
The expanding spectrum of diketopiperazine natural product biosynthetic pathways containing cyclodipeptide synthases
 *a
*a
Abstract
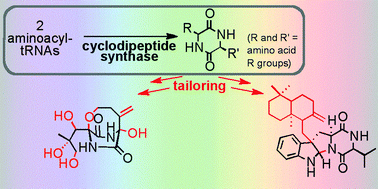
Microorganisms are remarkable chemists, with enzymes as their tools for executing multi-step syntheses to yield myriad natural products. Microbial synthetic aptitudes are illustrated by the structurally diverse 2,5-diketopiperazine (DKP) family of bioactive nonribosomal peptide natural products. Nonribosomal peptide synthetases (NRPSs) have long been recognized as catalysts for formation of DKP scaffolds from two amino acid substrates. Cyclodipeptide synthases (CDPSs) are more recently recognized catalysts of DKP assembly, employing two aminoacyl-tRNAs (aa-tRNAs) as substrates. CDPS-encoding genes are typically found in genomic neighbourhoods with genes encoding additional biosynthetic enzymes. These include oxidoreductases, cytochrome P450s, prenyltransferases, methyltransferases, and cyclases, which equip the DKP scaffold with groups that diversify chemical structures and confer biological activity. These tailoring enzymes have been characterized from nine CDPS-containing biosynthetic pathways to date, including four during the last year. In this review, we highlight these nine DKP pathways, emphasizing recently characterized tailoring reactions and connecting new developments to earlier findings. Featured pathways encompass a broad spectrum of chemistry, including the formation of challenging C–C and C–O bonds, regioselective methylation, a unique indole alkaloid DKP prenylation strategy, and unprecedented peptide-nucleobase bond formation. These CDPS-containing pathways also provide intriguing models of metabolic pathway evolution across related and divergent microorganisms, and open doors to synthetic biology approaches for generation of DKP combinatorial libraries. Further, bioinformatics analyses support that much unique genetically encoded DKP tailoring potential remains unexplored, suggesting opportunities for further expansion of Nature's biosynthetic spectrum. Together, recent studies of DKP pathways demonstrate the chemical ingenuity of microorganisms, highlight the wealth of unique enzymology provided by bacterial biosynthetic pathways, and suggest an abundance of untapped biosynthetic potential for future exploration.
- This article is part of the themed collections: Chemical Biology in OBC and Biosynthesis
 Please wait while we load your content...
Please wait while we load your content...

