recently, Schrader and coworkers also introduced terpenes as starting materials for the halohydroxylation reaction63,64 (Fig. 3).
As seen in Table 2, enzyme turnover numbers are very convincing in comparison to other non-enzymatic reactions, however, for preparative application the final product concentrations must be increased.
As mentioned above, aqueous reaction mixtures favour halohydroxylation reactions due to the abundance of the nucleophile (H2O or OH−). An interesting exemption arises with substrates containing a nucleophile in a suitable position for intramolecular attack of the halonium ion. Deska and coworkers converted a broad range of allenic alcohols using LfCPO into the corresponding halogenated furans (Scheme 8).66 Starting from γ,δ-unsaturated acids the corresponding halogenated butyrolactones are accessible (Scheme 8).48 Interestingly, the high water solubility of the substrate also enabled higher reagent payloads.
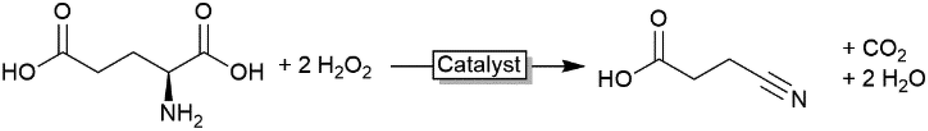
Oxidative decarboxylation
In all examples mentioned above the haloperoxidase-generated hypohalites function as stoichiometric reagents and are incorporated into the starting materials. There are, however, also some examples where the hypohalites themselves function as catalysts. Particularly, the oxidative decarboxylations of α-amino acids is worth mentioning here. If exposed to hypohalites, amino acids undergo oxidative decarboxylation to the corresponding (C1-shortened) nitriles or aldehydes (Scheme 6). In the light of a biobased chemical industry, this reaction could become relevant transforming (waste) amino acids into nitrile building blocks.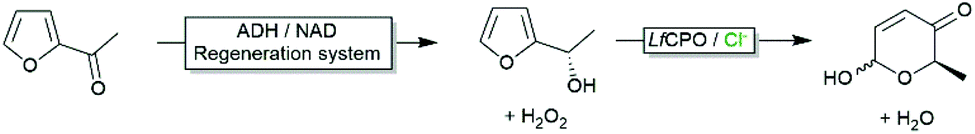 | ||
| Scheme 6 Chemoenzymatic Achmatowicz reaction with biocatalytically generated chiral furanols. | ||
Scott and coworkers explored the chemoenzymatic variant, i.e. using CiVCPO as catalyst to in situ generate hypohalites and thereby use the corresponding halide in catalytic amounts (also circumventing stoichiometric salt wastes).49,67 The aldehyde/nitrile selectivity of the oxidative decarboxylation reaction depends on the amino acid used as well as on the reaction conditions. While the enzymatic reaction presents higher turnover numbers than the W-catalysed reaction (Table 2), in order to achieve economic benefits the substrate loading must be improved (Scheme 7).
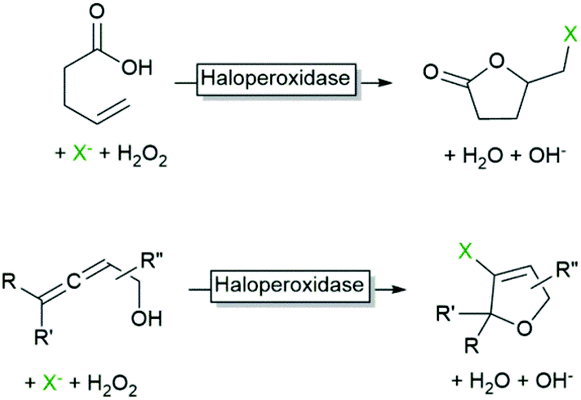 | ||
| Scheme 7 Halocyclisation reactions initiated by haloperoxidases. | ||
 | ||
| Scheme 8 Oxidative decarboxylation of amino acids using hypohalites. | ||
Achmatowicz reaction
The oxidative ring expansion of furans (Achmatowicz reaction) is another example of the catalytic versatility of haloperoxidases. Pioneered by Deska and coworkers68 the biocatalytic Achmatowicz reaction, especially if combined with further stereoselective biocatalytic reactions gives access to a range of building blocks e.g. for natural product synthesis (Scheme 7). Probably due to the rather early stage of development, so far no mechanistic studies elucidating the catalytic mechanism are reported.In their original contribution Deska and coworkers used the Heme-dependent haloperoxidase from Leptoxyphium fumago (LfCPO). Due to the high sensitivity of this enzyme, an in situ H2O2 generation system (glucose-oxidase-catalysed oxidation of glucose) had to be applied to maintain the in situ H2O2 concentration at low levels. Later, an aza-variant of this reaction was reported.69 Since the highly robust vanadium chloroperoxidase from Curvularia inaequalis (CiVCPO) was used, H2O2 could be added to the reaction mixture in stoichiometric amounts.
We expect that this interesting alternative to the chemical Achmatowicz reaction utilising elementary halogens will gain more interest in the near future and that the synthetic scope will be broadened significantly.
Conclusion and outlook
A broad range of hypohalite mediated transformations exist in organic synthesis. To alleviate selectivity and environmental issues arising from stoichiometric addition of hypohalite salts, a range of catalytic methods generating hypohalites in situ through oxidation of the corresponding halides have been developed (Table 2). Using haloperoxidases is one of many options. Still, the excellent catalytic performance of haloperoxidases (in terms of turnover frequency and turnover numbers) are very convincing compared to ‘classical’ chemical catalysts. As shown in Table 2, the catalyst loadings (expressed indirectly as turnover numbers, TN = molproduct × molcatalyst−1) needed for significant conversion are several orders of magnitude lower in case of haloperoxidases than using ‘traditional’ chemical catalysts. We believe that these numbers are so convincing that they should motivate organic chemists to evaluate haloperoxidases as catalysts (we are more than happy to share our expression systems and enzymes!).Currently, only a fraction of hypohalite-based chemistry has been realised using haloperoxidases as catalysts. We are therefore convinced that haloperoxidases are going to play an increasingly important role in this class of oxidation chemistry!
Nevertheless, some issues still need to be addressed to render haloperoxidases truly practical catalysts for large-scale organic synthesis.
First and foremost, the substrate loadings must be increased dramatically into the upper 100 mM or, preferentially, into the molar range. Dilute aqueous reaction mixtures as they are still common in biocatalytic practice are not attractive neither from an economical nor an environmental point-of-view.76 For this, a range of reaction engineering concepts are available. Multiple-phase reactions, for example, either based on slurry-to-slurry or utilising the two-liquid-phase concept are well-suited to dramatically increase the reagent concentration.
Another challenge to be addressed in the near future will be to tackle the selectivity issue. Today, most haloperoxidase reactions rely on diffusible hypohalites and thereby miss an important property of biocatalysis: selectivity. Some promising bacterial haloperoxidases, which exhibit selectivity in their oxidation reactions, are known. However, their potential is far from being extensively exploited. More, in-depth investigations aiming at a molecular understanding of the selectivity of these enzymes and transfer of this knowledge to engineer other haloperoxidases will certainly boost this class of biocatalysts.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Kolvari, N. Koukabi, A. Khoramabadi-zad, A. Shiri and M. A. Zolfigol, Curr. Org. Synth., 2013, 10, 837–863 CrossRef CAS.
- V. Conte and B. Floris, Inorg. Chim. Acta, 2010, 363, 1935–1946 CrossRef CAS.
- A. Schallmey and M. Schallmey, Appl. Microbiol. Biotechnol., 2016, 100, 7827–7839 CrossRef CAS PubMed.
- J. Dong, E. Fernández-Fueyo, F. Hollmann, C. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed.
- V. Weichold, D. Milbredt and K.-H. van Pée, Angew. Chem., Int. Ed., 2016, 55, 6374–6389 CrossRef CAS PubMed.
- C. Leblanc, H. Vilter, J. B. Fournier, L. Delage, P. Potin, E. Rebuffet, G. Michel, P. L. Solari, M. C. Feiters and M. Czjzek, Coord. Chem. Rev., 2015, 301–302, 134–146 CrossRef CAS.
- D. R. M. Smith, S. Gruschow and R. J. M. Goss, Curr. Opin. Chem. Biol., 2013, 17, 276–283 CrossRef CAS PubMed.
- K.-H. van Pee, Curr. Org. Chem., 2012, 16, 2583–2597 CrossRef CAS.
- C. Wagner, M. El Omari and G. M. Konig, J. Nat. Prod., 2009, 72, 540–553 CrossRef CAS PubMed.
- D. G. Fujimori and C. T. Walsh, Curr. Opin. Chem. Biol., 2007, 11, 553–560 CrossRef CAS PubMed.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2010, 15, 266–274 CrossRef.
- M. Hofrichter and R. Ullrich, Appl. Microbiol. Biotechnol., 2006, 71, 276–288 CrossRef CAS PubMed.
- R. Wever and M. A. van der Horst, Dalton Trans., 2013, 42, 11778–11786 RSC.
- R. Wever, in Vanadium: Biochemical and Molecular Biological Approaches, Springer, 2012, pp. 95–125 Search PubMed.
- R. Wever and R. Renirie, in Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology, and Physiology, 2010, pp. 363–385 Search PubMed.
- R. Wever, in Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology, and Physiology, 2010 Search PubMed.
- J. W. P. M. Van Schijndel, P. Barnett, J. Roelse, E. G. M. Vollenbroek and R. Wever, Eur. J. Biochem., 1994, 225, 151–157 CrossRef CAS PubMed.
- J. N. Carter-Franklin, J. D. Parrish, R. A. Tschirret-Guth, R. D. Little and A. Butler, J. Am. Chem. Soc., 2003, 125, 3688–3689 CrossRef CAS PubMed.
- J. Latham, E. Brandenburger, S. A. Shepherd, B. R. K. Menon and J. Micklefield, Chem. Rev., 2017, 118, 232–269 CrossRef PubMed.
- P. Bernhardt, T. Okino, J. M. Winter, A. Miyanaga and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 4268–4270 CrossRef CAS PubMed.
- L. Kaysser, P. Bernhardt, S.-J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2012, 134, 11988–11991 CrossRef CAS PubMed.
- Z. D. Miles, S. Diethelm, H. P. Pepper, D. M. Huang, J. H. George and B. S. Moore, Nat. Chem., 2017, 9, 1235 CrossRef CAS PubMed.
- D. R. Morris and L. P. Hager, J. Biol. Chem., 1966, 241, 1763–1768 CAS.
- F. van de Velde, M. Bakker, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2001, 72, 523–529 CrossRef CAS PubMed.
- B. O. O. Burek, S. Bormann, F. Hollmann, J. Bloh and D. Holtmann, Green Chem., 2019, 21, 3232–3249 RSC.
- S. Bormann, A. Gomez Baraibar, Y. Ni, D. Holtmann and F. Hollmann, Catal. Sci. Technol., 2015, 5, 2038–2052 RSC.
- R. Ullrich and M. Hofrichter, FEBS Lett., 2005, 579, 6247–6250 CrossRef CAS PubMed.
- R. Ullrich, J. Nüske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- H. S. Soedjak and A. Butler, Biochemistry, 1990, 29, 7974–7981 CrossRef CAS PubMed.
- J. N. Carter-Franklin and A. Butler, J. Am. Chem. Soc., 2004, 126, 15060–15066 CrossRef CAS PubMed.
- M. Sandy, J. N. Carter-Franklin, J. D. Martin and A. Butler, Chem. Commun., 2011, 47, 12086–12088 RSC.
- E. E. Coupe, M. G. Smyth, A. Fosberry, R. M. Hall and J. A. Littlechild, Protein Expression Purif., 2007, 52, 265–272 CrossRef CAS PubMed.
- K. Kaneko, K. Washio, T. Umezawa, F. Matsuda, M. Morikawa and T. Okino, Biosci. Biotechnol. Biochem., 2014, 78, 1310–1319 CrossRef CAS PubMed.
- N. Itoh, L. Y. Cheng, Y. Izumi and H. Yamada, J. Biotechnol., 1987, 5, 29–38 CrossRef CAS.
- H. Vilter, Phytochemistry, 1984, 23, 1387–1390 CrossRef CAS.
- N. Itoh, Y. Izumi and H. Yamada, J. Biol. Chem., 1986, 261, 5194–5200 CAS.
- A. Frank, C. J. Seel, M. Groll and T. Gulder, ChemBioChem, 2016, 17, 2028–2032 CrossRef CAS PubMed.
- H. Plat, B. E. Krenn and R. Wever, Biochem. J., 1987, 248, 277–279 CrossRef CAS PubMed.
- J. van Schijndel, E. Vollenbroek and R. Wever, Biochim. Biophys. Acta, 1993, 1161, 249–256 CrossRef CAS.
- R. Wever, B. E. Krenn and R. Renerie, in Methods in Enzymology, 2018, vol. 605 Search PubMed.
- E. Fernández-Fueyo, M. van Wingerden, R. Renirie, R. Wever, Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2015, 7, 4035–4038 CrossRef.
- R. Renirie, C. Pierlot, R. Wever and J. M. Aubry, J. Mol. Catal. B: Enzym., 2009, 56, 259–264 CrossRef CAS.
- R. Renirie, C. Pierlot, J.-M. Aubry, A. F. Hartog, H. E. Schoemaker, P. L. Alsters and R. Wever, Adv. Synth. Catal., 2003, 345, 849–858 CrossRef CAS.
- E. Deboer and R. Wever, J. Biol. Chem., 1988, 263, 12326–12332 CAS.
- G. P. Rai, S. Sakai, A. M. Flórez, L. Mogollon and L. P. Hager, Adv. Synth. Catal., 2001, 343, 638–645 CrossRef CAS.
- F. Tieves, S. J.-P. Willot, M. M. C. H. van Schie, M. C. R. Rauch, S. H. H. Younes, W. Zhang, P. G. de Santos, J. M. Robbins, B. Bommarius, M. Alcalde, A. Bommarius and F. Hollmann, Angew. Chem., Int. Ed., 2019, 58, 7873–7877 CrossRef CAS PubMed.
- A. But, A. van Noord, F. Poletto, J. P. M. Sanders, M. C. R. Franssen and E. L. Scott, Mol. Catal., 2017, 443, 92–100 CrossRef CAS.
- W. Zhang, E. Fernández-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55–62 CrossRef CAS PubMed.
- C. J. Seel, A. Králík, M. Hacker, A. Frank, B. König and T. Gulder, ChemCatChem, 2018, 10, 3960–3963 CrossRef CAS.
- D. I. Perez, M. Mifsud Grau, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2009, 44, 6848–6850 RSC.
- L. Getrey, T. Krieg, F. Hollmann, J. Schrader and D. Holtmann, Green Chem., 2014, 16, 1104–1108 RSC.
- P. L. Alsters, W. Jary, V. Nardello-Rataj and J. M. Aubry, Org. Process Res. Dev., 2010, 14, 259–262 CrossRef CAS.
- V. Nardello, J. Marko, G. Vermeersch and J. M. Aubry, Inorg. Chem., 1995, 34, 4950–4957 CrossRef CAS.
- V. Nardello, J. Barbillat, J. Marko, P. T. Witte, P. L. Alsters and J. M. Aubry, Chem. – Eur. J., 2003, 9, 435–441 CrossRef CAS PubMed.
- N. Itoh, A. Hasan, Y. Izumi and H. Yamada, Eur. J. Biochem., 1988, 172, 477–484 CrossRef CAS PubMed.
- D. Wischang and J. Hartung, Tetrahedron, 2012, 68, 9456–9463 CrossRef CAS.
- D. Wischang, O. Brucher and J. Hartung, Coord. Chem. Rev., 2011, 255, 2204–2217 CrossRef CAS.
- D. Wischang and J. Hartung, Tetrahedron, 2011, 67, 4048–4054 CrossRef CAS.
- D. Wischang, M. Radlow and J. Hartung, Dalton Trans., 2013, 42, 11926–11940 RSC.
- H. Yamada, N. Itoh and Y. Izumi, J. Biol. Chem., 1985, 260, 1962–1969 Search PubMed.
- B. A. Kaup, U. Piantini, M. Wust and J. Schrader, Appl. Microbiol. Biotechnol., 2007, 73, 1087–1096 CrossRef CAS PubMed.
- U. Piantini, J. Schrader, A. Wawrzun and M. Wust, Food Chem., 2011, 129, 1025–1029 CrossRef CAS PubMed.
- J. J. Dong, E. Fernandez-Fueyo, J. Li, Z. Guo, R. Renirie, R. Wever and F. Hollmann, Chem. Commun., 2017, 53, 6207–6210 RSC.
- J. Naapuri, J. D. Rolfes, J. Keil, C. Manzuna Sapu and J. Deska, Green Chem., 2017, 19, 447–452 RSC.
- A. But, J. Le Nôtre, E. L. Scott, R. Wever and J. P. M. Sanders, ChemSusChem, 2012, 5, 1199–1202 CrossRef CAS PubMed.
- D. Thiel, D. Doknić and J. Deska, Nat. Commun., 2014, 5, 5278 CrossRef CAS PubMed.
- E. Fernández-Fueyo, S. H. H. Younes, S. v. Rootselaar, R. W. M. Aben, R. Renirie, R. Wever, D. Holtmann, F. P. J. T. Rutjes and F. Hollmann, ACS Catal., 2016, 6, 5904–5907 CrossRef.
- M. Narender, M. S. Reddy, Y. V. D. Nageswar and K. R. Rao, J. Mol. Catal. A: Chem., 2006, 258, 10–14 CrossRef CAS.
- M. Andersson, V. Conte, F. Di Furia and S. Moro, Tetrahedron Lett., 1995, 36, 2675–2678 CrossRef CAS.
- V. Conte, B. Floris, P. Galloni and A. Silvagni, Pure Appl. Chem., 2005, 77, 1575–1581 CAS.
- M. R. Maurya, B. Uprety, F. Avecilla, P. Adão and J. Costa Pessoa, Dalton Trans., 2015, 44, 17736–17755 RSC.
- L. Claes, R. Matthessen, I. Rombouts, I. Stassen, T. De Baerdemaeker, D. Depla, J. A. Delcour, B. Lagrain and D. E. De Vos, ChemSusChem, 2015, 8, 345–352 CrossRef CAS PubMed.
- L. Claes, J. Verduyckt, I. Stassen, B. Lagrain and D. E. De Vos, Chem. Commun., 2015, 51, 6528–6531 RSC.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |