Crotonols A and B, two rare tigliane diterpenoid derivatives against K562 cells from Croton tiglium†
Received
11th October 2018
, Accepted 5th December 2018
First published on 6th December 2018
Abstract
Crotonols A and B (1 and 2), two tigliane diterpenoids featuring a rare C-7/C-14 cyclized and novel 5/7/7-fused carbon skeleton, along with the known tigliane wallichiioid A, were isolated from the leaves of Croton tiglium. Their structures were determined through spectroscopic methods, X-ray crystallography and ECD analysis. To the best of our knowledge, crotonol B (2) represents the first example of 13,14-seco-tigliane diterpenoids. Crotonols A and B displayed strong cytotoxic activities against the K562 cell line with IC50 values of 0.20 and 0.21 μM, respectively. Furthermore, crotonol A promoted the apoptosis of K562 cells through the cleavage of PARP and the accumulation of bax as well as the degradation of bcl-2.
Introduction
Tigliane diterpenoids have a unique architecture of a 5/7/6/3-tetracyclic ring system (Tigliane skeleton, Fig. 1), and are reported mainly from Euphorbiaceae and Thymelaeaceae families.1,2 To date, about 248 tigliane diterpenoids have been isolated from the plants of 23 genera, and the majority have been found in plants belonging to the Euphorbiaceae family.1 It is now known that Tigliane diterpenoids have many biological activities including immunomodulatory,3 anti-HIV,4,5 and anti-cancer.6,7 Phorbol 12-myristate 13-acetate is a tigliane diterpenoid currently in phase II clinical trials for treatment of acute myeloid leukaemia.1 The chemical complexity of bioactive tigliane diterpenoids stimulated many synthesis endeavors.8–10
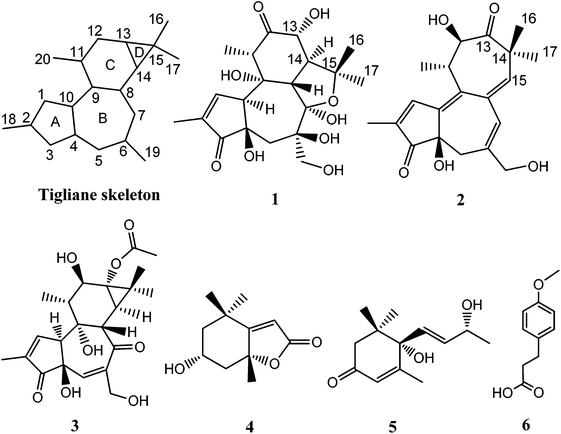 |
| | Fig. 1 Structures of compounds. | |
Croton tiglium belongs to the Euphorbiaceae family, and its seeds and leaves are known as traditional medicinal plants in China.5,7 In our continuing investigation of its constituents, two new tigliane diterpenoid derivatives, crotonols A and B (1 and 2) (Fig. 1), exhibiting a rare C-7/C-14 cyclized and 5/7/7-fused ring core (Tigliane skeleton, Fig. 1), along with a known tigliane and three non-tiglianes (Fig. 1), were isolated from its leaves. Herein, we report the isolation, structural elucidation, and cytotoxic activities of 1 and 2, as well as the apoptosis induction of 1 in K562 cells.
Results and discussion
Crotonol A (1) was obtained as a white crystal. Its molecular formula C20H28O9 was deduced from the [M − H]− ion at m/z 411.1649 (calcd 411.1661) in the HREIMS spectrum, which was in accordance with the 1H and 13C NMR spectroscopic data (Table 1), corresponding to 7 degrees of unsaturation. The IR spectrum showed the existence of hydroxyl groups (3469 and 3419 cm−1) and carbonyl groups (1715 and 1632 cm−1). In the 1H NMR spectrum, the most salient signals were shown for one olefinic proton signal at δH 7.62 (1H, s), one oxygenated methine at δH 4.07 (1H, d, J = 11.1 Hz), one oxygen-bearing methylene at δH 3.84 (1H, d, J = 10.6 Hz), 3.60 (1H, d, J = 10.6 Hz), one methylene at δH 2.23 (1H, d, J = 14.9 Hz), 1.78 (1H, d, J = 14.9 Hz) and four methyl signals at δH 1.79 (3H, s), 1.46 (3H, s), 1.32 (3H, s), and 1.11 (3H, d, J = 6.7 Hz). The 13C NMR (Table 1), DEPT, and HSQC spectra revealed the presence of 20 carbons, namely four methyls, two sp3 methylenes, four sp3 methines, five sp3 quarternary carbons, one sp2 methine, one sp2 quarternary carbon, one hemiketal, and two carbonyl groups. The molecular formula requires seven degrees of unsaturation, but only two carbonyls resonating at δC 212.7 (C, C-12) and 208.0 (C, C-3), and two olefinic carbons resonating at δC 160.0 (CH, C-1) and 136.8 (C, C-2) were detected, indicating the tetracyclic nature of compound 1. The HMBC correlations from H-1 to C-2, C-3, C-4, and C-10, H3-18 to C-1, C-2, and C-3, and the 1H–1H COSY correlation of H-1/H-10 suggested the presence of a 2-methyl-2-cyclopentenone moiety (A ring) (Fig. 1). The HMBC correlations from H2-5 to C-4, C-6, C-7, C-10, and C-19, H-8 to C-6, C-7, C-9, and C-10, H2-19 to C-5, C-6 and C-7 suggested the presence of a seven-membered ring (B ring). Besides, a hydroxymethyl (C-19) was linked to the B ring at C-6 (Fig. 1). The key HMBC correlations from H-10 to C-1, C-2, C-4, C-5, and C-8, and H2-5 to C-4 indicated the junction of the A/B ring via the bridge of C-4–C-10. Furthermore, the HMBC spectrum showed the following correlations: H-8 with C-9, C-11, C-13 and C-14, H-11 with C-8, C-9, and C-12, H-13 with C-12 and C-14. This evidence, as well as two proton spin systems deduced from the 1H–1H COSY correlations H-11/H3-20 and H-8/H-14/H-13, led to the establishment of a 2-methylcyclohexanone moiety (C ring). The 1-(hydroxymethyl)cycloheptanol (B ring) and 2-methylcyclohexanone moieties (C ring) were found to be connected via the bridge of C-8–C-9, which was further supported by COSY correlation of H-8/H-14 and by the key HMBC correlations from H-8 to C-11, H-14 to C-7, and H-14 to C-9 (Fig. 2). An oxygenated isopropyl unit located at C-14 was observed, which was further supported by key HMBC correlations from H3-16/H3-17 to C-14 and C-15, H-14 to C-15, C-16 and C-17, H-8/H-13 to C-15. Taking the molecular formula and the above NMR spectroscopic data into consideration, the 5/7/6-fused ring core, one double bond, and two carbonyls consumed six of the seven degrees of unsaturation. Therefore, the remaining one unsaturation unit required that compound 1 possessed another ring system. However, the 2D NMR spectra did not provide sufficient information to unambiguously elucidate the attachment pattern between C-7 and C-15. Connecting C-7 and C-15 via an oxygen bridge would be consistent with the degrees of unsaturation, molecular formula, and the downfield shift of the sp3 quarternary carbon C-7 (δC 109.1). In the NOESY data of 1, NOE correlations of H-10 (δH 3.66) with H-5 (δH 1.78) and H-19 (δH 3.84) determined these protons to be on the same face of the ring system, whereas NOE correlations of H-8 (δH 3.56) with H-11 (δH 2.97), H-13 (δH 4.07), and H3-16 (δH 1.32) suggested that these protons were on the other face of the molecule (Fig. 3). In order to verify the proposed structure, compound 1 was subjected to single crystal X-ray diffraction analysis (Fig. 4). Bearing on nine oxygen atoms in the molecule, the final refinement on the Cu Kα data resulted in a Flack parameter0.0(5),11–13 which not only indicated the connection of C-7 to C-15 through an oxygen bridge but also unambiguously determined the absolute stereochemistry of 1 to be 4R,6R, 7R, 8S, 9R, 10S, 11S, 13R, and 14S.
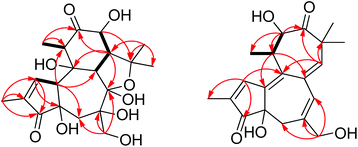 |
| | Fig. 2 Key 1H–1H COSY (bold), and 1H–13C HMBC (arrows) correlations of 1 and 2. | |
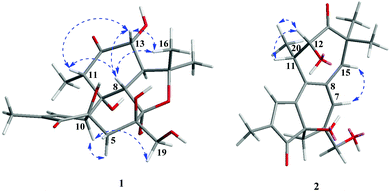 |
| | Fig. 3 Key NOESY (arrows) correlations of 1 and 2. | |
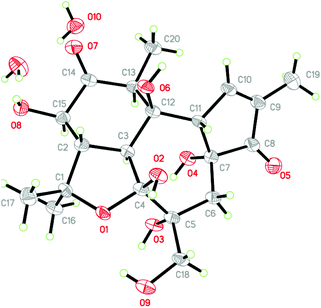 |
| | Fig. 4 ORTEP drawing of crotonol A (1) (Cu Kα) (ellipsoids shown at the 50% probability level). | |
Table 1
1H and 13C NMR data for 1 and 2 (600, 150 MHz, TMS, δ ppm, in CD3OD)
| Position |
1
|
2
|
|
δ
C type |
δ
H (J in Hz) |
δ
C type |
δ
H (J in Hz) |
| 1 |
160.0, CH |
7.62, s |
149.7, CH |
7.82, d (1.3) |
| 2 |
136.8, C |
|
140.1, C |
|
| 3 |
208.0, C |
|
210.2, C |
|
| 4 |
76.8, C |
|
75.9, C |
|
| 5 |
40.4, CH2 |
2.23, d (14.9); |
36.4, CH2 |
2.33, d (18.2); |
| 1.78, d (14.9) |
2.07, dd (18.2, 2.1) |
| 6 |
76.1, C |
|
135.3, C |
|
| 7 |
109.1, C |
|
126.3, CH |
6.32, s |
| 8 |
50.1, CH |
3.56, d (13.4) |
135.6, C |
|
| 9 |
76.2, C |
|
136.9, C |
|
| 10 |
59.5, CH |
3.66, m |
140.7, C |
|
| 11 |
53.5, CH |
2.97, q (6.7) |
46.2, CH |
2.88, m |
| 12 |
212.7, C |
|
77.0, CH |
4.53, d (8.7) |
| 13 |
78.2, CH |
4.07, d (11.1) |
216.4, C |
|
| 14 |
51.7, CH |
2.78, dd (13.6,11.2) |
50.0, C |
|
| 15 |
83.2, C |
|
139.1, CH |
5.73, s |
| 16 |
23.6, CH3 |
1.32, s |
24.0, CH3 |
1.13, s |
| 17 |
31.7, CH3 |
1.46, s |
28.3, CH3 |
1.26, s |
| 18 |
10.5, CH3 |
1.79, s |
10.6, CH3 |
1.90, d (1.3) |
| 19 |
68.4, CH2 |
3.84, d (10.6); |
69.0, CH2 |
3.98, d (13.3); |
| 3.60, d (10.6) |
3.94, d (13.3) |
| 20 |
10.6, CH3 |
1.11, d (6.7) |
21.3, CH3 |
1.24, d (6.8) |
Crotonol B (2) was obtained as yellow oil with the molecular formula C20H24O5 as determined by HRESIMS indicating 9 degrees of unsaturation. The 1H NMR spectroscopic data of 2 (Table 1) exhibited signals of four methyl groups [δH 1.90 (d, J = 1.3 Hz, H3-18), 1.26 (s, H3-17), 1.24 (d, J = 6.8 Hz, H3-20), 1.13 (s, H3-16)], one oxymethylene [δH 3.98 (d, J = 13.3 Hz, H-19a), 3.94 (d, J = 13.3 Hz, H-19b)], one methylene [δH 2.33 (d, J = 18.2 Hz, H-5a), 2.07 (dd, J = 18.2, 2.1 Hz, H-5b)], one methine [δH 2.88 (m, H-11)], one oxygenated methine [δH 4.53 (d, J = 8.7 Hz, H-12)], and three olefinic protons [δH 7.82 (d, J = 1.3 Hz, H-1), 6.32 (s, H-7), 5.73 (s, H-15)]. The 13C NMR spectroscopic data of 2 showed 20 carbon resonances classified by DEPT and HSQC experiments as four methyls, two sp3 methylenes [one oxygenated (δC 69.0)], two sp3 methines [one oxygenated (δC 77.0)], two sp3 quarternary carbons, three sp2 methine, five sp2 quarternary carbons, and two carbonyl groups (δC 216.4, 210.2). Taking the molecular formula and the above NMR data into consideration, four double bonds and two carbonyl groups consumed six of the nine degrees of unsaturation. Therefore, the remaining three unsaturation units required that compound 2 possessed a tricyclic ring system. Its 1H and 13C NMR spectroscopic data resembled those of phorbol.14 The structural differences between them were occurring on the B, C, and D rings. The HMBC correlations from H-1 to C-4, C-9, and C-10, H2-5 to C-3, C-7, C-10, and C-19, H-7 to C-5, C-9, and C-19 indicated that the dehydration of 9-OH in phorbol formed a double bond between C-9 and C-10 in the B ring of compound 2. Furthermore, the HMBC spectrum showed the following correlations: H-11 with C-8, C-10, and C-12, H-12 with C-11, C-13, and C-20, H-15 with C-7, C-9, and C-13, H3-16/H3-17 with C-13, C-14, and C-15. This evidence, as well as the one proton spin system deduced from 1H–1H COSY correlations, H-12/H-11/H3-20, led to the establishment of a novel seven-membered ring, replacing the C and D rings of the carbon skeleton of tigliane diterpenoid (Fig. 1 and 2). In the NOESY data of 2, the NOE correlation of H-7 with H-15 determined the (E)-geometry for the Δ6(7) and the (Z)-geometry for the Δ8(15) double bonds, whereas the NOE correlations observed between H-12, H-11, and H3-20 could not establish the relative stereochemistry at C-11 and C-12. Previous studies on the Euphorbiaceae and Thymelaeaceae families suggest that the essential elements of the tigliane carbon skeleton have the same stereochemistry (4R, 11S, 12R).1,2,8 To determine its absolute configuration, a computational modeling study was conducted using the Gaussian 09 program package. The conformers of 2 were used as the input for the structural optimization by the density functional theory method at the B3LYP/6-31G(d) level in the Gaussian 09. The calculated ECD spectrum of (4R, 11S, 12R)-2 was consistent with the experimental one (Fig. 5), confirming the absolute configuration of 2 to be 4R, 11S, and 12R, respectively.
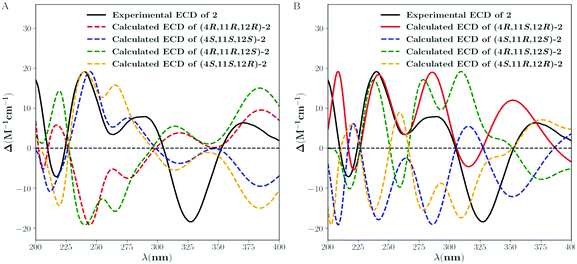 |
| | Fig. 5 Experimental and calculated ECD spectra of 2. | |
A hypothetical biosynthetic pathway for the unusual tigliane skeletons of Crotonols A and B (1 and 2) is proposed in Scheme 1, if these compounds arise from phorbol. Phorbol was oxygenated at the C-6, C-7 double bond to give the key intermediate 3, which possessed an epoxy group. A nucleophilic addition reaction of one molecule of water with the C6, C7 epoxide generates intermediate 4 under acidic conditions, which underwent an oxidation reaction at C-7 and C-12 to intermediate 5. And then, an oxidative cleavage between C-13 and C-15 was followed to give intermediate 6. Lastly, crotonol A (1) was formed by the intra molecular nucleophilic addition of OH-15 to the C-7 carbonyl in 6. On the other hand, crotonol B (2) featured a novel 13,14-seco-tigliane skeleton. Oxidation of the tigliane skeleton, which was followed by an oxidative cleavage between C-13 and C-14 to yield 7, was envisioned. Intermediate 7 further underwent dehydration to form 8, which was oxygenated at C-13 to give crotonol B (2).
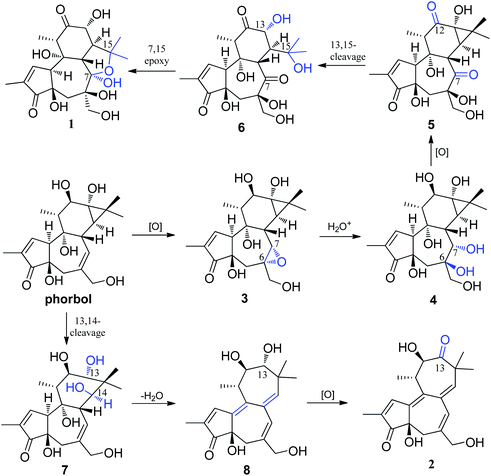 |
| | Scheme 1 Hypothetical biosynthetic pathway for 1 and 2. | |
The known compounds were identified as wallichiioid A (3),15 7-epiloliolide (4),16 vomifoliol (5),17 and p-methoxy phenylpropionic acid (6),18 respectively, by comparison of the spectroscopic data with those in the literature.
Prior to this study, 13,15-seco-tigliane diterpenoids with a C-7/C-14 ether linkage were rarely reported from nature, and include langduin A,19 curcusecons A–E,20 and neoglabrescins A and B.21 To the best of our knowledge, crotonol B (2) represents the first example of 13,14-seco-tigliane diterpenoids (Tigliane skeleton, Fig. 1). As aforementioned,1 tigliane diterpenoids have been viewed as promising leads in anti-cancer applications, and crotonols A (1) and B (2) were thus evaluated in vitro for their cytotoxicity against K562, MCF-7, and SGC-7901 human cancer cell lines with Paclitaxel used as the positive control. These compounds showed strong cytotoxic activities towards the K562 leukemia cell line, while exhibiting minimal cytotoxicity in the MCF-7 breast and SGC-7901 gastic cancer cell lines (Table 2).
Table 2 Cytotoxicities against tumor cells
| Cell lines |
IC50 (μM) |
|
1
|
2
|
Paclitaxel (nM) |
| K562 |
0.20 |
0.21 |
3.67 |
| MCF-7 |
17.60 |
>50 |
7.56 |
| SGC-7901 |
>50 |
>50 |
3.41 |
Since the occurrence of cancer is closely related to the reduced apoptosis of cells, we performed flow cytometry analyses to measure the apoptosis rate with crotonol A (1). Compared with the DMSO groups (22.0%), K562 cell apoptosis rates showed an increase to 46.3% with 0.1 μM crotonol A treatment, but they were relatively unchanged at higher concentrations of 0.2 and 0.4 μM (Fig. 6 and ESI, Table S4†). Moreover, the early apoptosis rate was significantly increased (35.6%) in K562 cells with 0.1 μM crotonol A treatment, compared with the control group (17.6%). These results indicated that crotonol A (1) could promote apoptosis especially early apoptosis in K562 cells at a low concentration. It is well known that apoptosis is a complex process regulated by various factors. For example, caspase-3 is generally considered as the main executor of apoptosis. One of the essential substrates cleaved by caspase-3 is PARP, an abundant DNA-binding enzyme that detects and signals DNA strand breaks.22 PARP is the key modulator of apoptosis, mainly through the cleavage of PARP. Moreover, bcl-2 and bax belong to the bcl-2 family, which play different roles in programmed cell death. On the one hand, overexpression of bax in cells accelerates apoptotic death, leading to its designation as a death agonist, while overexpression of bcl-2 leads to heterodimerization with bax, to repress cell death.23 Therefore, the ratio of bcl-2 to bax plays a vital role in determining cell susceptibility to apoptosis. In the present study, we detected expressions of proteins related to apoptosis in K562 cells by western blot assay. Compared with the DMSO group, the expression of cleaved-PARP was significantly increased in a concentration-dependent manner (Fig. 7A and B). Similarly, the expression of bax was also significantly elevated. However, the expression of bcl-2 and the ratio of bcl-2 to bax were gradually decreased when compared with the DMSO group (Fig. 7C–E). These results suggested that crotonol A (1) promoted the apoptosis of K562 cells through the cleavage of PARP and the accumulation of bax as well as the degradation of bcl-2. Taken together, crotonol A (1) has a dramatic effect on the expression of PARP, bax and bcl-2, which play an essential role in determining cell apoptosis and survival. Further studies are necessary to develop a deeper understanding of the anti-tumor effects of these compounds in the future.
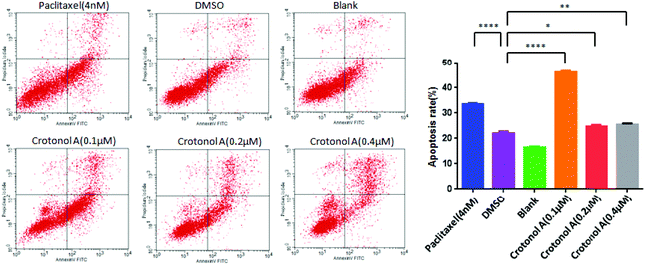 |
| | Fig. 6 Crotonol A (1) promoted K562 cell apoptosis. The apoptosis rates were detected by flow cytometry analyses. All results are expressed as mean ± S.D. from three independent experiments. *p < 0.1, **p < 0.01, ****p < 0.0001, vs. DMSO. | |
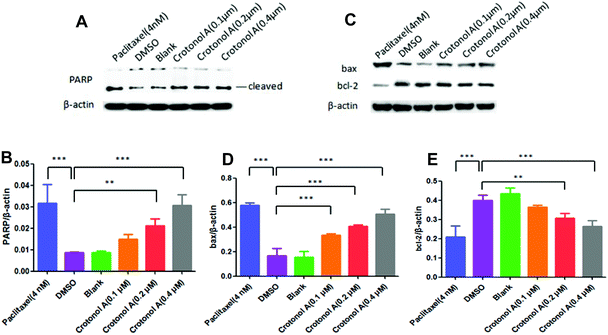 |
| | Fig. 7 The compound increased the expressions of cleaved-PARP and bax as well as decreased the expressions of bcl-2. The protein expressions of cleaved-PARP(A), bax and bcl-2(C) in K562 cells were measured by western blot assay. All results are expressed as mean ± S.D. from three independent experiments. **p < 0.01, ***p < 0.001 compared to DMSO. | |
Conclusions
In summary, we isolated and identified from the leaves of Croton tiglium two new tigliane diterpenoids featuring a C-7/C-14 ether linkage and a 5/7/7-fused carbon skeleton, along with a known tigliane and three non-tiglianes. To the best of our knowledge, crotonol B (2) represents the first example of 13,14-seco-tigliane diterpenoids. Meanwhile, we evaluated its bioactivity, and crotonols A (1) and B (2) displayed strong cytotoxic activities against the K562 cell line with IC50 values of 0.20 and 0.21 μM, respectively. Furthermore, crotonol A (1) promoted the apoptosis of K562 cells through the cleavage of PARP and the accumulation of bax as well as the degradation of bcl-2.
Experimental section
General experimental procedures
NMR spectra were recorded on a Bruker Avance III-600 MHz spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany). Optical rotations were recorded on a Horiba SEPA-300 polarimeter (Horiba, Tokyo, Japan). UV spectra were obtained on a Shimadzu UV-2401PC spectrophotometer (Shimadzu Corporation, Kyoto, Japan). IR spectra were obtained on a Tensor 27 (Bruker Optics Gmbh, Ettlingen, Germany) with KBr pellets. ESI-MS and HREIMS were determined with an API QSTAR Pulsar 1 spectrometer (MDS Sciex, Concord, ON, Canada). Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), RP-18 gel (40–63 μm, Daiso Co., Osaka, Japan), and Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) were used for column chromatography.
Plant material
The leaves of C. tiglium were collected from the Changning County of Sichuan Province, China, in August 2014, and authenticated by the corresponding author (X. J. Zhou). A voucher specimen (ZHXJ-0030) was deposited at our laboratory in Hunan University of Chinese Medicine.
Extraction and purification
The dried leaves of C. tiglium (30 kg) were extracted with 95% ethanol (2 × 100 L) to give a crude extract (2740 g), which was suspended in water and partitioned using petroleum ether and EtOAc (each 8 × 10 L), respectively. The EtOAc extracts (390 g) were subjected to column chromatography (CC) over silica gel (200–300 mesh, 5.0 kg) and eluted with a gradient of CHCl3/MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 98:2, 96:4, 93:7, 90:10, 85:15, 80:20, 70:30, 50:50) to give fractions A–F. Fraction D (78.1 g) was divided into five parts (D-1–D-5) using a MCI gel CHP 20P column eluted with a gradient of aqueous MeOH. Fraction D-1 (12.2 g) was subjected to column chromatography (CC) over silica gel and eluted with a gradient of petroleum ether/EtOAc (8:1, 6:1, 4:1, 3:1, 2:1, 1:1) to afford fractions D-1-1–D-1-4. Frs. D-1-1 (0.5 g) was first analysed using Sephadex LH-20 (MeOH), followed by purification by semi-preparative HPLC elution with 30% aqueous acetonitrile to produce compound 1 (23.6 mg). Frs. D-1-4 (2.3 g) was first submitted using a RP-18 gel column (MeOH–H2O 2:8–1:0), followed by Sephadex LH-20 (MeOH), and finally purified by semi-preparative HPLC (18% aqueous acetonitrile) to give compounds 2 (3.1 mg) and 3 (2.6 mg). Fraction D-5 (5.5 g) was subjected to column chromatography (CC) over silica gel and eluted with a gradient of n-hexane/EtOAc (6:1, 4:1, 3:1, 2:1, 1:1) to afford fractions D-5-1–D-5-4. Frs. D-5-1 (0.25 g) was purified by semi-preparative HPLC elution with 50% aqueous MeOH to produce compound 6 (8.7 mg). Frs. D-5-4 (0.15 g) was purified by semi-preparative HPLC elution with 30% aqueous MeOH to yield compounds 4 (5.4 mg) and 5 (4.8 mg).
:0, 98:2, 96:4, 93:7, 90:10, 85:15, 80:20, 70:30, 50:50) to give fractions A–F. Fraction D (78.1 g) was divided into five parts (D-1–D-5) using a MCI gel CHP 20P column eluted with a gradient of aqueous MeOH. Fraction D-1 (12.2 g) was subjected to column chromatography (CC) over silica gel and eluted with a gradient of petroleum ether/EtOAc (8:1, 6:1, 4:1, 3:1, 2:1, 1:1) to afford fractions D-1-1–D-1-4. Frs. D-1-1 (0.5 g) was first analysed using Sephadex LH-20 (MeOH), followed by purification by semi-preparative HPLC elution with 30% aqueous acetonitrile to produce compound 1 (23.6 mg). Frs. D-1-4 (2.3 g) was first submitted using a RP-18 gel column (MeOH–H2O 2:8–1:0), followed by Sephadex LH-20 (MeOH), and finally purified by semi-preparative HPLC (18% aqueous acetonitrile) to give compounds 2 (3.1 mg) and 3 (2.6 mg). Fraction D-5 (5.5 g) was subjected to column chromatography (CC) over silica gel and eluted with a gradient of n-hexane/EtOAc (6:1, 4:1, 3:1, 2:1, 1:1) to afford fractions D-5-1–D-5-4. Frs. D-5-1 (0.25 g) was purified by semi-preparative HPLC elution with 50% aqueous MeOH to produce compound 6 (8.7 mg). Frs. D-5-4 (0.15 g) was purified by semi-preparative HPLC elution with 30% aqueous MeOH to yield compounds 4 (5.4 mg) and 5 (4.8 mg).
Crotonol A
(1): White crystal; [α]19D +37.6 (c 0.2, MeOH); UV (MeOH) λmax (logε): 241 (3.90) nm; IR (KBr) νmax 3469, 3419, 1715, 1632, 1461, 1422, 1409, 1385, 1334, 1314, 1294, 1205, 1147, 1131, 1089, 1054, 1030, 994 cm−1; 1H NMR and 13C NMR data, see Table 1; (−)-HRESIMS m/z 411.1649 [M − H]− (calcd for C20H27O8, 411.1661).
Crotonol B
(2): Yellow oil; [α]19D +50.4 (c 0.3, MeOH); UV (MeOH) λmax (logε): 204 (3.99), 220 (3.98), 321 (3.84) nm; IR (KBr) νmax 3356, 3339, 2967, 2926, 2868, 1703, 1641, 1585, 1445, 1379, 1362, 1341, 1287, 1225, 1205, 1113, 1084, 1030, 907 cm−1; 1H NMR and 13C NMR data, see Table 1; (+)-HRESIMS m/z 367.1515 [M + Na]+ (calcd for C20H24NaO5, 367.1516).
X-ray crystallographic data of 1
Crotonol A (1) was crystallized from methanol to give colorless crystals. Crystal data: monoclinic, space group P2(1)2(1)2(1) with a = 6.9268(4) Å, b = 7.6212(4) Å, c = 39.6430(3) Å, V = 2092.8(2) Å3, Z = 4, Dcalc = 1.423 g cm−3, R = 0.1013, wR2 = 0.1902. The absolute configuration was determined on the basis of a Flack parameter of −0.0(5), refined using 3638 Friedel pairs. Crystallographic data (excluding structural factors) for structure 1 in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1823042.†
Computation section
Conformational analysis.
Conformational analysis was initially performed using Confab with a systematic search at MMFF94 force field for all possible configurations of compound 2.24 RMSD and energy filtration thresholds were set at 0.5 Å and 50 kcal mol−1. As a result, only two distinct conformers were obtained for each configuration. To avoid missing the real dominative conformers, all conformers were delivered to the subsequent quantum mechanics (QM) calculations.
ECD calculation.
Theoretical calculations were carried out using Gaussian 09. At first, conformers were optimized at B3LYP/6-311G(d,p) in methanol. Vibrational frequency analysis confirmed the stable structures. Under the same conditions, the ECD calculation was conducted using time-dependent density functional theory (TD-DFT). The ECD spectrum was simulated in SpecDis by overlapping Gaussian functions for each transition. Average spectra were obtained for each configuration by Boltzmann weighting.25
Biological assays
Cytotoxic assay.
Three human cancer cell lines (K562, MCF-7, SGC-7901) were used in the cytotoxic activity assay. All cells were purchased from Shanghai Cell Bank, Chinese Academy of Sciences. Cells were routinely grown and maintained in medium RPMI or DMEM with 10% FBS and with 1% penicillin/streptomycin. All cell lines were incubated in a Thermo/Forma Scientific CO2 Water Jacketed Incubator with 5% CO2 in air at 37 °C. Cell viability assay was determined by the CCK-8 (Dojindo, Japan) assay. Cells were seeded at a density of 400–800 cells per well in 384 well plates and treated with various concentrations of compounds or solvent control. After 72 h incubation, CCK-8 reagent was added, and absorbance was measured at 450 nm using an Envision 2104 multi-label Reader (PerkinElmer, USA). Dose response curves were plotted to determine the IC50 values using Prism 5.0 (GraphPad Software Inc., USA). Paclitaxel was used as the positive control with IC50 values of 3.67, 7.56, and 3.41 nM, respectively.
Cell culture and treatment
K562 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 50 U ml−1 penicillin and 50 μg ml−1 streptomycin. All the cells were maintained at 37 °C in a 95% air–5% CO2 humidified incubator. K562 cells were induced by a compound (0.1 μmol L−1, 0.2 μmol L−1, 0.4 μmol L−1) and treated with paclitaxel (4 nmol L−1).
Western blot analysis
Western blot analysis was performed as previously described.26 Briefly, cell lysates were centrifuged at 12000 rpm for 15 min at 4 °C. The concentrations of protein samples were determined using a BCA protein assay kit. The proteins were separated on 10% SDS-PAGE (Sigma, USA), and transferred to a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% skimmed milk for 1 h, PVDF membranes were incubated with anti-PARP, anti-bax, anti-bcl-2 or anti-β-actin (Proteintech, USA) antibodies overnight at 4 °C, respectively. And then the antibodies were incubated with a secondary antibody (Proteintech, USA) for 1 h. Specific proteins were visualized using an enhanced chemiluminescence kit (ECL, Thermo pierce, USA).
Flow cytometry
Flow cytometry was performed as previously described.27 Apoptotic cells were analyzed with fluorescein isothiocyanate (FITC)-conjugated annexin V staining (1:20, BioLegend) together with propidium iodide (PI) dead cell counterstain according to the manufacturer's recommendations. The percentage of non-viable cells was the sum of the late apoptotic cells (Annexin V+/PI+) and the early apoptotic cells (Annexin V+/PI+). Flow cytometry was performed using FlowJo, Microsoft Excel and Prism. All flow cytometry data were analyzed using FlowJo software v8.8.6.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31670362), the Hunan Provincial Key Research and Development Program, China (No. 2017NK2261), the Hunan Provincial special projects for tackling key scientific and technological achievements, China (No. 2018GK4027), and the National standardization construction project of traditional Chinese Medicine, China (No. ZYBZH-Y-HUN-24).
Notes and references
- H. B. Wang, X. Y. Wang, L. P. Liu, G. W. Qin and T. G. Kang, Chem. Rev., 2015, 115, 2975–3011 CrossRef CAS PubMed.
- A. Vasas and J. Hohmann, Chem. Rev., 2014, 114, 8579–8612 CrossRef CAS PubMed.
- N. Isakov and A. Altman, Front. Immunol., 2013, 4, 384 Search PubMed.
- L. N. Mckernan, D. Momjian and J. Kulkosky, Adv. Virol., 2012, 12, 805347–805354 Search PubMed.
- S. El-Mekkawy, M. R. Meselhy, N. Nakamura, M. Hattori, T. Kawahata and T. Otake, Phytochemistry, 2000, 53, 457–464 CrossRef CAS PubMed.
- H. J. Mackay and C. J. Twelves, Nat. Rev. Cancer, 2007, 7, 554–562 CrossRef CAS PubMed.
- D. D. Zhang, B. Zhou, J. F. Yu, C. H. Xu, J. Ding, H. Zhang and J. M. Yue, Tetrahedron, 2015, 71, 9638–9644 CrossRef CAS.
- S. H. Kawamura, H. Chu, J. Felding and P. S. Baran, Nature, 2016, 532, 90–93 CrossRef CAS PubMed.
- K. Lee and J. K. Cha, J. Am. Chem. Soc., 2001, 123, 5590–5591 CrossRef CAS PubMed.
- L. Wang, J. Yang, L. M. Kong, J. Deng, Z. J. Xiong, J. P. Huang, J. Y. Luo, Y. J. Yan, Y. K. Hu, X. N. Li, Y. Li, Y. Zhao and S. X. Huang, Org. Lett., 2017, 19, 3911–3914 CrossRef CAS PubMed.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- H. D. Flack, Crystallogr., 1983, A39, 876–881 CAS.
- R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96–103 CrossRef CAS PubMed.
- B. Q. Zhao, S. Peng, W. J. He, Y. H. Liu, J. F. Wang and X. J. Zhou, Bioorg. Med. Chem. Lett., 2016, 26, 4996–4999 CrossRef CAS PubMed.
- D. S. Yang, W. B. Peng, Y. P. Yang, K. C. Liu, X. L. Li and W. L. Xiao, J. Asian Nat. Prod. Res., 2015, 17, 946–951 CrossRef CAS PubMed.
- C. Lee, S. Lee and S. Y. Park, Nat. Prod. Sci., 2013, 19, 355–359 CAS.
- S. Hammami, H. Ben Jannet, A. Bergaoui, L. Ciavatta, G. Cimino and Z. Mighri, Molecules, 2004, 9, 602–608 CrossRef CAS PubMed.
- W. Li, W. L. Mei, W. J. Zuo, C. H. Cai, W. H. Dong and H. F. Dai, J. Trop. Subtrop. Bot., 2016, 24, 342–347 CAS.
- Q. G. Ma, W. Z. Liu, X. Y. Wu, T. X. Zhou and G. W. Qin, Phytochemistry, 1997, 44, 663–666 CrossRef CAS.
- J. Q. Liu, Y. F. Yang, X. Y. Li, E. Q. Liu, Z. R. Li, L. Zhou, Y. Li and M. H. Qiu, Phytochemistry, 2013, 96, 265–272 CrossRef CAS PubMed.
- A. T. Tchinda, A. Tsopmo, M. Tene, P. Kamnaing, D. Ngnokam, P. Tane, J. F. Ayfor, J. D. Connolly and L. J. Farrugia, Phytochemistry, 2003, 64, 575–581 CrossRef CAS PubMed.
- H. Wang, W. Ge, W. Jiang, D. Li and X. Ju, Mol. Med. Rep., 2018, 17, 2070–2076 CAS.
- T. F. Zhao, Y. X. Fu, H. Sun and X. Q. Liu, IUBMB Life, 2018, 70, 60–70 CrossRef CAS PubMed.
- N. M. O'Boyle, T. Vandermeersch, C. J. Flynn, A. R. Maguire and G. R. Hutchison, J. Cheminf., 2011, 3, 3–8 Search PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Spec Dis: Chirality, 2013, 25, 243–249 CAS.
- L. Qin, Y. B. Yang, Y. X. Yang, N. Zhu, Z. Lin, Y. G. Ni, S. X. Li, X. L. Zheng and D. F. Liao, Clin. Exp. Pharmacol. Physiol., 2016, 43, 182–192 CrossRef CAS PubMed.
- A. Canfrán-Duque, N. Rotllan, X. Zhang, M. Fernández-Fuertes, C. Ramírez-Hidalgo, E. Araldi, L. Daimiel, R. Busto, C. Fernández-Hernando and Y. Suárez, EMBO Mol. Med., 2017, 9, 1244–1266 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full spectroscopic data for new compounds 1 and 2, and computational details of 2. CCDC 1823042. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c8ob02519c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Li
Qin‡
a,
Biqing
Zhao‡
a,
Liang
Cai
c,
Zhenping
Zhong
d,
Yonghong
Liu
b and
Xiaojiang
Zhou
ab,
Li
Qin‡
a,
Biqing
Zhao‡
a,
Liang
Cai
c,
Zhenping
Zhong
d,
Yonghong
Liu
b and
Xiaojiang
Zhou







