
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn atom efficient synthesis of tamoxifen†
Dorus
Heijnen‡
,
Milan
van Zuijlen‡
,
Filippo
Tosi‡
and
Ben L.
Feringa
 *
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
First published on 28th January 2019
Abstract
The direct carbolithiation of diphenylacetylenes and their cross-coupling procedure taking advantage of the intermediate alkenyllithium reagents are presented. By employing our recently discovered highly active palladium nanoparticle based catalyst, we were able to couple an alkenyllithium reagent with a high (Z/E) selectivity (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and good yield to give the breast cancer drug tamoxifen in just 2 steps from commercially available starting materials and with excellent atom economy and reaction mass efficiency.
:1) and good yield to give the breast cancer drug tamoxifen in just 2 steps from commercially available starting materials and with excellent atom economy and reaction mass efficiency.
The continuous improvement of synthetic routes towards medicinally relevant and/or biologically active compounds has drawn the attention of synthetic chemists for decades.1 In order to reduce waste and increase yields and cost efficiency or to simplify the procedures towards relevant structures, transition metal catalysis has been a game changer in the field.2 Since the emergence of the Suzuki (B), Stille (Sn), Negishi (Zn) and Hiyama–Denmark (Si) reactions, the trend in the cross-coupling methodology3 has been to transmetallate highly polar (but straightforward to synthesize) organometallic reagents (RMgX, RLi) to softer nucleophiles in order to gain stability and functional group tolerance and reduce the overall sensitivity of the reaction. Despite their major role in our modern synthetic toolbox, the drawbacks of these additional synthetic steps are longer reaction times, the production of stoichiometric (sometimes toxic) waste, and a decrease in cost efficiency.4 Nonetheless, the direct coupling of organometallic reagents arising from a deprotonation or umpolung reaction has shown great advances in recent years.2,5 Since these reagents have an intrinsically higher reactivity, the corresponding cross-coupling reactions generally require a shorter reaction time, and can be performed at significantly lower temperatures.4 As part of our effort to expand the synthetic application of our recently reported organolithium cross-coupling reactions,5d,f,g,i–k we envisioned the direct carbolithation–cross coupling to be a very valuable alternative. The carbolithiation of (diphenyl)acetylenes has been well-studied and has led to several useful applications in the field of synthetic organic chemistry.6–10 The quenching of the formed nucleophilic sp2 vinyllithium reagent with an electrophile provides a direct approach to substituted diarylalkenes (stilbenes). Transmetallation of such anions to magnesium, boron, zinc or even aluminum yields an active cross-coupling partner, but drastically lowers the atom economy and E-factor.11,12 The direct cross-coupling of the formed organolithium reagent is therefore a highly desired synthetic shortcut, but remains unreported to the best of our knowledge. Tetrasubstituted alkenes and triphenylethylenes in particular, i.e. tamoxifen, make up a class of highly potent and valuable drugs with (potential) application in the treatment of a variety of conditions, including (breast) cancer, dyspareunia and osteoporosis (Fig. 1).13 Structural variations in the triphenylethylene scaffold are found in the alkyl–ether substituent (mostly consisting of an amine, shown in blue) and para phenylene functionality (shown in red) as well as in the alkyl fragment (shown in green) on the remaining alkene position.
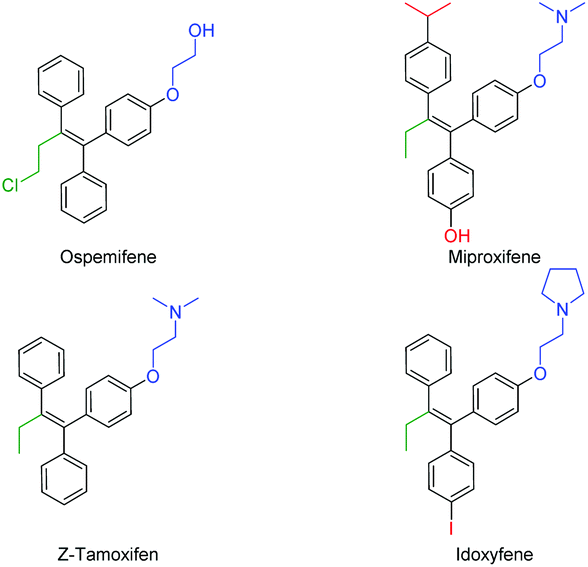 | ||
| Fig. 1 Examples of members of the triphenylethylene family of drugs. | ||
Because of its medicinal importance, a plethora of syntheses have been described for (Z)-tamoxifen (Fig. 2).15–37 McMurry coupling of two ketones is a well-established method for the synthesis of (hindered) alkenes, and as such has proven to be capable of constructing the alkene fragment in tamoxifen with reagents 1 and 2.21 Alternatively, 1,2-addition to ketone 3 with Grignard reagent 4 followed by elimination yielding an alkene is a valuable option, however it is common that both isomers (E/Z) are isolated via this approach.19,24 The transmetallation of the lithium intermediate that is the product of carbolithation of the corresponding acetylene yields the alkenyl–boronic acid/ester, or organozinc reagent 5.20,22 The cross-coupling of these reagents with bromide 6 provides a viable route towards the final drug. The transmetallation, however, generates extra synthetic steps and/or stoichiometric waste. We therefore reasoned that the direct coupling of the alkenyllithium reagent 7, which is obtained upon carbolithiation of diphenylacetylene, would be an important atom efficient alternative to these methods.
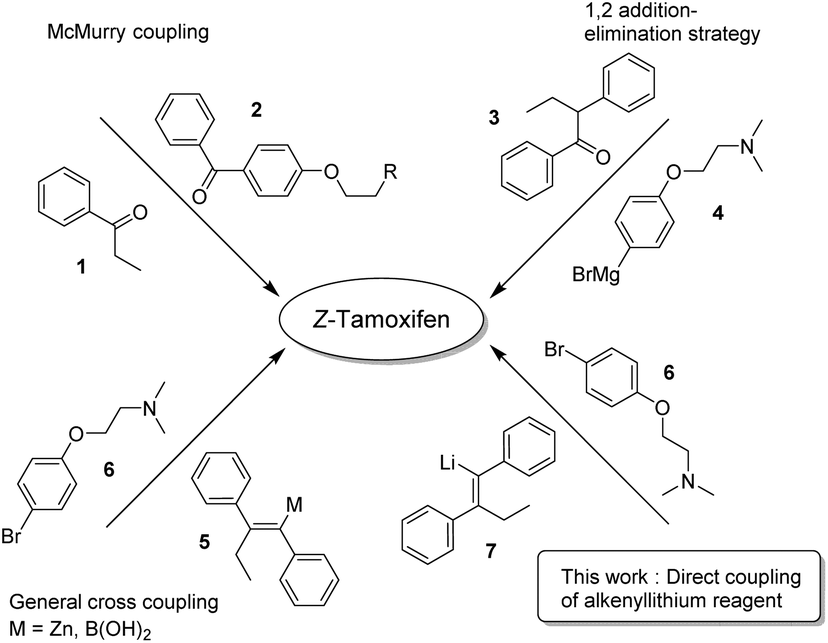 | ||
| Fig. 2 Synthetic approaches to (Z)-tamoxifen. | ||
In order to optimize the sequential synthetic steps, the carbolithiation of acetylene 8 was optimized separately, and the product 10 resulting from MeI quenching was subjected to GC-MS analysis (Table 1). With a high selectivity for the (Z)-alkenes for several solvents and solvent mixtures being investigated, we tried to avoid the use of THF (Table 1, entry 1) due to expected difficulties in the cross-coupling step arising from unwanted side reactions, such as lithium–halogen exchange.5b–g,i–l However, toluene/TMEDA mixtures (entry 2) or other ethereal solvents (entries 3–5) did not prove equally efficient as the reaction medium compared to THF due to a lower extent of lithiation and an increased amount of the (E)-alkene. Attempts to minimize waste production by neat carbolithiation (except for the solvent of the n-BuLi solution) resulted in mere recovery of the starting material (entry 6). Reducing the amount of THF by using a THF–toluene mixture resulted in incomplete conversion to the carbolithiated intermediate (entry 7). Despite the attempts to omit THF as the solvent, we found significantly better results for the carbolithiation in its presence, and therefore decided to use it as the solvent for further optimization.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | 1 h Conv.a | 2 h Conv.a | Selectivitya,b |
| Reaction conditions: 1.1 eq. of n-BuLi were added dropwise to a stirred 0.9 M solution of 8 in THF at 0 °C. At the end of the addition, the reaction mixture was quickly allowed to warm to room temperature and left to react for 2 h before 1 eq. of MeI was added.a As determined by GC-MS analysis after MeI quenching.b (Z/E) selectivity determined by GC-MS analysis.c 1 eq. of TMEDA. | ||||
| 1 | THF | 91% | 91% | 96% |
| 2 | Toluene/TMEDAc | 32% | 32% | 75% |
| 3 | 2-Me-THF | 52% | 70% | 99% |
| 4 | MTBE | 0% | 6% | — |
| 5 | Ether | 0% | 4% | — |
| 6 | Neat | 0% | 0% | — |
| 7 | Toluene/THF 3:1 |
48% | 65% | 96% |
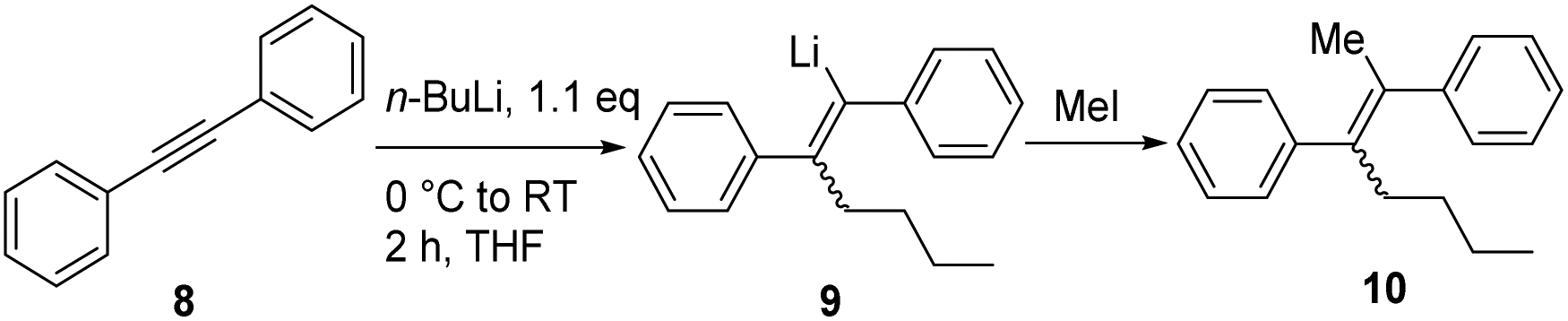
Having established the optimized conditions for the carbolithiation, the cross-coupling with 1-bromonaphthalene provided the test reaction in pursuit of the best catalyst. Our oxygen activated palladium nanoparticle catalytic system5k proved to be highly active in the coupling of alkenyllithium 9 and arylbromides (Table 2, entry 1), being only slightly outperformed by the commercial Pd-PEPPSI–Ipent complex (entry 4).
|
|
||
|---|---|---|
| Entry | Cat. (5%) | Conversion to 11a |
| Reaction conditions: 2 eq. of alkenyllithium reagent were added over 20 min to a stirred solution of arylbromide and (pre-oxidized) catalyst in toluene at 35 °C.a As determined by GCMS analysis. (Z/E) > 9:1. |
||
| 1 | Pd(PtBu3)2 + O2 | 82% (68% yield) |
| 2 | Pd-dppf | — |
| 3 | Ni-dppp | — |
| 4 | PEPPSI-Ipent | 88% |
| 5 | C1 | 51% |
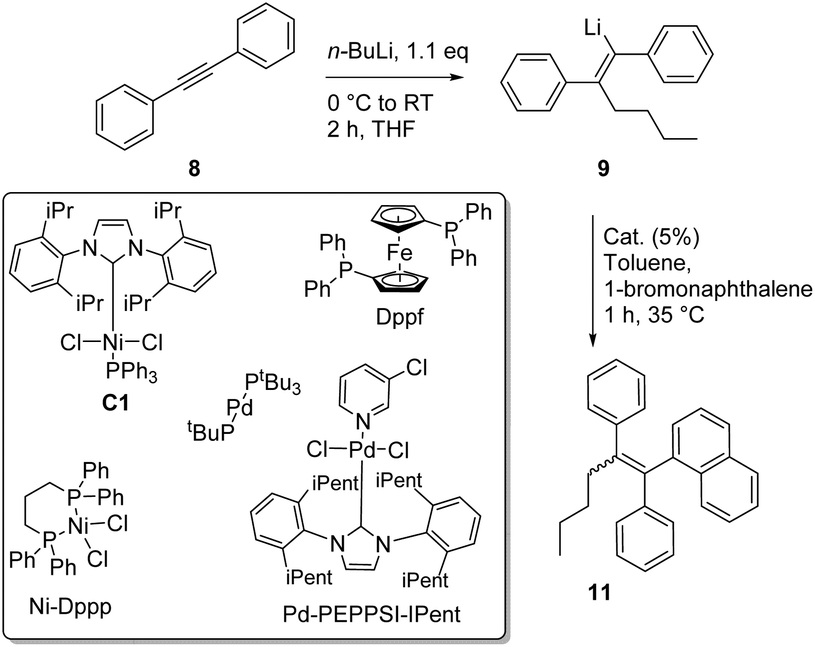
Nickel and palladium bisphosphine complexes (Pd-dppf and Ni-dppp) did not show conversion to the desired product (entries 2 and 3), but the nickel carbene complex C15j did provide the triarylethylene target 11 (entry 5), albeit in a reduced yield (for further attempts to use different electrophiles, see the ESI†). In order to see if this cross-coupling methodology could be applicable for the cross-coupling of other arylbromides, we decided to test a few further substrates (Table 3). The cross-coupling methodology proceeds smoothly with both electron rich arylbromides (R = Me, OMe, entries 1 and 2) and electron poor substrates (R = CF3, entry 3).
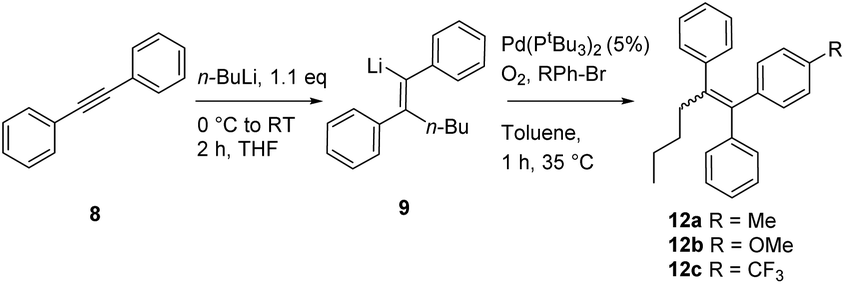
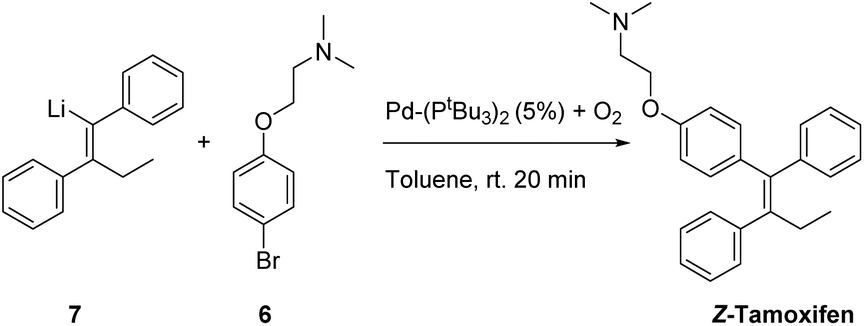
Having tested a small variety of catalysts for the cross coupling with different substrates, the optimized conditions were employed to synthesize the desired pharmaceutical (Z)-tamoxifen via our new methodology. Changing the nucleophile for the acetylene carbolithiation from n-butyllithium to ethyllithium gave identical results albeit with a slightly longer reaction time. We were pleased to see that the oxygenated Pd(PtBu3)2 catalyst gave (Z)-tamoxifen in only a slightly lower yield than with the naphthalene test substrate, but with very good (Z/E) selectivity (Table 4, entry 1, (Z/E) > 9:1). On pursuing a cheaper catalyst, with a more abundant metal, the attempted nickel complex gave only small amounts of the desired product (entry 2).
|
|
||
|---|---|---|
| Entry | Cat. (5%) | NMR yielda |
| Reaction conditions: 2 eq. of alkenyllithium reagent were added over 20 min to a stirred solution of arylbromide and (pre-oxidized) catalyst in toluene at 35 °C.a Yield determined by 1H-NMR with 1,1,2,2-tetrachloroethane as the internal standard, accompanied by <10% of (E)-tamoxifen relative to (Z)-tamoxifen.b Isolated yield up to 57%.c 2.5 mol% of the Pd dimer was used. | ||
| 1 | Pd(PtBu3)2 + O2 | 50–65%b |
| 2 | C1 | 17% |
| 3 | PEPPSI-IPent | 0% |
| 4 | Pd2dba3/Xphos | 36% |
| 5 | C2 | 60% |
| 6 | C3 | 59% |
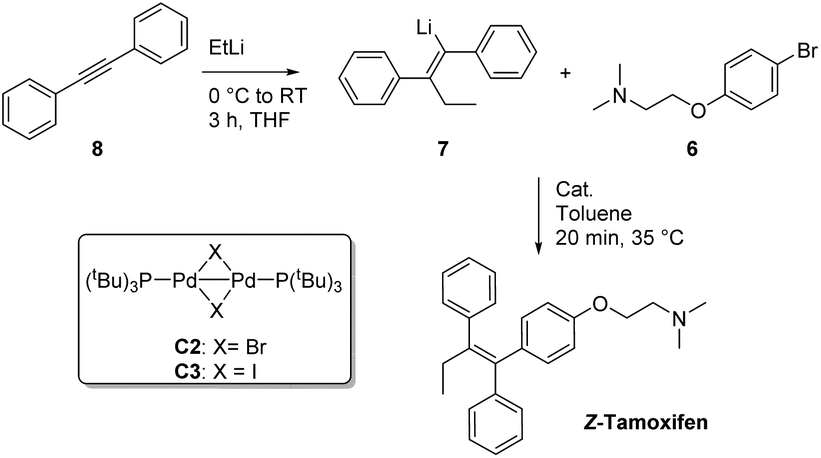
Much to our surprise, our “working horse” catalyst Pd-PEPPSI Ipent5g,i was completely inactive in the cross-coupling with the bromophenyl-aminoether electrophile 6 (Table 4, entry 3), whereas it proved to be the most active catalyst with the previously tested 1-bromonaphthalene electrophile (Table 2, entry 4). The chelating effect of the amino–ether moiety onto the metal center, overcrowding the palladium, might play a role making the catalyst inactive. Other dimeric palladium phosphine complexes, which have recently been shown to be active in related cross-coupling reactions, were also tested,5h,k and were found to have very similar reactivity compared to the catalyst used in entry 1. Being the cheapest of the three related structures (entries 1, 5 and 6), we decided to proceed our investigation with the bis(tri-tert-butylphosphine)palladium complex. The results of further optimization are shown in Table 5. Varying the temperature did not lead to an increased yield (entries 1 and 2) provided the temperature was kept above 30 °C, below which no conversion was observed. In an attempt to dissociate potential aggregates and activate the organolithium reagent, TMEDA was added, but this resulted in a sharp decline in yield (entry 3). The excess of organolithium reagent could be lowered to 1.3 equivalents without a significant loss in yield (entry 4). Further lowering of the catalyst loading (2.5 mol%) led to an inactive system, with no product formed (entries 6 and 7). This complete deactivation of the catalyst at 2.5 mol% has not been observed before, and it is potentially attributed to the chelating effect of the aminoether moiety that is present in the substrate. An attempt to prevent the chelating effect of the aminoether side chain by means of the addition of Lewis acids such as BF3 or MgCl2 did not prove beneficial for the reaction (entries 8 and 9). To minimize waste caused by the solvent, the reaction was performed in a minimal amount of solvent, with a 1 M concentration, which only led to a slight decrease in yield (entry 10). Lower loadings (1.25 mol%) of complexes C2 and C3 provided better conversion than our catalyst of choice at those concentrations (entries 6, 11 and 12), but still providing lower overall yields (14% and 28%, respectively).
|
|
||
|---|---|---|
| Entry | Modificationsa | Yieldb |
| a Reaction conditions: 2 eq. of alkenyllithium reagent were added over 20 min to a stirred solution of arylbromide (2 mmol) and pre-oxidized catalyst in toluene (2 ml) at room temperature. b Yield determined by 1H-NMR with 1,1,2,2-tetrachloroethane as the internal standard. c Different batches of catalyst. d Initial concentration of arylbromide. e The complexes C2 and C3 were used with the same Pd concentration as entries 6 and 7. | ||
| 1 | Temp 50 °C | 60 |
| 2 | Temp 35 °C | 68 |
| 3 | TMEDA (1 eq.) | 25 |
| 4 | 1.3 eq. of 7 | 65 |
| 5 | 10% cat., 1.3 eq. of 7 | 65 |
| 6 | 2.5% cat. | — |
| 7 | 2.5% cat.c | — |
| 8 | BF3 | — |
| 9 | MgCl2 | 58 |
| 10 | Concentrated (1 M)d | 54 |
| 11 | C2 1.25% cat. | 14 |
| 12 | C3 1.25% cat. | 28 |
Having a setup that produces this pharmaceutical compound in good yield and with minimal waste (LiBr being the only stoichiometric waste in the last step), we compared our procedure with other (recent) reported syntheses of tamoxifen6,15–37 focusing on atom economy and Reaction Mass Efficiency (RME).12bFig. 3 shows a large range in atom economy (shown in blue) between different reported syntheses of tamoxifen. The method described by Larock in 200537 is the closest to the reported route in this work in terms of atom economy (48 vs. 67%), but due to a large excess of some reagents, it scores much lower on RME (shown in red).
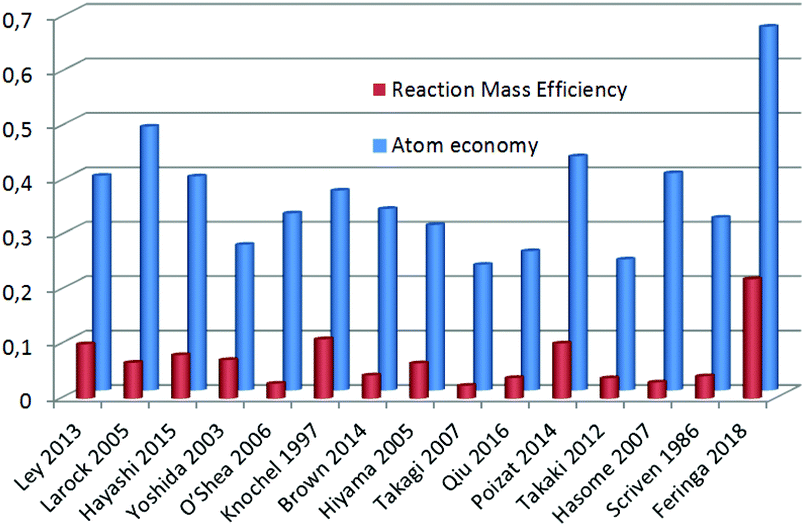 | ||
| Fig. 3 Atom economy and RME comparison for each reported tamoxifen synthesis. | ||
With our current setup, by employing commercially available starting materials, a total atom economy of 0.67 is achieved, and the resulting RME (22%) is almost twice as high as that of the runner-up (Knochel, 1997, 11%).20 We believe that the currently reported methodology presents additional relevant advantages, since LiBr, NaCl and HCl are the only stoichiometrically produced waste sources, and the reaction can be performed at a slightly elevated temperature in a minimal amount of solvent.
To establish the optimal isolation method, the synthesized (Z)-tamoxifen was purified by means of crystallization, extraction, column chromatography and distillation. The excess organolithium reagent (protonated after reaction quenching, 12-H) and the formed lithium bromide pose no difficulty in the separation from the product (Fig. 4). Acid–base extraction and column chromatography were both suitable means to achieve purification.
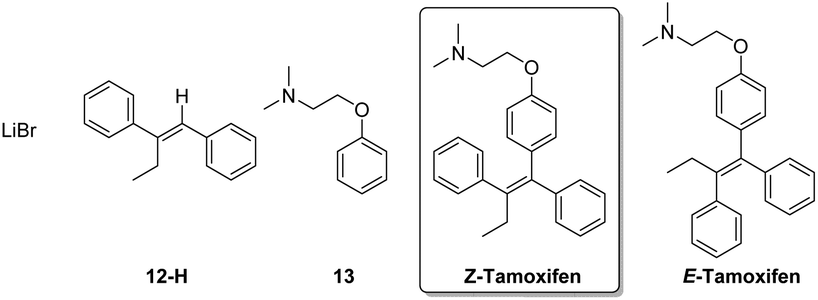 | ||
| Fig. 4 (Z)-Tamoxifen and side products. | ||
The remaining impurity of dehalogenated starting material 13 exhibits near identical behavior compared to the product, but flash chromatography was able to yield the pure (E/Z)-tamoxifen mixture. If desired, RP-preparative HPLC in water/acetonitrile/TFA effectively separates the (E) from the (Z) isomer of the product.
In conclusion, the carbolithiation of diphenylacetylene and the consecutive cross-coupling with the appropriate 4-bromo-dimethylamine-ethylether (6) yields (Z)-tamoxifen with a good (Z/E) selectivity (10:1) and with yields up to 65%. The reaction mixture was purified by flash chromatography to obtain the pure (E/Z)-tamoxifen mixture. Further optimization could lead to a lowering of the catalyst loading and suppression of the lithium halogen exchange or E–Z isomerisation that lead to the undesired side products which have proven to be a challenge in the purification of this pharmaceutical. The method distinguishes itself from previously reported syntheses by its high atom economy, reaction mass efficiency, non-toxic waste production, step count and ease of reaction setup. The organolithium cross-coupling is also an attractive strategy for the coupling of less reactive electrophiles (chlorides, fluorides and ethers)5j and future studies might further enhance the efficiency towards triarylethylenes and related products.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
B. L. F. acknowledges financial support from the Netherlands Organisation for Scientific Research, the European Research Council (ERC Advanced Grant 227897), the Royal Netherland Academy of Arts and Sciences (KNAW, Gravitation program 024.601035), and the Ministry of Education, Culture and Science.References
- (a) X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047–9050 CrossRef CAS PubMed; (b) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (c) J. J. Li and E. J. Corey, Total Synthesis of Natural Products: At the Frontiers of Organic Chemistry, Springer, Heidelberg, 2012 CrossRef; (d) L. Guo, C.-C. Hsiao, H. Yue, X. Liu and M. Rueping, ACS Catal., 2016, 6, 4438–4442 CrossRef CAS; (e) M. Tobisu, T. Takahira, T. Morioka and N. Chatani, J. Am. Chem. Soc., 2016, 138, 6711–6714 CrossRef CAS PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, New York, 1998 CrossRef; (c) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed; (d) C. E. I. Knappke and A. J. von Wangelin, Chem. Soc. Rev., 2011, 40, 4948–4962 RSC.
- E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- E. B. Pinxterhuis, M. Giannerini, V. Hornillos and B. L. Feringa, Nat. Commun., 2016, 7, 11698 CrossRef PubMed.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (b) F. Leroux, M. Schlosser, E. Zohar and I. Marek, The Preparation of Organolithium Reagents and Intermediates in Patai's Chemistry of Functional Groups, 2009 Search PubMed; (c) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (d) V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Org. Lett., 2013, 15, 5114–5117 CrossRef CAS PubMed; (e) R. Luisi and V. Capriati, Lithium Compounds in Organic Synthesis, Wiley VCH, Weinheim, 2014 CrossRef; (f) M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667–672 CrossRef CAS PubMed; (g) L. M. Castelló, V. Hornillos, C. Vila, M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Org. Lett., 2015, 17, 62–65 CrossRef PubMed; (h) M. Aufiero, T. Scattolin, F. Proutiere and F. Schoenebeck, Organometallics, 2015, 34, 5191–5195 CrossRef CAS; (i) J. Buter, D. Heijnen, C. Vila, V. Hornillos, E. Otten, M. Giannerini, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 3620–3624 CrossRef CAS PubMed; (j) D. Heijnen, J.-B. Gualtierotti, V. Hornillos and B. L. Feringa, Chem. – Eur. J., 2016, 22, 3991–3995 CrossRef CAS PubMed; (k) D. Heijnen, F. Tosi, C. Vila, M. C. A. Stuart, P. H. Elsinga, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 3354–3359 CrossRef CAS PubMed; (l) M. Busch, M. D. Wodrich and C. Corminboeuf, ACS Catal., 2017, 7, 5643–5653 CrossRef CAS.
- (a) N. F. McKinley and D. F. O'Shea, J. Org. Chem., 2006, 71, 9552–9555 CrossRef CAS PubMed; (b) S. Murahashi, M. Yamamura, K. Yanagisawa, N. Mita and K. Kondo, J. Org. Chem., 1979, 44, 2408–2417 CrossRef CAS.
- C. Fressigné, A.-L. Girard, M. Durandetti and J. Maddaluno, Angew. Chem., Int. Ed., 2008, 47, 891–893 CrossRef PubMed.
- G. Wu, F. E. Cederbaum and E. Negishi, Tetrahedron Lett., 1990, 31, 493–496 CrossRef CAS.
- C. Fressigné, R. Lhermet, A.-L. Girard, M. Durandetti and J. Maddaluno, J. Org. Chem., 2013, 78, 9659–9669 CrossRef PubMed.
- E. Shirakawa, D. Ikeda, T. Ozawa, S. Watanabe and T. Hayashi, Chem. Commun., 2009, 1885–1887 RSC.
- (a) R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC; (b) B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 CrossRef CAS PubMed.
- (a) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS; (b) A. P. Dicks and A. Hent, Green Chemistry Metrics: a Guide to Determining and Evaluating Process Greenness, Springer, 2015 Search PubMed.
- C. Avendano and C. Menendez, Medicinal Chemistry of Anticancer Drugs, Elsevier Science, 2015 Search PubMed.
- For further optimization studies, see the Experimental section.
- K. Itami, T. Kamei and J. Yoshida, J. Am. Chem. Soc., 2003, 125, 14670–14671 CrossRef CAS PubMed.
- P. E. Tessier, A. J. Penwell, F. E. S. Souza and A. G. Fallis, Org. Lett., 2003, 5, 2989–2992 CrossRef CAS PubMed.
- R. K. Pandey, R. D. Wakharkar and P. Kumar, Synth. Commun., 2005, 35, 2795–2800 CrossRef CAS.
- G. Cahiez, A. Moyeux and M. Poizat, Chem. Commun., 2014, 50, 8982–8984 RSC.
- P. R. D. Murray, D. L. Browne, J. C. Pastre, C. Butters, D. Guthrie and S. V. Ley, Org. Process Res. Dev., 2013, 17, 1192–1208 CrossRef CAS.
- T. Stiidemann and P. Knochel, Angew. Chem., Int. Ed. Engl., 1997, 36, 93–95 CrossRef.
- P. L. Coe and C. E. Scriven, J. Chem. Soc., Perkin Trans. 1, 1986, 475–477 RSC.
- R. B. Miller and M. I. Al-hassan, J. Org. Chem., 1985, 50, 2121–2123 CrossRef CAS.
- S. D. Brown and R. W. Armstrong, J. Org. Chem., 1997, 62, 7076–7077 CrossRef CAS PubMed.
- D. W. Robertson and J. A. Katzenellenbogen, J. Org. Chem., 1982, 47, 2387–2393 CrossRef CAS.
- M. Shimizu, C. Nakamaki, K. Shimono, M. Schelper, T. Kurahashi and T. Hiyama, J. Am. Chem. Soc., 2005, 127, 12506–12507 CrossRef CAS PubMed.
- J. Chen, S. Chen, X. Xu, Z. Tang, C.-T. Au and R. Qiu, J. Org. Chem., 2016, 81, 3246–3255 CrossRef CAS PubMed.
- K. Matsumoto and M. Shindo, Adv. Synth. Catal., 2012, 354, 642–650 CrossRef CAS.
- I. Shiina, M. Suzuki and K. Yokoyama, Tetrahedron Lett., 2004, 45, 965–967 CrossRef CAS.
- I. Shiina, Y. Sano, K. Nakata, M. Suzuki, T. Yokoyama, A. Sasaki, T. Orikasa, T. Miyamoto, M. Ikekita, Y. Nagahara and Y. Hasome, Bioorg. Med. Chem., 2007, 15, 7599–7617 CrossRef CAS.
- K. Shimizu, M. Takimoto, M. Mori and Y. Sato, Synlett, 2006, 3182–3184 CAS.
- Y. Takemoto, H. Yoshida and K. Takaki, Chem. – Eur. J., 2012, 18, 14841–14844 CrossRef CAS PubMed.
- Y. Zhou, W. You, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 3475–3479 CrossRef CAS PubMed.
- M. Pichette Drapeau, I. Fabre, L. Grimaud, I. Ciofini, T. Ollevier and M. Taillefer, Angew. Chem., Int. Ed., 2015, 54, 10587–10591 CrossRef CAS PubMed.
- Y. Nishihara, M. Miyasaka, M. Okamoto, H. Takahashi, E. Inoue, K. Tanemura and K. Takagi, J. Am. Chem. Soc., 2007, 129, 12634–12635 CrossRef CAS PubMed.
- C. Zhou, D. E. Emrich and R. C. Larock, Org. Lett., 2003, 5, 1579–1582 CrossRef CAS PubMed.
- F. Xue, J. Zhao, T. S. A. Hor and T. Hayashi, J. Am. Chem. Soc., 2015, 137, 3189–3192 CrossRef CAS PubMed.
- C. Zhou and R. C. Larock, J. Org. Chem., 2005, 70, 3765–3777 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02977f |
| ‡ These authors made equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2019 |