
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective inhibition of APOBEC3 enzymes by single-stranded DNAs containing 2′-deoxyzebularine†
Fareeda M.
Barzak
 a,
Stefan
Harjes
a,
Maksim V.
Kvach
a,
Harikrishnan M.
Kurup
ab,
Geoffrey B.
Jameson
ab,
Vyacheslav V.
Filichev
*ab and
Elena
Harjes
*ab
a,
Stefan
Harjes
a,
Maksim V.
Kvach
a,
Harikrishnan M.
Kurup
ab,
Geoffrey B.
Jameson
ab,
Vyacheslav V.
Filichev
*ab and
Elena
Harjes
*ab
aSchool of Fundamental Sciences, Massey University, Private Bag 11 222, Palmerston North 4442, New Zealand
bMaurice Wilkins Centre for Molecular Biodiscovery, Auckland 1142, New Zealand. E-mail: e.harjes@massey.ac.nz; v.filichev@massey.ac.nz
First published on 11th October 2019
Abstract
To restrict pathogens, in a normal human cell, APOBEC3 enzymes mutate cytosine to uracil in foreign single-stranded DNAs. However, in cancer cells, APOBEC3B (one of seven APOBEC3 enzymes) has been identified as the primary source of genetic mutations. As such, APOBEC3B promotes evolution and progression of cancers and leads to development of drug resistance in multiple cancers. As APOBEC3B is a non-essential protein, its inhibition can be used to suppress emergence of drug resistance in existing anti-cancer therapies. Because of the vital role of APOBEC3 enzymes in innate immunity, selective inhibitors targeting only APOBEC3B are required. Here, we use the discriminative properties of wild-type APOBEC3A, APOBEC3B and APOBEC3G to deaminate different cytosines in the CCC-recognition motif in order to best place the cytidine analogue 2′-deoxyzebularine (dZ) in the CCC-motif. Using several APOBEC3 variants that mimic deamination patterns of wild-type enzymes, we demonstrate that selective inhibition of APOBEC3B in preference to other APOBEC3 constructs is feasible for the dZCC motif. This work is an important step towards development of in vivo tools to inhibit APOBEC3 enzymes in living cells by using short, chemically modified oligonucleotides.
Introduction
The human APOBEC3 (A3A-H) protein family attacks retroviruses and other pathogens by hypermutating cytosine to uracil in single-stranded (ss)DNA (Fig. 1A).1 Deaminase activity is dependent on a zinc-mediated hydrolytic mechanism, in which a conserved zinc-coordinating motif (His-X-Glu-X25–31-Pro-Cys-X2–4-Cys, X indicates any amino acid) serves to coordinate the zinc ion into the active site of the enzyme.2–4 Besides targeting viruses1 and retrotransposons,5 several A3 enzymes, particularly A3A, A3B, and A3G, can deaminate cytosine in human nuclear and mitochondrial genomes.6 This mutational activity of A3 proteins is recruited by viruses and cancer cells to increase the rates of mutagenesis, escape adaptive immune responses, and become drug resistant.7–11 A3B is the major source of genetic mutations in multiple cancers (such as breast, bladder, cervix, lung, ovarian, head, and neck), and A3A and A3H are responsible for additional A3-dependent mutations.9–15 Because A3B is not an essential protein,16 its inhibition may be used to supplement existing anticancer and retroviral therapies.17 Due to the important role of A3 proteins for immune defence against pathogens, it is vital to specifically inhibit only one family member, in particular A3B, leaving others intact and active. The relative contribution of A3A and A3B to carcenogenesis is not clarified yet for many cancers.15,18 However, A3A is not expressed significantly and its activity is not detectable in breast cancer cell lines.10,19,20 In contrast, A3B is the major source of mutations in multiple breast cancer cell lines.19,20 Therefore, selective inhibition of A3B is important at least for treatment of breast cancers. In addition, selective inhibitors may provide in vitro and in vivo tools to address the mutagenic role of A3A and A3B in other cancers.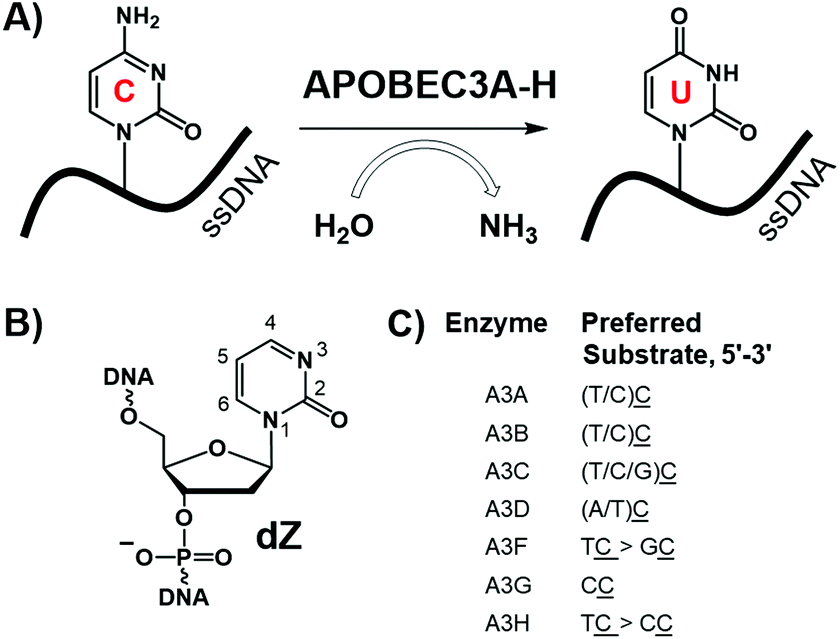 | ||
| Fig. 1 (A) Deamination of dC in ssDNA by A3 enzymes; (B) inhibitor of cytidine deamination used in this work: 2′-deoxyzebularine (dZ) incorporated in the ssDNA; (C) preferred ssDNA substrates of A3 enzymes, where the underlined C is the preferred target (adapted from ref. 25). | ||
We have recently developed the first substrate-like competitive A3 inhibitors,21 in which the target 2′-deoxycytidine (dC) in ssDNA (e.g., 5′-ATTTCATTT) was substituted by 2′-deoxyzebularine (dZ, Fig. 1B), a known inhibitor for cytidine deamination by cytidine deaminases that accept only single nucleosides.22,23 Protonation of N3 in dZ by the conserved glutamic acid present in the active site of A3s (and single nucleoside cytidine deaminase, CDA) activates C4 in dZ to accept nucleophilic OH−/H2O coordinated to the Zn2+, thereby converting dZ into a transition-state analogue of cytidine deamination.24 Low micromolar inhibition constants were observed for several A3 variants.21 However, because similar intrinsic preferences for deaminating cytosines preceded by thymine were observed for all A3 enzymes apart from A3G (Fig. 1C), there is uncertainty as to whether, or not, dZ-containing ssDNAs can selectively inhibit A3B in the context that no selective A3B inhibitors of any kind have been reported. Here we show that neighbouring nucleotides in ssDNA containing the inhibitor dZ strongly influence selectivity, allowing, in particular, selective targeting of A3B. This provides a platform for further development of oligonucleotide-inhibitors of A3 inhibitors as valuable probes for mechanistic and cellular studies, and, potentially, as adjuvants that suppress A3 mutagenesis in anti-cancer and anti-viral therapies.
Results and discussion
Hypothesis and methodology of this investigation
Although A3A and A3B favour deamination of the T![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) motif (underlined, bold C is deaminated) where the target dC is preceded by a thymidine, these enzymes also process cytidine in ssDNA with other adjacent nucleobases, but with lower efficiency (Fig. 1C).25,26 For example, A3A and A3B are also capable of deaminating dC in the CCC sequence motif,27 which is the most favoured motif for A3G. A3G and A3A preferentially deaminate dC at the 3′-end, denoted CC,25,28–31 with, respectively, at least 100 times and 5 times faster initial rate of deamination relative to other cytosines in the CCC-motif.27 This is reflected in Km values for catalytically active C-terminal domain of A3G (A3GCTD, see explanation below): Km (5′-ATT CC AATT) ≈ 570 μM compared to Km (5′-ATT CdU AATT) ≈ 3.6 mM, where dU is 2′-deoxyuridine.32 In contrast, A3B prefers dC at the 5′-end of this motif, denoted CC,26,27 with at least six-fold faster initial rate of deamination for this dC in comparison to the other cytosines.27 Therefore, we hypothesized that replacing dC by dZ at the 5′ end of the CC motif (dZCC-Oligo, Table 1) would lead to a selective inhibitor of A3B, and conversely CCdZ-Oligo would inhibit A3A and A3G but not A3B.
motif (underlined, bold C is deaminated) where the target dC is preceded by a thymidine, these enzymes also process cytidine in ssDNA with other adjacent nucleobases, but with lower efficiency (Fig. 1C).25,26 For example, A3A and A3B are also capable of deaminating dC in the CCC sequence motif,27 which is the most favoured motif for A3G. A3G and A3A preferentially deaminate dC at the 3′-end, denoted CC,25,28–31 with, respectively, at least 100 times and 5 times faster initial rate of deamination relative to other cytosines in the CCC-motif.27 This is reflected in Km values for catalytically active C-terminal domain of A3G (A3GCTD, see explanation below): Km (5′-ATT CC AATT) ≈ 570 μM compared to Km (5′-ATT CdU AATT) ≈ 3.6 mM, where dU is 2′-deoxyuridine.32 In contrast, A3B prefers dC at the 5′-end of this motif, denoted CC,26,27 with at least six-fold faster initial rate of deamination for this dC in comparison to the other cytosines.27 Therefore, we hypothesized that replacing dC by dZ at the 5′ end of the CC motif (dZCC-Oligo, Table 1) would lead to a selective inhibitor of A3B, and conversely CCdZ-Oligo would inhibit A3A and A3G but not A3B.
| Abbreviation | DNA sequence | Enzymes | ||
|---|---|---|---|---|
| A3A-mimic | A3BCTD-DM | A3GCTD | ||
| K m, μM (kcat, s−1) | K m, μM (kcat, s−1) | K m, μM (kcat, s−1) | ||
| a A3A-mimic (A3BCTD-QM-ΔL3-AL1swap, 50 nM) in pH 5.5 buffer and A3BCTD-DM (2 μM) in pH 7.5 buffer consisting of 50 mM citrate-phosphate buffer, 200 mM NaCl and 2 mM β-mercaptoethanol. A3GCTD (850 nM) in pH 6.0 buffer consisting of 50 mM sodium phosphate, 1 mM citrate, 100 mM NaCl, 2 mM β-mercaptoethanol, 50 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS). In all NMR assays, buffers contained 10% D2O. Km value for A3GCTD for CCC-oligo is stated for comparison with other enzymes and substrates. Uncertainties were calculated using standard error-propagation method. The cytidines that are not in bold and underlined were not deaminated in dZ-containing oligos over the time of the experiment. b “Not a substrate” means that none of dCs is deaminated under the experimental conditions reported. c “Not a preferred substrate” means that other dCs are also deaminated but at later time points when the concentration of the substrate with the preferred dC in the sequence decreased significantly due to deamination. | ||||
| TCA-oligo | 5′-ATTTATTT |
200 ± 30 (0.28 ± 0.04) | 320 ± 60 (0.0080 ± 0.0014) | Not a substrateb |
| CCC-oligo | 5′-ATTCCAATT |
440 ± 80 (0.38 ± 0.06) | Not a preferred substratec | 570 ± 90 (0.10 ± 0.04) (ref. 32) |
| 5′-ATTCCAATT |
Not a preferred substrate | 590 ± 90 (0.009 ± 0.001) | Not a preferred substrate | |
| dZCC-oligo | 5′-ATTdZCAATT |
520 ± 120 (0.44 ± 0.10) | Not a substrate | ≈1 mM (≈0.1) |
| CCdZ-oligo | 5′-ATTCdZAATT |
Not a substrate | 1370 ± 270 (0.0099 ± 0.0019) | Not a substrate |
To test our hypothesis we initially characterized kinetics of cytosine deamination by several A3 variants for a suboptimal substrate having the CCC-motif (CCC-oligo, Table 1). Here, we demonstrate that our A3 variants, A3A-mimic, A3BCTD-DM and A3GCTD, have the same deamination pattern on the CCC-motif reported previously for the wild-type enzymes (see Chart S2 in the ESI† for enzyme sequences). To evaluate the inhibitory potential of dZ-containing oligos we monitored the deamination of 5′-ATTTATTT by these A3 enzymes in the presence of potentially inhibitory oligos using the direct NMR-based assay.21,32,33
Evaluation of deamination of CCC-motif by various A3 constructs
GST-A3GCTD has a wild-type sequence of the catalytically active C-terminal domain of A3G. A3BCTD-DM is a double mutant of the catalytically active C-terminal domain of A3B where two hydrophobic residues on the protein surface are replaced by lysines (L230K and F308K) to enhance solubility (see Chart S3, ESI†).21,34 Because these two mutations are located on the opposite side of the protein relative to the active site and do not belong to loop 1 and loop 7 responsible for substrate specificity,27,29,34,35 they are unlikely to affect specificity, but some conflicting examples for other enzymes are known.36–40 A3BCTD-QM-ΔL3-AL1swap contains four mutations with loop 3 truncated and loop 1 of A3BCTD swapped with loop 1 of A3A, which constitute the main differences between amino acid sequences of A3A and A3BCTD (see Chart S2, ESI†).21,34 This construct has the substrate specificity of A3A and provides a good contrast to A3BCTD in the context of the CCC motif.27 That is why we refer to this enzyme as A3A-mimic.Initially, we evaluated activity of our A3A-mimic and A3BCTD-DM variants at different pH as very different optimum pH was reported for A3A and A3B.27,41 In our hands, the optimum pH for A3A-mimic was 5.5 consistent with Ito et al.41 However, A3BCTD-DM is barely active at pH 5.5 but its activity increases with pH and reaches reasonable activity at pH 7.5 (see Chart S4 in ESI†). The optimum pH for A3GCTD used in our work was 6.0, which is consistent with previous reports.27,32,41 Because even the remote mutations can sometimes affect the substrate specificity,36–40 we then monitored the deamination of 5′-ATTCCCAATT (CCC-oligo, Fig. 2 and Table 1) by our A3 variants at the pH defined above. In line with previous observations,27 A3A-mimic favoured the deamination of the third cytosine in the CC motif (dC labelled 3, Fig. 2A). Product formation as a result of deamination of individual cytosines is easily detectable using 1H NMR21,32,33 because there is a good dispersion of H5 doublets (J = 8.5 Hz) of product uracils in corresponding sequences (see Chart S5 in the ESI†). This deamination pattern is similar to that reported previously for CC-preferring A3GCTD, but with the qualifications that A3GCTD does not deaminate dC at the 5′ position (dC labelled 1, Fig. 2A) of the CCC-motif and has little enthusiasm for the middle C of the CCC motif: Km (5′-ATT CC AATT) ≈ 570 μM compared to Km (5′-ATT CdU AATT) ≈ 3.6 mM.32 Deamination of only one dC at the beginning of the reaction is observed, indicating that dissociation of the enzyme from the oligo occurs between deamination events, since the concentration of substrate greatly exceeds concentration of enzyme.
 | ||
| Fig. 2
1H-NMR spectra of preferential deamination of the CCC-oligo (800 μM) by A3A and A3B variants. Note that there is no signal in the uracil region of the 1H-NMR spectra at the beginning of the reaction for the substrate. Spectra shown are recorded every 15 min. (A) A3BCTD-QM-ΔL3-AL1swap (A3A-mimic, 2 μM) first deaminates dC at the 3′ position of the CC in pH 5.5 buffer. Deamination of the resultant CCU species is nearly random, leading to multiple peaks at intermediate deamination for CUU, UCU and UUU species. (B) A3BCTD-DM (2 μM) first deaminates dC at the 5′ position of the CC motif in pH 7.5 buffer. dC at position 2 is next to be deaminated, followed by dC at position 3. Buffer is 50 mM citrate-phosphate, 200 mM NaCl, 2 mM β-mercaptoethanol, 200 μM DSS, 10% deuterium oxide. | ||
In contrast to A3A-mimic, another enzyme variant, A3BCTD-DM deaminated the first dC at the 5′ position of the CC motif (Fig. 2B). The deamination products of other dCs were observed at later time-points and were always less intense in comparison with the signal of dU in position 1 indicating that deamination of non-preferred cytosines was incomplete during the experiment (2 h). The similar behaviour was observed previously for the wild type A3BCTD.27
To exclude the possibility that substrate structure rather than sequence determines deamination patterns by A342,43 we evaluated our CCC-oligo using m-Fold44 for possible formation of stem-loop structures. Two possible structures were proposed by m-Fold, both with positive unfavourable Gibbs free energy of formation (Chart S6, ESI†). Moreover, circular dichroism (CD) spectra recorded at pH 5.0, 6.0 and 7.2 showed no features indicating DNA secondary structure (Chart S7, ESI†). This establishes that the deamination patterns observed for the CCC-oligo with the different A3 enzymes are determined by the DNA sequence and not by the structure.
The Michaelis–Menten constants for the enzymes and ssDNA substrates used in this study are summarized in Table 1. Notably, the apparent binding affinity of A3BCTD-DM and A3A-mimic for their preferred cytosines in the CCC motif (respectively, Km ≈ 600 μM on CC and Km ≈ 400 μM on CC) are weaker by a factor of 2 in comparison to the preferred TA motif as previously reported (A3BCTD-DM Km ≈ 300 μM, A3A-mimic Km ≈ 200 μM, Table 1).21
With respect to the substrate 5′-ATT CCC AATT, we can conclude that our A3 variants mimic the deamination pattern of wild-type A3A and A3BCTD in DNA substrates containing several cytosines. A3BCTD prefers a thymine in the 5′-position next to the target dC, whereas A3A preferentially accepts dC that is directly followed by purine (dA or dG) at the 3′-end. Nucleotides around the target dC are known to substantially influence substrate recognition by A3 enzymes.18,27,45 Thus, we have a panel of A3 variants to test our hypothesis that we can selectively inhibit the catalytic activity of A3 enzymes by introducing dZ into different positions in the CCC sequence (dZCC and CCdZ-oligos, Tables 1 and 2).
| Abbreviation | DNA sequence | Enzymes | ||
|---|---|---|---|---|
| A3A (mimic) | A3BCTD-DM | A3GCTD | ||
| a See conditions reported in Table 1. b See ref. 21, Ki values are provided for comparison; all experiments were repeated multiple times in the same laboratory and the same time interval. | ||||
| TdZA-Oligo | 5′-ATTTdZATTT | 7.5 ± 1.7b | 11.4 ± 2.6b | Not an inhibitorb |
| CCdZ-Oligo | 5′-ATTCCdZAATT | 12.3 ± 1.8 | Not an inhibitor | 53 ± 10b |
| dZCC-Oligo | 5′-ATTdZCCAATT | Not an inhibitor | 18.5 ± 5.5 | Not an inhibitor |
Evaluation of inhibitors
Next, we turned our attention to inhibitors based on the CCC-motif. Very recently, we had developed a micromolar inhibitor of A3 enzymes by incorporating dZ into their preferred deamination motif.21 The TdZA-oligo21 (Table 2) did not significantly inhibit A3GCTD. However, it targeted both A3A-mimic and A3BCTD-DM with similar inhibition constants. To determine if we can selectively inhibit A3A-mimic, A3BCTD-DM and A3GCTD enzymes, we synthesized CCdZ and dZCC oligos using our previously established protocol21 (see ESI† for experimental details). We used the TCA-oligo as a substrate to monitor the residual enzyme activity by NMR-based assay21,32,33 in the presence of dZ-containing oligos because of its faster deamination than that of the CCC-oligo (Table 1). The NMR-based assay allows to obtain the initial velocity of deamination of the DNA substrates by A3 and consequently use the Michaelis–Menten kinetic model to characterize substrates and inhibitors of A3. To the best of our knowledge, all other in vitro A3 assays are indirect and rely on other enzymes with which DNA-based inhibitors may interact and provide false-positive or false-negative results. Moreover, dZ degrades in strong alkaline conditions,46 which precludes evaluation of dZ-containing oligos as substrates of A3 in some indirect assays that use strong heat and base addition after the second enzymatic reaction using uracil-DNA glucosylase (UDG). Therefore, currently, the NMR-based direct assay is best suited to study DNA-based inhibitors of A3.For an initial check, we analyzed deamination velocity by A3A-mimic and A3BCTD-DM at specific concentration of inhibitors, which provides qualitative information about A3-selectivity of our inhibitors. Consistent with our hypothesis, CCdZ-oligo significantly inhibits deamination of substrate 5′-ATTTATTT by A3A-mimic (Fig. 3A), as well as by A3GCTD (as previously reported in ref. 21), but not A3BCTD-DM (Fig. 3A and Table 2). In contrast, dZCC-oligo inhibits substrate deamination by A3BCTD-DM variant (Fig. 3B) but has no measureable effect on the initial velocity of A3A-mimic (Fig. 3A, dZCC) or A3GCTD. To ensure that the inhibitor is not processed by the inhibited enzyme we checked NMR spectrum of CCdZ-oligo in the presence of A3A-mimic, which is the most active deaminase in our set of A3. No changes in NMR spectrum were observed over 3.7 hours and, most importantly, no signals from uridine appeared in the range of 5.5–6.0 ppm (see Chart S18, ESI†). This confirms that A3A specifically recognizes dZ in CCdZ-oligo and does not deaminate other dCs in this motif in excess of oligo relative to the enzyme.
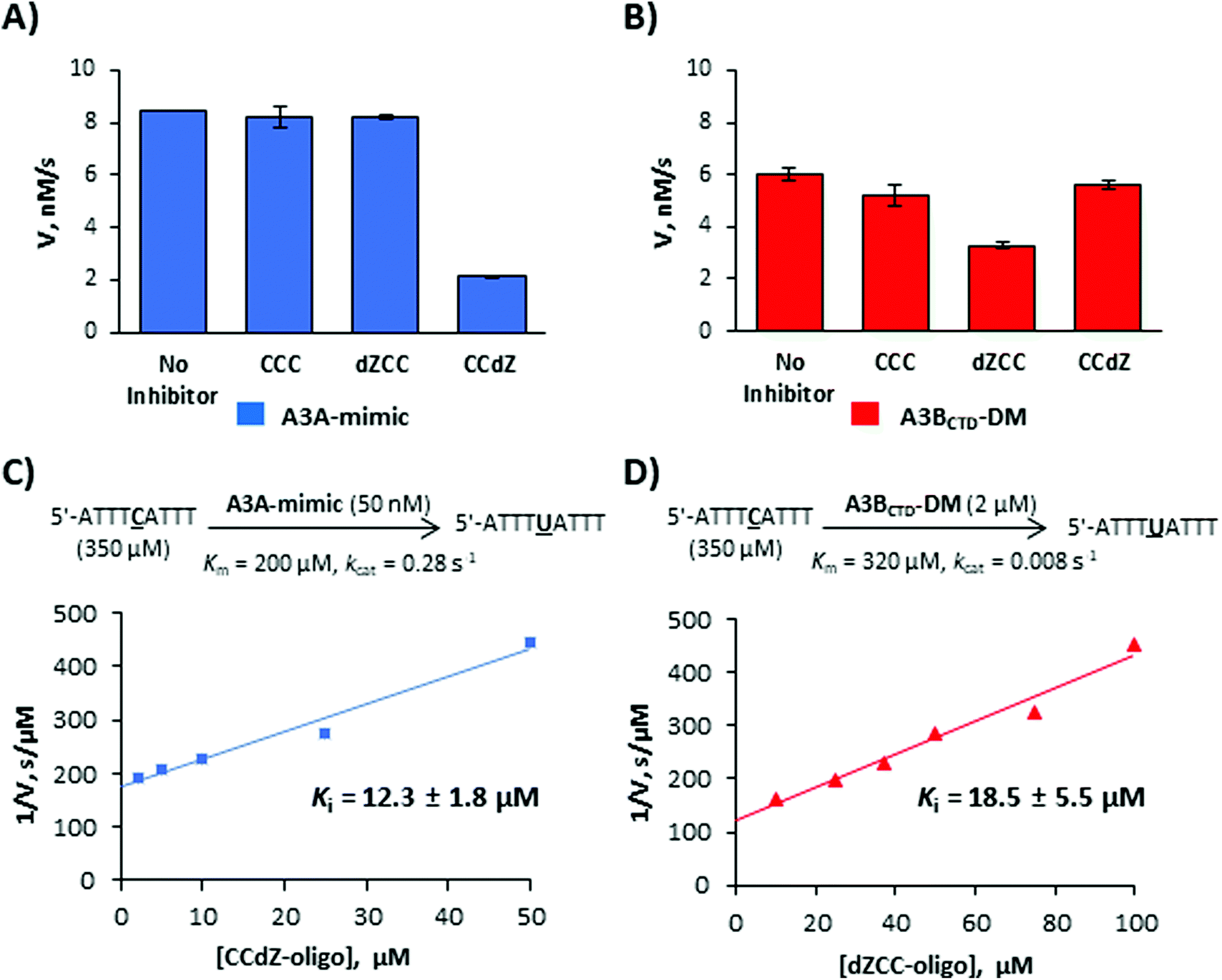 | ||
| Fig. 3 Selective inhibition of A3 enzymes by dZ-containing ssDNAs. (A) and (B) Deamination of 5′-ATTTATTT (TCA-oligo, 350 μM) in the absence and presence of potential inhibitors (50 μM ssDNA CCC-oligo, dZCC-oligo and CCdZ-oligo). (A) A3A-mimic (A3BCTD-QM-ΔL3-AL1swap, 50 nM). (B) A3BCTD-DM (2 μM). (C) Inhibition of deamination of the TA substrate by A3A-mimic in the presence of various concentrations of CCdZ-oligo. (D) Inhibition of deamination of the TA substrate by A3BCTD-DM in the presence of various concentrations of dZCC-oligo. | ||
To quantitatively characterize the extent of inhibition we analyzed the linear dependence of inverse deamination speed on inhibitor concentration and obtained inhibition constants (Ki, Fig. 3C, D and Table 2) based on a competitive mode of inhibition.21Ki values of the selective dZ-oligos on A3A-mimic and A3BCTD-DM variants (Fig. 3C, D and Table 2) indicate approximately two-fold weaker inhibition than the Ki of our previously reported essentially non-selective TdZA inhibitor.21 This is consistent with the two-fold weaker Km of the CCC oligo to these enzymes in comparison with Km's for the preferred TA-oligo (Table 1). Nevertheless, we see a thirty-fold increase in apparent affinity (Km/Ki) because dZ is placed in the ssDNA CCC sequence instead of the preferred dC.
To evaluate the scenario where dZCC/CCdZ-inhibitors are deaminated by other A3 enzymes, we used dZCC- and CCdZ-oligos as substrates and determined Km values for non-preferred enzymes A3A-mimic/A3GCTD and A3BCTD-DM, respectively. The dZCC-oligo, which is an inhibitor of A3BCTD-DM but not of A3A-mimic and A3GCTD, is a worse substrate for these latter two enzymes than TCA-oligo (Table 1, Km = 520 ± 90 μM for A3A-mimic and Km ≈ 1 mM for A3GCTD). More markedly, CCdZ-oligo, which inhibits A3A-mimic and A3GCTD, is a very poor substrate for A3BCTD-DM with Km ≈ 1370 ± 270 μM (Table 1). By comparing the Km value for dZCC-oligo deaminated by A3A-mimic (Km = 520 μM, which is the lowest Km value for dZ-oligos as substrates to A3A-mimic reported here) with the Ki value for A3BCTD-DM (Ki = 18.5 μM) by the same oligo (Km/Ki = 30), we can conclude that inhibition of A3BCTD-DM will prevail over deamination by A3A-mimic.
Conclusions
Taking advantage of the discrimination pattern for deamination of the CCC motif by A3A/A3G compared to A3B,27 we incorporated our previously reported inhibitor of cytidine deamination, 2′-deoxyzebularine (dZ), in different positions in the CCC-motif. Incorporation of dZ in place of preferentially deaminated dC led to the first inhibitors capable of selectively targeting the catalytic activity of individual members of the A3 family, especially cancer-associated A3B variant. Comparison of the A3BCTD-DM inhibition constant (Ki) for dZCC (Table 2) to Km values for dZC deamination of the target dC by A3A-mimic and A3GCTD (Table 1) indicates at least 30-fold higher apparent binding to A3BCTD-DM than to other enzymes. This means that dZCC-oligo is an inhibitor of A3BCTD-DM rather than a substrate of A3A-mimic and A3GCTD. dZCC-oligo should not be deaminated by A3BCTD-DM, which it inhibits, because Ki ≪ Km for the remaining cytidines. Our data illustrate this argument because CCdZ-oligo is an inhibitor of A3A-mimic and it is not deaminated by the same enzyme, which indicates that A3A-mimic strongly prefers to bind to dZ rather than to either dC in the oligo. To our knowledge, dZCC-oligo is the first reported selective inhibitor of A3BCTD, although the selective small-molecule A3G inhibitors have been reported.47,48 Inhibition of full-length two-domain A3B and A3G enzymes and the cellular effect of our inhibitors still need to be determined, but we expect longer oligonucleotides to maintain similar inhibition patterns since binding to the catalytically inactive N-terminal domains has been established as non-specific.49–51 The use of more powerful inhibitors of cytidine deamination than dZ is expected to increase inhibition to the level required for the cellular studies. For CCdZ-oligo, different inhibition constants against A3A-mimic and A3GCTD indicate potential for further discrimination between these enzymes. Other DNA sequences that have been shown to be deaminated differently by A3A and A3B18,35,43,52 can also be evaluated as alternative starting points for the design of A3-selective inhibitors in future. Our dZ-oligos do not inhibit CDA as CDA accepts only individual nucleosides not oligonucleotides.53 Similarly, we do not expect our inhibitors to inhibit DNA methyltransferases54 because these enzymes recognise flipped out cytosines in double-stranded DNA not in single-stranded DNA.55 Nevertheless, we plan to test our inhibitors in normal cells as control for unanticipated cytotoxicity at later stage. Overall, our work provides a platform for development of more potent and selective A3-inhibitors with the ultimate goal to be used for cellular studies and as possible adjuvants in anti-viral and anti-cancer treatments.
Material and methods
Detailed methods are provided in the ESI.†Synthesis of oligonucleotides containing 2′-deoxyzebularine (dZ)
2′-Deoxyzebularine phosphoramidite was prepared as described previously.21 Synthetic procedures are provided in ESI.†Protein expression and purification
Expression and purification procedures are provided in ESI.† A3B C-terminal domain (residues 187 to 378) protein variants A3BCTD-QM-ΔL3-AL1swap (termed A3A-mimic) and A3BCTD-DM, and GST-fused A3G C-terminal domain (residues 191 to 384) protein variant A3GCTD were prepared as described previously.32,34Kinetic characterization of deamination activity using NMR-based assay
Kinetic characterization of A3 protein variants (A3BCTD-QM-ΔL3-AL1swap, A3BCTD-DM, and A3GCTD) on preferred cytidine in 5′-ATTCCCAATT, 5′-ATTdZCCAATT, and 5′-ATTCCdZAATT substrates were evaluated using an established NMR-based assay21,32,33 as described in ESI.†Evaluation of inhibitors in NMR-based assay
The NMR-based inhibition assay as described previously21,32 was conducted on 350 μM 5′-ATTTATTT substrate with A3 variants in the presence of dZ-containing oligodeoxynucleotides. Procedures and inhibition constants calculation are provided in ESI.†
Conflicts of interest
There are no conflicts to declare.Funding
We are grateful for the financial support provided by the Worldwide Cancer Research (grant 16-1197), Massey University Research Fund (MURF 2015, 7003) and School of Fundamental Sciences, Massey University. Funding for open access charge: Worldwide Cancer Research (grant APC-19-0007).Acknowledgements
NMR and mass-spectrometry facilities at Massey University and the assistance of Dr P. J. B. Edwards and D. Lun are gratefully acknowledged. We thank Prof. Reuben S. Harris (University of Minnesota, USA) and members of his cancer research program for helpful discussions.References
- R. S. Harris and J. P. Dudley, Virology, 2015, 479–480, 131–145 CrossRef CAS PubMed.
- S. G. Conticello, C. J. F. Thomas, S. K. Petersen-Mahrt and M. S. Neuberger, Mol. Biol. Evol., 2005, 22, 367–377 CrossRef CAS.
- R. S. Harris and M. T. Liddament, Nat. Rev. Immunol., 2004, 4, 868–877 CrossRef CAS PubMed.
- R. S. LaRue, V. Andrésdóttir, Y. Blanchard, S. G. Conticello, D. Derse, M. Emerman, W. C. Greene, S. R. Jónsson, N. R. Landau, M. Löchelt, H. S. Malik, M. H. Malim, C. Münk, S. J. O'Brien, V. K. Pathak, K. Strebel, S. Wain-Hobson, X.-F. Yu, N. Yuhki and R. S. Harris, J. Virol., 2009, 83, 494–497 CrossRef CAS PubMed.
- T. Izumi, K. Shirakawa and A. Takaori-Kondo, Mini-Rev. Med. Chem., 2008, 8, 231–238 CrossRef CAS PubMed.
- R. Suspene, M.-M. Aynaud, D. Guetard, M. Henry, G. Eckhoff, A. Marchio, P. Pineau, A. Dejean, J.-P. Vartanian and S. Wain-Hobson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4858–4863 CrossRef CAS PubMed.
- C. Swanton, N. McGranahan, G. J. Starrett and R. S. Harris, Cancer Discovery, 2015, 5, 704–712 CrossRef CAS PubMed.
- E.-Y. Kim, R. Lorenzo-Redondo, S. J. Little, Y.-S. Chung, P. K. Phalora, I. Maljkovic Berry, J. Archer, S. Penugonda, W. Fischer, D. D. Richman, T. Bhattacharya, M. H. Malim and S. M. Wolinsky, PLoS Pathog., 2014, 10, e1004281 CrossRef PubMed.
- S. Venkatesan, R. Rosenthal, N. Kanu, N. McGranahan, J. Bartek, S. A. Quezada, J. Hare, R. S. Harris and C. Swanton, Ann. Oncol., 2018, 29, 563–572 CrossRef CAS PubMed.
- E. K. Law, A. M. Sieuwerts, K. LaPara, B. Leonard, G. J. Starrett, A. M. Molan, N. A. Temiz, R. I. Vogel, M. E. Meijer-van Gelder, F. C. G. J. Sweep, P. N. Span, J. A. Foekens, J. W. M. Martens, D. Yee and R. S. Harris, Sci. Adv., 2016, 2, e1601737 CrossRef PubMed.
- J. Zou, C. Wang, X. Ma, E. Wang and G. Peng, Cell Biosci., 2017, 7, 29 CrossRef PubMed.
- A. M. Sieuwerts, S. Willis, M. B. Burns, M. P. Look, M. E. Meijer-Van Gelder, A. Schlicker, M. R. Heideman, H. Jacobs, L. Wessels, B. Leyland-Jones, K. P. Gray, J. A. Foekens, R. S. Harris and J. W. M. Martens, Horm. Cancer, 2014, 5, 405–413 CrossRef CAS PubMed.
- P. J. Vlachostergios and B. M. Faltas, Nat. Rev. Clin. Oncol., 2018, 15, 495–509 CrossRef CAS PubMed.
- L. Galluzzi and I. Vitale, Trends Genet., 2017, 33, 491–492 CrossRef CAS PubMed.
- M. B. Burns, N. A. Temiz and R. S. Harris, Nat. Genet., 2013, 45, 977–983 CrossRef CAS PubMed.
- J. M. Kidd, T. L. Newman, E. Tuzun, R. Kaul and E. E. Eichler, PLoS Genet., 2007, 3, 584–592 CAS.
- M. E. Olson, R. S. Harris and D. A. Harki, Cell Chem. Biol., 2018, 25, 36–49 CrossRef CAS PubMed.
- K. Chan, S. A. Roberts, L. J. Klimczak, J. F. Sterling, N. Saini, E. P. Malc, J. Kim, D. J. Kwiatkowski, D. C. Fargo, P. A. Mieczkowski, G. Getz and D. A. Gordenin, Nat. Genet., 2015, 47, 1067–1072 CrossRef CAS PubMed.
- M. B. Burns, L. Lackey, M. A. Carpenter, A. Rathore, A. M. Land, B. Leonard, E. W. Refsland, D. Kotandeniya, N. Tretyakova, J. B. Nikas, D. Yee, N. A. Temiz, D. E. Donohue, R. M. McDougle, W. L. Brown, E. K. Law and R. S. Harris, Nature (London, U. K.), 2013, 494, 366–370 CrossRef CAS PubMed.
- B. Leonard, J. L. McCann, G. J. Starrett, L. Kosyakovsky, E. M. Luengas, A. M. Molan, M. B. Burns, R. M. McDougle, P. J. Parker, W. L. Brown and R. S. Harris, Cancer Res., 2015, 75, 4538–4547 CrossRef CAS PubMed.
- M. V. Kvach, F. M. Barzak, S. Harjes, H. A. M. Schares, G. B. Jameson, A. M. Ayoub, R. Moorthy, H. Aihara, R. S. Harris, V. V. Filichev, D. A. Harki and E. Harjes, Biochemistry, 2019, 58, 391–400 CrossRef CAS PubMed.
- S. Xiang, S. A. Short, R. Wolfenden and C. W. Carter, Biochemistry, 1995, 34, 4516–4523 CrossRef CAS PubMed.
- J. J. Barchi, A. Haces, V. E. Marquez and J. J. McCormack, Nucleosides Nucleotides, 1992, 11, 1781–1793 CrossRef CAS.
- C. H. Borchers, V. E. Marquez, G. K. Schroeder, S. A. Short, M. J. Snider, J. P. Speir and R. Wolfenden, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15341–15345 CrossRef CAS PubMed.
- M. D. Stenglein, M. B. Burns, M. Li, J. Lengyel and R. S. Harris, Nat. Struct. Mol. Biol., 2010, 17, 222–229 CrossRef CAS PubMed.
- I.-J. L. Byeon, J. Ahn, M. Mitra, C.-H. Byeon, K. Hercik, J. Hritz, L. M. Charlton, J. G. Levin and A. M. Gronenborn, Nat. Commun., 2013, 4, ncomms2883 CrossRef PubMed.
- I.-J. L. Byeon, C.-H. Byeon, T. Wu, M. Mitra, D. Singer, J. G. Levin and A. M. Gronenborn, Biochemistry, 2016, 55, 2944–2959 CrossRef CAS PubMed.
- M. A. Carpenter, E. Rajagurubandara, P. Wijesinghe and A. S. Bhagwat, DNA Repair, 2010, 9, 579–587 CrossRef CAS PubMed.
- A. Rathore, M. A. Carpenter, O. Demir, T. Ikeda, M. Li, N. M. Shaban, E. K. Law, D. Anokhin, W. L. Brown, R. E. Amaro and R. S. Harris, J. Mol. Biol., 2013, 425, 4442–4454 CrossRef CAS PubMed.
- R. M. Kohli, S. R. Abrams, K. S. Gajula, R. W. Maul, P. J. Gearhart and J. T. Stivers, J. Biol. Chem., 2009, 284, 22898–22904 CrossRef CAS PubMed.
- R. M. Kohli, R. W. Maul, A. F. Guminski, R. L. McClure, K. S. Gajula, H. Saribasak, M. A. McMahon, R. F. Siliciano, P. J. Gearhart and J. T. Stivers, J. Biol. Chem., 2010, 285, 40956–40964 CrossRef CAS PubMed.
- S. Harjes, W. C. Solomon, M. Li, K.-M. Chen, E. Harjes, R. S. Harris and H. Matsuo, J. Virol., 2013, 87, 7008–7014 CrossRef CAS.
- A. Furukawa, T. Nagata, A. Matsugami, Y. Habu, R. Sugiyama, F. Hayashi, N. Kobayashi, S. Yokoyama, H. Takaku and M. Katahira, EMBO J., 2009, 28, 440–451 CrossRef CAS.
- K. Shi, M. A. Carpenter, K. Kurahashi, R. S. Harris and H. Aihara, J. Biol. Chem., 2015, 290, 28120–28130 CrossRef CAS.
- K. Shi, M. A. Carpenter, S. Banerjee, N. M. Shaban, K. Kurahashi, D. J. Salamango, J. L. McCann, G. J. Starrett, J. V. Duffy, O. Demir, R. E. Amaro, D. A. Harki, R. S. Harris and H. Aihara, Nat. Struct. Mol. Biol., 2017, 24, 131–139 CrossRef CAS PubMed.
- V. Weinreb, L. Li, S. N. Chandrasekaran, P. Koehl, M. Delarue and C. W. Carter Jr., J. Biol. Chem., 2014, 289, 4367–4376 CrossRef CAS.
- G. Yang, N. Hong, F. Baier, C. J. Jackson and N. Tokuriki, Biochemistry, 2016, 55, 4583–4593 CrossRef CAS.
- B. Song, Y. Yue, T. Xie, S. Qian and Y. Chao, Mol. Biotechnol., 2014, 56, 232–239 CrossRef CAS.
- M. Schmidt, D. Hasenpusch, M. Kähler, U. Kirchner, K. Wiggenhorn, W. Langel and U. T. Bornscheuer, ChemBioChem, 2006, 7, 805–809 CrossRef CAS.
- P. Oelschlaeger, S. L. Mayo and J. Pleiss, Protein Sci., 2005, 14, 765–774 CrossRef CAS.
- F. Ito, Y. Fu, S. A. Kao, H. Yang and X. S. Chen, J. Mol. Biol., 2017, 429, 1787–1799 CrossRef CAS.
- C. M. Holtz, H. A. Sadler and L. M. Mansky, Nucleic Acids Res., 2013, 41, 6139–6148 CrossRef CAS.
- T. V. Silvas, S. Hou, W. Myint, E. Nalivaika, M. Somasundaran, B. A. Kelch, H. Matsuo, N. Kurt Yilmaz and C. A. Schiffer, Sci. Rep., 2018, 8, 7511 CrossRef PubMed.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed.
- J. W. Rausch, L. Chelico, M. F. Goodman and S. F. J. Le Grice, J. Biol. Chem., 2009, 284, 7047–7058 CrossRef CAS PubMed.
- M. Vives, R. Eritja, R. Tauler, V. E. Marquez and R. Gargallo, Biopolymers, 2004, 73, 27–43 CrossRef CAS.
- M. Li, S. M. D. Shandilya, M. A. Carpenter, A. Rathore, W. L. Brown, A. L. Perkins, D. A. Harki, J. Solberg, D. J. Hook, K. K. Pandey, M. A. Parniak, J. R. Johnson, N. J. Krogan, M. Somasundaran, A. Ali, C. A. Schiffer and R. S. Harris, ACS Chem. Biol., 2012, 7, 506–517 CrossRef CAS PubMed.
- M. E. Olson, M. Li, R. S. Harris and D. A. Harki, ChemMedChem, 2013, 8, 112–117 CrossRef CAS PubMed.
- Y. Iwatani, H. Takeuchi, K. Strebel and J. G. Levin, J. Virol., 2006, 80, 5992–6002 CrossRef CAS PubMed.
- F. Navarro, B. Bollman, H. Chen, R. Konig, Q. Yu, K. Chiles and N. R. Landau, Virology, 2005, 333, 374–386 CrossRef CAS PubMed.
- J. D. Salter and H. C. Smith, Trends Biochem. Sci., 2018, 43, 606–622 CrossRef CAS.
- M. Liu, A. Mallinger, M. Tortorici, Y. Newbatt, M. Richards, A. Mirza, R. L. M. van Montfort, R. Burke, J. Blagg and T. Kaserer, ACS Chem. Biol., 2018, 13, 2427–2432 CrossRef CAS PubMed.
- C. B. Yoo, S. Jeong, G. Egger, G. Liang, P. Phiasivongsa, C. Tang, S. Redkar and P. A. Jones, Cancer Res., 2007, 67, 6400–6408 CrossRef CAS PubMed.
- V. E. Marquez, J. A. Kelley, R. Agbaria, T. Ben-Kasus, J. C. Cheng, C. B. Yoo and P. A. Jones, Ann. N. Y. Acad. Sci., 2005, 1058, 246–254 CrossRef CAS.
- S. S. Smith, B. E. Kaplan, L. C. Sowers and E. M. Newman, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4744–4748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob01781j |
| This journal is © The Royal Society of Chemistry 2019 |