
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vinylboronic acid-caged prodrug activation using click-to-release tetrazine ligation†
Lianne P. W. M.
Lelieveldt
,
Selma
Eising
,
Abel
Wijen
and
Kimberly M.
Bonger
 *
*
Department of Biomolecular Chemistry and Synthetic Organic Chemistry, Radboud University Nijmegen, The Netherlands. E-mail: k.bonger@science.ru.nl
First published on 17th September 2019
Abstract
Bioorthogonal reactions can be performed selectively in the presence of any biological functional group and are widely used to achieve site-selective chemical modifications of biomolecules. The click-to-release reaction is a bioorthogonal bond-cleavage variant that has gained much interest over the last few years. The bioorthogonal reaction between tetrazines and trans-cyclooctenes or vinyl ethers, for example, initiates the release of a small molecule immediately after the cycloaddition with tetrazines. Recently, our group reported that vinylboronic acids (VBAs) give exceptionally high reaction rates in the bioorthogonal inverse electron-demand Diels–Alder reaction with tetrazines that are substituted with boron-coordinating ligands. In the present study, we show that VBAs can be used in a click-to-release variant and demonstrate its bioorthogonality with a VBA-protected doxorubicin prodrug. We show that the cytotoxicity of doxorubicin is silenced by the attachment of the VBA, and activity can be largely restored upon the reaction with a tetrazine, inducing cell death.
Introduction
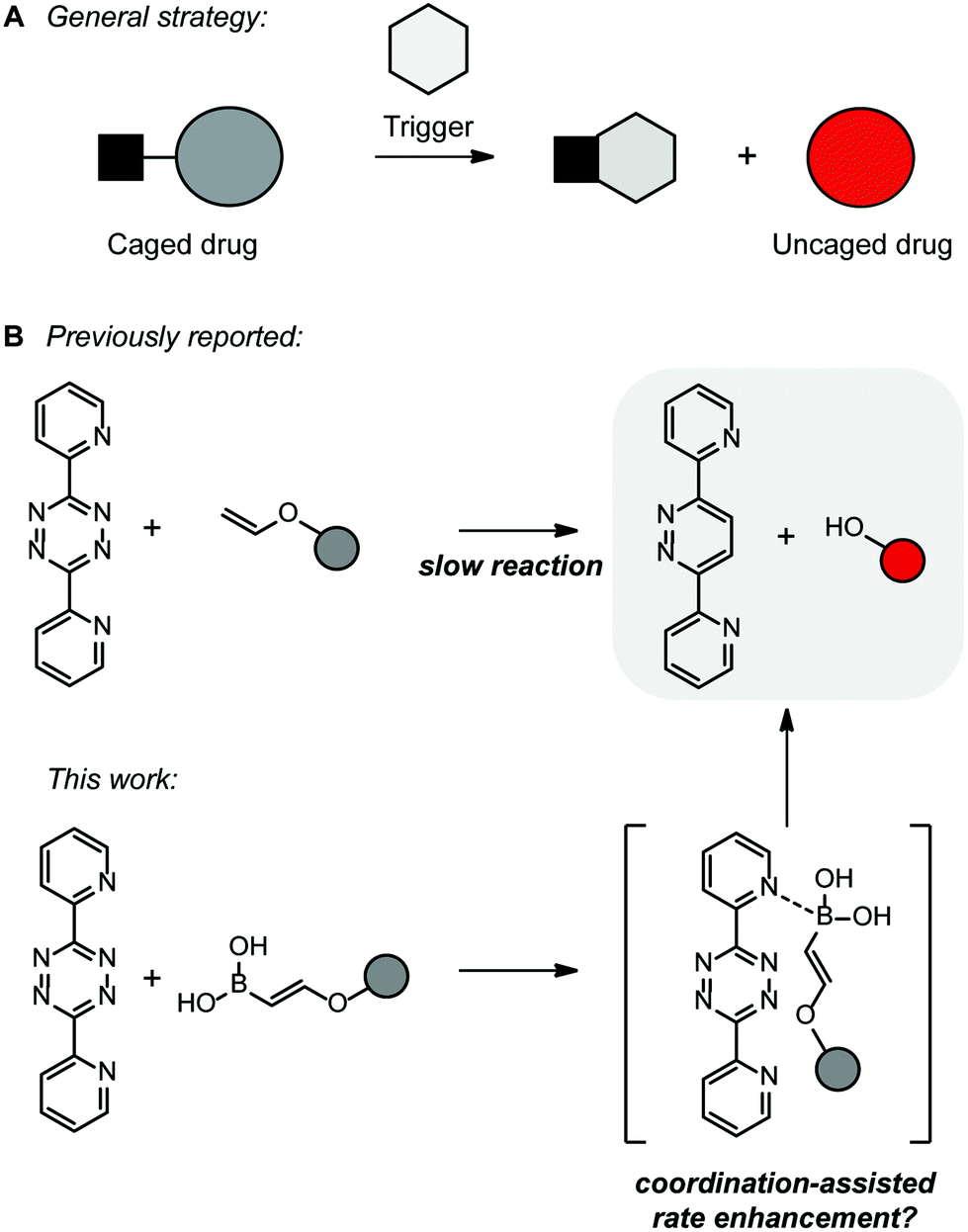
Chemical site-selective protein modifications have become increasingly popular for modification and control over protein functions in vitro and in living systems. Several reactions have been developed to decorate biomolecules with a desired functionality such as fluorophores, affinity probes or reactive tags. All these reactions have some properties in common as they must be selective over other functional groups in biomolecules, have fast reaction rates and proceed in aqueous media around physiological pH.1 Reactions such as the strain-promoted alkyne–azide cycloaddition (SPAAC)2 or the inverse electron-demand Diels–Alder reaction with tetrazines3 have been used extensively for modifying purposes. Our group has recently reported a new bioorthogonal tetrazine ligation utilizing vinylboronic acids (VBAs).4–7 We showed that by introducing a boronic acid moiety on an alkene, reaction rates of an inverse electron-demand Diels–Alder (iEDDA) reaction with pyridyl-containing tetrazines improved by several orders of magnitude compared to the non-modified linear alkene.4 The hydrophilic properties and the small size of VBAs compared to other bioorthogonal reactants make them attractive for use in biomolecular labeling experiments. VBAs were shown to be biocompatible, non-toxic, and highly stable in aqueous media and cell lysates. In addition, while the number of bioorthogonal reactions used in situ and in vivo is limited due to the need for toxic reagents (e.g. copper, in copper(I)-catalyzed alkyne–azide cycloaddition8) or possible side reactions of the reactants (e.g. strained alkenes or alkynes can react with cysteines9), VBAs appeared to be useful in cellular experiments.7 Reactions with vicinal diols, present in carbohydrates for example, were not observed when performing reactions in living cells.The reactivity of VBAs depends on the tetrazine and its substituents. Since the boronic acid coordinates to the pyridyl ring of dipyridyl-s-tetrazine, reaction rates were enhanced in comparison with tetrazines lacking such a potential Lewis base.6 This unique feature allows one to perform orthogonal bioorthogonal reactions with two different tetrazines, one with and one without substituents allowing for coordination, and using a VBA and a strained alkene.
The interest in bioorthogonal bond-cleavage reactions for activation of protected biologically relevant molecules in vivo has increased significantly (Fig. 1A). A substantial number of so-called ‘click-to-release’ reactions have been developed in the last few years.10–12 These reactions enable new applications in vitro and in vivo, such as the activation of prodrugs or fluorogenic compounds. Most reactions are based on the iEDDA reaction between a tetrazine and a (strained) alkene.11
 | ||
| Fig. 1 (A) General scheme of a click-to-release reaction to uncage a prodrug. (B) Overview of the inverse electron-demand Diels–Alder cycloaddition with a tetrazine and a vinyl ether or vinylboronic acid to release an alcohol functionality. | ||
In 2013, the group of Robillard reported the first bioorthogonal click-to-release reaction based on the reaction of tetrazines with trans-cyclooctenes (TCOs) that possess a carbamate substituent at the allylic position (Fig. 1A).13 After the iEDDA reaction of the tetrazine and TCO, the 4,5-dihydropyridazine eliminates the leaving group to liberate both CO2 and the amine. Unfortunately, part of the dihydropyridazine tautomerizes to the 1,4-dihydropyridazine, which does not lead to a release reaction and therefore this click-to-release process did not result in complete elimination. Over the last few years, the reaction has been optimized14,15 and the scope was further expanded16 and applied in biological systems for activation of prodrugs,17–23 RNA24 or caged proteins.14,25,26 Recently, it was noted that this click-to-release reaction is environmentally sensitive and improvements on both the TCO and tetrazine resulted in ultrafast click-to-release reaction rates and the complete elimination of the leaving group.27
In 2016 and 2017, the groups of Devaraj and Bernardes reported both the bioorthogonal click-to-release reactions of tetrazines and vinyl ethers (Fig. 1A).28,29 These alkenes bear their leaving group directly on the alkene and therefore this results in immediate full release of the alcohol after the iEDDA reaction. The vinyl ethers appeared to be stable in aqueous solutions for at least 8 hours and were suitable for the release of a prodrug in living cells.30 The vinyl ethers have the advantage of being small in size, but have disappointingly low rate constants when reacting with tetrazines.
Since VBAs have proven to be useful in bioorthogonal conjugation, we envisioned that by introducing a boronic acid group on the vinyl ethers, we could increase the rate of these click-to-release reactions. In addition, similar to vinyl ethers, we expect that VBA ethers fully release the alcohol, or the amine when linked via a carbamate, after the iEDDA reaction. The proposed iEDDA click-to-release reaction mechanism of vinylboronic acids with tetrazines is shown in Fig. 1B. Upon cycloaddition and retro-Diels–Alder reaction, N2, the boric acid and the uncaged drug are released. We here explore the applicability of the VBA click-to-release strategy and develop a synthetic route to VBA protected prodrugs, which can be released using a tetrazine in the presence of cells.
Results and discussion
We first aimed to compare the reactivity of the VBA ether with the unsubstituted vinyl ethers and synthesized VBA 6 and its primary alkene derivative 9 (Scheme 1). The synthesis of VBA 6 started with the etherification of 4-hydroxybenzyl alcohol 1 with trichloroethylene and subsequent silyl protection of the primary alcohol. Next, dichlorovinyl ether 3 was treated with n-BuLi to eliminate hydrogen chloride and to provide alkynyl ether 4. Deprotection of the primary alcohol and hydroboration of alkyne 5 with pinacolborane yielded VBA pinacol ester 6. In our kinetic experiments, we used the pinacol ester as a precursor for the free boronic acid as it hydrolyzes quickly in aqueous media (see Fig. SI-1†). The synthesis of vinyl ether 9 was performed following a literature procedure.31 Here, 4-hydroxybenzaldehyde (7) was first reacted with 1,2-dibromoethane to yield compound 8 which was subsequently eliminated using t-BuOK and the aldehyde was reduced using NaBH4 to yield the corresponding benzyl alcohol 9.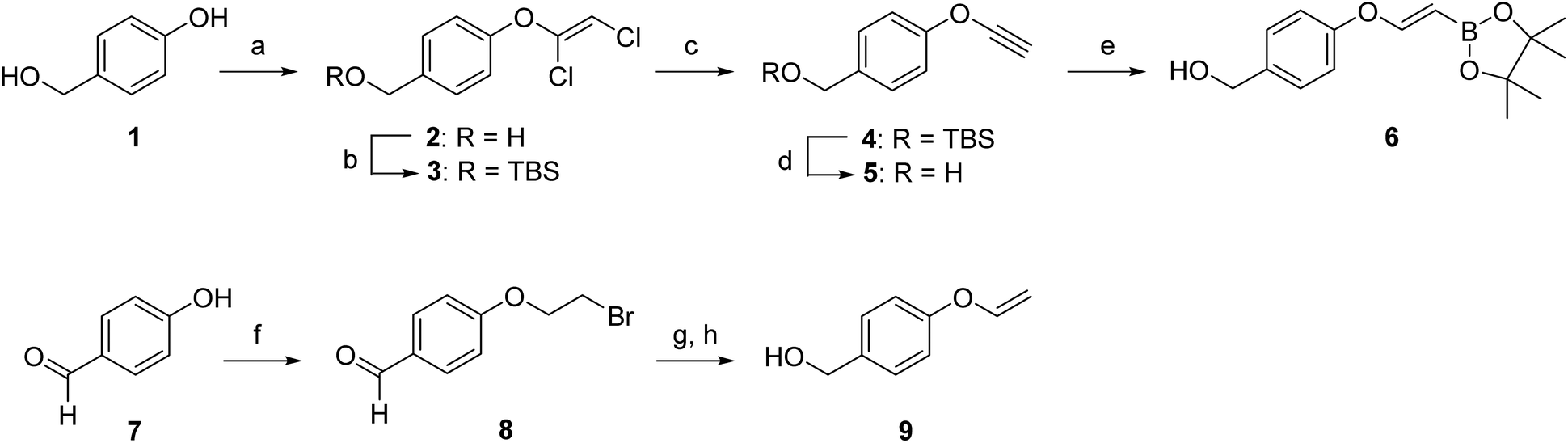 | ||
| Scheme 1 Synthesis of VBA ether 6 and vinyl ether 9. (a) Trichloroethylene, K2CO3, DMF, 70 °C, 16 h, 87%. (b) TBSCl, imidazole, DMF, 2 h, quant. (c) n-BuLi, Et2O, −78 to −40 °C, 87%. (d) TBAF, THF, 0 °C, 91%. (e) Pinacolborane, RuHClCO(PPh3)3, toluene, 50 °C, 16 h, 76%. (f) Dibromoethane, K2CO3, MeCN, reflux, 20 h, 79%. (g) t-BuOK, DMSO, r.t., 10 min. (h) NaBH4, MeOH, r.t., 1.5 h, 18%. | ||
Having alkenes 6 and 9 in hand, we examined the second-order rate constants with 3,6-dipyridyl-s-tetrazine 10 in 75% MeOH in PBS at a controlled temperature of 20 °C (Fig. 2A). As we used the pinacol ester as a precursor, we incubated ester 6 for 2 h in the solvent mixture to achieve full hydrolysis to the free boronic acid before measuring its k2 value. Second-order rate constants were obtained by following the decrease of the tetrazine UV absorbance in the visible region. The pseudo first-order constant kobs was first determined by plotting the decay of tetrazine absorbance against time (minutes) for five different amounts of excess alkene (see also the ESI†). Next, the kobs was plotted against the five different alkene concentrations and a linear regression fit gave the linear function, of which the slope corresponded to the k2 (Fig. SI-2†). The second-order rate constant for VBA 6 was 3.3 × 10−3 M−1 s−1 and 4-fold higher than the rate constant of vinyl ether 9, clearly indicating a positive effect of the boronic acid on the reactivity of the alkene.
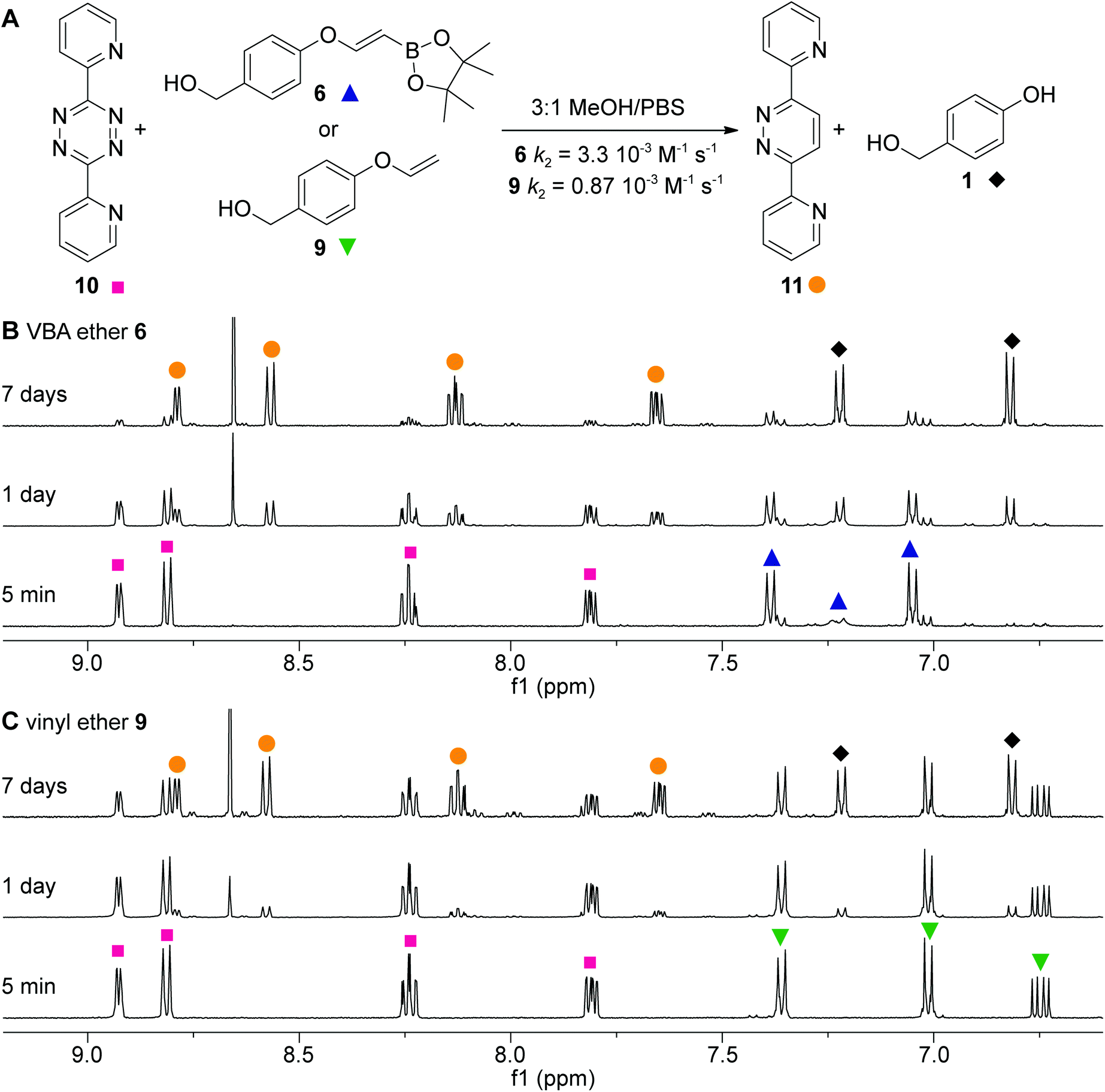 | ||
Fig. 2 Click-to-release reactions and reaction rates. (A) Scheme of the reaction of 3,6-dipyridyl-s-tetrazine Tz with VBA ether 6 and vinyl ether 9. k2 values were measured in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/PBS; (B) 1H-NMR study of the reaction between tetrazine 10 ( :1 MeOH/PBS; (B) 1H-NMR study of the reaction between tetrazine 10 ( ) and VBA ether 6 ( ) and VBA ether 6 ( ) yielding pyridazine 11 ( ) yielding pyridazine 11 ( ) and 4-hydroxybenzyl alcohol 1 ( ) and 4-hydroxybenzyl alcohol 1 ( ) in 3:1 CD3OD/deuterated PBS; (C) similar to (B) with the reaction of Tz with vinyl ether 9 ( ) in 3:1 CD3OD/deuterated PBS; (C) similar to (B) with the reaction of Tz with vinyl ether 9 ( ). ). | ||
The observed rate constant of VBA ether 6 was lower than expected as we previously observed rate enhancements of up to 100-fold compared to the unsubstituted alkene derivatives.4 We hypothesize that the moderate rate enhancement is due to the ether on the vinylic position. Since this alcohol is slightly electron-donating, we assume that the alkene becomes more electron-rich, thereby lowering the acidity of the boronic acid, resulting in less favorable coordination to the pyridyl substituents of the tetrazine.32
Next, we looked in more detail into the click-to-release reaction of 3,6-dipyridyl-s-tetrazine 10 and alkenes 6 and 9 by 1H NMR spectroscopy in 75% CD3OD in deuterated PBS over time (Fig. 2B and C). A near to complete conversion of VBA ether 6 was observed after 7 days, while vinyl ether 9 gave around 50% conversion. Both reactions resulted in the clean formation of pyridazine 11 and 4-hydroxybenzyl alcohol 1. No intermediates were observed showing that the initial cycloaddition step is rate limiting for both VBA ether 6 and vinyl ether 9.
To further examine the click-to-release reaction with a VBA ether in a more complex system, we synthesized a VBA-caged cytotoxin doxorubicin and explored the effect on HeLa cells upon addition of a water soluble tetrazine 12 (Fig. 3A). For this, we aimed to couple doxorubicin to VBA alcohol 6, which acts as a self-cleavable linker by 1,6 elimination upon release (Fig. 3B).33 We commenced the synthesis from VBA alcohol 6 and activated the alcohol using bis(pentafluorophenyl)carbonate and Et3N as a base, yielding successfully VBA-PFP carbonate 13. Doxorubicin was subsequently added and reacted overnight to obtain VBA-caged doxorubicin 14 (VBA-Dox) in reasonable yields.
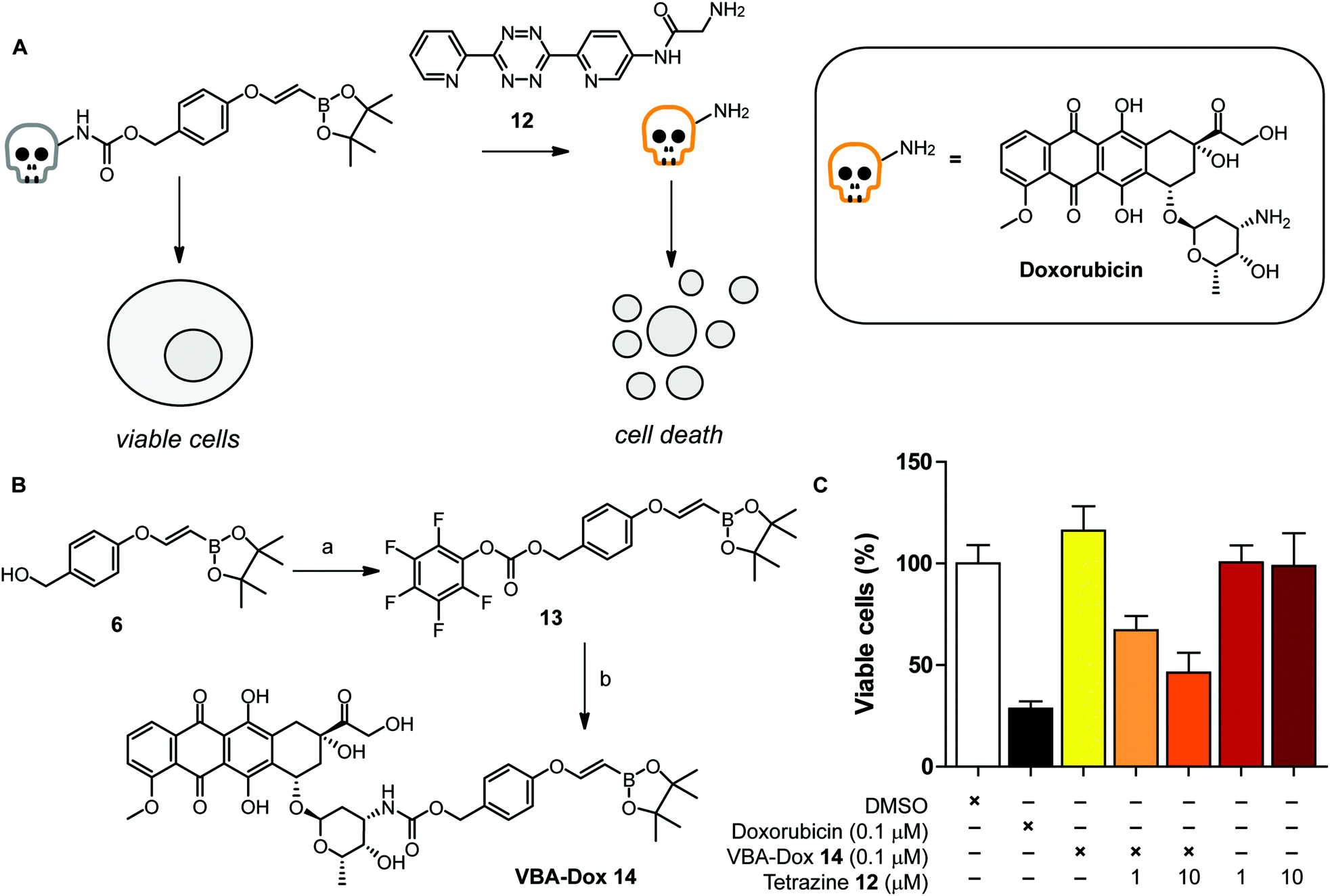 | ||
| Fig. 3 Click-to-release reaction of VBA-Dox 14 to yield the free doxorubicin. (A) General overview of the caging strategy. (B) Synthetic route of VBA-Dox. (a) Bis(pentafluorophenyl)carbonate, Et3N, r.t., 1 h, 84%. (b) Doxorubicin HCl, Et3N, DMF, r.t., 24 h, 40%. (C) Evaluation of cytotoxicity of VBA-Dox 14 incubated with tetrazine 12 (1 or 10 μM, respectively, 10 equiv. or 100 equiv.) for 72 h with HeLa cells. | ||
First, we tested the stability of the VBA-Dox 14 at 100 μM in PBS at 37 °C before performing the click-to-release reactions with cells. As shown in Fig. SI-3,† hydrolysis of the pinacol ester was observed, as expected, while minor decomposition (around 10%) of VBA-Dox 14 was observed after 48 hours. Decomposition likely results from protodeboronation of the VBA cage.34 Importantly, no free doxorubicin was observed after incubating VBA-Dox 14 in PBS for up to 7 days. Encouraged by these results we next evaluated the cycloaddition with a water soluble tetrazine 12 and followed the subsequent release over time in a buffer system with LCMS. Deprotection of 14 at 100 μM in PBS at 37 °C, using 1 equivalent of tetrazine 12 resulted in decaging and release of doxorubicin (Fig. SI-4†). No intermediate products were observed, indicating that the cleavage of the linker occurred instantaneously after the cycloaddition.
Next, we tested the VBA release and toxicity of doxorubicin on living HeLa cells (Fig. 3A). We first incubated HeLa cells with free doxorubicin and observed a dose dependent toxic effect, as expected (Fig. SI-5†). In addition, we incubated VBA-caged doxorubicin in the absence of a tetrazine. No toxicity was observed with 100 nM VBA-doxorubicin 14 while 1 μM VBA-Dox did show some toxicity after 3 days of incubation. As we observed more than 70% cell death with free doxorubicin at the lower concentration of 100 nM we used these conditions to test the efficiency of the VBA click-to-release strategy with tetrazine 12.
We incubated VBA-doxorubicin 14 with 10 and 100 equivalents of tetrazine 12 with HeLa cells for 3 days. To our delight, we observed a dose-dependent toxicity of tetrazine-activated doxorubicin and an almost similar toxicity level was obtained to that when incubated with free doxorubin (Fig. 3C). Using higher amounts of tetrazine 12 also showed signs of toxicity of the tetrazine itself, so we omitted this concentration in further cell studies (Fig. SI-5†).
Conclusion
In this work we have shown that VBAs can be used in bioorthogonal click-to-release tetrazine ligations. The release was instantaneous after the tetrazine cycloaddition as no reaction intermediates were observed by NMR experiments. We assume that the slight electron-donating nature of the ether at the vinylic position of the boronic acid reduces the reaction rate as compared to VBAs lacking the ether functionality. The boronic acid moiety increases the reactivity of vinyl ethers in the click-to-release tetrazine ligation several folds when tetrazines bearing boron-coordinating ligands are used.The use of VBA-caged prodrugs may be of interest as we have recently shown that this moiety can orthogonally react with tetrazines bearing a coordinating phenol substituent. These tetrazines are too electron rich to react with vinyl ethers.5 It would further be interesting to explore different substituents on the VBA cage, such as carbamates, to study the electronic effects in the click-to-release reaction with different coordinating tetrazines. These studies are currently ongoing in our laboratory.
Conflicts of interest
No conflict declared.Acknowledgements
This research was funded by the NWO gravity program ‘Institute for Chemical Immunology’ NWO-024.002.009 and the Institute for Molecules and Materials at Radboud University. We thank Selina Thijssen for her pioneering work on this project and Professor Floris Rutjes for fruitful discussions.References
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 1–20 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- S. Eising, F. Lelivelt and K. M. Bonger, Angew. Chem., Int. Ed., 2016, 55, 12243–12247 CrossRef CAS PubMed.
- S. Eising, A. H. J. Engwerda, X. Riedijk, F. M. Bickelhaupt and K. M. Bonger, Bioconjugate Chem., 2018, 29, 3054–3059 CrossRef CAS PubMed.
- S. Eising, B. T. Xin, F. Kleinpenning, J. Heming, B. I. Florea, H. S. Overkleeft and K. M. Bonger, ChemBioChem, 2018, 19, 1648–1165 CrossRef CAS PubMed.
- S. Eising, N. G. A. van der Linden, F. Kleinpenning and K. M. Bonger, Bioconjugate Chem., 2018, 29, 982–986 CrossRef CAS PubMed.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- R. Van Geel, G. J. M. Pruijn, F. L. Van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392–398 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- X. Ji, Z. Pan, B. Yu, L. Kimberly, D. La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC.
- S. Davies, B. J. Stenton, G. J. L. Bernardes, S. Davies and B. Stenton, Chimia, 2018, 72, 771–776 CrossRef CAS PubMed.
- R. M. Versteegen, R. Rossin, W. Ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- X. Fan, Y. Ge, F. Lin, Y. Yang, G. Zhang, W. S. C. Ngai, Z. Lin, S. Zheng, J. Wang, J. Zhao, J. Li and P. R. Chen, Angew. Chem., Int. Ed., 2016, 55, 14046–14050 CrossRef CAS PubMed.
- J. C. T. Carlson, H. Mikula and R. Weissleder, J. Am. Chem. Soc., 2018, 140, 3603–3612 CrossRef CAS PubMed.
- R. M. Versteegen, W. Hoeve, R. Rossin, M. A. R. De Geus, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2018, 57, 10494–10499 CrossRef CAS PubMed.
- J. M. M. Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476–482 CrossRef PubMed.
- R. Rossin, S. M. J. Van Duijnhoven, W. Ten Hoeve, H. M. Janssen, L. H. J. Kleijn, F. J. M. Hoeben, R. M. Versteegen and M. S. Robillard, Bioconjugate Chem., 2016, 27, 1697–1706 CrossRef CAS PubMed.
- R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. Hoeve, H. M. Janssen, A. H. A. M. Van Onzen, P. J. Hudson and M. S. Robillard, Nat. Commun., 2018, 9, 1484–1494 CrossRef PubMed.
- S. Davies, L. Qiao, B. L. Oliveira and C. D. Navo, ChemBioChem, 2019, 20, 1541–1546 CAS.
- I. Khan, P. F. Agris, M. V. Yigit and M. Royzen, Chem. Commun., 2016, 52, 6147–6177 Search PubMed.
- S. Davies, B. L. Oliveira and G. J. L. Bernardes, Org. Biomol. Chem., 2019, 17, 5725–5730 RSC.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Li, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed.
- E. Agustin, P. N. Asare Okai, I. Khan, M. R. Miller, R. Wang, J. Sheng and M. Royzen, Chem. Commun., 2016, 52, 1405–1408 RSC.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003–1005 CrossRef CAS PubMed.
- G. Zhang, J. Li, R. Xie, X. Fan, Y. Liu, S. Zheng, Y. Ge and P. R. Chen, ACS Cent. Sci., 2016, 2, 325–331 CrossRef CAS PubMed.
- J. C. T. Carlson, H. Mikula and R. Weissleder, J. Am. Chem. Soc., 2018, 140, 3603–3612 CrossRef CAS PubMed.
- H. Wu, S. C. Alexander, S. Jin and N. K. Devaraj, J. Am. Chem. Soc., 2016, 138, 11429–11432 CrossRef CAS PubMed.
- E. Jiménez-Moreno, Z. Guo, B. L. Oliveira, I. S. Albuquerque, A. Kitowski, A. Guerreiro, O. Boutureira, T. Rodrigues, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2017, 56, 243–247 CrossRef PubMed.
- K. Neumann, S. Jain, A. Gambardella, S. E. Walker, E. Valero, A. Lilienkampf and M. Bradley, ChemBioChem, 2017, 18, 91–95 CrossRef CAS PubMed.
- J. Jiang, W. Liu, J. Cheng, L. Yang, H. Jiang, D. Bai and W. Liu, Chem. Commun., 2012, 48, 8371–8373 RSC.
- D. G. Hall, Boronic acids, Preparation and Applications in Organic Synthesis, Medicine and Materials, WILEY-VCH, Weinheim, 2011 Search PubMed.
- C. A. Blencowe, A. T. Russell, F. Greco, W. Hayes and D. W. Thornthwaite, Polym. Chem., 2011, 2, 773–790 RSC.
- P. A. Cox, A. G. Leach, A. D. Campbell, G. C. Lloyd-jones, P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob01881f |
| This journal is © The Royal Society of Chemistry 2019 |