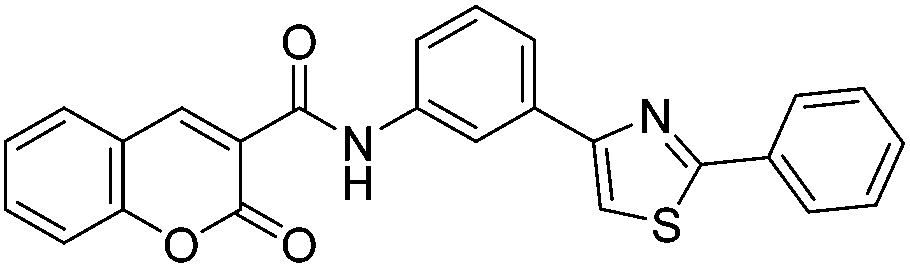
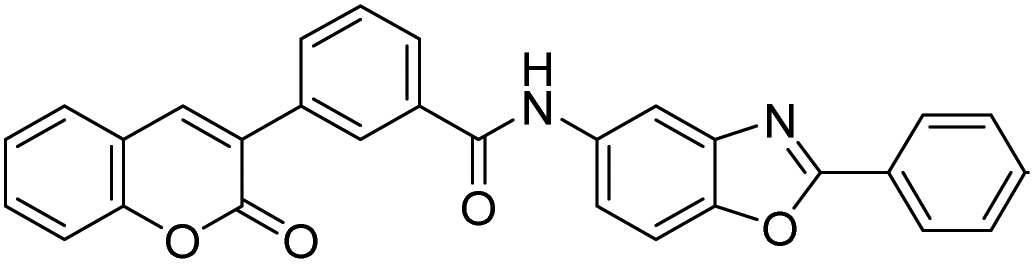
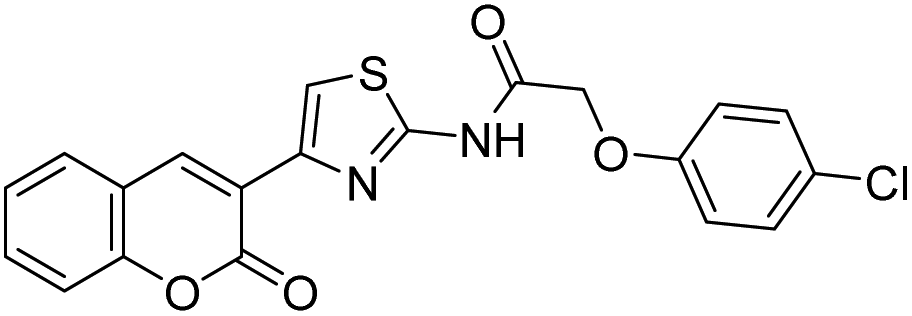
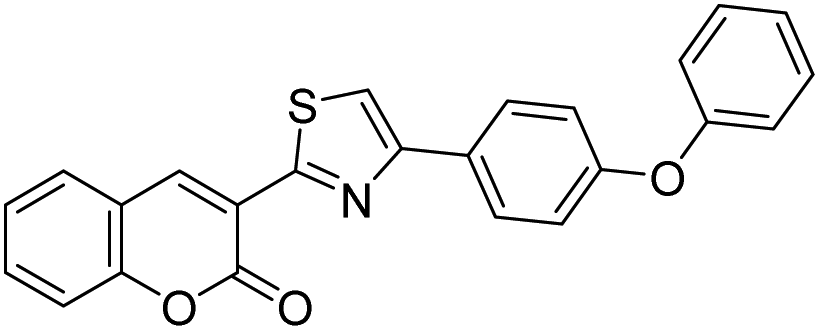
Discovery of fluorescent 3-heteroarylcoumarin derivatives as novel inhibitors of anaplastic lymphoma kinase†
Received
17th November 2018
, Accepted 5th December 2018
First published on 6th December 2018
Abstract
Altered expression or hyperactivation of anaplastic lymphoma kinase (ALK), as a consequence of translocations or point mutations, is one of the main oncogenic drivers in non-small cell lung cancer. Using structure-based design and in vitro enzyme assays, we identified 3-heteroarylcoumarin as a new template for the development of novel fluorescent ALK inhibitors. Molecular simulation provided structural insights for the design of 3-heteroarylcoumarin derivatives, which were easily prepared through efficient synthetic approaches including direct C–H cross coupling. Importantly, these coumarin-based ALK inhibitors can be tracked using microscopy techniques: we illustrated the use of the most potent compound in this series, 5a, (ALK/IC50 = 0.51 μM, λemi = 500 nm, ϕF = 0.29) to monitor its subcellular distribution pattern by confocal fluorescence microscopy.
Introduction
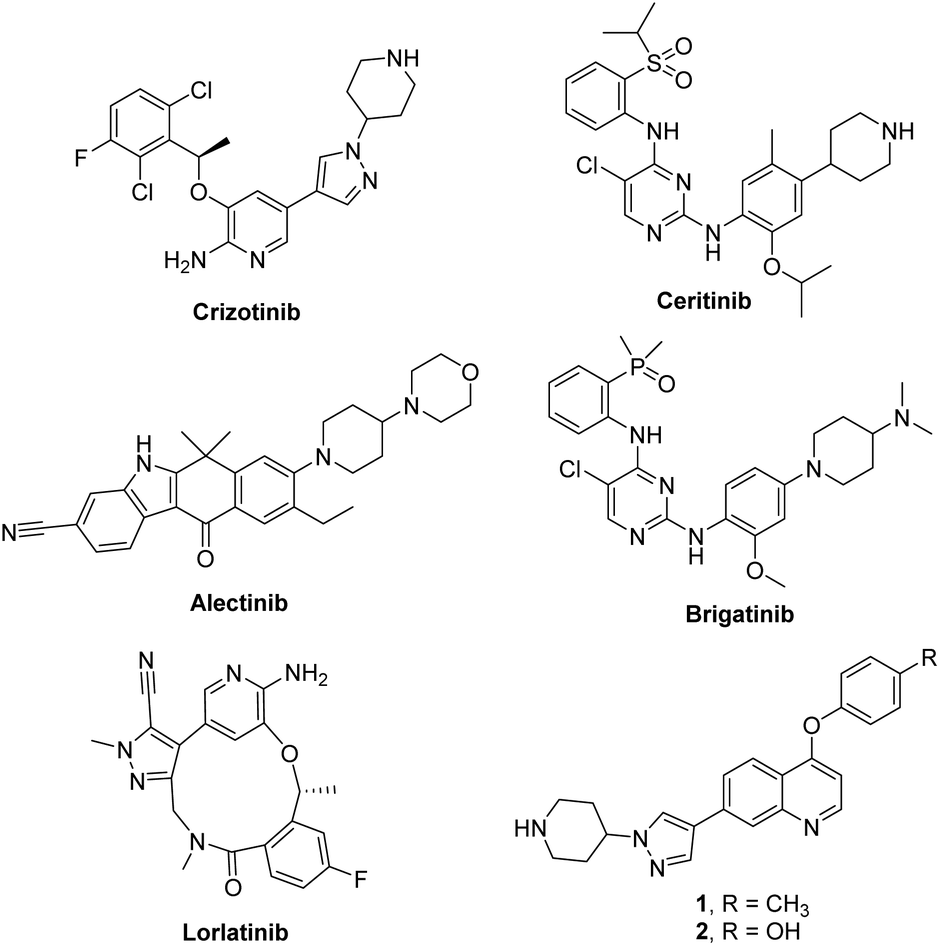
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase that belongs to the insulin receptor superfamily and normally plays a critical role in the development and maintenance of the nervous system.1 Constitutive activation of ALK mainly caused by translocations or point mutations has been highly associated with various human cancers such as anaplastic large cell lymphoma (ALCL),2 non-small cell lung cancer (NSCLC),3 squamous cell carcinoma,4 and diffuse large B-cell lymphoma.5 In particular, an oncogenic EML4-ALK protein, generated from the fusion of the N-terminal part of echinoderm microtubule-associated protein-like-4 (EML4) and the kinase domain of ALK, has been intensively studied as a therapeutic target in NSCLC. Accordingly, numerous ALK inhibitors have been reported as recently reviewed in a comprehensive fashion,6,7 and five ALK inhibitors including crizotinib, ceritinib, alectinib, brigatinib and Lorlatinib were approved by the US FDA (Fig. 1).8,9 Patients have shown excellent response rates to these ALK inhibitors; however, they inevitably relapse due to the emergence of resistance.10–14 Therefore, identification of structurally diverse ALK inhibitors is important for providing novel therapeutic options for patients harboring oncogenic ALK.15 Recently, our group reported new 4-phenoxyquinoline-based inhibitors targeting ALK WT and the L1196M mutant through a structure-based design strategy (1 and 2, Fig. 1).16
 |
| | Fig. 1 Chemical structures of representative ALK inhibitors. | |
Understanding of intracellular drug behavior such as cell membrane penetration, distribution in cells, and target engagement in biological systems is of enormous value. A tagged drug conjugated with a fluorescent dye can be broadly used to monitor the time course of drug distribution and its fate, and study biological processes in cells, animals, and humans using a noninvasive and nonradioactive procedure.17–20 However, tethering an additional fluorescent dye in the drug molecule often causes negative effects on binding affinity, cell permeability, toxicity, and in vivo activity. For this reason, inherently autofluorescent drugs are more beneficial than fluorophore-conjugated compounds and can be considered as an ideal species for tracking their translocation and disposition in cells.21–28 Coumarins constitute a major class of naturally occurring compounds, and various synthetic derivatives exhibit a broad range of biological activities as well as photophysical properties.29–34 Therefore, significant efforts have been directed toward constructing and designing a coumarin structural motif having valuable optical properties and biological activities.35–40 In this study, we aim to identify novel fluorescent ALK inhibitors in which the coumarin fluorophore was modified to be a pharmacophore capable of inhibiting ALK. Herein, we report studies on the identification of new coumarin-based fluorescent ALK inhibitors and demonstrate visualization of subcellular localization in living cells by fluorescence microscopy.
Results and discussion
Identification of 3-heteroarylcoumarin derivatives as fluorescent ALK inhibitors
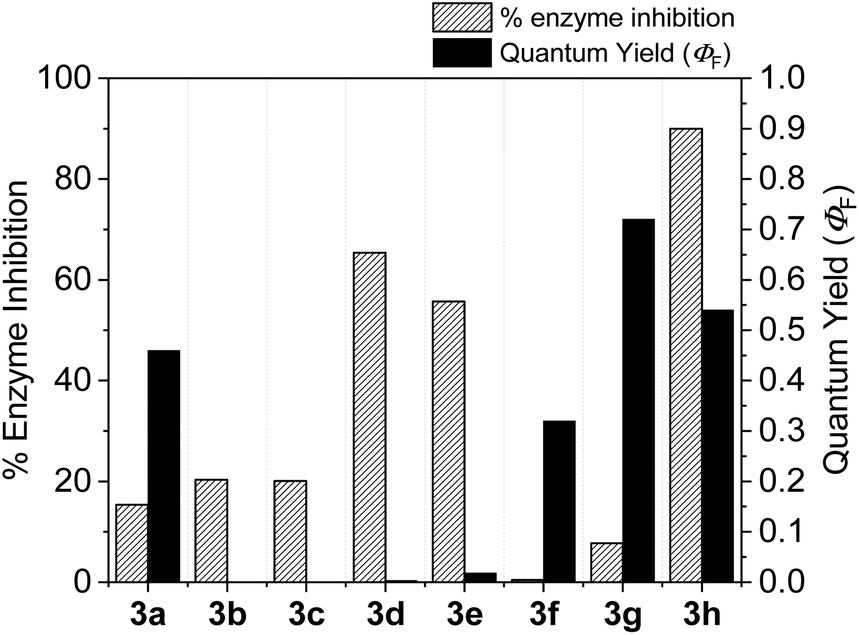
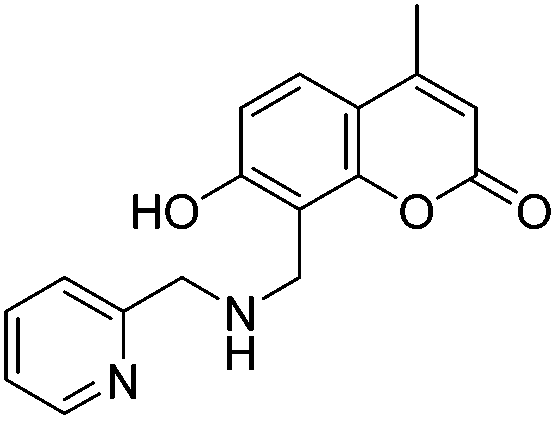
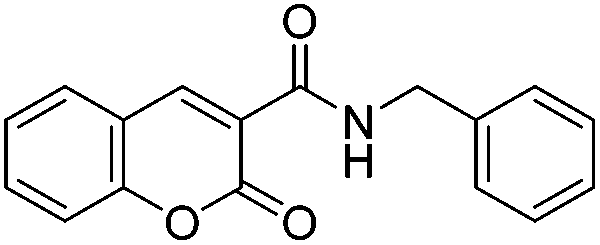
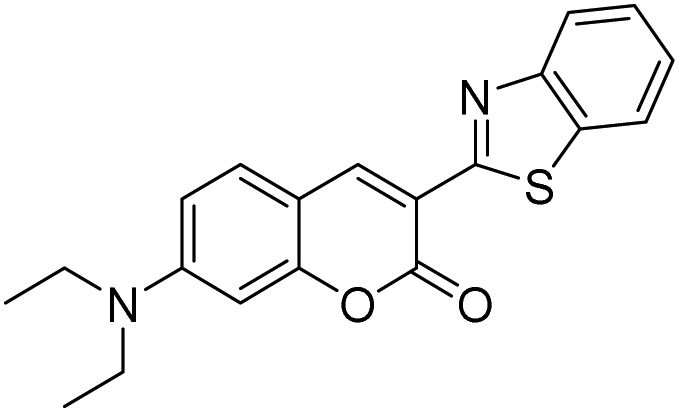
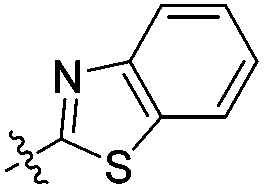
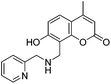
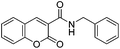
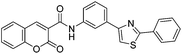
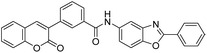
With the goal of developing novel fluorescent ALK inhibitors, we initiated our study by testing fluorophore derivatives in our in-house coumarin-based library which was constructed by the direct cross-coupling of a coumarin core and various heteroarenes.30 To identify the suitable structures having both inhibitory activity and fluorescence, percent enzyme activity at 100 μM against ALK and photophysical properties were examined as a primary screen, summarized in Fig. 2 and Table 1. Among these coumarin derivatives that exhibited a broad emission range over 400–600 nm, 4-methylcoumarin derivative 3a showed desirable fluorescent efficiency but weak ALK inhibitory activity (ϕF = 0.46, enzyme inhibition = 15%). Compared to the 4,7,8-substituents (3a), substitution of an amide or phenyl group at the C3-position of coumarins led to weakening of optical properties (3b–3d), albeit 3-phenylcoumarin derivative 3d displayed enhanced ALK inhibition. A comparison of 3d with 3e–3h, the C3 thiazole or benzothiazole substituted analogs, showed that installation of these groups induces a red-shift in the emission wavelength and a significant increase in quantum yield (3f: ϕF = 0.32, 3g: ϕF = 0.72, 3h: ϕF = 0.72). Furthermore, the addition of 7-alkoxy or 7-amino substituents influences the pathway of radiative decay, resulting in an increased value of quantum yield.41 It is remarkable to note that the installation of the 7-amino substituent improved the inhibitory activities with respect to ALK. Because 3h displayed promising potency against ALK and good photophysical properties among tested coumarin analogs, 3h was selected as a promising starting point for the development of new fluorescent ALK inhibitors.
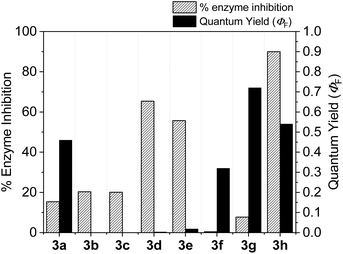 |
| | Fig. 2 Percent enzyme activity at 100 μM and fluorescence quantum yield of coumarin derivatives. | |
Table 1 Structures and absorption/excitation wavelength of coumarin derivatives
| Compd |
Structure |
λ
abs
(nm) |
λ
emi
(nm) |
|
Only the longest absorption maxima are shown.
Excited at the maximum excitation wavelength.
N.D. = not detected.
|
|
3a
|
|
324 |
435 |
|
3b
|
|
295 |
N.D.c |
|
3c
|
|
256 |
N.D.c |
|
3d
|
|
326 |
403 |
|
3e
|
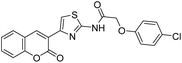
|
305 |
460 |
|
3f
|
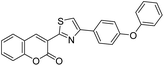
|
383 |
538 |
|
3g
|
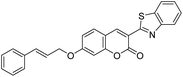
|
394 |
469 |
|
3h
|
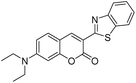
|
446 |
494 |
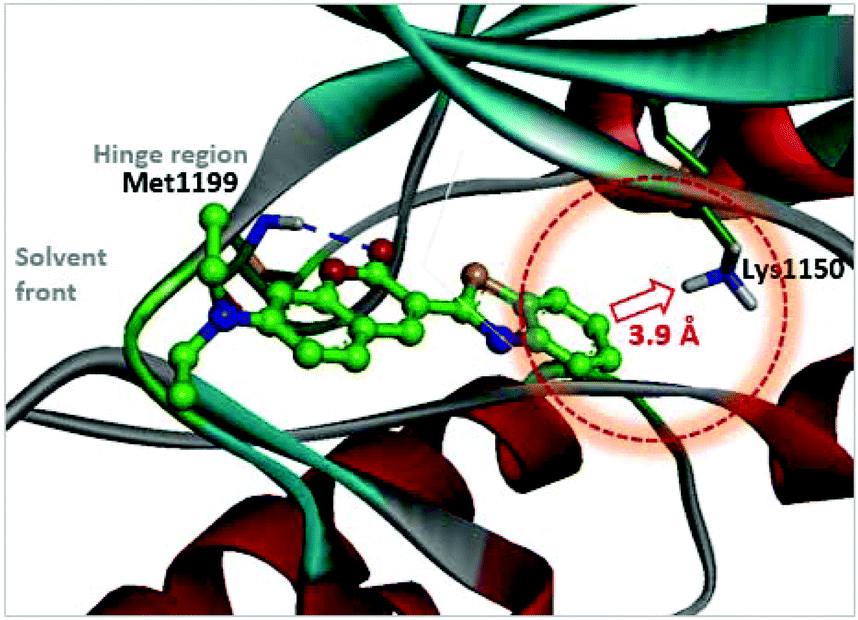
As a strategy to increase potency, we initially explored the C3 substituent space on the coumarin and designed several 3-heteroarylcoumarin derivatives. To gain structural insight into the inhibitory activities, the interaction patterns of coumarins in the ATP-binding sites of ALK were investigated in a comparative fashion. Fig. 3 shows the lowest energy conformation of 3h in the ATP binding pocket of ALK (PDB ID: 2XP2)42 as calculated with the Discovery Studio 4.5 software. Molecular simulation of the 3h-ALK complex revealed that the carbonyl oxygen of the coumarin core establishes a hydrogen bond with the backbone amide group of a residue (Met1199) in the hinge region of ALK. As a check for the presence of a peripheral binding pocket nearby a catalytic lysine, the calculated binding modes in this site were analyzed in detail. Because Lys1150 is located adjacent to the 6-position of the benzothiazole subunit, maintaining a distance within 3.9 Å, we hypothesized that the introduction of a suitable hydrogen bond acceptor at the C6-position of benzothiazole might improve the overall ALK inhibitory activity. To assess this hypothesis, we installed a methoxy group into this position (4d), which led to a slight enhancement of inhibitory activity against ALK (3h, IC50 = 2.63 μM; 4d, IC50 = 1.60 μM), indicating that this enhancement of potency most likely stemmed from the strengthening of hydrogen bonds between the inhibitor and Lys1150.
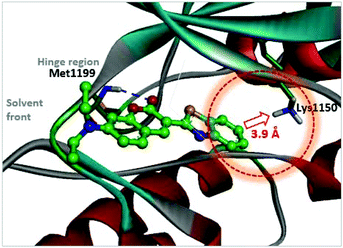 |
| | Fig. 3 Calculated binding mode of 3h in the ATP binding pocket of ALK (PDB ID: 2XP2). The blue broken line indicates a hydrogen bond. A red dotted circle shows the proximity of the C6-position of benzothiazole to Lys1150. | |
Optimization of ALK inhibitory activity via structure-based drug design
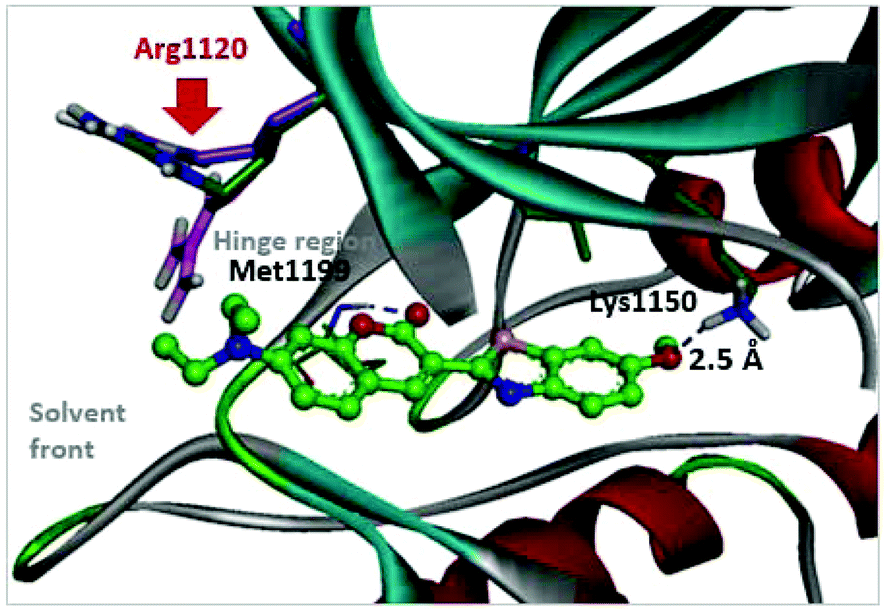
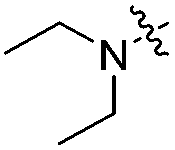
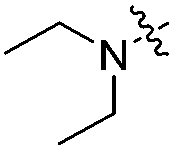
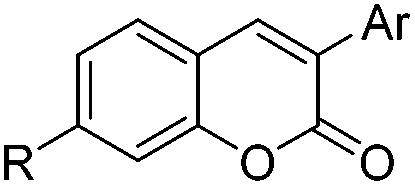
With respect to improving the inhibitory activity, we investigated the effect of 3-heteroaryl derivatives and substituents attached to the C7-position. For the structure–activity relationship (SAR) analysis, a survey of C3-benzothiazole analogs was conducted and IC50 values were determined. Conversion of benzothiazole (3h) into benzoxazole (4a) showed a subtle effect on inhibitory activity but a slightly negative effect on fluorescence. Substitution of the phenyl group at the C6-position of benzothiazole (4b, IC50 = 70.9 μM) was detrimental to the potency compared with the parent molecule (3h, IC50 = 2.63 μM). A caffeine-based analog (4c), which also provides intense photonic luminescence, exhibited weak ALK inhibitory activity with an IC50 value of 39.2 μM. The negative results of 4b and 4c could be due to steric hindrance between the inhibitor and amino acid residues in the ALK active site. To improve the ALK inhibitory activity of 4d, we further analyzed the structural features of the neighboring amino acids in the solvent front of the ATP binding site. The calculated binding mode of 4d (Fig. 4) revealed that the 7-diethylamino group of the coumarin extends toward the solvent-exposed region and is surrounded by several amino acid residues. For example, Arg1120 is located at the upper lip of the ATP binding pocket and stretches out into the solvent exposed exterior. Interestingly, the side chain of Arg1120 shows conformational variation in several X-ray crystal structures of the ALK kinase domain: the downstretched mode of Arg1120 (Fig. 4)43,44 could cause a putative steric clash with the rotatable diethylamine group. Based on our structural analysis, the diethylamine group was replaced with cyclic functionalities to evaluate the substituent space of the C7 position of the coumarin.
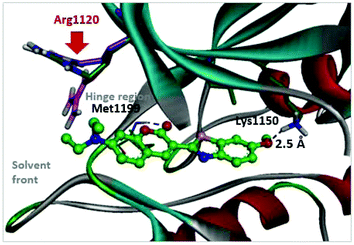 |
| | Fig. 4 Predicted binding mode of compound 4d in the ATP binding pocket of ALK (PDB ID: 2XP2). Two possible locations of the Arg1120 side chain reported in PDB 3AOX43 were superimposed. | |
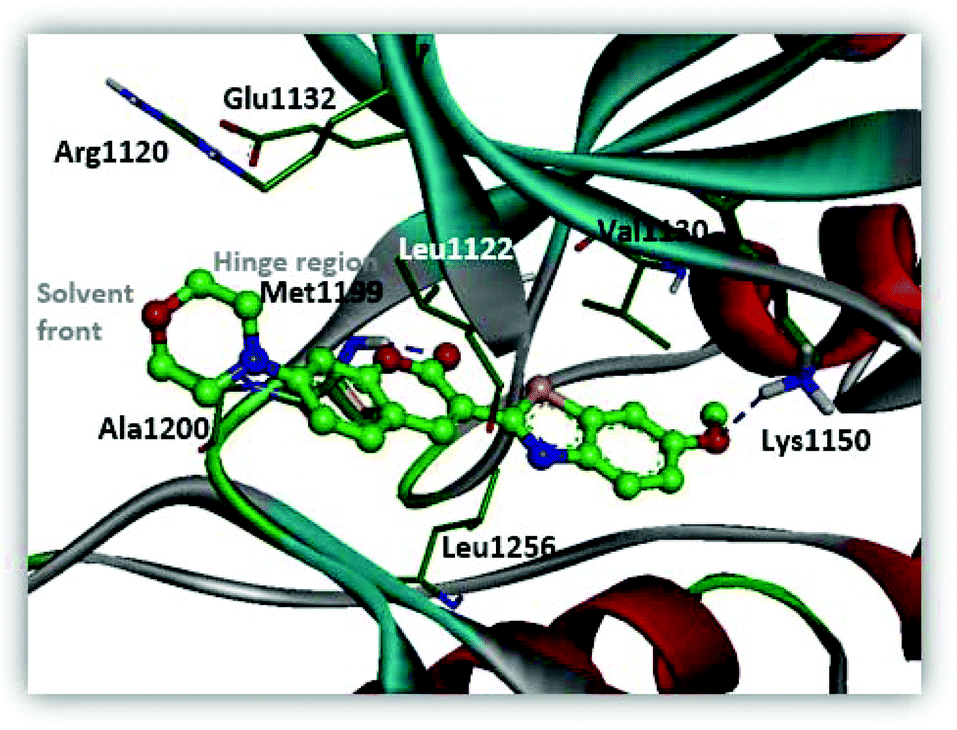
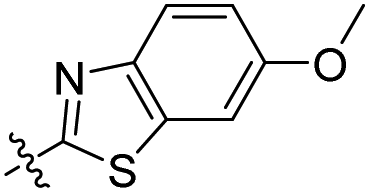
The installation of the morpholine or piperazine moieties was intended to improve the aqueous solubility of these derivatives, which is required as a probe for real-time monitoring of the cellular distribution. Of particular significance is the observation that the substitution with the morpholine group yielded more potent inhibitor 5a with submicromolar activity (5a, IC50 = 0.51 μM). According to the molecular docking study as depicted in Fig. 5, 5a appears to be stabilized by multiple hydrophobic interactions with various residues including Leu1122, Val1130, and Leu1256. The improved binding affinity can be rationalized by additional hydrophobic interaction of the cyclic fragment and Ala1200, Arg1120 and Glu1132 that stabilizes ligand binding. Moreover, compound 5a showed promising fluorescence emission (5a, λemi = 500 nm, ϕF = 0.29) and Stokes shift (Δλ ≥ 70 nm), which can be beneficial as a biological imaging study tool.21 The enzymatic inhibitory activity of the derivatives was decreased when more sterically hindered heterocycles such as the N-methyl or N-Boc piperazine moiety were introduced (5b, IC50 = 4.1 μM; 5c, IC50 = 8.8 μM), indicating that the morpholine group is most suitable for this position in the benzothiazole core. Replacement of the morpholine with a methoxy group resulted in reduction of inhibitory activity against ALK (6a, IC50 = 1.75 μM). The binding energy of 6a in docking simulation was much lower than that of 5a (5a, -CDocker energy = 21.8214; 6a, -CDocker energy = 17.6135, calculated by using Discovery Studio 4.5), revealing that loss of van der Waals interaction between morpholine and a small pocket located in the solvent front inevitably culminates in the weakening of the binding affinity of 6a. Next, importance of the hydrogen bond network in 6-methoxybenzothiazole was further demonstrated by removal of the methoxy group (6b, IC50 = 22.1 μM). Investigation of alternate moieties such as the 3-methoxyphenyl-thiazole group revealed that 6-methoxybenzothiazole was optimal for this series, which is in turn responsible for weaker biochemical potency of 6c (IC50 > 100 μM). These results confirm the importance of forming a stable hydrogen bond of 6-methoxybenzothiazole with Lys1150 to enhance the inhibitory activity (Fig. 5).
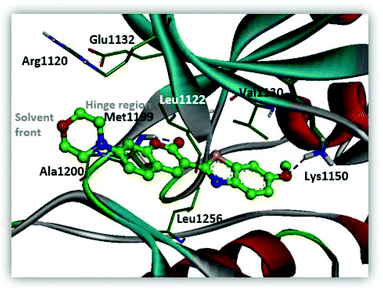 |
| | Fig. 5 Predicted binding mode of compound 5a in the ATP binding pocket of ALK (PDB ID: 2XP2). | |
Next, the representative compounds 5a and 6a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 were further tested against the most common secondary ALK mutants such as L1196M, G1269A and G1202R (Table 3). Compound 5a showed potent inhibitory activities against three ALK mutants (L1196M IC50 = 0.27 μM; G1269A IC50 = 0.30 μM; G1202R IC50 = 0.59 μM). Compound 6a displayed an IC50 value of 0.45 μM for L1196M and compatible enzymatic inhibitory activity against G1269A and G1202R with that against WT ALK.
45 were further tested against the most common secondary ALK mutants such as L1196M, G1269A and G1202R (Table 3). Compound 5a showed potent inhibitory activities against three ALK mutants (L1196M IC50 = 0.27 μM; G1269A IC50 = 0.30 μM; G1202R IC50 = 0.59 μM). Compound 6a displayed an IC50 value of 0.45 μM for L1196M and compatible enzymatic inhibitory activity against G1269A and G1202R with that against WT ALK.
Table 2 Structure–activity relationship information of 3-heteroarylcoumarin derivatives
Table 3 IC50 values of compounds against ALK WT and mutants
| Compd |
ALK mutant IC50a (μM) |
| WT |
L1196M |
G1269A |
G1202R |
|
The IC50 measurements were performed at the Reaction Biology Corp. (Malvern, PA, USA).
|
|
5a
|
0.51 |
0.27 |
0.30 |
0.59 |
|
6a
|
1.75 |
0.45 |
2.30 |
1.82 |
Monitoring molecular behavioral variability in cells by confocal microscopy
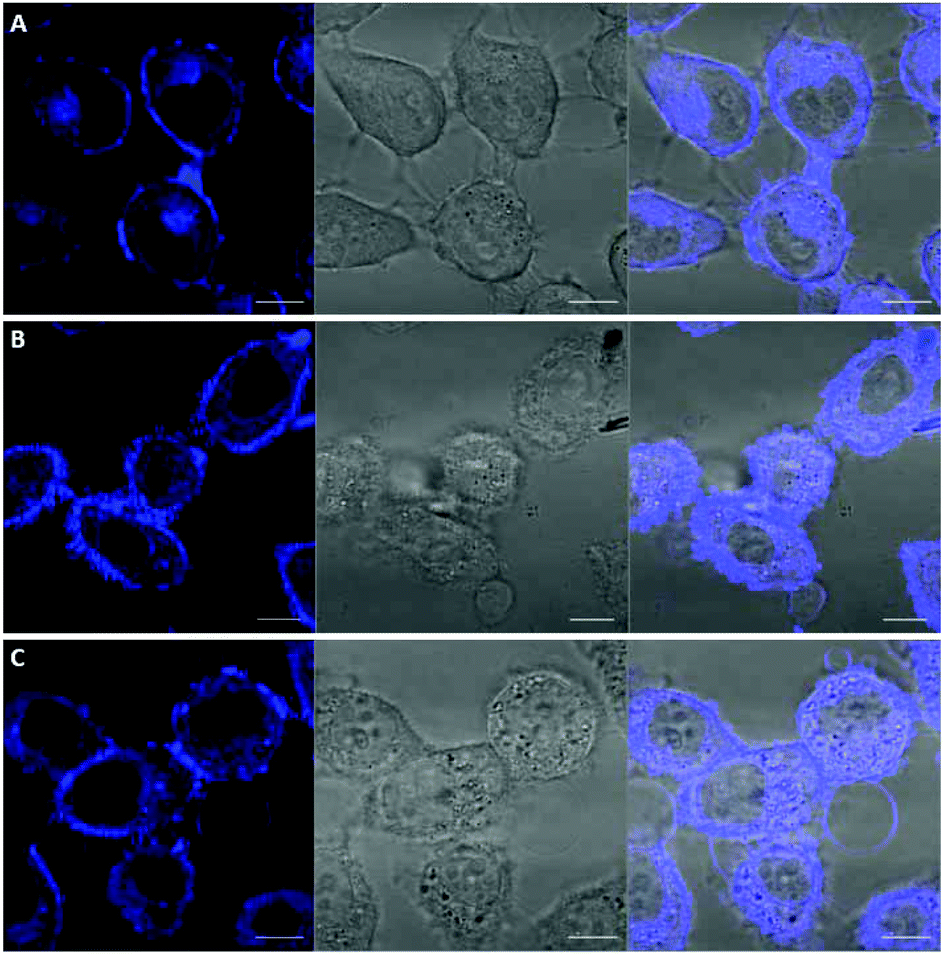
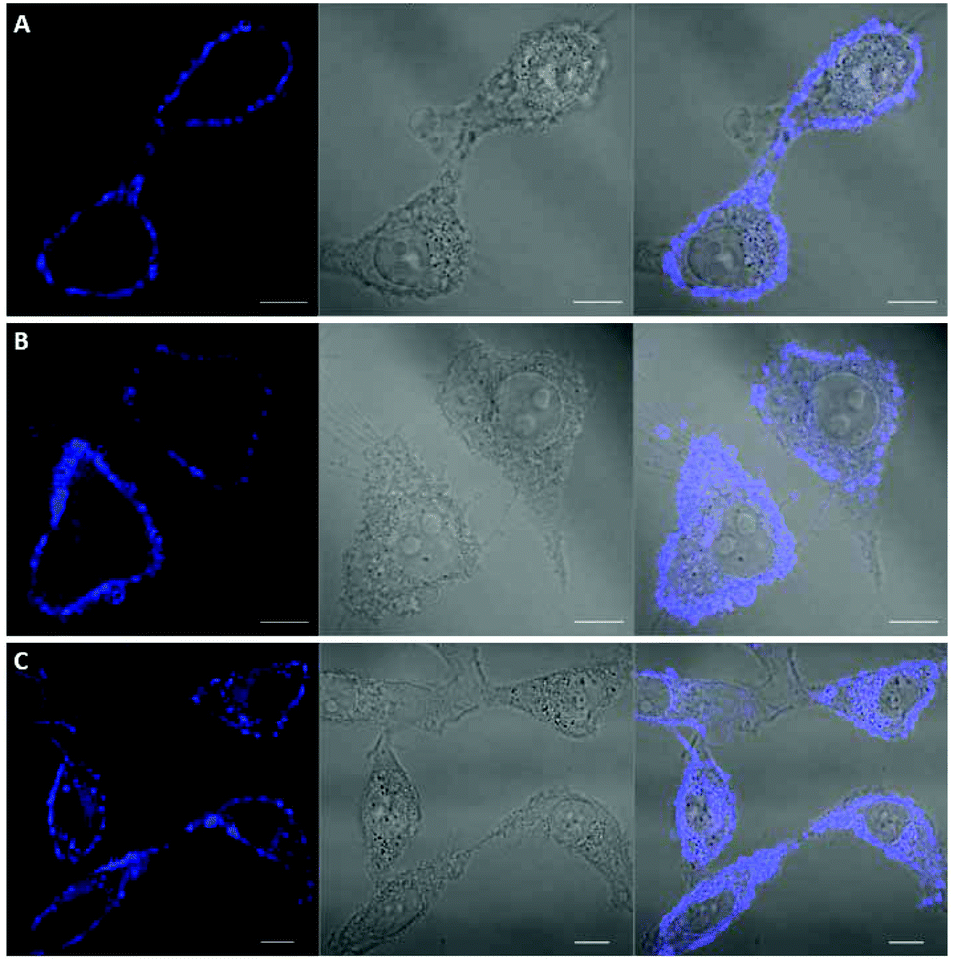
Among the fluorescent ALK inhibitors with desirable inhibitory activities, two leading compounds 5a and 6a were selected for further investigation of the imaging ability of their molecular behavior. Compound 5a seems to possess more favorable physicochemical properties than 6a based on calculated AlogP values (5a, ALogP = 3.70; 6a, ALogP = 3.84, predicted by using Discovery Studio 4.5 Client). In the present study, HeLa cells were used to monitor the general behavior of our fluorescent inhibitors such as cell penetration and cellular distribution. HeLa cells were incubated with 5a or 6a at 10 μM concentration for 1 h. Unbound compounds were washed, and the resulting cells were further incubated for 4 h and 12 h. After treatment of 5a for 1 h, visible fluorescence was clearly detected in both the plasma membrane and cytoplasm of HeLa cells, which was maintained even after 12 h incubation (Fig. 6). To our delight, we also observed the fluorescence in the membrane region that might be evidence of membrane penetration that provided the feasibility of monitoring a time-dependent morphology change of the membrane (Fig. 6B and C). On the other hand, 6a seems to be attached to the cell surface plasma membrane after 1 h treatment due to the high lipophilicity. Therefore, the cell membrane penetration of 6a appears to be inferior to that of 5a. When the fluorescence of 6a was monitored after 4 h or 12 h treatment, the compound was gradually distributed into the cytoplasm (Fig. 7B and C). These results provide a visible insight of molecular behavior such as cell membrane penetration and intracellular distribution of fluorescent compounds as well as a way to monitor the cell morphology changes. The present 5a can be used to not only functionally alter ALK activity but also monitor drug distributions, which are known to be important indications of various cancerous cells.
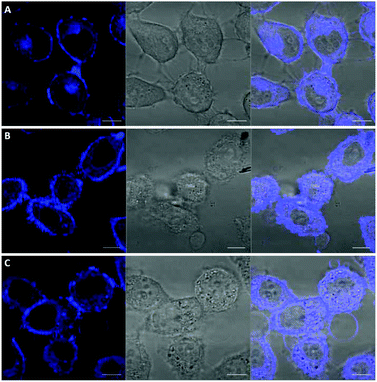 |
| | Fig. 6 Confocal imaging of the intracellular disposition of compound 5a in HeLa cells. Cells were incubated with a 10 μM solution of compound for 1 h. (A) Cells after 1 h upon treatment of 5a. (B) Cells after 4 h upon treatment of 5a. (C) Cells after 12 h upon treatment of 5a. Left: transmitted light image; middle: fluorescence microscopy image; right: overlaid image. Scale bar, 10 μm. | |
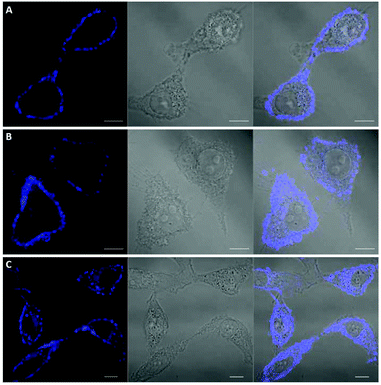 |
| | Fig. 7 Confocal imaging of the intracellular disposition of compound 6a in HeLa cells. Cells were incubated with 10 μM solution of compound for 1 h. (A) Cells after 1 h upon treatment of 6a. (B) Cells after 4 h upon treatment of 6a. (C) Cells after 12 h upon treatment of 6a. Left: transmitted light image; middle: fluorescence microscopy image; right: overlaid image. Scale bar, 10 μm. | |
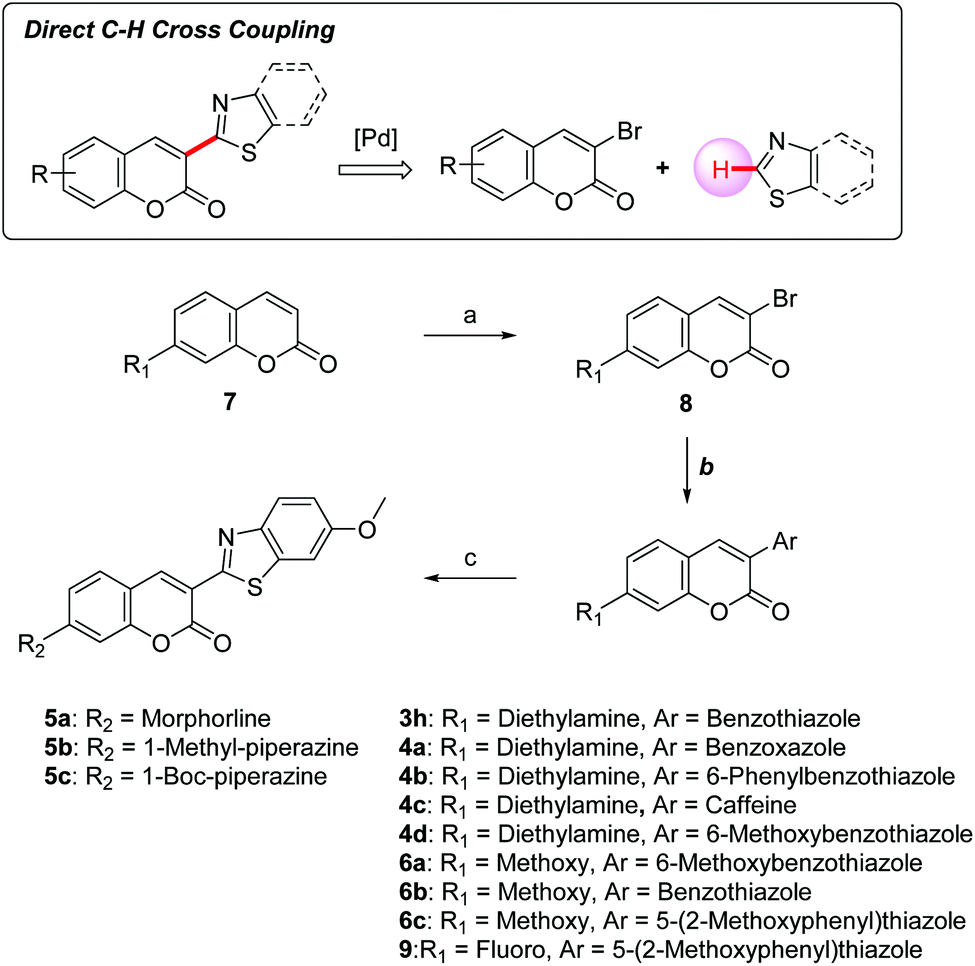
Synthesis of coumarin derivatives
Our group reported an efficient direct C–H cross-coupling approach to heteroaryl coumarins using a Pd/Cu catalytic system, which enabled the straightforward preparation of 3-heteroarylcoumarin derivatives.30 The focused compound library was established by this synthetic method allowing for rapid exploration of the SAR profile of 3-heteroarylcoumarin derivatives (Scheme 1). To install heteroaromatic rings, the C3 position of coumarins 7 was first brominated using a mixture of oxone and hydrobromic acid in dichloromethane at room temperature, and then treating the product with Et3N. Subsequently, various heteroaryl groups were installed at the C3 position through a Pd-catalyzed direct cross-coupling of 3-bromocoumarins with heteroarenes to afford the desired products. For compounds bearing morpholine or piperazine rings at the C7 position, the 7-fluoro-coumarin intermediate 9 was further converted to target compounds (5a–5c) using the SNAr reactions with appropriate amines.
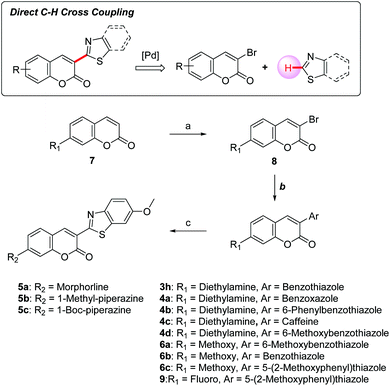 |
| | Scheme 1 Synthetic route for 3-heteroarylcoumarins. Reagents and conditions: (a) oxone, 2N HBr, CH2Cl2, rt, 2 h, then TEA, rt, 2 h; (b) heteroarenes, Pd(PPh3)4, CuI, Cs2CO3, 1,4-dioxane, 110 °C, 6 h; (c) DIPEA, acetonitrile, 90 °C, 12 h. | |
Conclusions
In summary, we discovered a family of coumarin-based ALK inhibitors serving a dual function in exhibiting fluorescence and inhibiting ALK. Guided by structure-based design and analysis of photophysical properties, we successfully identified compound 5a with potent activity and significant fluorescence properties (ALK IC50 = 0.51 μM, λemi = 500 nm, ϕF = 0.29). Cell imaging after treatment of 5a and 6a showed the insight of molecular behavior such as membrane penetration and intracellular distribution and provided a way to monitor cellular morphology changes such as deconstruction of the lipid bilayer. The combined features of newly discovered fluorescent ALK inhibitors would be useful in biological or pharmaceutical research enabling visualization of subcellular localization in living cells by fluorescence microscopy.
Experimental
General
Unless otherwise stated, reactions were performed in flame-dried glassware and monitored by TLC. Analytical thin layer chromatography (TLC) was performed on pre-coated silica gel 60 F254 plates and compounds were visualized by UV light (254 and 365 nm). Flash column chromatography was performed on silica gel (400–630 mesh) and ethyl acetate and n-hexane were used as eluents for product purification. 1H and 13C NMR spectra were recorded in CDCl3 or CD2Cl2 solution on a 400 or 600 MHz instrument. All chemical shifts were quoted in parts per million (ppm) referenced to the appropriate solvent peak or 0.0 ppm for tetramethylsilane. The following abbreviations were used to describe peak splitting patterns when appropriate: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, td = doublet of triplet, ddd = doublet of doublet of doublet. Coupling constant, J values were reported in hertz unit (Hz). Mass spectral data were obtained from KAIST Basic Science Institute by using the ESI method. Commercial grade reagents and solvents were used without further purification except as indicated below.
General procedure I (GP I) for 3-bromocoumarins
The coumarin derivative (1 equiv.) and oxone (1.2 equiv.) were dissolved in CH2Cl2 (0.25 M). 2 N HBr solution (HBr 2.2 equiv.) was added and the reaction mixture was stirred at room temperature. After 2 h, Et3N (3 equiv.) was added and stirred for additional 2 h. The reaction mixture was diluted with CH2Cl2 and water, and then the organic compounds were extracted by using CH2Cl2. The organic layer was dried over Na2SO4 and evaporated. The residue was purified by flash column chromatography on silica gel (hexanes:EtOAc = 6:1) to give the desired product.
General procedure II (GP II) for 3-heteroarylcoumarins
3-Bromocoumarin derivative (1 equiv.), heteroarene (2 equiv.), Pd(PPh3)4 (2.5 mol%), CuI (3 equiv.), and Cs2CO3 (2 equiv.) were combined in 1,4-dioxane under air. The reaction mixture was heated to 110 °C with stirring, and monitored by TLC using ethyl acetate and n-hexane as the mobile phase. After disappearance of the starting material, the reaction mixture was diluted and filtered through Celite with CH2Cl2. After removal of the solvent, the residue was purified by flash column chromatography on silica gel to provide the desired product.
General procedure III (GP III) for 7-substituted 3-heteroarylcoumarins
7-Fluoro-3-(6-methoxy-1,3-benzothiazol-2-yl)chromen-2-one (1 equiv.) was dissolved in acetonitrile at room temperature under air. To this solution, cyclic aliphatic amines (2 equiv.) and DIPEA (3 equiv.) were added. The reaction mixture was stirred at 90 °C for 12 h. After completion of the reaction, the mixture was cooled to rt. Then, the precipitate was collected by filtration with cold acetonitrile and water to give the desired product.
3-(1,3-Benzothiazol-2-yl)-7-(diethylamino)chromen-2-one (3h).
Compound 3h was prepared (yellowish orange solid, 8.0 mg, 2 steps – 21% overall yields) according to GP I and GP II. 1H NMR (600 MHz, chloroform-d) δ 8.92 (d, J = 1.9 Hz, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.49 (dd, J = 7.3, 4.1 Hz, 2H), 7.36 (t, J = 7.6 Hz, 1H), 6.68 (dt, J = 9.0, 2.2 Hz, 1H), 6.57 (d, J = 2.3 Hz, 1H), 3.47 (q, J = 7.4 Hz, 4H), 1.26 (t, J = 7.2 Hz, 6H). 13C NMR (150 MHz, chloroform-d) δ 161.8, 161.0, 157.0, 152.6, 152.1, 142.0, 136.3, 130.8, 126.0, 124.4, 122.1, 121.6, 112.5, 109.9, 108.7, 97.0, 45.1, 12.5. HRMS (ESI+) m/z calcd for C20H18N2NaO2S+ [M + Na]+: 373.0981, found: 373.0984.
3-(1,3-Benzoxazol-2-yl)-7-(diethylamino)chromen-2-one (4a).
Compound 4a was prepared (yellow solid, 8.4 mg, 2 steps – 24% overall yields) according to GP I and GP II. 1H NMR (600 MHz, methylene chloride-d2) δ 8.60 (s, 1H), 7.73 (dd, J = 5.9, 3.2 Hz, 1H), 7.59 (dd, J = 5.9, 3.3 Hz, 1H), 7.45 (d, J = 8.9 Hz, 1H), 7.34 (dd, J = 6.0, 3.2 Hz, 2H), 6.67 (dd, J = 8.9, 2.4 Hz, 1H), 6.53 (d, J = 2.3 Hz, 1H), 3.46 (q, J = 7.1 Hz, 4H), 1.23 (t, J = 7.2 Hz, 6H). 13C NMR (150 MHz, methylene chloride-d2) δ 160.1, 157.8, 157.6, 152.6, 150.4, 145.4, 142.1, 130.6, 124.8, 124.4, 119.7, 110.3, 110.0, 109.7, 108.1, 96.7, 45.1, 12.2. HRMS (ESI+) m/z calcd for C20H18N2NaO3+ [M + Na]+: 357.1210, found: 357.1219.
7-(Diethylamino)-3-(6-phenyl-1,3-benzothiazol-2-yl)chromen-2-one (4b).
Compound 4b was prepared (dark red solid, 8.1 mg, 2 steps – 18% overall yields) according to GP I and GP II. 1H NMR (600 MHz, methylene chloride-d2) δ 8.93 (s, 1H), 8.15 (d, J = 1.7 Hz, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.73 (dd, J = 8.6, 1.7 Hz, 1H), 7.71–7.65 (m, 2H), 7.52 (d, J = 8.9 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.36 (t, J = 7.4 Hz, 1H), 6.71 (dd, J = 8.9, 2.3 Hz, 1H), 6.57 (d, J = 2.3 Hz, 1H), 3.47 (q, J = 7.2 Hz, 4H), 1.24 (t, J = 7.1 Hz, 6H). 13C NMR (150 MHz, methylene chloride-d2) δ 162.0, 160.9, 157.1, 152.3, 151.9, 142.0, 140.7, 137.6, 136.9, 130.8, 128.8, 127.3, 127.2, 125.7, 122.0, 119.6, 112.1, 110.0, 108.6, 96.8, 45.1, 12.2. HRMS (ESI+) m/z calcd for C26H23N2O2S+ [M + H]+: 427.1475, found: 427.1488.
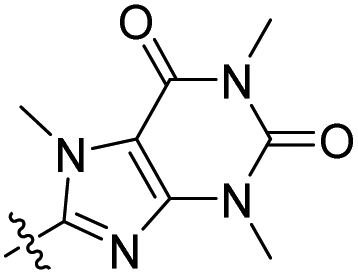
8-[7-(Diethylamino)-2-oxo-chromen-3-yl]-1,3,7-trimethyl-purine-2,6-dione (4c).
Compound 4c was prepared (yellow solid, 14.5 mg, 2 steps – 34% overall yields) according to GP I and GP II. 1H NMR (400 MHz, methylene chloride-d2) δ 8.02 (s, 1H), 7.39 (d, J = 8.9 Hz, 1H), 6.67 (dd, J = 8.9, 2.5 Hz, 1H), 6.54 (d, J = 2.4 Hz, 1H), 3.91 (s, 3H), 3.55 (s, 3H), 3.47 (q, J = 7.1 Hz, 4H), 3.36 (s, 3H), 1.24 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, methylene chloride-d2) δ 159.7, 158.0, 155.7, 152.6, 152.0, 148.6, 148.4, 147.7, 130.5, 110.0, 109.0, 108.4, 97.2, 45.4, 34.0, 29.8, 28.0, 12.6. HRMS (ESI+) m/z calcd for C21H23N5NaO4+ [M + Na]+: 432.1642, found: 432.1667.
7-(Diethylamino)-3-(6-methoxybenzo[d]thiazol-2-yl)-2H-chromen-2-one (4d).
Compound 4d was prepared (yellow solid, 9.1 mg, 2 steps – 17% overall yields) according to GP I and GP II. 1H NMR (400 MHz, methylene chloride-d2) δ 8.83 (s, 1H), 7.86 (d, J = 8.9 Hz, 1H), 7.50 (d, J = 8.9 Hz, 1H), 7.41 (d, J = 2.5 Hz, 1H), 7.08 (dd, J = 8.9 Hz, 2.5 Hz, 1H), 6.70 (dd, J = 8.9, 2.5 Hz, 1H), 6.57 (d, J = 2.4 Hz, 1H), 3.89 (s, 3H), 3.47 (q, J = 7.1 Hz, 4H), 1.25 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, methylene chloride-d2) δ 161.4, 159.6, 157.8, 157.3, 152.5, 147.7, 141.6, 138.1, 131.0, 123.1, 116.1, 112.9, 110.3, 108.9, 104.0, 97.2, 56.2, 45.4, 12.6. HRMS (ESI+) m/z calcd for C21H20N2NaO3S+ [M + Na]+: 403.1087, found: 403.1092.
3-(6-Methoxy-1,3-benzothiazol-2-yl)-7-morpholino-chromen-2-one (5a).
Compound 5a was prepared (yellowish orange solid, 15.0 mg, 3 steps – 34% overall yields) according to GP I, GP II and GP III. 1H NMR (400 MHz, chloroform-d) δ 8.89 (s, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.55 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 2.5 Hz, 1H), 7.12 (dd, J = 8.9, 2.5 Hz, 1H), 6.88 (dd, J = 8.8, 2.4 Hz, 1H), 6.78 (d, J = 2.3 Hz, 1H), 3.91 (s, 3H), 3.90–3.84 (m, 4H), 3.41–3.33 (m, 4H). 13C NMR (150 MHz, chloroform-d) δ 160.7, 158.5, 157.6, 156.1, 154.4, 147.2, 140.7, 138.0, 130.2, 123.0, 116.2, 115.4, 111.8, 111.0, 103.6, 100.1, 66.4, 55.8, 47.3. HRMS (ESI+) m/z calcd for C21H18N2NaO4S+ [M + Na]+: 417.0880, found: 417.0872.
3-(6-Methoxy-1,3-benzothiazol-2-yl)-7-(4-methylpiperazin-1-yl)chromen-2-one (5b).
Compound 5b was prepared (yellowish orange solid, 12.0 mg, 3 steps – 26% overall yields) according to GP I, GP II and GP III. 1H NMR (600 MHz, chloroform-d) δ 8.84 (d, J = 1.3 Hz, 1H), 7.91 (dd, J = 8.9, 1.3 Hz, 1H), 7.50 (dd, J = 8.9, 1.4 Hz, 1H), 7.38 (dd, J = 2.5, 1.4 Hz, 1H), 7.10 (ddd, J = 8.9, 2.6, 1.3 Hz, 1H), 6.86 (ddd, J = 8.9, 2.5, 1.4 Hz, 1H), 6.75 (t, J = 1.7 Hz, 1H), 3.90 (d, J = 1.4 Hz, 3H), 3.51–3.21 (m, 4H), 2.67–2.42 (m, 4H), 2.36 (d, J = 1.4 Hz, 3H). 13C NMR (150 MHz, chloroform-d) δ 160.8, 158.7, 157.5, 156.2, 154.3, 147.2, 140.8, 137.9, 130.2, 122.9, 116.1, 114.8, 112.0, 110.5, 103.6, 100.0, 55.8, 54.5, 47.1, 46.1. HRMS (ESI+) m/z calcd for C22H22N3O3S+ [M + H]+: 408.1376, found: 408.1383.
tert-Butyl 4-[3-(6-methoxy-1,3-benzothiazol-2-yl)-2-oxo-chromen-7-yl]piperazine-1-carboxylate (5c).
Compound 5c was prepared (yellowish orange solid, 13.0 mg, 3 steps – 24% overall yields) according to GP I, GP II and GP III. 1H NMR (400 MHz, chloroform-d) δ 8.87 (s, 1H), 7.92 (d, J = 8.9 Hz, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 2.6 Hz, 1H), 7.11 (dd, J = 9.0, 2.6 Hz, 1H), 6.87 (dd, J = 9.0, 2.3 Hz, 1H), 6.76 (d, J = 2.4 Hz, 1H), 3.91 (s, 3H), 3.70–3.55 (m, 4H), 3.48–3.39 (m, 4H), 1.50 (s, 9H). 13C NMR (150 MHz, methylene chloride-d2) δ 160.6, 158.5, 157.6, 156.2, 154.4, 154.2, 147.2, 140.8, 137.8, 130.3, 122.9, 120.1, 115.9, 114.8, 112.0, 110.5, 103.5, 99.8, 79.8, 55.7, 47.0, 28.1. HRMS (ESI+) m/z calcd for C26H27N3NaO5S+ [M + Na]+: 516.1564, found: 516.1552.
7-Methoxy-3-(6-methoxy-1,3-benzothiazol-2-yl)chromen-2-one (6a).
Compound 6a was prepared (yellow solid, 11.0 mg, 2 steps – 33% overall yields) according to GP I and GP II. 1H NMR (600 MHz, chloroform-d) δ 8.94 (s, 1H), 7.94 (d, J = 8.9 Hz, 1H), 7.61 (d, J = 8.6 Hz, 1H), 7.40 (d, J = 2.5 Hz, 1H), 7.13 (dd, J = 8.9, 2.5 Hz, 1H), 6.95 (dd, J = 8.6, 2.4 Hz, 1H), 6.92 (d, J = 2.4 Hz, 1H), 3.93 (s, 3H), 3.91 (s, 3H). 13C NMR (150 MHz, chloroform-d) δ 164.0, 160.2, 157.9, 157.7, 155.8, 147.2, 140.7, 138.1, 130.3, 123.2, 117.1, 116.3, 113.8, 112.8, 103.5, 100.6, 56.0, 55.8. HRMS (ESI+) m/z calcd for C18H13NNaO4S+ [M + Na]+: 362.0458, found: 362.0464.
3-(Benzo[d]thiazol-2-yl)-7-methoxy-2H-chromen-2-one (6b).
Compound 6b was prepared (yellow solid, 15.5 mg, 2 steps – 53% overall yields) according to GP I and GP II. 1H NMR (400 MHz, chloroform-d) δ 9.03 (s, 1H), 8.06 (d, J = 8.2 Hz, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.52–7.48 (m, 1H), 7.41–7.37 (m, 1H), 6.93 (dd, J = 8.8, 2.4 Hz, 1H), 6.89 (d, J = 2.4 Hz, 1H), 3.90 (s, 3H). 13C NMR δ (100 MHz, chloroform-d) δ 164.7, 160.9, 160.1, 156.2, 151.2, 142.7, 135.9, 130.9, 126.7, 125.4, 122.2, 121.8, 115.9, 114.1, 112.7, 100.6, 56.1. HRMS (ESI+) m/z calcd for C17H11NNaO3S+ [M + Na]+: 332.0352, found: 332.0371.
7-Methoxy-3-[5-(2-methoxyphenyl)thiazol-2-yl]chromen-2-one (6c).
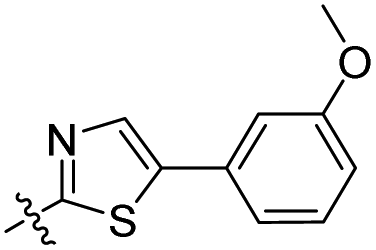
Compound 6c was prepared (yellow solid, 17.0 mg, 2 steps – 19% overall yields) according to GP I and GP II. 1H NMR (600 MHz, chloroform-d) δ 8.99 (s, 1H), 7.69 (s, 1H), 7.68 (d, 1H), 7.62 (t, 1H), 7.59 (d, 1H), 7.39 (t, 1H), 6.98 (dd, 1H), 6.95 (d, 1H). 6.93 (dd, 1H), 3.93 (s, 3H), 3.91 (s, 3H). 13C NMR δ (150 MHz, chloroform-d) δ 193.2, 163.9, 160.1, 159.3, 155.5, 154.9, 139.6, 135.7, 130.2, 129.7, 118.6, 116.8, 115.9, 113.6, 112.8, 112.0, 110.0, 100.5, 56.0, 55.3. HRMS (ESI+) m/z calcd for C20H15NNaO4S+ [M + Na]+: 388.0614, found: 388.0626.
Virtually screened fluorophore derivatives including 3a–3g were purchased from a commercial source (InterBioScreen Ltd, Chernogolovka, Russia).
Fluorescence quantum yield measurement
Fluorescence quantum yields (ϕF) were measured by a combination system of a spectrophotometer (UV-2600 UV/Vis Spectrophotometer, SHIMADZU Corp. and V-530 UV/Vis Spectrophotometer, Jasco, Inc.) with a fluorimeter (RF-6000 Spectro-fluorophotometer, SHIMADZU Corp. and RF-5301PC Spectro-fluorophotometer, SHIMADZU Corp.). Samples were prepared as solutions by dilution of the fluorescent compound in DMSO (Table 1 and Fig. 2) or CH2Cl2 (Table 2). Absorption λmax was measured with a spectrophotometer scanning the 700–250 range wavelength, while fluorescence emission λmax and integrated intensity were analyzed with MATLAB R2014b and Origin2018b software. The sample absorbance was maintained <0.1 to minimize internal absorption. The absolute quantum yield of a known fluorescent dye – fluorescein and norharmane – was obtained to determine those of the sample precisely (fluorescein: ϕF = 0.925 ± 0.015 in 0.1 N NaOH aqueous solution, norharmane: ϕF = 0.58 in 0.1 N H2SO4 aqueous solution).
Intracellular monitoring study
Human cervical cancer (HeLa) cells from ATCC with fewer than 15 passages were cultured as monolayers in complete growth media, DMEM (Dulbecco's Modified Eagle's medium, Gibco, USA) supplemented with 10% FBS (Fetal Bovine Serum, Gibco, USA) and 1% penicillin–streptomycin (Gibco, USA), at 37 °C under 5% CO2. Cells were treated with 10 μM fluorescent compounds for 1 h and washed 3 times with DPBS. Treated cells were further incubated for 4 h and 12 h in complete growth media and fixed with 4% paraformaldehyde for confocal imaging. Fluorescence images were obtained with a LSM 800 laser scanning confocal microscope (Carl Zeiss, LSM 800) using 100× oil objective lens. Optimized laser lines for 5a (λex 426 nm/λem 500 nm) and 6a (λex 407 nm/λem 489 nm) were used.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported financially by the Institute for Basic Science (IBS-R010-G1).
Notes and references
- T. Iwahara, J. Fujimoto, D. Wen, R. Cupples, N. Bucay, T. Arakawa, S. Mori, B. Ratzkin and T. Yamamoto, Oncogene, 1997, 14, 439–449 CrossRef CAS PubMed.
- R. H. Palmer, E. Vernersson, C. Grabbe and B. Hallberg, Biochem. J., 2009, 420, 345–361 CrossRef CAS PubMed.
- M. Soda, Y. L. Choi, M. Enomoto, S. Takada, Y. Yamashita, S. Ishikawa, S.-i. Fujiwara, H. Watanabe, K. Kurashina and H. Hatanaka, Nature, 2007, 448, 561–567 CrossRef CAS PubMed.
- F. R. Jazii, Z. Najafi, R. Malekzadeh, T. P. Conrads, A. A. Ziaee, C. Abnet, M. Yazdznbod, A. A. Karkhane and G. H. Salekdeh, World J. Gastroenterol., 2006, 12, 7104–7112 CrossRef CAS PubMed.
- R. D. Gascoyne, L. Lamant, J. I. Martin-Subero, V. S. Lestou, N. L. Harris, H.-K. Müller-Hermelink, J. F. Seymour, L. J. Campbell, D. E. Horsman and I. Auvigne, Blood, 2003, 102, 2568–2573 CrossRef CAS PubMed.
- R. Roskoski Jr., Pharmacol. Res., 2013, 68, 68–94 CrossRef PubMed.
- R. Crescenzo and G. Inghirami, Curr. Opin. Pharmacol., 2015, 23, 39–44 CrossRef CAS PubMed.
- Y.-J. Bang, Arch. Pathol. Lab. Med., 2012, 136, 1201–1204 CrossRef CAS PubMed.
- W.-C. Wang, H.-Y. Shiao, C.-C. Lee, K.-S. Fung and H.-P. Hsieh, MedChemComm, 2014, 5, 1266–1279 RSC.
- Y. L. Choi, M. Soda, Y. Yamashita, T. Ueno, J. Takashima, T. Nakajima, Y. Yatabe, K. Takeuchi, T. Hamada and H. Haruta, N. Engl. J. Med., 2010, 363, 1734–1739 CrossRef CAS PubMed.
- T. Sasaki, K. Okuda, W. Zheng, J. Butrynski, M. Capelletti, L. Wang, N. S. Gray, K. Wilner, J. G. Christensen and G. Demetri, Cancer Res., 2010, 70, 10038–10043 CrossRef CAS PubMed.
- T. Sasaki, J. Koivunen, A. Ogino, M. Yanagita, S. Nikiforow, W. Zheng, C. Lathan, J. P. Marcoux, J. Du and K. Okuda, Cancer Res., 2011, 71, 6051–6060 CrossRef CAS PubMed.
- R. C. Doebele, A. B. Pilling, D. Aisner, T. G. Kutateladze, A. T. Le, A. J. Weickhardt, K. L. Kondo, D. J. Linderman, L. E. Heasley and W. A. Franklin, Clin. Cancer Res., 2012, 18, 1472–1482 CrossRef CAS PubMed.
- J. F. Gainor and A. T. Shaw, J. Clin. Oncol., 2013, 31, 3987–3996 CrossRef CAS PubMed.
- S. Shin, S. Mah, S. Hong and H. Park, J. Chem. Inf. Model., 2016, 56, 802–810 CrossRef CAS PubMed.
- S. Mah, J. H. Park, H.-Y. Jung, K. Ahn, S. Choi, H. S. Tae, K. H. Jung, J. K. Rho, J. C. Lee and S.-S. Hong, J. Med. Chem., 2017, 60, 9205–9221 CrossRef CAS PubMed.
- M. Kawaguchi, T. Okabe, S. Okudaira, K. Hanaoka, Y. Fujikawa, T. Terai, T. Komatsu, H. Kojima, J. Aoki and T. Nagano, J. Am. Chem. Soc., 2011, 133, 12021–12030 CrossRef CAS PubMed.
- L. C. Kenmogne, R. Maltais and D. Poirier, Bioorg. Med. Chem. Lett., 2016, 26, 2179–2183 CrossRef CAS PubMed.
- J. J. Yim, M. Tholen, A. Klaassen, J. Sorger and M. Bogyo, Mol. Pharm., 2017, 15, 750–758 CrossRef PubMed.
- H. Yao, G. Wei, Y. Liu, H. Yao, Z. Zhu, W.-C. Ye, X. Wu, J. Xu and S. Xu, ACS Med. Chem. Lett., 2018, 9, 1030–1034 CrossRef CAS PubMed.
- J. C. McGrath, S. Arribas and C. J. Daly, Trends Pharmacol. Sci., 1996, 17, 393–399 CrossRef CAS PubMed.
- J. G. Baker, I. P. Hall and S. J. Hill, Br. J. Pharmacol., 2003, 139, 232–242 CrossRef CAS PubMed.
- X.-h. Liu and G. D. Prestwich, Bioorg. Med. Chem. Lett., 2004, 14, 2137–2140 CrossRef CAS PubMed.
- C. M. Crane, J. Kaiser, N. L. Ramsden, S. Lauw, F. Rohdich, W. Eisenreich, W. N. Hunter, A. Bacher and F. Diederich, Angew. Chem., Int. Ed., 2006, 45, 1069–1074 CrossRef CAS PubMed.
- J. Son, J.-J. Lee, J.-S. Lee, A. Schüller and Y.-T. Chang, ACS Chem. Biol., 2010, 5, 449–453 CrossRef CAS PubMed.
- D. Kim, H. Lee, H. Jun, S.-S. Hong and S. Hong, Bioorg. Med. Chem., 2011, 19, 2508–2516 CrossRef CAS PubMed.
- J. Dhuguru, W. Liu, W. G. Gonzalez, W. M. Babinchak, J. Miksovska, R. Landgraf and J. N. Wilson, J. Org. Chem., 2014, 79, 4940–4947 CrossRef CAS PubMed.
- V. M. Yenugonda, T. B. Deb, S. C. Grindrod, S. Dakshanamurthy, Y. Yang, M. Paige and M. L. Brown, Bioorg. Med. Chem., 2011, 19, 2714–2725 CrossRef CAS PubMed.
- M. Min and S. Hong, Chem. Commun., 2012, 48, 9613–9615 RSC.
- M. Min, B. Kim and S. Hong, Org. Biomol. Chem., 2012, 10, 2692–2698 RSC.
- D. Kim, M. Min and S. Hong, Chem. Commun., 2013, 49, 4021–4023 RSC.
- M. Min, Y. Kim and S. Hong, Chem. Commun., 2013, 49, 196–198 RSC.
- Y. Moon, Y. Kim, H. Hong and S. Hong, Chem. Commun., 2013, 49, 8323–8325 RSC.
- Y. Kim, Y. Moon, D. Kang and S. Hong, Org. Biomol. Chem., 2014, 12, 3413–3422 RSC.
- J. Hoult and M. Paya, Gen. Pharmacol., 1996, 27, 713–722 CrossRef CAS PubMed.
- I. Kostova, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 29–46 CrossRef CAS PubMed.
- R. Dayam, R. Gundla, L. Q. Al-Mawsawi and N. Neamati, Med. Res. Rev., 2008, 28, 118–154 CrossRef CAS PubMed.
- P. T. Thuong, T. M. Hung, T. M. Ngoc, D. T. Ha, B. S. Min, S. J. Kwack, T. S. Kang, J. S. Choi and K. Bae, Phytother. Res., 2010, 24, 101–106 CrossRef CAS PubMed.
- S.-Y. Bai, X. Dai, B.-X. Zhao and J.-Y. Miao, RSC Adv., 2014, 4, 19887–19890 RSC.
- S. Emami and S. Dadashpour, Eur. J. Med. Chem., 2015, 102, 611–630 CrossRef CAS PubMed.
- B. D. Wagner, Molecules, 2009, 14, 210–237 CrossRef CAS PubMed.
- J. J. Cui, M. Tran-Dubé, H. Shen, M. Nambu, P.-P. Kung, M. Pairish, L. Jia, J. Meng, L. Funk and I. Botrous, J. Med. Chem., 2011, 54, 6342–6363 CrossRef CAS PubMed.
- H. Sakamoto, T. Tsukaguchi, S. Hiroshima, T. Kodama, T. Kobayashi, T. A. Fukami, N. Oikawa, T. Tsukuda, N. Ishii and Y. Aoki, Cancer Cell, 2011, 19, 679–690 CrossRef CAS PubMed.
- T. W. Johnson, P. F. Richardson, S. Bailey, A. Brooun, B. J. Burke, M. R. Collins, J. J. Cui, J. G. Deal, Y.-L. Deng and D. Dinh, J. Med. Chem., 2014, 57, 4720–4744 CrossRef CAS PubMed.
- When cell viability assay against Ba/F3 EML4-ALK WT (EA WT) was conducted, representative compounds 5a and 6a inhibited the cell growth in a dose-dependent manner (5a: IC50 = 6.8 μM, 6a: IC50 = 28.2 μM).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/c8ob02874e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 bc,
Yongwon
Jung
*a and
Sungwoo
Hong
bc,
Yongwon
Jung
*a and
Sungwoo
Hong