
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective deconjugation of macrocyclic α,β-unsaturated esters by sequential amidation and olefin transposition: application to enantioselective phase-transfer catalysis†‡
Alexandre
Homberg
 a,
Radim
Hrdina
a,
Mahesh
Vishe
a,
Laure
Guénée
b and
Jérôme
Lacour
*a
a,
Radim
Hrdina
a,
Mahesh
Vishe
a,
Laure
Guénée
b and
Jérôme
Lacour
*a
aDepartment of Organic Chemistry, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: jerome.lacour@unige.ch
bLaboratory of Crystallography, University of Geneva, Quai Ernest Ansermet 24, 1211 Geneva 4, Switzerland
First published on 26th June 2019
Abstract
The stereoselective synthesis of chiral macrocycles bearing two aliphatic amide functional groups is reported. After the amidation mediated by TBD, a guanidine derivative, the olefin transposition step is performed with a slight excess of t-BuOK. The products are afforded in moderate to good combined yields (up to 59%) and with an excellent syn diastereoselectivity (dr > 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Introducing enantiopure α-branched substituents was possible and it resulted in mixtures of diastereomers, which could be tested as phase-transfer catalysts using the formation of a phenylalanine analog as a test reaction (up to 43% ee). A clear matched–mismatched situation was observed in the two diastereomeric series.
:1). Introducing enantiopure α-branched substituents was possible and it resulted in mixtures of diastereomers, which could be tested as phase-transfer catalysts using the formation of a phenylalanine analog as a test reaction (up to 43% ee). A clear matched–mismatched situation was observed in the two diastereomeric series.
Introduction
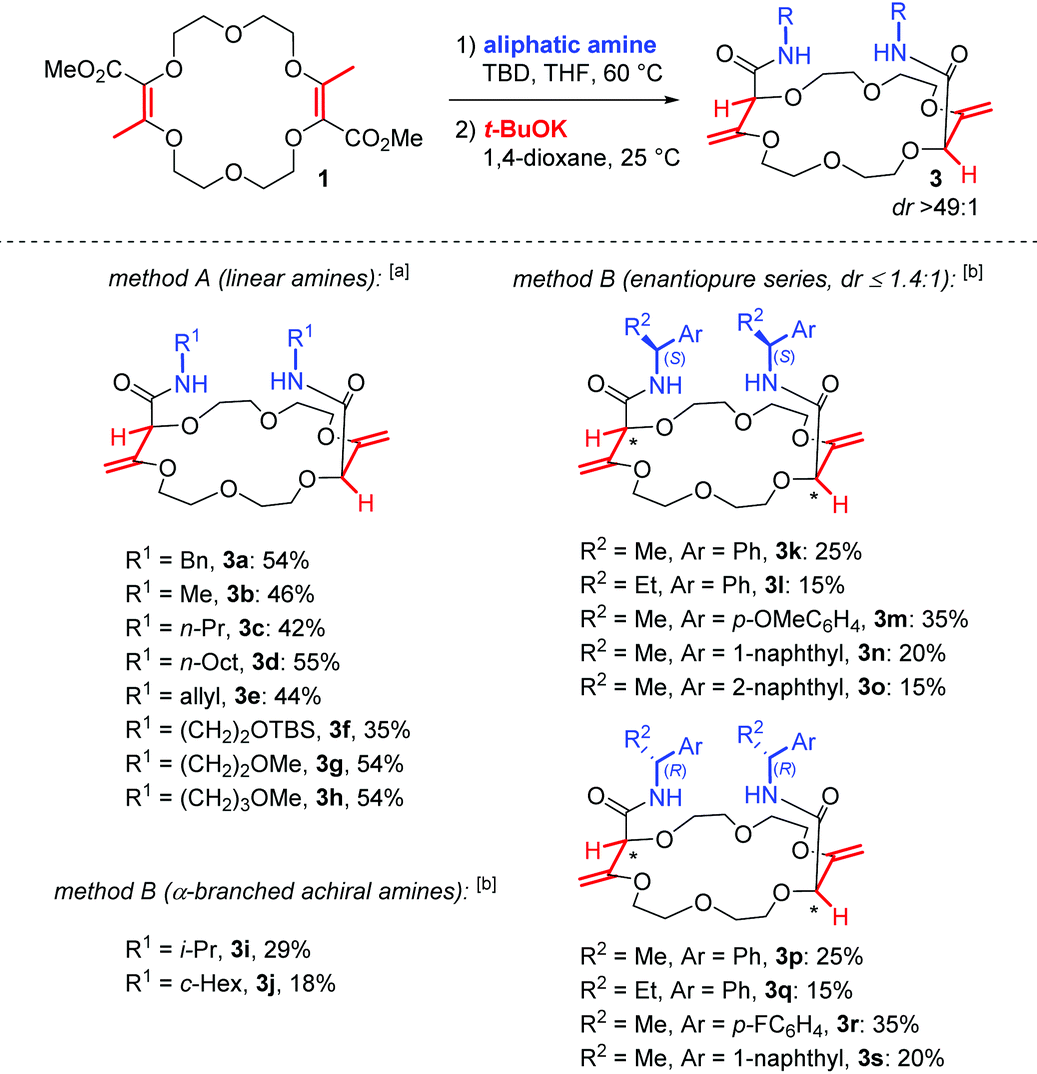
In unsaturated esters, olefin transposition from the α,β to β,γ-position is thermodynamically disfavored. To promote this deconjugation, various protocols are usually employed which utilize photochemical1 or strongly basic2 conditions to generate dienol(ate) intermediates (Scheme 1A). Subsequent (enantioselective) protonation in the α position leads to β,γ-unsaturated esters.3 In this context, our group recently reported the remote stereoselective deconjugations of bis-α,β-unsaturated macrocycle 1 in the presence of an excess of aromatic amines and t-BuOK.4 In a single step, chiral polyether macrocycles of type 2 are formed by a double tandem [amidation + olefin transposition] process (Scheme 1B, left). Mechanistically, it is believed that the ester functions are first transformed into amide groups. Then, irreversible olefin transpositions occur to yield macrocycles 2 as single stereoisomers (racemic, dr > 49:1). Various applications have been developed for compounds 2. In fact, such bis(aromatic) derivatives have been used as pH-independent nanosensors,5 heteroditopic receptors for salts,6 ratiometric luminescent or reversible chiroptical switches7 and circularly-polarized electrochemiluminescent emitters.8 Herein, the stereoselective synthesis of macrocycles of type 3 bearing two aliphatic amide functional groups is reported (Scheme 1B, right). A two-step process is this time necessary. Amine additions are mediated by TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene), a guanidine derivative that helps in the formation of the α,β-unsaturated amide derivatives 4. Then, olefin transpositions under basic conditions (t-BuOK) afford the corresponding chiral macrocycles 3 with high syn diastereoselectivity (dr > 49:1, yields up to 59% for the combined steps). Chiral enantiopure amines can be utilized and after the preparation of the resulting diastereomers, macrocycles were used as phase-transfer catalysts. In the enantioselective alkylation of a protected glycine (ee up to 43%), a clear matched–mismatched situation is observed in the two diastereomeric series.
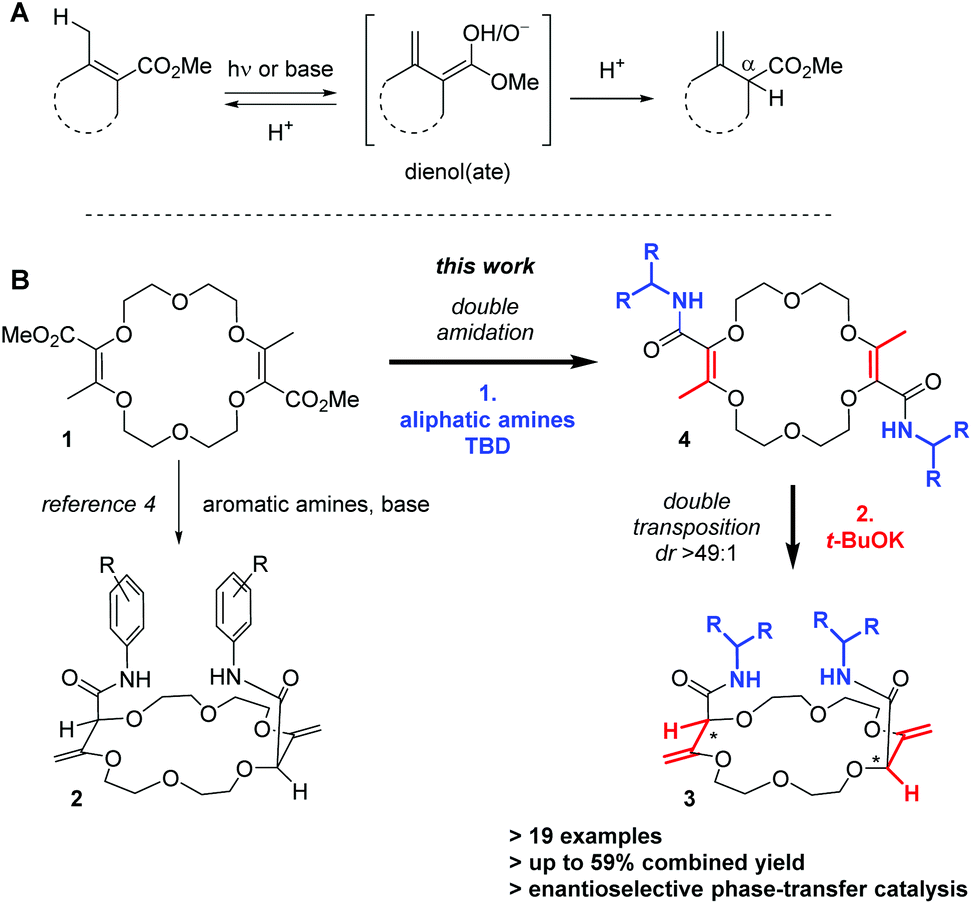 | ||
| Scheme 1 Typical conditions for the deconjugation of α,β-unsaturated esters (A). Remote stereoselective synthesis of chiral polyether macrocycles (B, syn transposition, dr > 49:1). | ||
Results and discussion
Initial attempts
Previously, it was shown that methyl α-diazo-β-ketoester reacts with 1,4-dioxane under dirhodium catalysis in a formal [3 + 6 + 3 + 6] multi-component condensation. The process is mild and affords the resulting unsaturated macrocycle 1 on a multi-gram scale (up to 20 grams) while using a low catalyst loading (0.01–0.001 mol%).9,10 As mentioned earlier, compound 1 reacts with excesses of ArNH2 and t-BuOK (>3 equiv. each) to yield unsaturated bis(aromatic) derivatives 2. This reaction tolerates a large variety of aromatic amines.4–8,11 However, despite major efforts by the group, it was never possible to achieve the analogous 1 → 3 transformation with aliphatic amines instead of anilines. Of the two consecutive steps, it was clearly the first one that was problematic under the previous conditions. Care was thus taken to decouple the two steps of the process and perform the amide formation first, prior to the olefin transposition.Optimization of the two-step process
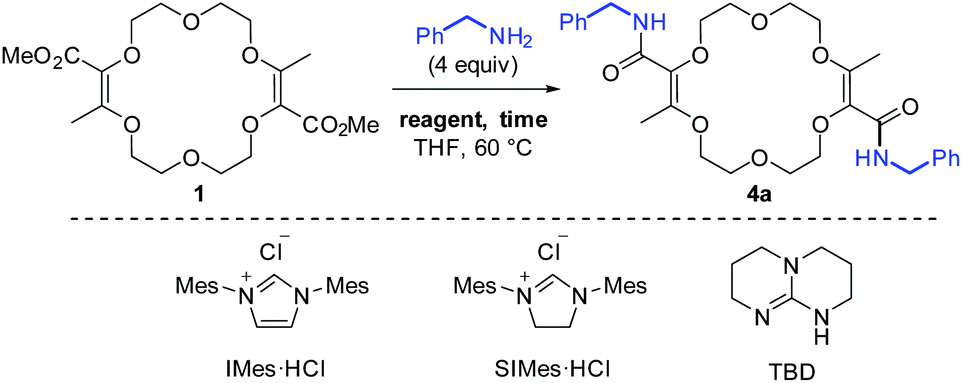
For the transformation of macrocycle 1 into the corresponding α,β-unsaturated bis(aliphatic) macrocycles 4, mild conditions of amidation were looked for. With regular non-activated esters,12 the use of additives such as (Lewis) acids,13 bases,14 metal salts and complexes,15 enzymes16 or organocatalysts is required to promote the reaction.17 Many such conditions were tested using macrocycle 1 and benzyl amine as the model substrate and reagent, respectively (Table 1). Heating the macrocycle and the amine in THF at 60 °C did not induce a conversion (entry 1). Strongly basic conditions, t-BuOK or n-BuLi, led either to a total degradation or a partial 30% conversion of macrocycle 1 respectively (entries 2 and 3). Lewis acids (Yb(OTf)3, BF3·OEt2) and N-heterocyclic carbenes were also tested (entries 4–10). In many instances, a lack of reactivity was observed. Otherwise, it was a full degradation of 1. Previously, for the transformation of esters into amides, Mioskowski and collaborators used TBD as the catalyst.17g,18,19,20 Using 20 mol% of this cyclic guanidine base, an amidation could not be observed (entry 11).21 However, with a stoichiometric amount of it (2 equiv.), macrocycle 4a was obtained in a satisfactory yield (47%) after 4 hours of reaction and a simple filtration (entry 12). Increasing the reaction time to 15 hours afforded 4a in 75% yield (entry 10). The structure of 4a was confirmed by NMR spectroscopy and X-ray diffraction analysis (see Fig. 1A).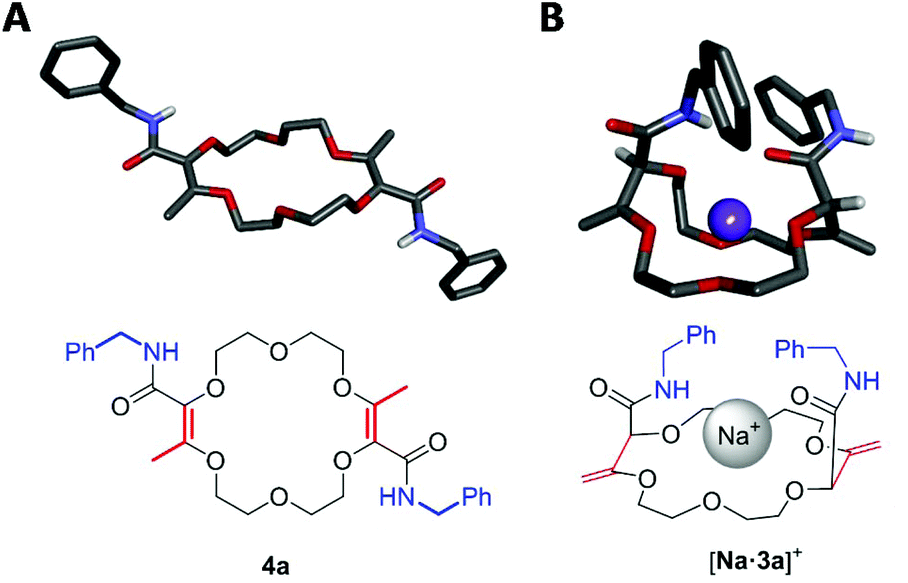 | ||
| Fig. 1 Stick view of the crystal structure of 4a (A) and [Na·3a][BArF] (B) (BArF anion and most hydrogen atoms are hidden for clarity). | ||
|
|
|||
|---|---|---|---|
| Entry | Reagent (equiv.) | Time (h) | Isolated yield |
| a −100 °C (1 min), then 25 °C instead of 60 °C. b The isolation of 4a was attempted but failed; abbreviations: Mes = mesityl. | |||
| 1 | — | 15 | No conv. |
| 2a | t-BuOK (4) | 3 | Degradation |
| 3a | n-BuLi (4) | 3 | <30% conv.b |
| 4 | Yb(OTf)3 (0.1) | 15 | No conv. |
| 5 | Yb(OTf)3 (2) | 15 | No conv. |
| 6 | BF3·OEt2 (0.2) | 15 | No conv. |
| 7 | BF3·OEt2 (2) | 15 | No conv. |
| 8 | IMes·HCl/t-BuOK (0.05) | 15 | No conv. |
| 9 | IMes·HCl/t-BuOK (2) | 15 | Degradation |
| 10 | SIMes·HCl/t-BuOK (2) | 15 | Degradation |
| 11 | TBD (0.2) | 15 | No conv. |
| 12 | TBD (2) | 4 | 47% |
| 13 | TBD (2) | 15 | 75% |
Next, the olefin transposition of compound 4a was considered under basic conditions.4 After optimization (Table S1‡), it was found that the combination of t-BuOK (2.2 equiv.) and 1,4-dioxane as the solvent afforded the best conditions.22 Macrocycle 3a was isolated in good yield (65%, Scheme 2) as a single diastereoisomer (dr > 49:1, 1H NMR monitoring). Once again, an effective remote stereoselectivity is noticed in this type of process.4 The relative configuration of 3a was determined by the solid state structure of the sodium BArF (tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) adduct (Fig. 1B). Only the chiral cis-diastereoisomer (racemic) is formed during the reaction and traces of the achiral (meso, trans) stereoisomer could not be detected. This result indicates that the mechanism for the olefin transposition(s) is similar to that previously reported;4 the potassium ion acts as a template and helps to relay the second reprotonation on the same prochiral face as the first one.
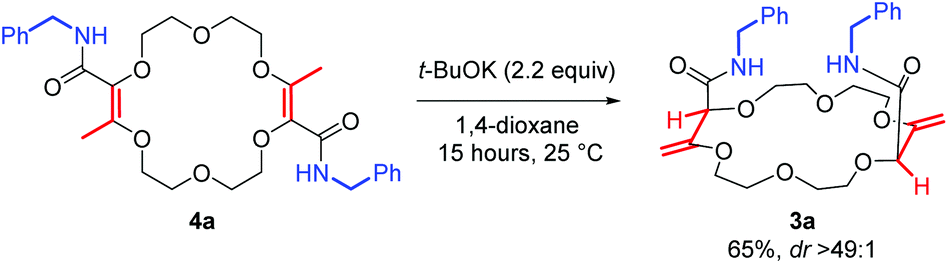 | ||
| Scheme 2 Base-induced olefin transposition of 4a to form 3a. | ||
In solution, compounds 3 differ from the bis-anilide derivatives 2. In fact, in terms of the simple host–guest chemistry, it had been previously observed that a water molecule is usually complexed inside the cavity of macrocycles 2.4 Herein, in chloroform-d, there is little of interaction of 3a with water as monitored by 1H NMR spectroscopy at different concentrations (8–116 mM, see the ESI‡). In comparison with the aniline series (compounds 2), the proton signals of the amide groups or of the water molecule are only weakly perturbed.
2-Step process: scope and (lack of) asymmetric induction with enantiopure amines
With the optimal conditions for the amidation and the olefin transposition in hand, various linear and α-branched amines were introduced. In Table 2, the yields are reported for the two steps. In general, with linear primary amines, moderate to good combined yields were obtained (35–55%); the nature of the side chains has little influence on the outcome. For the standard reaction, macrocycle 3a was obtained in 54% overall yield. Methyl, propyl, octyl and allyl substituted derivatives 3b to 3e were obtained in similar yields (42%–55%). Silyl or methyl protected amino alcohols were used and afforded macrocycles 3f to 3h in 35% to 54% yield.23|
a Method A: 1 (0.25 mmol), linear amine (4.0 equiv.), TBD (2.0 equiv.), THF, 60 °C, 15 hours; then t-BuOK (2.2 equiv.), dioxane, 25 °C, 15 hours.
b Method B: 1 (0.25 mmol), α-branched amine (4.0 equiv.), TBD (4.0 equiv.), THF, 60 °C, 10 days; then t-BuOK (2.2 equiv.), dioxane, 25 °C, 15 hours. Macrocycles 3k to 3s are obtained as mixtures of diastereoisomers (1:1 ≤ dr ≤ 1.4:1, see the ESI).
|
|---|
|
However, when the introduction of α-branched amines was investigated under the above conditions, only poor conversions were observed for the first amidation step, the sterically-hindered amines being much less reactive. It was necessary to increase both the amount of TBD (from 2 to 4 equiv.) and the reaction time (15 hours to 10 days) to reach full conversion of 1. Then, upon transposition with t-BuOK, macrocycles 3i (i-Pr) and 3j (c-Hex) were obtained in moderate combined yields (29% and 18%, respectively). Similarly, with enantiopure α-alkyl substituted benzyl or methylnaphthyl amines, we could isolate the corresponding macrocycles 3k to 3s (15–35%). In all these reactions, the critical step in terms of yield is not the transposition but the amidation. In fact, a slow competitive degradation of the starting macrocycle 1 occurs during the amidation that disfavors the overall process.24
Finally, care was taken to study the stereochemical outcome of the reaction in the presence of enantiopure α-branched amines.25 Satisfactorily, the double transposition still occurs with a syn stereoselectivity but it leads, this time, to two different products. Indeed, the enantiopure amine residues exert little influence on the newly created stereogenic centers inside the macrocyclic skeleton and two diastereoisomers are formed. For instance, with (S)-phenylethylamine, product 3k exists in two different (S,S,S,S) and (S,R,R,S) configurations with a low overall stereoselectivity (dr 1.3:1).26 Several other amines were tested and the selectivity has remained low in all cases (dr ≤ 1.4:1). In Table 2, the yields are given for the two steps and both diastereoisomers together; the stereoisomers were separated in only one instance (see the next paragraph).
Application: enantioselective phase-transfer catalysis
Crown ethers, thanks to their ability to complex alkali metal salts and render them soluble in both polar and apolar solutions, are efficient phase-transfer catalysts (PTCs). Several chiral non-racemic versions of these derivatives have been developed and applied in various enantioselective reactions such as alkylations, 1,4-additions or oxidations.27 It was then logical to test compounds 3k to 3s bearing α-branched enantiopure substituents as PTCs. We selected the alkylation of protected glycine 5 with benzyl bromide which affords chiral phenylalanine derivative 6 as the product as the benchmark; this reaction occurs only in the presence of catalysts (Table 3, entry 1).28 The different macrocycles were reacted under conditions that are standard for phase-transfer catalysis with chiral crown ethers (Tables S2–S5‡). Overall and not surprisingly, it was found that sodium and potassium salts afforded the best conversions (Table S3‡). A rather large screening of solvents was also performed and CH2Cl2 was the most favorable followed by 1,1-dichloroethane and 1,1,1-trichloroethane, similar ee values however being observed (Table S5‡). With derivative 3k as the catalyst (5 mol%, Table 3, entry 2), amino acid (+)-6 was obtained in good yield (86%) and low enantioselectivity (17% ee). The increase of catalyst loading to 10 mol% improved slightly the enantiomeric excess to 21% (entry 3). With toluene as the solvent, a longer reaction time, lower enantioselectivity and reversal of the stereoinduction in favor of the antipodal enantiomer (−)-6 were observed (entry 4). Using macrocycle 3p (with an inverted configuration on the amide side chain), analogous results to 3k were obtained in favor of the levorotatory enantiomer. It was then shown that 1-naphthyl derivative 3n is the most efficient in the series (see the ESI‡). Using 3n as the PTC (10 mol%, dr 1.4:1) at 25 °C, amino acid (+)-6 was obtained in good yield (86%) and moderate enantioselectivity (32% ee) (Table 3, entry 6). Decreasing the temperature to 10 °C improved slightly the enantiomeric excess (39% ee, entry 7). The two diastereoisomers of 3n, namely (S,S,S,S)-3n (major) and (S,R,R,S)-3n (minor), could be separated by column chromatography (SiO2, CH2Cl2/MeOH gradient) and used independently in the phase-transfer reaction. The configuration of (S,S,S,S)-3n was ascertained by the X-ray structural analysis of the NaBArF salt. With (S,S,S,S)-3n, the dextrorotatory enantiomer of 6 was again favored with a slightly improved enantiomeric excess (43% ee, entry 8). Higher catalyst loading (20 mol%) or temperature decrease (0 °C) did not lead to a larger enantioinduction (entries 9 and 10). Interestingly, using the minor diastereoisomer (S,R,R,S)-3n and after a longer reaction time (48 hours), the antipodal enantiomer (−)-6 was obtained in 86% yield and only a 13% ee value (entry 11). The difference in reactivity and selectivity between the two diastereoisomers of 3n illustrates thus a clear matched–mismatched situation in the enantioselective alkylation of glycine 5.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Temp. (°C) | Time (h) | ee (%) |
| a For all catalyzed reactions, isolated yields are consistent in the range of 85–87%. b No conversion of 5. c In toluene instead of CH2Cl2. | ||||
| 1 | — (Blank) | 25 | 15 | —b |
| 2 | 3k (5) | 25 | 15 | 17 |
| 3 | 3k (10) | 25 | 15 | 21 |
| 4c | 3k (5) | 25 | 96 | −12 |
| 5 | 3p (5) | 25 | 15 | −17 |
| 6 | 3n (10) | 25 | 15 | 32 |
| 7 | 3n (10) | 10 | 15 | 39 |
| 8 | (S,S,S,S)-3n (10) | 10 | 15 | 43 |
| 9 | (S,S,S,S)-3n (20) | 10 | 15 | 42 |
| 10 | (S,S,S,S)-3n (10) | 0 | 15 | 42 |
| 11 | (S,R,R,S)-3n (10) | 10 | 48 | −13 |
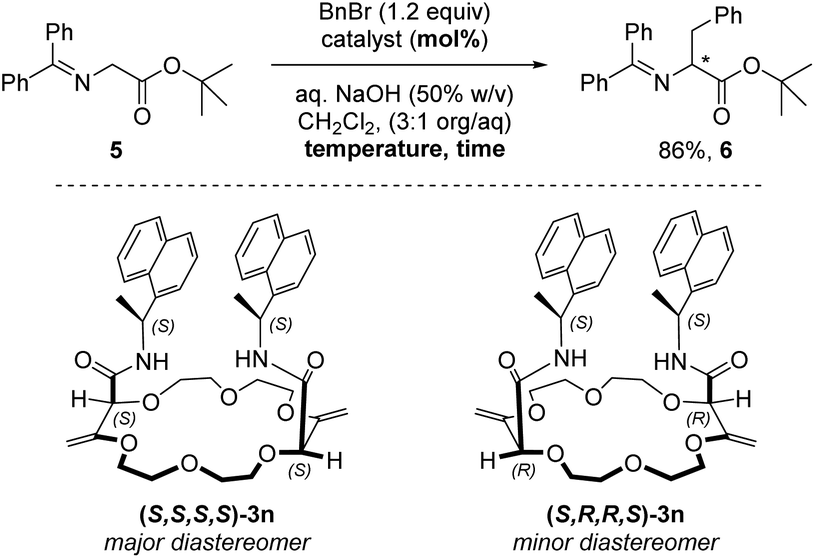
Conclusion
Starting from the readily prepared unsaturated macrocyclic precursor 1, chiral crown ethers bearing two aliphatic amide functional groups were synthesized. It was necessary to separate the amidation (mediated by TBD) and the olefin transposition step (induced by a slight excess of t-BuOK). The products were afforded in moderate to good combined yields (up to 59%) and with an excellent syn diastereoselectivity (dr > 49:1). Introducing enantiopure α-branched substituents was possible and resulted in mixtures of diastereomers, which could be tested as phase-transfer catalysts using the formation of enantioenriched phenylalanine analogs as a benchmark (up to 43% ee). In this reaction, a clear matched–mismatched situation was observed in the two diastereomeric series.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation for financial support (SNF 200020-172497 and 200020-184843). We acknowledge the contributions of the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences, University of Geneva. We thank Professor Dr Klaus Ditrich (BASF) for generous gifts of the chiral enantiopure amines.Notes and references
- (a) F. Bargiggia and O. Piva, Tetrahedron: Asymmetry, 2003, 14, 1819–1827 CrossRef CAS; (b) T. Bach and F. Höfer, J. Org. Chem., 2001, 66, 3427–3434 CrossRef CAS PubMed; (c) O. Piva, J. Org. Chem., 1995, 60, 7879–7883 CrossRef CAS; (d) O. Piva and J.-P. Pete, Tetrahedron: Asymmetry, 1992, 3, 759–768 CrossRef CAS; (e) R. Mortezaei, D. Awandi, F. Henin, J. Muzart and J. P. Pete, J. Am. Chem. Soc., 1988, 110, 4824–4826 CrossRef CAS; (f) S. L. Eng, R. Ricard, C. S. K. Wan and A. C. Weedon, J. Chem. Soc., Chem. Commun., 1983, 236–238 RSC; (g) J. R. Scheffer and B. A. Boire, J. Am. Chem. Soc., 1971, 93, 5490–5495 CrossRef CAS; (h) M. J. Jorgenson, J. Am. Chem. Soc., 1969, 91, 198–200 CrossRef CAS; (i) G. Buechi and S. H. Feairheller, J. Org. Chem., 1969, 34, 609–612 CrossRef CAS; (j) R. R. Rando and W. V. E. Doering, J. Org. Chem., 1968, 33, 1671–1673 CrossRef CAS; (k) M. J. Jorgenson and T. Leung, J. Am. Chem. Soc., 1968, 90, 3769–3774 CrossRef CAS; (l) M. J. Jorgenson and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 1698–1699 CrossRef CAS.
- (a) J. Martin, J.-C. Plaquevent, J. Maddaluno, J. Rouden and M.-C. Lasne, Eur. J. Org. Chem., 2009, 5414–5422 CrossRef CAS; (b) E. Vedejs, A. W. Kruger, N. Lee, S. T. Sakata, M. Stec and E. Suna, J. Am. Chem. Soc., 2000, 122, 4602–4607 CrossRef CAS; (c) C. Fehr, Angew. Chem., Int. Ed. Engl., 1996, 35, 2566–2587 CrossRef; (d) A. C. Weedon, Can. J. Chem., 1984, 62, 1933–1939 CrossRef CAS.
- (a) C. Fehr, N. Chaptal-Gradoz and J. Galindo, Chem. – Eur. J., 2002, 8, 853–858 CrossRef CAS PubMed; (b) C. Fehr and J. Galindo, Angew. Chem., Int. Ed. Engl., 1994, 33, 1888–1889 CrossRef; (c) C. Fehr, I. Stempf and J. Galindo, Angew. Chem., Int. Ed. Engl., 1993, 32, 1042–1044 CrossRef; (d) C. Fehr, I. Stempf and J. Galindo, Angew. Chem., Int. Ed. Engl., 1993, 32, 1044–1046 CrossRef; (e) C. Fehr and J. Galindo, J. Am. Chem. Soc., 1988, 110, 6909–6911 CrossRef CAS.
- M. Vishe, R. Hrdina, A. I. Poblador-Bahamonde, C. Besnard, L. Guénée, T. Bürgi and J. Lacour, Chem. Sci., 2015, 6, 4923–4928 RSC.
- Z. Jarolímová, M. Vishe, J. Lacour and E. Bakker, Chem. Sci., 2016, 7, 525–533 RSC.
- S. K. Ray, A. Homberg, M. Vishe, C. Besnard and J. Lacour, Chem. – Eur. J., 2018, 24, 2944–2951 CrossRef CAS PubMed.
- (a) A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Górecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052 RSC; (b) S. Sinn, F. Biedermann, M. Vishe, A. Aliprandi, C. Besnard, J. Lacour and L. De Cola, ChemPhysChem, 2016, 17, 1829–1834 CrossRef CAS PubMed.
- F. Zinna, S. Voci, L. Arrico, E. Brun, A. Homberg, L. Bouffier, T. Funaioli, J. Lacour, N. Sojic and L. Di Bari, Angew. Chem., Int. Ed., 2019, 58, 6952–6956 CrossRef CAS PubMed.
- (a) D. Poggiali, A. Homberg, T. Lathion, C. Piguet and J. Lacour, ACS Catal., 2016, 6, 4877–4881 CrossRef CAS; (b) M. Vishe, R. Hrdina, L. Guénée, C. Besnard and J. Lacour, Adv. Synth. Catal., 2013, 355, 3161–3169 CrossRef CAS; (c) W. Zeghida, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2010, 49, 7253–7256 CrossRef CAS PubMed.
- Macrocycles of different scaffolds or sizes can be obtained using similar Rh(II)-catalyzed procedures by condensation of α-diazo-β-ketoesters with other small cyclic ethers such as THP, THF, oxetane, oxepane or morpholines. See ref. 9.
- M. Vishe, T. Lathion, S. Pascal, O. Yushchenko, A. Homberg, E. Brun, E. Vauthey, C. Piguet and J. Lacour, Helv. Chim. Acta, 2018, 101, e1700265 CrossRef.
- For examples of amidation without the help of external reagents or catalysts, see: (a) S. Furukawa, T. Fukuyama, A. Matsui, M. Kuratsu, R. Nakaya, T. Ineyama, H. Ueda and I. Ryu, Chem. – Eur. J., 2015, 21, 11980–11983 CrossRef CAS PubMed; (b) N. D. Karis, W. A. Loughlin and I. D. Jenkins, Tetrahedron, 2007, 63, 12303–12309 CrossRef CAS; (c) F.-Z. Zradni, J. Hamelin and A. Derdour, Synth. Commun., 2002, 32, 3525–3531 CrossRef CAS.
- (a) A. Mondal, M. Subaramanian, A. Nandakumar and E. Balaraman, Org. Lett., 2018, 20, 3381–3384 CrossRef CAS PubMed; (b) D. T. Nguyen, D. C. Lenstra and J. Mecinovic, RSC Adv., 2015, 5, 77658–77661 RSC; (c) H. Morimoto, R. Fujiwara, Y. Shimizu, K. Morisaki and T. Ohshima, Org. Lett., 2014, 16, 2018–2021 CrossRef CAS PubMed; (d) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (e) V. M. de Oliveira, R. Silva de Jesus, A. F. Gomes, F. C. Gozzo, A. P. Umpierre, P. A. Z. Suarez, J. C. Rubim and B. A. D. Neto, ChemCatChem, 2011, 3, 1911–1920 CrossRef CAS; (f) A. Novak, L. D. Humphreys, M. D. Walker and S. Woodward, Tetrahedron Lett., 2006, 47, 5767–5769 CrossRef CAS; (g) R. Arora, S. Paul and R. Gupta, Can. J. Chem., 2005, 83, 1137–1140 CrossRef CAS; (h) C. Han, J. P. Lee, E. Lobkovsky and J. A. Porco, J. Am. Chem. Soc., 2005, 127, 10039–10044 CrossRef CAS PubMed; (i) B. C. Ranu and P. Dutta, Synth. Commun., 2003, 33, 297–301 CrossRef CAS; (j) K. Ishihara, Y. Kuroki, N. Hanaki, S. Ohara and H. Yamamoto, J. Am. Chem. Soc., 1996, 118, 1569–1570 CrossRef CAS.
- (a) N. Caldwell, C. Jamieson, I. Simpson and A. J. B. Watson, Chem. Commun., 2015, 51, 9495–9498 RSC; (b) N. Caldwell, P. S. Campbell, C. Jamieson, F. Potjewyd, I. Simpson and A. J. B. Watson, J. Org. Chem., 2014, 79, 9347–9354 CrossRef CAS PubMed; (c) N. Caldwell, C. Jamieson, I. Simpson and A. J. B. Watson, ACS Sustainable Chem. Eng., 2013, 1, 1339–1344 CrossRef CAS; (d) T. Ohshima, Y. Hayashi, K. Agura, Y. Fujii, A. Yoshiyama and K. Mashima, Chem. Commun., 2012, 48, 5434–5436 RSC; (e) V. R. Chintareddy, H.-A. Ho, A. D. Sadow and J. G. Verkade, Tetrahedron Lett., 2011, 52, 6523–6529 CrossRef CAS; (f) S. A. Rzhevskiy, A. A. Ageshina, G. A. Chesnokov, P. S. Gribanov, M. A. Topchiy, M. S. Nechaev and A. F. Asachenko, RSC Adv., 2019, 9, 1536–1540 RSC.
- (a) Y.-L. Zheng and S. G. Newman, ACS Catal., 2019, 9, 4426–4433 CrossRef CAS; (b) B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682–1685 CrossRef CAS PubMed.
- (a) K. P. Dhake, Z. S. Qureshi, R. S. Singhal and B. M. Bhanage, Tetrahedron Lett., 2009, 50, 2811–2814 CrossRef CAS; (b) L. Couturier, D. Taupin and F. Yvergnaux, J. Mol. Catal. B: Enzym., 2009, 56, 29–33 CrossRef CAS; (c) N. Aoyagi, S. Kawauchi and T. Izumi, Tetrahedron Lett., 2003, 44, 5609–5612 CrossRef CAS.
- (a) J. Lauberteaux, D. Pichon, O. Baslé, M. Mauduit, R. M. de Figueiredo and J.-M. Campagne, ChemCatChem, 2019, 11, 1–19 CrossRef; (b) L.-Y. Chen and M.-F. Wu, Synthesis, 2019, 1595–1602 CrossRef CAS; (c) H. Guo, Y. Wang, G.-F. Du, B. Dai and L. He, Tetrahedron, 2015, 71, 3472–3477 CrossRef CAS; (d) F. J. Weiberth, Y. Yu, W. Subotkowski and C. Pemberton, Org. Process Res. Dev., 2012, 16, 1967–1969 CrossRef CAS; (e) X. Yang and V. B. Birman, Org. Lett., 2009, 11, 1499–1502 CrossRef CAS PubMed; (f) K. E. Price, C. Larrivée-Aboussafy, B. M. Lillie, R. W. McLaughlin, J. Mustakis, K. W. Hettenbach, J. M. Hawkins and R. Vaidyanathan, Org. Lett., 2009, 11, 2003–2006 CrossRef CAS PubMed; (g) C. Sabot, K. A. Kumar, S. Meunier and C. Mioskowski, Tetrahedron Lett., 2007, 48, 3863–3866 CrossRef CAS; (h) M. Movassaghi and M. A. Schmidt, Org. Lett., 2005, 7, 2453–2456 CrossRef CAS PubMed; (i) S. Karthik, K. Muthuvel and T. Gandhi, J. Org. Chem., 2019, 84, 738–751 CrossRef CAS PubMed.
- TBD is usually used as an organocatalyst for transesterification or ester polymerization reactions. See: (a) R. Yuan, Q. Shou, Q. Mahmood, G. Xu, X. Sun, J. Wan and Q. Wang, Synlett, 2019, 928–931 CAS; (b) S. Park and J. G. Kim, Beilstein J. Org. Chem., 2019, 15, 963–970 CrossRef CAS PubMed; (c) M. K. Kiesewetter, M. D. Scholten, N. Kirn, R. L. Weber, J. L. Hedrick and R. M. Waymouth, J. Org. Chem., 2009, 74, 9490–9496 CrossRef CAS PubMed.
- For the TBD mediated synthesis of carbamate and urea, see: (a) S. Carloni, D. E. De Vos, P. A. Jacobs, R. Maggi, G. Sartori and R. Sartorio, J. Catal., 2002, 205, 199–204 CrossRef CAS; (b) R. Ballini, D. Fiorini, R. Maggi, P. Righi, G. Sartori and R. Sartorio, Green Chem., 2003, 5, 396–398 RSC.
- For the TBD mediated formylation of amines, see: H.-W. Noh, Y. An, S. Lee, J. Jung, S. U. Son and H.-Y. Jang, Adv. Synth. Catal., 2019, 361, 1–7 CrossRef.
- In addition, increasing the reaction time and/or temperature led only to partial mono addition or to degradation products.
- The use of 0.2 equivalents of t-BuOK led only to a partially-induced isomerization (Table S1‡). This indicates that the secondary amide functions are too acidic and it is necessary to deprotonate them quantitatively prior to the isomerization.
- The isomerization does not proceed with unprotected alcohol functional groups. See the ESI.‡.
- Full conversion of 1 is always reached. The degradation products could unfortunately neither be isolated nor identified.
- It has been previously shown that TBD does not racemize this type of chiral amine. See ref. 17g.
- Both products present a local C2-symmetry in solution (1H and 13C NMR spectroscopy monitoring).
- (a) R. Schettini, M. Sicignano, F. De Riccardis, I. Izzo and G. Della Sala, Synthesis, 2018, 4777–4795 CAS; (b) Z. Rapi, T. Nemcsok, P. Bagi, G. Keglevich and P. Bakó, Tetrahedron, 2019 DOI:10.1016/j.tet.2019.06.020.
- (a) J. Vachon and J. Lacour, Chimia, 2006, 60, 266–275 CrossRef CAS; (b) T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222–4266 CrossRef CAS PubMed; (c) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed; (d) N. Patel, R. Sood and P. V. Bharatam, Chem. Rev., 2018, 118, 8770–8785 CrossRef CAS PubMed.
Footnotes |
| † The dataset for this article can be found at the following DOI: 10.26037/yareta:fp326563trgt3gp3l3ofsyfiem. It will be preserved for 10 years. |
| ‡ Electronic supplementary information (ESI) available: Experimental conditions, full characterization, 1H NMR, 13C NMR and 19F NMR spectra of all new compounds (PDF), and CSP-HPLC traces. CCDC 1923025–1923027. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob01355e |
| This journal is © The Royal Society of Chemistry 2019 |