
Synthesis of modified β-methoxyphenylalanines via diazonium chemistry and their incorporation in desoxycyclomarin analogues†
Alexander
Kiefer
and
Uli
Kazmaier
 *
*
Institute of Organic Chemistry, Saarland University, P.O. Box 151150, 66041 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
First published on 6th December 2018
Abstract
The marine natural products cyclomarins have remarkable anti-mycobacterial and antiplasmodial activities. The heptapeptic structure of this compound class comprisis four highly interesting non-canonical amino acids, including a rather unusual syn β-methoxyphenylalanine. To get a deeper insight into the structure–activity realtionship of cyclomarines, a straightforward protocol for the stereoselective synthesis of this building block was developed, based on diazonium chemistry.
Introduction
Cyclomarins A–C are unique all-(S) cyclic heptapeptides isolated from marine bacteria by Clardy et al. in 1999 (Fig. 1).1 Cyclomarin A, the major metabolite of the Streptomycetes strain CNB-982, inhibits the growth of Plasmodium falciparum (Pfalcp), the pathogen of Malaria tropica, in the blood stages with an IC50 value of 40 nM.2 In addition to the antiplasmodial activity, cyclomarin A also shows a significant antibacterial effect against Mycobacterium tuberculosis (Mtb)3 associated with an entirely new mode of action.4 Both biological activities have also been recently demonstrated for cyclomarin C.5 The fact that several representatives of these natural products treat two different pathogens simultaneously turns them into an extremely rare and highly interesting compound class for antiinfective research.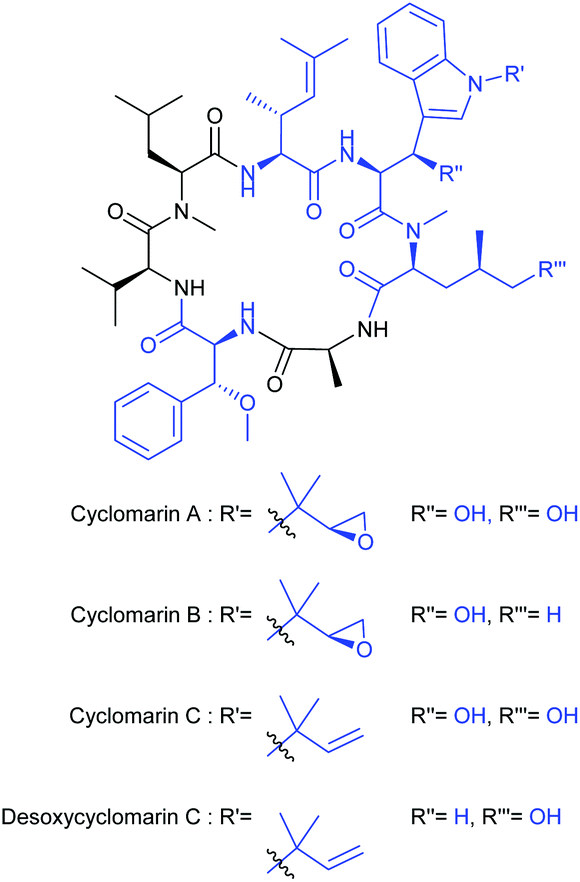 | ||
| Fig. 1 Structures of cyclomarin A–C and the simplified desoxycyclomarin C. | ||
Furthermore, a metabolite similar to cyclomarin C, M10709, was isolated from Streptomyces sp. IFM10709.6 In the latter, the unique structural motif of the aminohexenoic acid is replaced by an L-valine. In complement to the three proteinogenic amino acids (Ala, Val, N-MeLeu), the cyclopeptides differ slightly in the methylation and oxidation pattern of the non-canonical amino acids (blue).7,8 The β-hydroxytryptophan unit can be incorporated either as a N′-(1,1-dimethyl-2S,3-epoxypropyl)- or a N′-tert-prenyl residue.9 Interestingly, this structural motif and the δ-hydroxyleucine fragment are also found in another class of anti-tuberculosis substances, the ilamycins.10–14 The β-methoxyphenylalanine component derivatives thereof is also found in a number of natural products e.g. cytotoxic discokiolides15 or the antiviral papuamide16 depsipeptides, to mention some of them.
Due to their interesting biological activities, it is not surprising that there is also a great interest in the synthesis of these natural products and their derivatives. During the last years several interesting protocols especially for the preparation of syn-β-methoxyphenylalanines have been developed (Scheme 1). Yokokawa and co-workers17 used a classical Schöllkopf auxiliary approach to generate the corresponding amino alcohol A. In principle, this approach is also suitable for the synthesis of modified syn-β-methoxyphenylalanines, but starts from the rather expensive bislactim ether. In addition, the O-methylation was found to be a critical step, giving only moderate yields (51%) of the desired product. Yao et al.18 started from phthalate-protected phenylalanine, which was converted into the desired β-hydroxylated amino acid B in the course of a Wohl-Ziegler bromination and subsequent silver(I) mediated SN2 reaction using water. This approach is limited to the cyclomarine building block. Joullié et al.,19 on the other hand, have chosen a Grignard addition to a chiral serinaldehyde C.20 Despite the satisfactory diastereoselectivity in the addition step, the yield of D was unfortunately rather low. As an alternative, Kazmaier and Barbie used a titaniumaryl species generated from PhMgBr and Ti(OiPr)4 to obtain the desired amino alcohol E in high yield and excellent diastereoselectivity.21–23 Nevertheless, running the reaction in large-scale was infeasible. For structure–activity relationship (SAR) studies of natural products such as the cyclomarins it is important to have access not only to the core structures itself, but also to derivatives thereof.24–27 Here, we report on the synthesis of novel (S)-syn-β-methoxyphenylalanine derivatives and their incorporation into the cyclomarin skeleton.
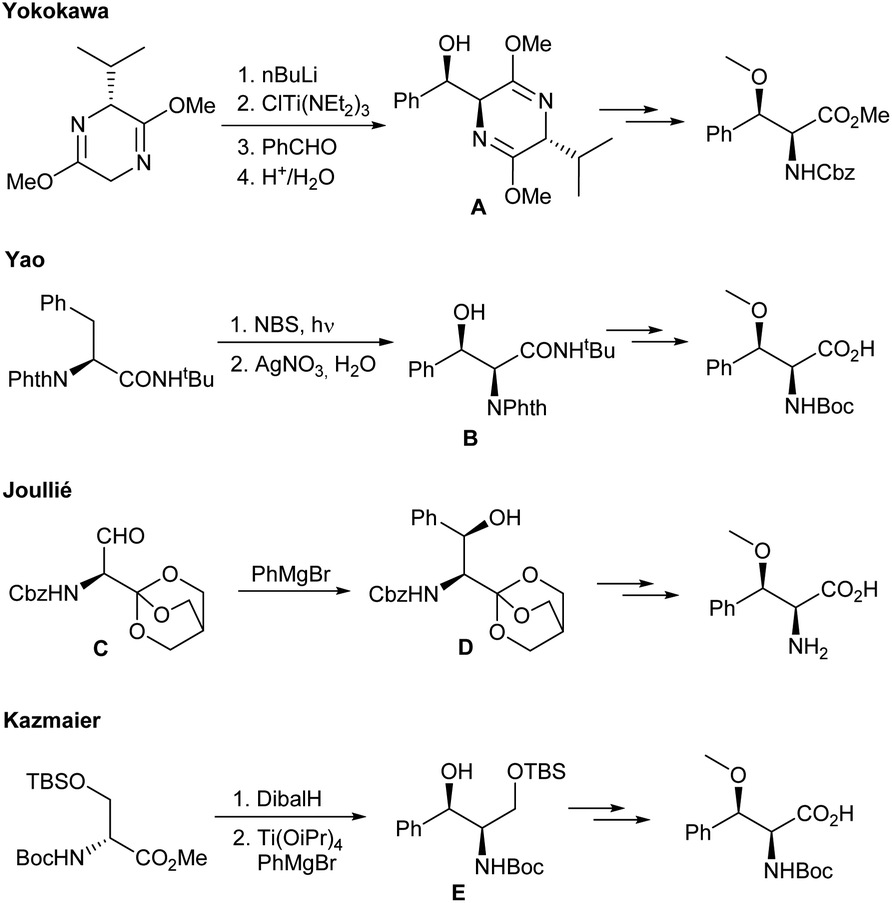 | ||
| Scheme 1 Established methods for the synthesis of syn-β-methoxyphenylalanines. | ||
Results and discussion
We commenced our work in an ex-chiral pool strategy with the commercially available chloramphinicol base 1 (Scheme 2). Thus, amino diol 1 was first converted stepwise with Boc2O and TBS-Cl in the presence of imidazole into the protected derivative 2. Subsequent methylation using methyl iodide and LHMDS at −15 °C quantitatively resulted in the O-methylated product 3a. No side reactions, such as the formation of N-methylated or double methylated product, were observed. Nitro compound 3a could easily be reduced to the corresponding aniline derivative 3b, which is an ideal candidate for further structural variations via diazonium chemistry. In an initial attempt, using tBuONO for diazotation, we used pTsOH to generate the diazonium salt, which should then be reacted with KBr.28 However, this approach led to complete decomposition of the starting material already during the addition of pTsOH. On the other hand, utilizing stoichiometric amounts of copper(II) bromide at 60 °C provided the desired product, although contaminated with a byproduct which could not be separated.29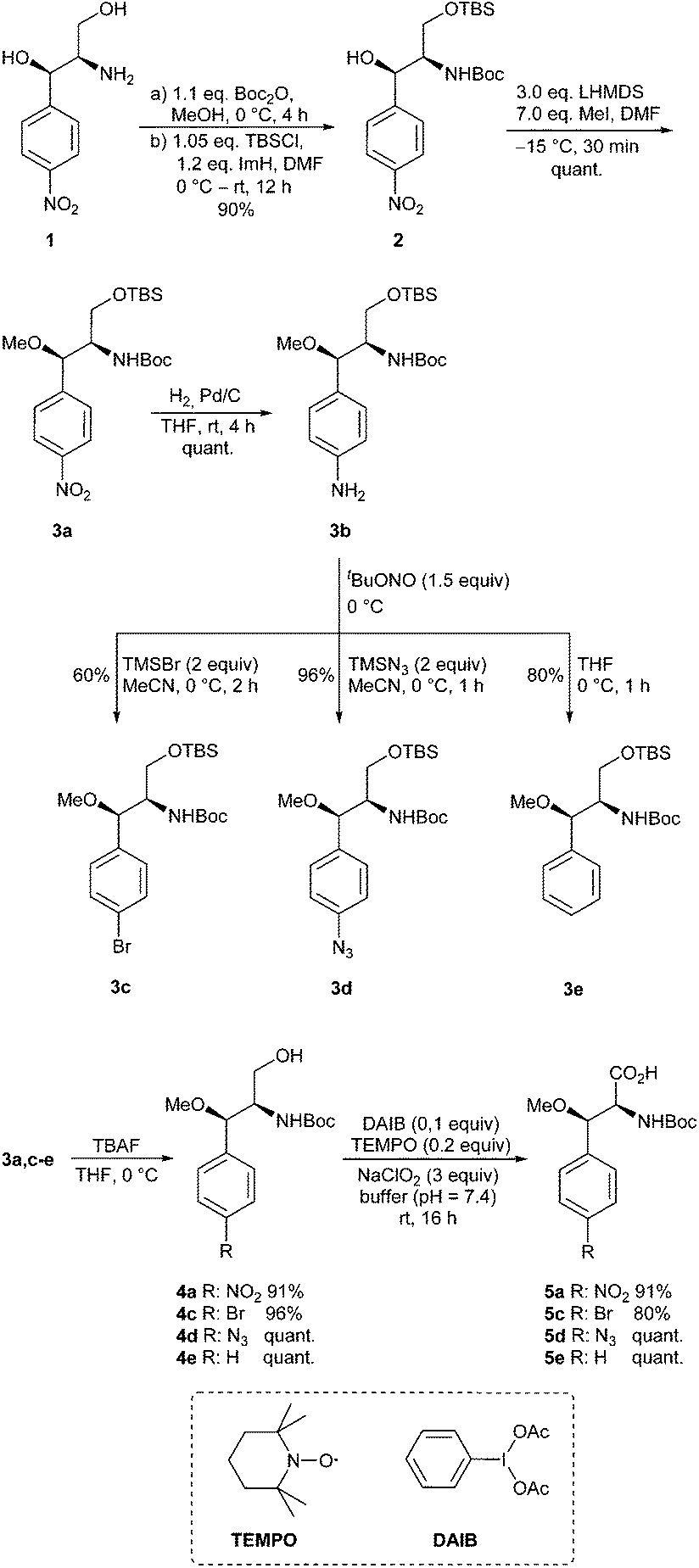 | ||
| Scheme 2 Synthesis of the modified β-methoxyphenylalanines. | ||
Reducing the reaction temperature resulted predominantly in deaminated product, while catalytic amounts of copper(II) bromide in combination with TBAB gave the product only in traces.30 Finally, TMS-Br led to success and the desired product 3c could be isolated in satisfactory yield. Formation of the corresponding azide 3d was easily achieved in 96% according to a literature procedure by Moses et al.31 For the unsubstituted β-methoxyphenylalanine found in cyclomarines, aniline 3b also served as a precursor. Therefore, the deamination using tBuONO without additives had to be optimized.32,33 It is well known that a proper choice of the solvent is essential for good yields.34,35 First attempts in DMF, either at 60 or 0 °C, gave only moderate yields (25–28%). By changing the solvent to the less polar acetonitrile 3e was obtained in 55% yield, but by far the best results were obtained with the even less polar THF, which serves as a hydrogen source.32,33
In the following sequence, the primary alcohol functionalities of 3 were liberated with TBAF and a modified Epp-Widlanski oxidation36,37 provided the desired carboxylic acids in excellent isolated yields over two steps.
With these building blocks in hand we turned to the successive assembly of the peptide backbone of the desired desoxycyclomarin analogues (Scheme 3). Starting with the formation of the tripeptides 7, the N-terminus of dipeptide 622 was released by hydrogenolytic cleavage of the Cbz group. To avoid the formation of the corresponding dioxopiperazine, the reaction was carried out in the presence of HCl. For the coupling step, the modified β-methoxyphenylalanines 5 were activated at −20 °C with isobutyl chloroformate (IBCF)38 as mixed anhydrides and reacted with the dipeptide salt to the tripeptides 7. Removal of the N-terminal Boc-protecting group and coupling with N-Boc-L-Ala-OH 8 yielded the tetrapeptides 9, which were further converted into the desired pentapeptides 11. To avoid side reactions with the functionalized β-methoxyphenylalanine units, the N-terminal Alloc-protecting group was gently removed utilizing Pd-catalysis.39 Within one hour the pentapeptides 11 were completely deprotected without the formation of by-products.
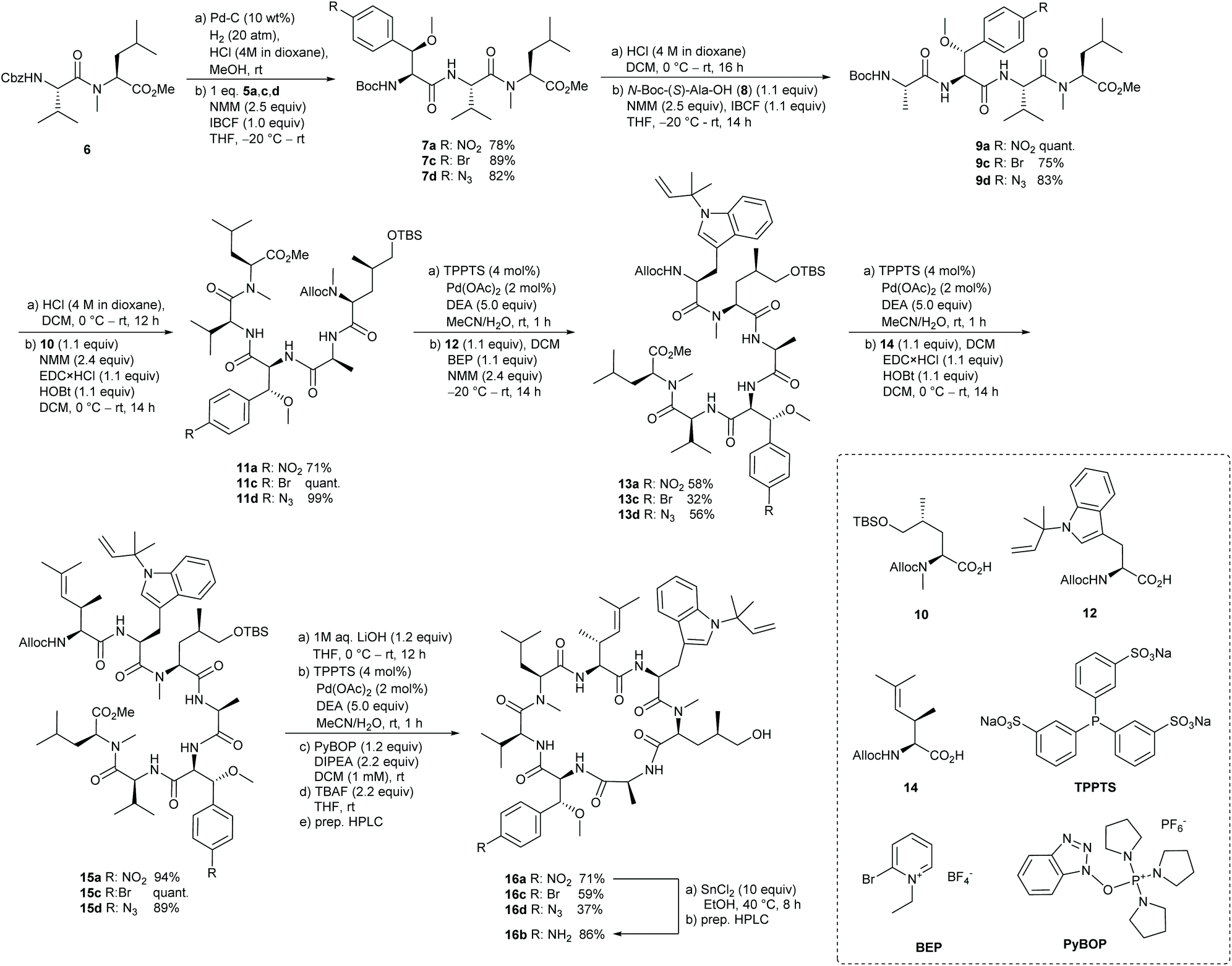 | ||
| Scheme 3 Synthesis of modified desoxycyclomarin analogues. | ||
Subsequent 2-bromo-1-ethyl-pyridinium tetrafluoroborate (BEP)40,41 mediated coupling with N′-tert-prenyl tryptophan 1242,43 afforded the desired hexapeptides 13 in satisfactory yield. Assembly to the linear heptapeptides 15 with γ,δ-unsaturated amino acid 14 was realized by utilizing EDC/HOBt as activating agents.23 The desired precursors could be obtained in yields >89% over two steps. To complete the synthesis of the cyclopeptide analogues, the C-terminal ester function was saponified with lithium hydroxide, before the N-termini were deprotected under mild Pd-catalysis. The macrolactamization was carried out according to Yao et al. with two equivalents PyBOP and DIPEA as base under high dilution conditions (1 mM).44 In the final step, the TBS ether was cleaved with TBAF providing desoxycyclomarin derivatives 16a,c,d in good yields after preparative HPLC. Furthermore, the nitro group of derivative 16a could be selectively reduced to the corresponding amine using tin(II) chloride in ethanol, to afford 16b.45
With the four generated analogues, cyclomarin C, desoxycyclomarin C (16e) and isoniazid as standard in hand, they were tested for their biological activities against Mtb wild-type strain Erdman. The in vitro growth inhibition was determined by a resazurin reduction microtiter assay (REMA)46 and the minimal inhibitory concentrations (MIC) of the compounds are in submicromolar range (Table 1).
| Entry | Compound | MIC [μM] |
|---|---|---|
| 1 | Isoniazid | 0.90 |
| 2 | Cyclomarin C | 0.25 |
| 3 | Desoxycyclomarin C (16e) | 0.93 |
| 4 | 16a | 0.13 |
| 5 | 16b | 0.25 |
| 6 | 16c | 4.09 |
| 7 | 16d | 0.26 |
Desoxycyclomarin C (16e) was around 4 times less potent than cyclomarin C, but still as active as our reference compound isoniazid. The amino- and azido-derivatives 16b and 16c were equipotent compared to cyclomarin C, and nitrocompound 16a was even twice as active. Only in case of bromo-derivative 16c a significant drop in the activity was observed. The reason for that is not clear so far and will be investigated in more detail.
Conclusions
We have synthesized a small library of novel desoxycyclomarin C analogues via diazonium ion chemistry and have evaluated their antitubercular activities to deepen our understanding of the structure–activity relationship. While the bioactivity is mostly in the same range like desoxycyclomarin C, the synthetic protocol might provide a good option for further side chain modifications to develop more potent anti-mycobacterial drugs.Experimental
General experimental details
All air- or moisture-sensitive reactions were carried out in dried glassware (>100 °C) under an atmosphere of nitrogen or argon. Dried solvents were distilled before use. The products were purified by flash chromatography on silica gel columns (Macherey-Nagel 60, 0.04–0.063 mm). Analytical TLC was performed on pre-coated silica gel plates (Macherey-Nagel, Polygram® SIL G/UV254). Visualization was accomplished with UV-light, KMnO4 or a ceric ammonium molybdate chamber. Preparative high performance liquid chromatography (HPLC) was performed on a Waters Autopurifier System (APS) with a Phenomenex Gemini C18 column (250 × 4.6 mm, particle size 5 μm) as an analytical column for method development and a Phenomenex Gemini C18 column (250 × 19 mm, particle size 5 μm) for preparative separation. Detection was performed using mass trigger. 1H and 13C spectra were recorded with a Bruker AV II 400 [400 MHz (1H), 100 MHz (13C)], a Bruker AV 500 [500 MHz (1H), 125 MHz (13C)] or a AV 700 [700 MHz (1H), 175 MHz (13C)] spectrometer in CDCl3 or methanol-d4 unless otherwise specified. NMR spectra were evaluated using Mestrec. Chemical shifts are reported in ppm relative to Si(CH3)4 and CHCl3 or CHD2OD was used as the internal standard [CHCl3: δ(1H) = 7.26 ppm, δ(13C) = 77.0 ppm, CHD2OD: δ(1H) = 3.31 ppm, δ(13C) = 49.05 ppm]. LC-MS analysis was performed with a Shimadzu system (LC: 10A-series with Autosampler, MS: LCMS-2020). High resolution mass spectra were recorded with a Finnigan MAT 95 spectrometer using the CI technique (M < 600 g mol−1) or with an Bruker maXis 4G UHR-TOF-spectrometer using the ESI technique (M > 600 g mol−1). Optical rotations were measured with a PerkinElmer polarimeter (model 341) in a thermostated (20 °C ± 1 °C) cuvette. The radiation source used was a sodium vapour lamp (λ = 589 nm). The concentrations are given in g per 100 mL. Melting points were determined with a melting point apparatus MEL-TEMP II by Laboratory devices and are uncorrected.General procedure A: TBAF deprotection of 3
To a solution of the TBS-protected methyl ether 3 in THF (0.15 M) TBAF (1.1 equiv., 1 M in THF) was added at 0 °C and the mixture was stirred for 4 h at room temperature. After full conversion (TLC), the reaction mixture was diluted with ethyl acetate and washed with 1 N aq. KHSO4 and brine. After drying (Na2SO4) and removal of the solvent, the residue was purified by column chromatography.General procedure B: oxidation of 4
The alcohol 4 was dissolved in a mixture of acetonitrile and 1 mM aq. phosphate buffer pH = 6.4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.25 M). To the resulting solution, PhI(OAc)2 (0.1 equiv.), TEMPO (0.2 equiv.) and NaClO2 (3.5 equiv., 80%) were added at 0 °C. The mixture was stirred overnight at room temperature and quenched by addition of a solution of 2 M aq. Na2CO3. After stirring for 10 min, the mixture was washed with ether and the aqueous layer was acidified with 1 N aq. HCl. After extraction with ethyl acetate (3 times), the combined organic layers were dried over Na2SO4 and the solvent was removed to obtain the desired acid 5.
:1, 0.25 M). To the resulting solution, PhI(OAc)2 (0.1 equiv.), TEMPO (0.2 equiv.) and NaClO2 (3.5 equiv., 80%) were added at 0 °C. The mixture was stirred overnight at room temperature and quenched by addition of a solution of 2 M aq. Na2CO3. After stirring for 10 min, the mixture was washed with ether and the aqueous layer was acidified with 1 N aq. HCl. After extraction with ethyl acetate (3 times), the combined organic layers were dried over Na2SO4 and the solvent was removed to obtain the desired acid 5.
General procedure C: synthesis of the tripeptides 7 using isobutyl chloroformate
To a solution of N-Cbz-(S)-Val-(S)-N-methyl-Leu-OMe 6 (1.1 equiv.) in methanol (0.1 M) HCl (2.2 equiv., 4 M in dioxane) and 10% wt. Pd–C (10% on charcoal) were added. The reaction mixture was stirred in an autoclave at 20 atm H2-atmosphere until complete conversion (TLC). After removing the solvent under reduced pressure, the crystalline hydrochloride was dissolved in dichloromethane (0.2 M). The corresponding β-methoxyphenylalanine 5 (1.0 equiv.) was dissolved in THF (0.1 M) and cooled down to −20 °C. Then NMM (2.5 equiv.) and isobutyl chloroformate (1.0 equiv.) were added dropwise. The reaction mixture was stirred at this temperature for 30 minutes and then the dipeptide hydrochloride solution was added. After the reaction mixture had reached room temperature, it was diluted with ether after full conversion (TLC). The organic phase was washed with 1 N KHSO4 solution, sat. NaHCO3 solution and brine, dried (Na2SO4) and the solvent was removed at the rotary evaporator. Purification by column chromatographic afforded the corresponding tripeptide 7.General procedure D: synthesis of the tetrapeptides 9 using isobutyl chloroformate
Tripeptide 7 (1 equiv.) was dissolved in dichloromethane (0.1 M) and treated at 0 °C with HCl (10.0 equiv., 4 M in dioxane). The mixture was warmed up to room temperature and after complete conversion (TLC) the solvent was removed under reduced pressure to give the crystalline hydrochloride remained, which was subsequently dissolved in dichloromethane (0.1 M). N-Boc-(S)-alanine 8 (1.1 equiv.) in THF (0.2 M) was cooled down to −20 °C and NMM (2.5 equiv.) and isobutyl chloroformate (2.1 equiv.) were added dropwise. The reaction mixture was stirred at this temperature for 30 minutes before the tripeptide hydrochloride solution was added. After the reaction mixture had reached room temperature (full conversion), it was diluted with ethyl acetate. The organic phase was washed with 1 N KHSO4 solution, sat. NaHCO3 solution and brine. Drying over Na2SO4, removal of the solvent in vacuo and purification by column chromatographic gave the corresponding tetrapeptide 9.General procedure E: synthesis of the pentapeptides 11 using EDC/HOBt coupling reagents
Boc deprotection of the corresponding tetrapeptide 9 (1.0 equiv.) was performed in dichloromethane (0.1 M) using HCl (10.0 equiv., 4 M in dioxane). After complete conversion (TLC) the solvent was removed under reduced pressure and the residue was dissolved in dichloromethane (0.1 M). Protected δ-hydroxyleucine 10 (1.2 equiv.) was added to this solution and the reaction mixture was cooled to 0 °C. Coupling was achieved by EDC·HCl (1.1 equiv.), HOBt (1.0 equiv.) and NMM (2.2 equiv.). The reaction mixture was warmed up to room temperature and after complete reaction (TLC) diluted with ethyl acetate and washed successively with 1 N KHSO4 solution, sat. NaHCO3 solution and brine. After drying over Na2SO4 and removing the solvent under reduced pressure, the residue was purified column chromatographically to give the desired product 11.General procedure F: synthesis of the hexapeptides 13 using BEP coupling reagent
The corresponding pentapeptide 11 was dissolved in acetonitrile/H2O (1:1, 0.1 M) and diethylamine (5.0 equiv.), TPPTS (4 mol%) and Pd(OAc)2 (2 mol%, 0.02 M in acetonitrile) were added at room temperature. After full conversion (LC-MS) the mixture was concentrated in vacuo and the residue was dissolved in dichloromethane (0.5 M), mixed with N′-tert-prenylated tryptophan 12 (1.05 equiv.) and cooled down to −20 °C. NMM (2.2 equiv.) and BEP (1.1 equiv.) were added successively at this temperature. The reaction mixture was slowly warmed up 0 °C. After complete conversion (TLC), the solution was diluted with dichloromethane and washed with water, sat. NaHCO3 solution and brine. The organic phase was dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by column chromatography.
General procedure G: synthesis of heptapetides 15 using EDC/HOBt coupling reagents
The corresponding hexapeptide 13 was dissolved in acetonitrile/H2O (1:1, 0.1 M) and diethylamine (5.0 equiv.), TPPTS (4 mol%) and Pd(OAc)2 (2 mol%, 0.02 M in acetonitrile) were added at room temperature. After one hour (LC-MS) the solvent was removed under reduced pressure and the residue was dissolved in dichloromethane (0.5 M). After cooling to 0 °C, the γ,δ-unsaturated amino acid 14 (1.10 equiv.), EDC·HCl (1.1 equiv.), HOBt (1.0 equiv.) and NMM (2.2 equiv.) were added. The reaction mixture was warmed up room temperature and after complete conversion (LC-MS) diluted with dichloromethane and washed successively with sat. NaHCO3 solution, 1 N KHSO4 solution and brine. The organic phase was dried over Na2SO4 and the solvent was removed under reduced pressure. Flash chromatography afforded the desired product.
General procedure H: cyclization and TBS-deprotection
LiOH (1.5 equiv., 1.0 M in H2O) was added to a solution of a heptapeptide 15 in THF (0.1 M) at 0 °C and then stirred overnight at room temperature. After complete conversion (LC-MS), the solvent was removed under reduced pressure and the residue was dissolved in acetonitrile/H2O (1:1, 0.1 M) and treated at room temperature with diethylamine (5.0 equiv.), TPPTS (4 mol%) and Pd(OAc)2 (2 mol%, 0.02 M in acetonitrile). After complete conversion (LC-MS) the solvent was removed and the residue was diluted in dichloromethane (2 mM). PyBOP (2.0 equiv.) and DIPEA (2.2 equiv.) were also dissolved in dichloromethane (4 mM). The peptide solution was transferred to the coupling reagent via transfer cannula within 6 hours. After 24 h stirring at room temperature, the solution was washed with 1 N KHSO4 solution, sat. NaHCO3 solution and brine. After drying the organic layer over Na2SO4 and evaporation of the solvent, the residue was dissolved in THF (0.5 M) and mixed with 2.2 equiv. TBAF (1 M in THF) at room temperature. After complete conversion (LC-MS), the solvent was removed under reduced pressure and the residue was filtered via a short silica gel column with ethyl acetate and then purified via preparative HPLC.
(2R,3R)-N-Boc-3-hydroxy-1-O-(tert-butyldimethylsilyl)-para-nitrophenylalaninol (2)
(2R,3R)-3-Hydroxy-para-nitrophenylalaninol 1 (4.0 g, 18.85 mmol) was suspended in methanol (10 mL) at 0 °C. After portion-wise addition of Boc2O (4.73 g, 20.73 mmol) the reaction mixture was stirred for 4 h at the same temperature. The crude mixture was concentrated and the residue was dissolved in DMF (20 mL). After cooling down to 0 °C imidazole (1.54 g, 22.62 mmol) and TBS-Cl (2.98 g, 19.79 mmol) were added. After stirring over night at room temperature the reaction mixture was taken up in ether (200 mL), washed twice with water, 1 N aq. KHSO4, sat. aq. NaHCO3 and brine. The solvent was removed after drying (Na2SO4). Column chromatography (silica gel, petroleum ether/ethyl acetate 8:2) yielded the protected para-nitrophenylalaninol 2 (7.27 g, 17.04 mmol, 90%) as colourless oil. TLC: Rf(2) = 0.35 (petroleum ether/ethyl acetate = 8:2). [α]20D = −99.3 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 0.11 (s, 6 H), 0.94 (s, 9 H), 1.32 (s, 9 H), 3.83 (m, 1 H), 3.88 (d, J = 3.2 Hz, 2 H), 4.10 (bs, 1 H, OH), 5.13 (d, J = 9.2 Hz, 1 H), 5.16 (bs, 1 H), 7.54 (d, J = 8.4 Hz, 2 H), 8.19 (d, J = 8.4 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = −5.6, 18.1, 25.8, 28.2, 55.8, 65.6, 74.5, 79.9, 123.4, 126.9, 147.3, 148.6, 155.9 ppm. HRMS (CI) m/z calculated for C20H35N2O6Si [M + 1]+ 427.2264; found, 427.2232.
(2R,3R)-N-Boc-3-methoxy-1-O-(tert-butyldimethylsilyl)-para-nitrophenylalaninol (3a)
To a solution of secondary alcohol 2 (5.67 g, 13.30 mmol) in DMF (130 mL) LHMDS (39.9 mL, 39.90 mmol, 1 M THF) was slowly added at −15 °C, followed by methyl iodide (5.82 mL, 93 mmol). After stirring for 30 min at this temperature, the mixture was quenched with water and extracted with ether (3 times). The combined organic layers were washed with 1 N aq. KHSO4, sat. aq. NaHCO3 and brine. After drying (Na2SO4) the solvent was removed under reduced pressure. Purification via column chromatography (silica gel, petroleum ether/ethyl acetate 8:2) gave the desired compound 3a (5.86, 13.30 mmol, quant.) as colourless oil. TLC: Rf(3a) = 0.50 (petroleum ether/ethyl acetate = 8:2). [α]20D = −125.2 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 0.08 (s, 3 H), 0.09 (s, 3 H), 0.02 (s, 9 H), 1.30 (s, 9 H), 3.29 (s, 3 H), 3.55 (dd, J = 9.4 Hz, J = 4.8 Hz, 1 H), 3.64 (dd, J = 9.4 Hz, J = 8.4 Hz, 1 H), 3.77 (m, 1 H), 4.62 (d, J = 1.9 Hz, 1 H), 4.80 (d, J = 9.4 Hz, 1 H), 7.47 (d, J = 8.5 Hz, 2 H), 8.20 (d, J = 8.4 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = −5.6, −5.4, 18.2, 25.8, 28.2, 56.9, 57.7, 61.7, 79.4, 79.9, 123.4, 126.9, 147.4, 147.5, 155.2 ppm. HRMS (CI) m/z calculated for C21H36N2O6Si [M]+ 440.2343; found, 440.2371.
(2R,3R)-N-Boc-3-methoxy-1-O-(tert-butyldimethylsilyl)-para-aminophenylalaninol (3b)
To the fully protected para-nitrophenylalaninol 3a (5.86 g, 13.30 mmol) in THF (133 mL) Pd–C (10% on charcoal) was added. The resulting mixture was stirred for 4 h at room temperature under H2 (1 atm), filtered over Celite® and concentrated under reduced pressure. The corresponding product 3b (5.30 g, 12.90 mmol, 97%) was obtained as white powder. TLC: Rf(3b) = 0.13 (petroleum ether/ethyl acetate = 8:2). [α]20D = −53.4 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 0.03 (s, 3 H), 0.05 (s, 3 H), 0.91 (s, 9 H), 1.39 (s, 9 H), 3.20 (s, 3 H), 3.42 (dd, J = 9.7 Hz, J = 3.4 Hz, 1 H), 3.64 (dd, J = 9.7 Hz, J = 6.9 Hz, 1 H), 3.77 (m, 2 H), 4.30 (d, J = 4.2 Hz, 1 H), 4.89 (d, J = 7.1 Hz, 1 H), 6.64 (d, J = 8.3 Hz, 2 H), 7.07 (d, J = 8.3 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = −5.6, −5.4, 18.2, 25.8, 28.3, 56.8, 61.9, 78.8, 80.7, 114.9, 128.2, 129.1, 145.8, 155.6 ppm. HRMS (CI) m/z calculated for C21H38N2O4 [M]+ 410.2601; found, 410.2595.
(2R,3R)-N-Boc-3-methoxy-1-O-(tert-butyldimethylsilyl)-para-bromophenylalaninol (3c)
Aniline 3b (100 mg, 244 μmol) was dissolved in acetonitrile (1 mL) and cooled to 0 °C. To this stirred mixture tert-BuONO (48 μL, 365 μmol) was added, followed by TMS-Br (66 μL, 487 μmol) dropwise. The resulting solution was stirred at room temperature for 2 h and diluted with ether (30 mL). After washing with water and brine, drying over Na2SO4, the solvent was removed. Purification via column chromatography (silica gel, petroleum ether/ethyl acetate 9:1) gave colourless oil 3c (69.0 mg, 145.0 μmol, 60%). TLC: Rf(3c) = 0.56 (petroleum ether/ethyl acetate = 8:2). [α]20D = −89.9 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 0.06 (s, 3 H), 0.07 (s, 3 H), 0.91 (s, 9 H), 1.35 (s, 9 H), 3.24 (s, 3 H), 3.46 (dd, J = 9.1 Hz, J = 4.0 Hz, 1 H), 3.62 (dd, J = 9.1 Hz, J = 7.9 Hz, 1 H), 3.72 (m, 1 H), 4.43 (d, J = 3.0, 1 H), 4.82 (d, J = 8.9 Hz, 1 H), 7.17 (d, J = 8.3 Hz, 2 H), 7.45 (d, J = 8.3 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = −5.6, −5.4, 18.2, 25.9, 28.3, 57.0, 57.3, 61.7, 79.2, 80.1, 121.4, 128.6, 131.4, 138.5, 155.5 ppm. HRMS (CI) m/z calculated for C21H37BrN1O4 [M + 1]+ 474.1675; found, 474.1637.
(2R,3R)-N-Boc-3-methoxy-1-O-(tert-butyldimethylsilyl)-para-azidophenylalaninol (3d)
Aniline 3b (100 mg, 244 μmol) was dissolved in acetonitrile (1 mL) and cooled to 0 °C. To this stirred mixture tert-BuONO (48 μL, 365 μmol) was added dropwise, followed by TMSN3 (68 μL, 487 μmol). The resulting solution was stirred at room temperature for 12 h and diluted with ether (30 mL). After washing with water and brine, drying over Na2SO4, the solvent was removed in vacuo. Purification via column chromatography (silica gel, petroleum ether/ethyl acetate 9:1) gave colourless oil 3d (102.0 mg, 234.0 μmol, 96%). TLC: Rf(3d) = 0.43 (petroleum ether/ethyl acetate = 8:2). [α]20D = −69.8 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 0.04 (s, 3 H), 0.05 (s, 3 H), 0.88 (s, 9 H), 1.34 (s, 9 H), 3.22 (s, 3 H), 3.47 (dd, J = 9.0 Hz, J = 3.7 Hz, 1 H), 3.64 (dd, J = 9.0 Hz, J = 7.7 Hz, 1 H), 3.69 (m, 1 H), 4.42 (m, 1 H), 4.83 (d, J = 8.9 Hz, 1 H), 6.97 (d, J = 8.3 Hz, 2 H), 7.28 (d, J = 8.3 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = −5.6, −5.4, 18.2, 25.9, 28.3, 57.1, 57.2, 61.8, 79.1, 80.2, 118.9, 128.4, 139.3, 155.5 ppm. HRMS (CI) m/z calculated for C21H37N4O4Si [M + 1]+ 437.2584; found, 437.2563.
(2R,3R)-N-Boc-3-methoxy-1-O-(tert-butyldimethylsilyl)-phenylalaninol (3e)
Aniline 3b (100.0 mg, 244.0 μmol) was dissolved in THF (2.5 mL) and cooled to 0 °C. To this stirred mixture tert-BuONO (58.0 μL, 487.0 μmol) was added dropwise. The resulting solution was warmed to room temperature for 3 h and diluted with ether (30 mL). After washing with water and brine, drying over Na2SO4, the solvent was removed in vacuo. Purification via column chromatography (silica gel, petroleum ether/ethyl acetate 9:1) gave colourless oil 3e (77.0 mg, 195.0 μmol, 80%). TLC: Rf(3e) = 0.62 (petroleum ether/ethyl acetate = 8:2). [α]20D = −52.0 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 0.02 (s, 3 H), 0.03 (s, 3 H), 0.88 (s, 9 H), 1.31 (s, 9 H), 3.22 (s, 3 H), 3.43 (dd, J = 9.7 Hz, J = 3.7 Hz, 1 H), 3.60 (dd, J = 9.7 Hz, J = 7.5 Hz, 1 H), 3.71 (m, 1 H), 4.42 (d, J = 3.3, 1 H), 4.85 (d, J = 8.3 Hz, 1 H), 7.20–7.31 (m, 5 H) ppm. 13C NMR (100 MHz, CDCl3): δ = −5.5, −5.4, 18.2, 25.9, 28.3, 57.3, 61.8, 79.0, 80.7, 126.9, 127.6, 128.2, 139.2, 155.5 ppm. HRMS (CI) m/z calculated for C21H38NO4Si [M + H]+ 396.2565; found, 396.2573.
(2R,3R)-N-Boc-3-methoxy-para-nitrophenylalaninol (4a)
According to general procedure A TBS protected methyl ether 3a (291.0 mg, 660.0 μmol) was reacted with TBAF (793.0 μL, 793.0 μmol, 1 M in THF) to the corresponding alcohol 4a. After purification via column chromatography (silica gel, dichloromethane/ethyl acetate 1:1) alcohol 4a (197.0 mg, 604.0 μmol, 91%) was obtained as colourless oil. TLC: Rf(4a) = 0.32 (dichloromethane/ethyl acetate = 1:1). [α]20D = −135.2 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 1.30 (s, 9 H), 2.57 (bs, 1 H), 3.30 (s, 3 H), 3.72–3.74 (m, 2 H), 3.76–3.77 (m, 1 H), 4.61 (bs, 1 H), 5.05 (bs, 1 H), 7.49 (d, J = 8.6 Hz, 2 H), 8.21 (d, J = 8.6 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 28.1, 57.6, 63.2, 79.8, 82.1, 123.6, 127.7, 146.4, 147.6, 155.6 ppm. HRMS (CI) m/z calculated for C15H23N2O6 [M + 1]+ 327.1556; found, 327.1550.
(2R,3R)-N-Boc-3-methoxy-para-bromophenylalaninol (4c)
According to general procedure A TBS protected methyl ether 3c (175.0 mg, 369.0 μmol) was reacted with TBAF (406.0 μL, 406.0 μmol, 1 M in THF) to the corresponding alcohol 4c. After purification via column chromatography (silica gel, petroleum ether/ethyl acetate 1:1) alcohol 4c (128 mg, 355 μmol, 96%) was obtained as colourless oil. TLC: Rf(4c) = 0.27 (petroleum ether/ethyl acetate = 1:1). [α]20D = −130.7 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 1.34 (s, 9 H), 2.63 (bs, 1 H), 3.25 (s, 3 H), 3.66–3.74 (m, 3 H), 4.44 (d, J = 2.7, 1 H), 5.0 (bs, 1 H), 7.18 (d, J = 8.4 Hz, 2 H), 7.47 (d, J = 8.4 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 28.2, 57.2; 63.5, 79.6, 82.5, 121.9, 128.7, 131.6, 137.5, 155.9 ppm. HRMS (CI) m/z calculated for C15H23BrN1O4 [M + 1]+ 360.0810; found, 360.0801.
(2R,3R)-N-Boc-3-methoxy-para-azidophenylalaninol (4d)
According to general procedure A TBS protected methyl ether 3d (100.0 mg, 229.0 μmol) was reacted with TBAF (275.0 μL, 275.0 μmol, 1 M in THF) to the corresponding alcohol 4d. After purification via column chromatography (silica gel, petroleum ether/ethyl acetate 1:1) the alcohol 4d (74 mg, 229 μmol, quant.) was obtained as colourless oil. TLC: Rf(4d) = 0.40 (petroleum ether/ethyl acetate = 1:1). [α]20D = −101.3 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 1.35 (s, 9 H), 3.24 (s, 3 H), 3.66–3.74 (m, 3 H), 4.42 (d, J = 3.2, 1 H), 5.0 (bs, 1 H), 7.01 (d, J = 8.4 Hz, 2 H), 7.29 (d, J = 8.4 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 28.2, 57.1, 63.6, 79.6, 82.6, 119.1, 128.4, 135.2, 139.7, 155.9 ppm. HRMS (CI) m/z calculated for C15H23N4O4 [M + 1]+ 323.1719; found, 323.1731.
(2R,3R)-N-Boc-3-methoxy-phenylalaninol (4e)
According to general procedure A TBS protected methyl ether 3e (387.0 mg, 977.0 μmol) was reacted with TBAF (1.075 mL, 1.075 mmol, 1 M in THF) to the corresponding alcohol 4e. After purification via column chromatography (silica gel, petroleum ether/ethyl acetate 1:1) alcohol 4e (275.0 mg, 977.0 μmol, quant.) was obtained as colourless oil. TLC: Rf(4e) = 0.35 (petroleum ether/ethyl acetate = 1:1). [α]20D = −42.3 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 1.34 (s, 9 H), 3.26 (s, 3 H), 3.67–3.79 (m, 3 H), 4.42 (d, J = 4.2, 1 H), 5.1 (bs, 1 H), 7.28–7.35 (m, 5 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 28.2, 57.1, 63.8, 79.5, 82.2, 126.9, 127.9, 128.4, 138.2 ppm. HRMS (CI) m/z calculated for C15H24NO4 [M + H]+ 282.1700; found, 282.1706.
(2S,3R)-N-Boc-3-methoxy-para-nitrophenylalanine (5a)
According to general procedure B alcohol 4a (181.0 mg, 555.0 μmol) was reacted with PhI(OAc)2 (18.0 mg, 55.0 μmol), TEMPO (17.0 mg, 110.0 μmol) and NaClO2 (219.0 mg, 1.94 mmol) to the nitrophenylalanine 5a (172.0 mg, 505.0 μmol, 91%) as an colourless oil. TLC: Rf(5a) = 0.42 (petroleum ether/ethyl acetate = 1:1). [α]20D = −37.4 (c = 1.0, methanol). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 1.29 (s, 9 H), 3.35 (s, 3 H), 4.60 (dd, J = 9.6 Hz, J = 2.4 Hz, 1 H), 4.99 (d, J = 2.2 Hz, 1 H), 5.31 (d, J = 9.6 Hz, 1 H), 7.53 (d, J = 8.5 Hz, 2 H), 8.24 (d, J = 8.5 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 28.1, 58.1, 58.5, 80.6, 81.8, 123.7, 127.8, 144.5, 147.9, 155.3, 174.3 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 1.16 (s, 9 H), 3.35 (s, 3 H), 4.41 (dd, J = 9.6 Hz, J = 2.4 Hz, 1 H), 4.92 (d, J = 2.2 Hz, 1 H), 5.91 (d, J = 9.6 Hz, 1 H), 7.57 (m, 2 H), 8.21 (m, 2 H) ppm. HRMS (ESI+) m/z calculated for C15H21N2O7 [M + 1]+ 341.1343; found, 341.128.
(2S,3R)-N-Boc-3-methoxy-para-bromophenylalanine (5c)
According to general procedure B alcohol 4c (105.0 mg, 291.0 μmol) was reacted with PhI(OAc)2 (9.4 mg, 29 μmol), TEMPO (9.1 mg, 58.0 μmol) and NaClO2 (115.0 mg, 1.02 mmol) to acid 5c (86.0 mg, 230.0 μmol, 80%) as an colourless oil. TLC: Rf(5c) = 0.40 (petroleum ether/ethyl acetate = 1:1). [α]20D = −47.3 (c = 1.0, methanol). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, methanol-d4): δ = 1.31 (s, 9 H), 3.25 (s, 3 H), 4.33 (d, J = 3.1 Hz, 1 H), 4.82 (d, J = 3.1 Hz, 1 H), 7.28 (d, J = 8.3 Hz, 2 H), 7.50 (d, J = 8.3 Hz, 2 H) ppm. 13C NMR (100 MHz, methanol-d4): δ = 28.6, 57.8, 60.4, 80.7, 83.7, 122.8, 130.3, 132.5, 138.5, 157.8, 173.4 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, methanol-d4): δ = 1.16 (s, 9 H), 4.30 (m, 1 H), 4.79 (m, 1 H), 7.27 (d, J = 8.3 Hz, 2 H), 7.53 (d, J = 8.3 Hz, 2 H) ppm. HRMS (CI) m/z calculated for C15H21BrNO5 [M + 1]+ 374.0603; found, 374.0588.
(2S,3R)-N-Boc-3-methoxy-para-azidophenylalanine (5d)
According to general procedure B alcohol 4d (65.0 mg, 202.0 μmol) was reacted with PhI(OAc)2 (6.5 mg, 20.0 μmol), TEMPO (6.3 mg, 40 μmol) and NaClO2 (80.0 mg, 706.0 μmol) to the corresponding acid 5d (68 mg, 202 μmol, quant.) as an colourless oil. TLC: Rf(5d) = 0.18 (petroleum ether/ethyl acetate = 1:1). [α]20D = −56.3 (c = 1.0, methanol). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, methanol-d4): δ = 1.35 (s, 9 H), 3.24 (s, 3 H), 4.32 (d, J = 2.9 Hz, 1 H, 5-H), 4.81 (d, J = 2.9 Hz, 1 H, 6-H), 7.05 (d, J = 8.3 Hz, 2 H), 7.39 (d, J = 8.3 Hz, 2 H) ppm. 13C NMR (100 MHz, methanol-d4): δ = 28.6, 57.8, 60.5, 80.6, 83.7, 119.9, 129.9, 136.1, 141.1, 157.8, 173.5 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, methanol-d4): δ = 1.18 (s, 9 H), 4.29 (m, 1 H, 5-H), 4.79 (m, 1 H, 6-H), 7.09 (m, 2 H) ppm. HRMS (CI) m/z calculated for C15H21N2O5 [M + 1]+ 337.1512; found, 337.1503.
(2S,3R)-N-Boc-3-methoxyphenylalanine (5e)
According to general procedure B alcohol 4e (275.0 mg, 977.0 μmol) was reacted with PhI(OAc)2 (31.0 mg, 97.0 μmol), TEMPO (31.0 mg, 194.0 μmol) and NaClO2 (387.0 mg, 3.42 mmol) to 3-methoxyphenylalanine 5e (289.0 mg, 977.0 μmol, quant.) as white solid, mp. 108–110 °C. TLC: Rf(5e) = 0.26 (petroleum ether/ethyl acetate = 1:1). [α]20D = −24.8 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 1.33 (s, 9 H), 3.32 (s, 3 H), 4.56 (dd, J = 9.4 Hz, J = 2.6 Hz, 1 H), 4.88 (d, J = 2.6 Hz, 1 H), 5.33 (d, J = 9.4 Hz, 1 H), 7.28–7.41 (m, 5 H), 10.21 (bs, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 28.2, 57.6, 59.0, 80.1, 82.4, 126.9, 128.2, 128.5, 136.8, 155.6, 175.2 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 1.16 (s, 9 H), 3.30 (s, 3 H), 4.56 (d, J = 8.0 Hz, 1 H), 4.80 (bs, 1 H), 5.85 (d, J = 8.0 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 27.8, 57.5, 60.6, 80.9, 82.7, 127.0 ppm. HRMS (CI) m/z calculated for C15H22NO5 [M + 1]+ 296.1492, found, 296.1495.
N-Boc-(2S,3R)-(3-methoxy-para-nitrophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (7a)
N-Cbz-(S)-Val-(S)-N-methyl-Leu-OMe 6 (150.0 mg, 382.0 μmol) was reacted with β-methoxyphenylalanine 5a (118.0 mg, 347.0 μmol) according to general procedure C to tripeptide 7a. Purification by column chromatographic (silica gel, petroleum ether/ethyl acetate 6:4) gave tripeptide 7a (158.0 mg, 272.0 μmol, 78%) as colourless foam. TLC: Rf(7a) = 0.31 (petroleum ether/ethyl acetate = 6:4). [α]20D = −172.6 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 0.91 (d, J = 6.3 Hz, 3 H), 0.92 (d, J = 6.3 Hz, 3 H), 0.94 (d, J = 6.7 Hz, 3 H), 1.00 (d, J = 6.8 Hz, 3 H), 1.33 (s, 9 H), 1.47 (m, 1 H), 1.76 (m, 2 H), 2.11 (m, 1 H), 3.00 (s, 3 H), 3.36 (s, 3 H), 3.69 (s, 3 H), 4.47 (dd, J = 8.0 Hz, J = 2.9 Hz, 1 H), 4.83 (dd, J = 8.8 Hz, J = 5.8 Hz, 1 H), 5.01 (d, J = 2.9 Hz, 1 H), 5.27–5.33 (m, 2 H), 7.29 (d, J = 8.7 Hz, 1 H), 7.46 (d, J = 8.7 Hz, 1 H), 8.16 (d, J = 8.7 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 17.0, 19.5, 21.4, 23.2, 24.9, 28.1, 31.3, 36.8, 52.2, 54.0, 54.7, 58.0, 59.0, 80.5, 81.2, 123.5, 127.7, 144.9, 147.7, 155.2, 168.3, 171.9, 172.0 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 1.29 (s, 9 H), 2.81 (s, 3 H), 3.35 (s, 3 H), 3.62 (s, 3 H), 4.41 (m, 1 H), 4.77 (m, 1 H), 5.05 (m, 1 H), 5.23 (d, = 8.7 Hz, 1 H), 7.12 (d, J = 8.7 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 123.6, 127.5 ppm. HRMS (CI) m/z calculated for C28H44N4O9 [M]+ 580.3108; found, 580.3082
N-Boc-(2S,3R)-(3-methoxy-para-bromphenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (7c)
In accordance to general procedure C, N-Cbz-(S)-Val-(S)-N-methyl-Leu-OMe 6 (81.0 mg, 206.0 μmol) was converted with β-methoxyphenylalanine 5c (70.0 mg, 187.0 μmol) to tripeptide 7c. After column chromatographic purification (silica gel, petroleum ether/ethyl acetate 6:4) tripeptide 7c (102.0 mg, 166.0 μmol, 89%) was isolated as colourless foam. TLC: Rf(7c) = 0.36 (petroleum ether/ethyl acetate = 6:4). [α]20D = −81.3 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 0.90 (d, J = 6.5 Hz, 3 H), 0.92 (d, J = 6.5 Hz, 3 H), 0.94 (d, J = 6.7 Hz, 3 H), 1.00 (d, J = 6.7 Hz, 3 H), 1.35 (s, 9 H), 1.48 (m, 1 H), 1.73 (m, 2 H), 2.11 (m, 1 H), 3.00 (s, 3 H), 3.31 (s, 3 H), 3.69 (s, 3 H), 4.39 (dd, J = 8.0 Hz, J = 2.7 Hz, 1 H), 4.83 (dd, J = 8.8 Hz, J = 6.3 Hz, 1 H), 4.85 (d, J = 2.0 Hz, 1 H), 5.23 (d, J = 8.0 Hz, 1 H), 5.31 (dd, J = 10.4 Hz, J = 5.2 Hz, 1 H), 7.13 (d, J = 8.2 Hz, 1 H), 7.28 (d, J = 8.5 Hz, 1 H), 7.42 (d, J = 8.2 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 17.2, 19.4, 21.4, 23.2, 24.9, 28.1, 31.4, 31.5, 36.9, 52.2, 53.9, 54.6, 57.6 59.3, 80.2, 81.3, 121.9, 128.5, 131.5, 136.3, 155.3, 168.9, 171.9, 172.0 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 0.82 (d, J = 6.5 Hz, 3 H), 1.31 (s, 9 H), 2.82 (s, 3 H), 3.20 (s, 3 H), 3.62 (s, 3 H), 4.33 (m, 1 H), 4.67 (m, 1 H), 4.90 (m, 1 H) ppm. HRMS (CI) m/z calculated for C28H45BrN3O7 [M + H]+ 614.2441; found, 614.2471.
N-Boc-(2S,3R)-(3-methoxy-para-azidophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (7d)
N-Cbz-(S)-Val-(S)-N-methyl-Leu-OMe 6 (64.0 mg, 164.0 μmol) was reacted with β-methoxyphenylalanine 5d (50.0 mg, 149.0 μmol) according to general procedure C to tripeptide 7d. Column chromatographic purification (silica gel, petroleum ether/ethyl acetate 1:1) resulted in tripeptide 7d (70.0 mg, 121.0 μmol, 82%) as colourless foam. TLC: Rf(7d) = 0.56 (petroleum ether/ethyl acetate = 1:1). [α]20D = −139.3 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 0.90 (d, J = 6.4 Hz, 3 H), 0.92 (d, J = 6.4 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H), 1.00 (d, J = 6.8 Hz, 3 H), 1.35 (s, 9 H), 1.47 (m, 1 H), 1.74 (m, 2 H), 2.11 (m, 1 H), 3.00 (s, 3 H), 3.30 (s, 3 H), 3.68 (s, 3 H), 4.39 (dd, J = 8.0 Hz, J = 2.7 Hz, 1 H), 4.83 (dd, J = 8.8 Hz, J = 6.1 Hz, 1 H), 4.86 (d, J = 2.1 Hz, 1 H), 5.25 (d, J = 8.0 Hz, 1 H), 5.31 (dd, J = 10.4 Hz, J = 5.2 Hz, 1 H), 6.96 (d, J = 8.2 Hz, 1 H), 7.25 (d, J = 8.2 Hz, 1 H), 7.29 (d, J = 8.8 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 17.0, 19.5, 21.4, 23.2, 24.9, 28.1, 31.3, 31.5, 36.9, 52.2, 53.9, 54.6, 59.4, 80.1, 81.3, 118.9, 128.3, 134.0, 139.7, 155.3, 168.9, 171.9, 172.0 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 1.31 (s, 9 H), 2.82 (s, 3 H), 3.29 (s, 3 H), 3.62 (s, 3 H), 4.31 (m, 1 H), 4.77 (m, 1 H), 7.07 (d, J = 8.7 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 119.1, 128.1 ppm. HRMS (CI) m/z calculated for C28H45N6O7 [M + H]+ 577.3344; found, 577.3345.
N-Boc-(S)-alanyl-(2S,3R)-(3-methoxy-para-nitrophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (9a)
According to general procedure D, tripeptide 7a (128.0 mg, 220.0 μmol) was converted with N-Boc-(S)-alanine 8 (46.0 mg, 242.0 μmol) to tetrapeptide 9a. After column chromatographic purification (silica gel, petroleum ether/ethyl acetate 6:4) tetrapeptide 9a (144.0 mg, 220.0 μmol, quant.) could be achieved as colourless foam. TLC: Rf(9a) = 0.16 (petroleum ether/ethyl acetate = 6:4). [α]20D = −102.8 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 0.92 (d, J = 6.8 Hz, 3 H), 0.94 (d, J = 6.5 Hz, 3 H), 0.96 (d, J = 6.5 Hz, 3 H), 0.99 (d, J = 6.8 Hz, 3 H), 1.31 (d, J = 7.1 Hz, 1 H), 1.45 (s, 9 H), 1.47 (m, 1 H), 1.76 (m, 2 H), 2.14 (m, 1 H), 2.99 (s, 3 H), 3.38 (s, 3 H), 3.70 (s, 3 H), 4.11 (m, 1 H), 4.75 (dd, J = 7.1 Hz, = 3.6 Hz, 1 H), 4.80 (dd, J = 8.7 Hz, J = 5.4 Hz, 1 H), 4.86 (d, J = 5.5 Hz, 1 H), 4.98 (d, J = 3.4 Hz, 1 H), 5.33 (dd, J = 10.5 Hz, J = 5.2 Hz, 1 H), 6.85 (d, J = 7.1 Hz, 1 H), 7.39 (d, J = 8.7 Hz, 1 H), 7.45 (d, J = 8.5 Hz, 1 H), 8.14 (d, J = 8.5 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 16.9, 17.9, 19.5, 21.3, 23.2, 24.9, 28.3, 31.1, 31.3, 36.9, 50.4, 52.1, 54.2, 54.7, 57.0, 58.0, 80.4, 123.5, 127.9, 144.4, 147.8, 155.4, 167.6, 171.9, 172.7 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 1.46 (s, 9 H), 2.00 (m, 1 H), 2.87 (s, 3 H), 3.64 (s, 3 H), 4.05 (m, 1 H), 4.69 (m, 1 H), 6.90 (d, J = 7.9 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 18.7 ppm. HRMS (ESI+) m/z calculated for C31H50N5O10 [M + H]+ 652.3552; found, 652.3559.
N-Boc-(S)-alanyl-(2S,3R)-(3-methoxy-para-bromphenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (9c)
According to general procedure D, tripeptide 7c (88.0 mg, 143.0 μmol) was transformed with N-Boc-(S)-alanine 8 (30.0 mg, 158.0 μmol) to tetrapeptide 9c. Tetrapeptide 9c (74.0 mg, 107.0 μmol, 75%) could be obtained as colourless foam after column chromatography (silica gel, petroleum ether/ethyl acetate 1:1). TLC: Rf(9c) = 0.30 (petroleum ether/ethyl acetate = 1:1). [α]20D = −75.0 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 0.91 (d, J = 6.7 Hz, 3 H), 0.92 (d, J = 6.4 Hz, 3 H), 0.94 (d, J = 6.7 Hz, 3 H), 0.98 (d, J = 6.4 Hz, 3 H), 1.30 (d, J = 7.1 Hz, 1 H), 1.45 (s, 9 H), 1.47 (m, 1 H), 1.74 (m, 2 H), 2.12 (m, 1 H), 2.99 (s, 3 H, 7-H), 3.32 (s, 3 H), 3.69 (s, 3 H), 4.12 (m, 1 H), 4.68 (dd, J = 7.1 Hz, = 3.5 Hz, 1 H), 4.78 (m, 1 H), 4.81 (d, J = 3.4 Hz, 1 H), 4.90 (m, 1 H), 5.33 (dd, J = 10.5 Hz, J = 5.2 Hz, 1 H), 6.78 (d, J = 7.2 Hz, 1 H), 7.10 (d, J = 8.2 Hz, 1 H), 7.36 (d, J = 8.4 Hz, 1 H), 7.39 (d, J = 8.2 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 17.1, 18.2, 19.5, 21.4, 23.3, 24.8, 28.3, 31.3 (q, d), 36.9, 50.4, 52.1, 54.1, 54.5, 57.3, 57.6, 80.5, 122.2, 128.7, 131.5, 135.8, 155.4, 168.0, 171.9, 172.0, 172.7 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 1.47 (s, 9 H), 2.00 (m, 1 H), 2.85 (s, 3 H), 3.62 (s, 3 H), 4.66 (m, 1 H), 4.88 (m, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 23.8, 50.3 ppm. HRMS (ESI+) m/z calculated for C31H50BrN4O8 [M + H]+ 685.2807; found, 685.2803.
N-Boc-(S)-alanyl-(2S,3R)-(3-methoxy-para-azidophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (9d)
Tripeptide 7d (45.0 mg, 78.0 μmol) was reacted with N-Boc-(S)-alanine 8 (16.0 mg, 86.0 μmol) as described by general procedure D to tetrapeptide 9d. Column chromatography (silica gel, petroleum ether/ethyl acetate 1:1) yielded tetrapeptide 9d (42.0 mg, 195.0 μmol, 83%) as colourless foam. TLC: Rf(9d) = 0.24 (petroleum ether/ethyl acetate = 1:1). [α]20D = −56.3 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (400 MHz, CDCl3): δ = 0.92 (d, J = 6.2 Hz, 3 H), 0.94 (d, J = 6.2 Hz, 3 H), 0.95 (d, J = 6.7 Hz, 3 H), 0.98 (d, J = 6.7 Hz, 3 H), 1.31 (d, J = 7.0 Hz, 1 H), 1.45 (s, 9 H), 1.47 (m, 1 H), 1.74 (m, 2 H), 2.12 (m, 1 H), 2.99 (s, 3 H), 3.32 (s, 3 H), 3.69 (s, 3 H), 4.12 (m, 1 H), 4.69 (dd, J = 6.9 Hz, J = 3.5 Hz, 1 H), 4.79 (m, 1 H), 4.82 (d, J = 3.6 Hz, 1 H), 4.91 (m, 1 H), 5.34 (dd, J = 10.4 Hz, J = 5.3 Hz, 1 H), 6.76 (d, J = 7.1 Hz, 1 H), 6.92 (d, J = 8.4 Hz, 1 H), 7.21 (d, J = 8.4 Hz, 1 H), 7.39 (d, J = 8.7 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 17.0, 18.3, 19.5, 21.4, 23.3, 24.8, 28.3, 31.2, 31.3, 36.9, 50.4, 52.1, 54.1, 54.5, 57.4, 57.5, 80.6, 118.9, 128.5, 133.4, 139.8, 155.3, 168.1, 171.9, 172.0, 172.7 ppm. Minor rotamer (selected signals). 1H NMR (400 MHz, CDCl3): δ = 1.47 (s, 9 H), 2.00 (m, 1 H), 2.85 (s, 3 H), 3.64 (s, 3 H), 4.62 (m, 1 H), 4.88 (m, 1 H), 6.81 (d, J = 7.1 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 23.8, 50.3 ppm. HRMS (ESI+) m/z calculated for C31H50N7O8 [M + H]+ 648.3715; found, 648.3720.
N-Alloc-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-nitrophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (11a)
Tetrapeptide 9a (128.0 mg, 196.0 μmol) was reacted with δ-hydroxyleucine 10 (78.0 mg, 216.0 μmol) as described by general procedure E to pentapeptide 11a. Column chromatography (silica gel, petroleum ether/ethyl acetate 1:1) yielded pentapeptide 11a (125.0 mg, 140.0 μmol, 71%) as colourless resin. TLC: Rf(11a) = 0.47 (dichloromethane/ethyl acetate = 1:1). [α]20D = −151.0 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.03 (s, 6 H), 0.86 (s, 9 H), 0.91–0.99 (m, 15 H), 1.30 (d, J = 6.0 Hz, 3 H), 1.48 (m, 1 H), 1.55–1.63 (m, 2 H), 1.75 (m, 2 H), 1.96 (m, 1 H), 2.13 (m, 1 H), 2.85 (s, 3 H), 2.99 (s, 3 H), 3.38 (s, 3 H), 3.44 (m, 1 H), 3.51 (m, 1 H), 3.70 (s, 3 H), 4.32 (m, 1 H), 4.60 (m, 2 H), 4.70 (m, 1 H), 4.74 (dd, J = 7.0 Hz, J = 3.6 Hz, 1 H), 4.78 (dd, J = 7.7 Hz, J = 5.3 Hz, 1 H), 4.94 (m, 1 H), 5.22 (m, 1 H), 5.29 (m, 1 H), 5.34 (m, 1 H), 5.93 (m, 1 H), 6.46 (bs, 1 H), 6.76 (bs, 1 H), 7.40 (d, J = 8.6 Hz, 1 H), 7.45 (d, J = 7.7 Hz, 2 H), 8.15 (d, J = 7.7 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 16.9, 17.4, 17.9, 18.3, 19.6, 21.3, 23.2, 24.9, 25.9, 29.8, 31.0, 31.2, 31.3, 32.2, 36.8, 49.3, 52.1, 54.2, 54.7, 56.7, 57.5, 58.1, 66.6, 67.1, 80.4, 117.6, 123.5, 127.9, 132.6, 144.2, 147.8, 157.1, 167.4, 170.7, 171.4, 171.9, 172.0 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = 1.24 (m, 3 H), 2.87 (s, 3 H), 3.62 (s, 3 H), 6.20 (bs, 1 H), 6.70 (bs, 1 H) ppm. HRMS (ESI+) m/z calculated for C43H73N6O12Si [M + H]+ 893.5050; found, 893.5049.
N-Alloc-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-bromophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (11c)
According to general procedure E, tetrapeptide 9c (70.0 mg, 102.0 μmol) was converted with δ-hydroxyleucine 10 (40.0 mg, 112.0 μmol) into pentapeptide 11c. After column chromatographic purification (silica gel, petroleum ether/ethyl acetate 1:1) pentapeptide 11c (95.0 mg, 102.0 μmol, quant.) could be achieved as colourless resin. TLC: Rf(11c) = 0.46 (dichloromethane/ethyl acetate = 1:1). [α]20D = −112.0 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.02 (s, 3 H), 0.03 (s, 3 H), 0.87 (s, 9 H), 0.91–0.99 (m, 15 H), 1.30 (d, J = 6.7 Hz, 3 H), 1.45 (m, 1 H), 1.55–1.63 (m, 2 H), 1.74 (m, 2 H), 1.94 (m, 1 H), 2.11 (m, 1 H), 2.83 (s, 3 H), 2.98 (s, 3 H), 3.31 (s, 3 H), 3.43 (m, 1 H), 3.50 (m, 1 H), 3.68 (s, 3 H), 4.35 (m, 1 H), 4.58 (m, 1 H), 4.61 (m, 2 H), 4.68 (dd, J = 7.3 Hz, J = 3.6 Hz, 1 H), 4.74 (m, 1 H), 4.80 (dd, J = 8.6 Hz, J = 5.7 Hz, 1 H), 5.20 (m, 1 H), 5.28 (m, 1 H), 5.33 (dd, J = 10.6 Hz, J = 5.0 Hz, 1 H), 5.92 (m, 1 H), 6.51 (bs, 1 H), 6.72 (bs, 1 H), 6.92 (d, J = 8.4 Hz, 2 H), 7.37 (d, J = 8.4 Hz, 1 H), 7.39 (d, J = 8.4 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.5, −5.4, 17.1, 17.5, 17.9, 18.3, 19.5, 21.4, 23.2, 24.8, 25.9, 29.6, 31.2, 32.2, 36.9, 49.1, 52.1, 54.1, 54.6, 56.7, 57.4, 57.6, 66.5, 67.0, 80.6, 117.5, 122.3, 128.7, 131.5, 132.6, 135.6, 156.8, 167.9, 170.9, 171.7, 171.9, 172.0 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = 1.24 (m, 3 H), 2.87 (s, 3 H), 3.64 (s, 3 H), 6.31 (bs, 1 H), 6.64 (bs, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.5, 25.9, 66.3, 117.3 ppm. HRMS (ESI+) m/z calculated for C43H73BrN5O10Si [M + H]+ 926.4305; found, 926.4303.
N-Alloc-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-azidophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (11d)
According to general procedure E, tetrapeptide 9d (31.0 mg, 48.0 μmol) was transformed with δ-hydroxyleucine 10 (19.0 mg, 53.0 μmol) into pentapeptide 11d (42.0 mg, 47.0 μmol, 99%), which could be obtained as colourless resin by column chromatography (silica gel, petroleum ether/ethyl acetate 1:1). TLC: Rf(11d) = 0.33 (dichloromethane/ethyl acetate = 1:1). [α]20D = −60.1 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.02 (s, 6 H), 0.87 (s, 9 H), 0.91–0.99 (m, 15 H), 1.30 (d, J = 5.9 Hz, 3 H), 1.48 (m, 1 H), 1.55–1.63 (m, 2 H), 1.75 (m, 2 H), 1.98 (m, 1 H), 2.11 (m, 1 H), 2.83 (s, 3 H), 2.99 (s, 3 H), 3.31 (s, 3 H), 3.43 (m, 1 H), 3.50 (m, 1 H), 3.68 (s, 3 H), 4.35 (m, 1 H), 4.58 (m, 1 H), 4.61 (m, 2 H), 4.68 (dd, J = 6.8 Hz, J = 3.6 Hz, 1 H), 4.74 (m, 1 H), 4.80 (dd, J = 7.9 Hz, J = 5.7 Hz, 1 H), 5.21 (m, 1 H), 5.27 (m, 1 H), 5.35 (dd, J = 10.3 Hz, J = 4.9 Hz, 1 H), 5.92 (m, 1 H), 6.51 (bs, 1 H), 6.69 (bs, 1 H), 6.92 (d, J = 7.6 Hz, 2 H), 7.16 (d, J = 7.6 Hz, 2 H), 7.40 (d, J = 8.6 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 17.0, 17.4, 17.6, 18.3, 19.5, 21.3, 23.2, 24.8, 25.9, 29.6, 31.2, 32.1, 36.8, 49.1, 52.1, 54.0, 54.5, 56.7, 57.4, 57.5, 66.5, 67.0, 80.6, 117.6, 118.9, 128.4, 132.6, 139.9, 157.0, 168.0, 171.5, 172.0 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = 1.24 (m, 3 H), 2.87 (s, 3 H), 3.64 (s, 3 H), 6.31 (bs, 1 H), 6.64 (bs, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 18.1, 22.6, 64.2, 70.5 ppm. HRMS (ESI+) m/z calculated for C43H73N8O10Si [M + H]+ 889.5213; found, 889.5217.
N-Alloc-(S)-[N′-tert-prenyl-tryptophanyl]-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)-oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-nitrophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (13a)
Pentapeptide 11a (113.0 mg, 127.0 μmol) was reacted with N′-tert-prenylated tryptophan 12 (47.0 mg, 133.0 μmol) as described by general procedure F to hexapeptide 13a. Column-chromatographic purification (silica gel, petroleum ether/ethyl acetate 1:1) yielded hexapeptide 13a (84.0 mg, 73.0 μmol, 58%) as colourless resin. TLC: Rf(13a) = 0.53 (dichloromethane/ethyl acetate = 1:1). [α]20D = −36.0 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.02 (s, 3 H), 0.03 (s, 3 H), 0.86 (s, 9 H), 0.89–1.00 (m, 15 H), 1.24 (d, J = 7.1 Hz, 3 H), 1.43–1.56 (m, 3 H), 1.68–1.79 (m, 8 H), 2.11 (m, 1 H), 2.27 (m, 1 H), 2.85 (s, 3 H), 2.97 (s, 3 H), 3.16 (m, 2 H), 3.34 (s, 3 H), 3.43 (m, 2 H), 3.70 (s, 3 H), 4.23 (m, 1 H), 4.49 (m, 2 H), 4.57 (m, 1 H), 4.69 (dd, J = 7.4 Hz, J = 3.3 Hz, 1 H), 4.73 (m, 1 H), 4.91 (m, 1 H), 4.95 (d, J = 3.4 Hz, 1 H), 5.09–5.32 (m, 4 H), 5.33 (dd, J = 10.7 Hz, J = 5.1 Hz, 1 H), 5.56 (d, J = 7.3 Hz, 1 H), 5.86 (m, 1 H), 6.09 (dd, J = 17.4 Hz, J = 10.7 Hz, 1 H), 6.52 (d, J = 6.4 Hz, 1 H), 6.89 (d, J = 7.4 Hz, 1 H), 7.05–7.13 (m, 2 H), 7.17 (s, 1 H), 7.42 (d, J = 8.7 Hz, 2 H), 7.46–7.50 (m, 2 H), 7.69 (d, J = 7.1 Hz, 1 H), 8.12 (d, J = 8.7 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 16.9, 17.4, 17.5, 18.2, 19.4, 21.3, 23.2, 24.9, 25.8, 27.8, 28.0, 29.1, 31.1, 31.3, 31.4, 32.2, 36.9, 49.7, 51.9, 52.1, 54.1, 54.2, 57.4, 58.0, 58.9, 65.6, 67.7, 81.1, 108.0, 113.6, 113.9, 117.5, 118.4, 119.1, 121.0, 123.4, 124.2, 127.8, 131.9, 132.7, 135.3, 143.9, 144.7, 147.7, 156.7, 167.6, 170.6, 171.8, 171.9, 172.0, 172.5 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = –0.58 (m, 1 H), −0.09 (s, 3 H), −0.07 (s, 3 H), 0.28 (d, J = 6.6 Hz, 3 H), 0.78 (s, 9 H), 0.89–1.00 (m, 12 H), 1.25 (d, J = 6.9 Hz, 3 H), 1.35–1.40 (m, 2 H), 1.68–1.79 (m, 8 H), 1.84 (m, 1 H), 2.10 (m, 1 H), 2.60 (m, 1 H), 2.74 (s, 3 H), 3.04 (s, 3 H), 3.31 (m, 2 H), 3.37 (s, 3 H), 3.69 (s, 3 H), 3.98 (m, 1 H), 4.41 (m, 2 H), 4.64 (m, 1 H), 4.76 (dd, J = 8.7 Hz, J = 3.3 Hz, 1 H), 4.73 (m, 1 H), 4.80 (m, 1 H), 4.99 (d, J = 3.4 Hz, 1 H), 5.09–5.32 (m, 4 H), 5.38 (dd, J = 10.6 Hz, J = 5.0 Hz, 1 H), 5.86 (m, 1 H), 6.09 (dd, J = 17.4 Hz, J = 10.7 Hz, 1 H), 6.99 (d, J = 7.8 Hz, 1 H), 7.05–7.13 (m, 2 H), 7.33 (d, J = 7.4 Hz, 1 H), 7.53 (d, J = 8.7 Hz, 2 H), 7.62 (m, 2 H), 7.69 (d, J = 7.1 Hz, 1 H), 7.88 (d, J = 6.4 Hz, 1 H), 8.18 (d, J = 8.7 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 15.3, 16.8, 17.3, 18.3, 19.5, 24.7, 25.9, 27.9, 28.0, 29.0, 30.9, 31.7, 31.8, 32.1, 36.9, 50.8, 57.2, 58.0, 66.3, 68.2, 107.4, 113.5, 113.8, 118.5, 118.5, 119.4, 121.3, 123.9, 123.4, 128.2, 156.4, 169.0 ppm. (Carbonyl signals are not distinguishable from the rotameric signals.) HRMS (ESI+) m/z calculated for C59H91N8O13Si [M + H]+ 1147.6469; found, 1147.6473.
N-Alloc-(S)-[N′-tert-prenyl-tryptophanyl]-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)-oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-bromophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (13c)
Pentapeptide 11c (80.0 mg, 86.0 μmol) was reacted with N′-tert-prenylated tryptophan 12 (32.0 mg, 91.0 μmol) as described by general procedure F to hexapeptide 13c. Column-chromatographic purification (silica gel, petroleum ether/ethyl acetate 1:1) resulted in hexapeptide 13c (32.0 mg, 27.0 μmol, 32%) as colourless resin. TLC: Rf(13c) = 0.46 (dichloromethane/ethyl acetate = 1:1). [α]20D = −65.8 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.02 (s, 3 H), 0.03 (s, 3 H), 0.86 (s, 9 H), 0.88–1.01 (m, 15 H), 1.24 (m, 3 H), 1.43–1.56 (m, 3 H), 1.65–1.79 (m, 8 H), 2.10 (m, 1 H), 2.18 (m, 1 H), 2.83 (s, 3 H), 2.98 (s, 3 H), 3.15 (m, 2 H), 3.32 (s, 3 H), 3.43 (m, 2 H), 3.69 (s, 3 H), 4.26 (m, 1 H), 4.49 (m, 2 H), 4.57 (m, 1 H), 4.61 (m, 1 H), 4.70 (m, 1 H), 4.80 (m, 1 H), 4.92 (m, 1 H), 5.09–5.25 (m, 4 H), 5.35 (m, 1 H), 5.59 (d, J = 7.4 Hz, 1 H), 5.85 (m, 1 H), 5.98 (bs, 1 H), 6.09 (dd, J = 17.5 Hz, J = 10.6 Hz, 1 H), 6.78 (d, J = 7.3 Hz, 1 H), 7.03–7.12 (m, 3 H), 7.13 (m, 1 H), 7.40 (d, J = 8.4 Hz, 1 H), 7.45–7.65 (m, 2 H), 7.86 (d, J = 6.5 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 17.1, 17.4, 17.7, 18.2, 19.4, 21.3, 23.2, 24.9, 25.8, 27.8, 28.0, 29.1, 31.1, 31.3, 31.4, 32.2, 36.9, 49.7, 51.7, 52.1, 54.1, 54.3, 54.5, 57.5, 57.7, 58.9, 65.6, 67.4, 81.0, 108.1, 113.6, 113.9, 117.5, 118.3, 119.1, 121.1, 124.2, 128.8, 131.5 132.0, 132.7, 135.4, 135.5, 143.9, 156.0, 156.7, 168.2, 170.5, 171.6, 171.8, 172.0, 172.1, 172.2, 172.5, 173.5 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = −0.54 (m, 1 H), −0.07 (s, 3 H), −0.05 (s, 3 H), 0.32 (d, J = 6.5 Hz, 3 H), 0.80 (s, 9 H), 0.89–1.00 (m, 12 H), 1.25 (m, 3 H), 1.65–1.79 (m, 8 H), 1.83 (m, 1 H), 2.19 (m, 1 H), 2.60 (m, 1 H), 2.74 (s, 3 H), 3.00 (s, 3 H), 3.27 (m, 2 H), 3.27 (s, 3 H), 3.68 (s, 3 H), 4.11 (m, 1 H), 4.45 (m, 2 H), 4.54 (m, 1 H), 4.65 (m, 1 H), 4.73–4.84 (m, 3 H), 5.09–5.24 (m, 4 H), 5.29 (m, 1 H), 5.86 (m, 1 H), 6.09 (dd, J = 17.5 Hz, J = 10.6 Hz, 1 H), 6.48 (d, J = 6.8 Hz, 1 H), 6.83 (d, J = 7.4 Hz, 1 H), 7.03–7.12 (m, 4 H), 7.12 (m, 1 H), 7.37 (d, J = 8.4 Hz, 1 H), 7.45–7.65 (m, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 15.3, 17.1, 17.3, 18.3, 19.6, 24.8, 25.9, 27.8, 27.9, 28.7, 31.2, 31.6, 31.8, 49.8, 50.8, 54.6, 57.3, 57.7, 66.3, 68.3, 80.6, 107.4, 113.5, 118.4, 118.5, 119.4, 121.3, 123.9, 128.6, 131.4, 136.0, 144.0, 156.7, 168.8 ppm. (Carbonyl signals are not distinguishable from the rotameric signals.) HRMS (ESI+) m/z calculated for C59H91BrN7O11Si [M + H]+ 1180.5724; found, 1180.5725.
N-Alloc-(S)-[N′-tert-prenyl-tryptophanyl]-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)-oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-azidophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (13d)
According to general procedure F, pentapeptide 11d (40.0 mg, 45.0 μmol) was converted with N′-tert-prenylated tryptophan 12 (17.0 mg, 47.0 μmol) into hexapeptide 13d. After column chromatography (silica gel, petroleum ether/ethyl acetate 1:1) hexapeptide 13d (29.0 mg, 25.0 μmol, 56%) could be obtained as colourless resin. TLC: Rf(13d) = 0.62 (dichloromethane/ethyl acetate = 1:1). [α]20D = −64.5 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.02 (s, 3 H), 0.03 (s, 3 H), 0.86 (s, 9 H), 0.87–1.00 (m, 15 H), 1.24 (d, J = 6.7 Hz, 3 H), 1.41–1.56 (m, 3 H), 1.65–1.79 (m, 8 H), 2.11 (m, 1 H), 2.30 (m, 1 H), 2.83 (s, 3 H), 2.96 (s, 3 H), 3.15 (m, 2 H), 3.32 (s, 3 H), 3.43 (m, 2 H), 3.70 (s, 3 H), 4.25 (m, 1 H), 4.46 (m, 2 H), 4.55 (m, 1 H), 4.62 (m, 1 H), 4.70 (m, 1 H), 4.81 (m, 1 H), 4.93 (m, 1 H), 5.09–5.25 (m, 4 H), 5.60 (m, 1 H), 5.56 (d, J = 6.7 Hz, 1 H), 5.85 (m, 1 H), 5.98 (bs, 1 H), 6.09 (dd, J = 17.5 Hz, J = 11.1 Hz, 1 H), 6.76 (d, J = 7.3 Hz, 1 H), 6.93 (d, J = 8.2 Hz, 1 H), 7.08 (m, 3 H), 7.13 (m, 1 H), 7.24 (m, 2 H), 7.45–7.51 (m, 2 H), 7.86 (d, J = 6.4 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 17.1, 17.4, 17.7, 18.2, 19.4, 21.3, 23.2, 24.9, 25.8, 27.8, 28.0, 29.1, 31.1, 31.3, 31.4, 32.2, 36.9, 49.7, 51.7, 52.1, 54.1, 54.3, 54.4, 57.5, 57.7, 58.9, 65.6, 67.7, 81.0, 108.1, 113.6, 113.9, 117.5, 118.3, 118.9, 119.1, 121.0, 124.2, 128.6, 132.0, 132.7, 133.7, 135.4, 139.8, 143.9, 156.7, 168.2, 170.6, 171.6, 171.7, 172.0, 172.1, 172.2, 172.5, 173.5 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = −0.52 (m, 1 H), −0.07 (s, 3 H), −0.05 (s, 3 H), 0.31 (d, J = 6.5 Hz, 3 H), 0.80 (s, 9 H), 0.89–1.00 (m, 12 H), 1.25 (d, J = 6.7 Hz, 3 H), 1.65–1.79 (m, 8 H), 1.86 (m, 1 H), 2.11 (m, 1 H), 2.60 (m, 1 H), 2.74 (s, 3 H), 3.00 (s, 3 H), 3.27 (m, 1 H), 3.27 (s, 3 H), 3.68 (s, 3 H), 4.12 (m, 1 H), 4.45 (m, 2 H), 4.58 (m, 1 H), 4.65 (m, 1 H), 4.73–4.84 (m, 3 H), 5.09–5.24 (m, 4 H), 5.28 (m, 1 H), 5.86 (m, 1 H), 6.09 (dd, J = 17.4 Hz, J = 10.7 Hz, 1 H), 6.48 (d, J = 6.4 Hz, 1 H), 6.83 (d, J = 7.8 Hz, 1 H), 6.88 (d, J = 8.2 Hz, 1 H), 7.08 (m, 3 H), 7.13 (m, 1 H), 7.36 (d, J = 8.9 Hz, 1 H), 7.51–7.64 (m, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 15.3, 17.2, 17.3, 18.3, 19.6, 24.8, 25.9, 27.8, 27.9, 28.7, 31.2, 31.6, 31.8, 49.8, 50.8, 54.5, 57.4, 57.4, 66.3, 68.2, 80.6, 107.4, 113.5, 118.4, 118.5, 118.8, 119.4, 121.3, 123.9, 128.5, 133.5, 139.6, 144.0, 168.8 ppm. (Carbonyl signals are not distinguishable from the rotameric signals.) HRMS (ESI+) m/z calculated for C59H91N10O11Si [M + H]+ 1143.6633; found, 1143.6639.
N-Alloc-(2S,3R)-(2-amino-3,5-dimethyl-hex-4-enoyl)-(S)-N′-tert-prenyl-tryptophanyl-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-nitrophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (15a)
According to general procedure G, hexapeptide 13a (72.0 mg, 63.0 μmol) was converted with γ,δ-unsaturated amino acid 14 (17.0 mg, 69.0 μmol) into heptapeptide 15a. Flash chromatography (silica gel, dichloromethane/ethyl acetate 1:1) provided the desired product 15a (76 mg, 59 μmol, 94%) as colourless resin. TLC: Rf(15a) = 0.55 (dichloromethane/ethyl acetate). [α]20D = −77.8 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.01 (s, 3 H), 0.02 (s, 3 H), 0.85 (s, 9 H), 0.86–1.00 (m, 18 H), 1.25 (d, J = 7.0 Hz, 3 H), 1.37 (m, 1 H), 1.43–1.52 (m, 2 H), 1.55 (s, 3 H), 1.66 (s, 3 H), 1.69 (s, 6 H), 1.71–1.80 (m, 3 H), 2.11 (m, 1 H), 2.79 (s, 3 H), 2.97 (s, 3 H), 3.01–3.20 (m, 3 H), 3.39 (s, 3 H), 3.42 (m, 2 H), 3.69 (s, 3 H), 4.11 (m, 1 H), 4.23 (m, 1 H), 4.53 (m, 2 H), 4.72 (dd, J = 6.7 Hz, J = 3.4 Hz, 1 H), 4.74–4.80 (m, 2 H), 4.89–4.96 (m, 3 H), 5.10–5.24 (m, 3 H), 5.26–5.37 (m, 3 H), 5.90 (m, 1 H), 6.08 (dd, J = 17.1 Hz, J = 10.6 Hz, 1 H), 6.46 (d, J = 6.8 Hz, 1 H), 6.76 (bs, 1 H), 6.81 (d, J = 6.7 Hz, 1 H), 6.85 (d, = 6.5 Hz, 1 H), 7.04–7.12 (m, 2 H), 7.14 (s, 1 H), 7.38–7.50 (m, 3 H), 7.63 (d, J = 7.2 Hz, 1 H), 8.13 (d, J = 8.1 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, −5.4, 16.9, 17.1, 17.2, 17.6, 18.0, 18.3, 19.6, 21.4, 23.2, 24.8, 25.8, 27.7, 27.8, 27.9, 31.0, 31.2, 31.3, 31.7, 32.3, 35.4, 36.9, 49.5, 49.8, 50.0, 52.1, 54.1, 54.6, 57.0, 57.7, 58.9, 59.0, 65.7, 67.2, 81.5, 107.4, 113.5, 113.6, 113.9, 117.7, 118.5, 119.1, 121.0, 123.4, 124.1, 124.5, 129.3, 132.7, 134.7, 135.4, 143.9, 144.1, 147.7, 155.7, 167.4, 167.6, 169.0, 170.2, 171.4, 171.7, 171.8, 171.9, 172.0, 172.8 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = −0.46 (m, 1 H), −0.09 (s, 3 H), −0.07 (s, 3 H), 0.32 (d, J = 6.1 Hz, 3 H), 0.78 (s, 9 H), 1.38 (m, 1 H, 26), 1.54 (s, 3 H), 1.63 (s, 3 H), 1.86 (m, 1 H), 2.11 (m, 1 H), 2.66 (m, 1 H), 2.73 (s, 3 H), 2.77 (m, 1 H), 2.97 (s, 3 H), 3.31 (m, 1 H), 3.34 (s, 3 H), 3.69 (s, 3 H), 4.14 (m, 1 H), 4.54 (m, 2 H), 4.68 (dd, J = 7.1 Hz, J = 4.5 Hz, 1 H), 4.93 (m, 1 H), 5.30 (m, 1 H), 5.90 (m, 1 H), 6.10 (dd, J = 17.4 Hz, J = 10.7 Hz, 1 H), 6.92 (bs, 1 H), 7.04–7.12 (m, 2 H), 7.38–7.50 (m, 3 H), 7.56 (d, J = 7.2 Hz, 1 H), 8.05 (d, J = 5.7 Hz, 1 H), 8.10 (d, J = 8.1 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 15.4, 18.1, 18.2, 19.5, 21.4, 23.3, 24.9, 25.9, 28.9, 29.1, 32.0, 35.5, 49.5, 52.1, 54.1, 54.7, 57.1, 57.9, 66.0, 68.2, 81.0, 108.0, 113.4, 113.9, 118.0, 118.3, 119.4, 121.3, 123.5, 123.7, 125.0, 129.0, 132.6, 135.5, 144.0, 144.5, 147.8 ppm. (Carbonyl signals are not distinguishable from the rotameric signals.) HRMS (ESI+) m/z calculated for C67H103N9NaO14Si [M + Na]+ 1308.7286; found, 1308.7299.
N-Alloc-(2S,3R)-(2-amino-3,5-dimethyl-hex-4-enoyl)-(S)-N′-tert-prenyl-tryptophanyl-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-bromophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (15c)
According to general procedure G, hexapeptide 13c (25.0 mg, 21.0 μmol) was converted with acid 14 (5.6 mg, 23.0 μmol) into heptapeptide 15c. Flash chromatography (silica gel, dichloromethane/ethyl acetate 1:1) gave the desired product 15c (30.0 mg, 21.0 μmol, quant.) as colourless resin. TLC: Rf(15c) = 0.56 (dichloromethane/ethyl acetate 1:1). [α]20D = −54.0 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.01 (s, 3 H), 0.02 (s, 3 H), 0.90 (s, 9 H), 0.87–1.00 (m, 18 H), 1.27 (d, J = 6.9 Hz, 3 H), 1.37 (m, 1 H), 1.43–1.52 (m, 1 H), 1.55 (s, 3 H), 1.62 (s, 3 H), 1.69 (s, 6 H), 1.71–1.78 (m, 2 H), 2.00 (m, 1 H), 2.11 (m, 1 H), 2.79 (s, 3 H), 2.81 (m, 1 H), 2.97 (s, 3 H), 3.14 (m, 2 H), 3.32 (s, 3 H), 3.64 (m, 2 H), 3.69 (s, 3 H), 4.07 (m, 1 H), 4.22 (m, 1 H), 4.55 (m, 2 H), 4.68 (dd, J = 7.1 Hz, J = 4.0 Hz, 1 H), 4.72–4.82 (m, 3 H), 4.87–4.96 (m, 2 H), 5.10–5.24 (m, 3 H), 5.26–5.36 (m, 2 H), 5.46 (d, J = 8.9 Hz, 1 H), 5.89 (m, 1 H), 6.09 (m, 1 H), 6.46 (d, J = 6.7 Hz, 1 H), 6.74 (d, J = 7.4 Hz, 1 H), 6.78 (d, J = 6.8 Hz, 1 H), 6.90 (bs, 1 H), 7.04–7.16 (m, 5 H), 7.39 (m, 2 H), 7.44–7.63 (m, 2 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, −5.4, 17.1, 17.1, 17.2, 17.6, 18.0, 18.3, 19.6, 21.4, 23.2, 24.8, 25.8, 27.7, 27.8, 27.9, 31.1, 31.2, 31.3, 31.7, 32.2, 35.4, 36.9, 49.3, 50.0, 52.1, 54.1, 54.6, 57.2, 57.8, 58.9, 65.7, 67.2, 80.6, 107.3, 113.4, 113.6, 117.7, 118.5, 119.1, 121.0, 122.1, 124.1, 124.6, 128.7, 129.4, 131.4, 132.7, 134.7, 135.3, 135.6, 143.9, 155.7, 167.9, 168.1, 168.8, 170.0, 170.1, 171.5, 171.6, 171.8, 172.0, 172.8 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = −0.44 (m, 1 H), −0.07 (s, 3 H), −0.05 (s, 3 H), 0.34 (d, J = 6.5 Hz, 3 H), 0.80 (s, 9 H), 1.47 (m, 1 H), 1.54 (s, 3 H), 1.60 (s, 3 H), 1.67 (s, 6 H), 1.99 (m, 1 H), 2.21 (m, 1 H), 2.66 (m, 1 H), 2.72 (s, 3 H), 2.97 (s, 3 H), 3.00 (m, 1 H), 3.31 (s, 3 H), 3.68 (s, 3 H), 4.08 (m, 1 H), 4.54 (m, 2 H), 4.65 (dd, J = 7.3 Hz, J = 3.6 Hz, 1 H), 5.90 (m, 1 H), 6.07 (dd, J = 17.6 Hz, J = 10.7 Hz, 1 H), 6.88 (bs, 1 H), 7.04–7.16 (m, 5 H), 7.39 (m, 2 H), 7.44–7.63 (m, 2 H), 8.06 (d, J = 8.1 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 15.5, 18.0, 18.2, 19.5, 21.4, 23.3, 24.9, 25.9, 29.1, 29.6, 31.8, 32.0, 35.5, 52.1, 54.1, 54.5, 57.5, 66.0, 68.2, 80.5, 108.0, 113.4, 113.6, 118.0, 118.3, 119.4, 121.3, 122.2, 123.7, 125.0, 128.9, 129.0, 131.5, 132.6, 135.4, 135.8, 144.1 ppm. (Carbonyl signals are not distinguishable from the rotameric signals.) HRMS (ESI+) m/z calculated for C67H104BrN8O12Si [M + H]+ 1319.6721; found, 1319.6730.
N-Alloc-(2S,3R)-(2-amino-3,5-dimethyl-hex-4-enoyl)-(S)-N′-tert-prenyl-tryptophanyl-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-azidophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine methyl ester (15d)
According to general procedure G, hexapeptide 13d (20.0 mg, 17.0 μmol) was converted with γ,δ-unsaturated amino acid 14 (4.6 mg, 19.0 μmol) into heptapeptide 15d. Flash chromatography (silica gel, dichloromethane/ethyl acetate 1:1) yielded product 15d (20.0 mg, 16.0 μmol, 89%) as colourless resin. TLC: Rf(15d) = 0.45 (dichloromethane/ethyl acetate). [α]20D = −80.4 (c = 1.0, CHCl3). Mixture of rotamers. Major rotamer. 1H NMR (500 MHz, CDCl3): δ = 0.02 (s, 3 H), 0.03 (s, 3 H), 0.87 (s, 9 H), 0.87–1.00 (m, 18 H), 1.27 (d, J = 7.0 Hz, 3 H), 1.37 (m, 1 H), 1.43–1.52 (m, 2 H), 1.56 (s, 3 H), 1.62 (s, 3 H), 1.69 (s, 6 H), 1.71–1.80 (m, 2 H), 2.02 (m, 1 H), 2.12 (m, 1 H), 2.79 (s, 3 H), 2.82 (m, 1 H), 2.97 (s, 3 H), 3.12 (m, 2 H), 3.30 (s, 3 H), 3.42 (m, 2 H), 3.69 (s, 3 H), 4.07 (m, 1 H), 4.24 (m, 1 H), 4.55 (m, 2 H), 4.69 (m, 1 H), 4.75–4.82 (m, 2 H), 4.87–4.98 (m, 3 H), 5.10–5.24 (m, 3 H), 5.26–5.38 (m, 2 H), 5.46 (d, J = 7.0 Hz, 1 H), 5.90 (m, 1 H), 6.09 (m, 1 H), 6.44 (d, J = 6.9 Hz, 1 H), 6.63 (bs, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.75 (bs, 1 H), 6.94 (d, J = 8.4 Hz, 2 H), 7.04–7.12 (m, 2 H), 7.14 (s, 1 H), 7.21 (m, 2 H), 7.45 (d, J = 7.8 Hz, 1 H), 7.63 (d, J = 7.3 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, −5.4, 16.9, 17.0, 17.2, 17.5, 18.0, 18.3, 19.6, 21.4, 23.2, 24.8, 25.8, 27.7, 27.8, 27.9, 31.0, 31.2, 31.3, 31.7, 32.2, 35.4, 36.9, 49.3, 50.0, 52.1, 54.1, 54.6, 57.3, 57.8, 58.9, 59.0, 65.7, 67.2, 80.6, 107.4, 113.4, 113.6, 117.7, 118.5, 118.9, 119.1, 121.0, 124.1, 124.6, 128.6, 129.4, 132.7, 133.4, 134.7, 135.4, 139.7, 144.1, 155.7, 167.9, 168.1, 168.8, 170.1, 170.2, 171.4, 171.7, 171.9, 172.0, 172.8 ppm. Minor rotamer (selected signals). 1H NMR (500 MHz, CDCl3): δ = −0.40 (m, 1 H), −0.07 (s, 3 H), −0.05 (s, 3 H), 0.36 (d, J = 6.4 Hz, 3 H), 0.80 (s, 9 H), 1.39 (m, 1 H, 26), 1.54 (s, 3 H), 1.60 (s, 3 H), 1.73 (s, 6 H), 1.86 (m, 1 H), 2.30 (m, 1 H), 2.66 (m, 1 H), 2.72 (s, 3 H), 2.97 (s, 3 H), 2.99 (m, 1 H), 3.12 (m, 2 H), 3.29 (s, 3 H), 3.69 (s, 3 H), 4.07 (m, 1 H), 4.55 (m, 2 H), 5.30 (m, 1 H), 5.90 (m, 1 H), 6.07 (dd, J = 17.4 Hz, J = 10.7 Hz, 1 H), 6.82 (bs, 1 H), 6.90 (d, J = 8.4 Hz, 2 H), 7.04–7.12 (m, 3 H), 7.21 (m, 2 H), 7.48 (d, J = 7.3 Hz, 1 H), 7.55 (d, J = 7.8 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = −5.4, 15.5, 18.0, 18.2, 19.5, 21.4, 23.3, 24.9, 25.9, 28.1, 29.7, 32.1, 35.5, 52.1, 54.1, 54.5, 57.4, 57.5, 68.3, 80.7, 108.0, 113.4, 113.6, 118.0, 118.3, 119.4, 121.3, 123.7, 125.0, 128.6, 129.0, 132.6, 135.5, 139.9, 144.1 ppm. (Carbonyl signals are not distinguishable from the rotameric signals.) Minor rotamer (selected signals). HRMS (ESI+) m/z calculated for C67H104N11O12Si [M + H]+ 1282.7630; found, 1282.7637.
cyclo-(2S,3R)-(2-Amino-3,5-dimethyl-hex-4-enoyl)-(S)-N′-tert-prenyl-tryptophanyl-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-nitrophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine (16a)
Linear heptapeptide 15a (52.0 mg, 40.0 μmol) was transformed into the cyclopeptide 16a according to general procedure H. Purification with preparative HPLC gave the desired cyclic heptapeptide 16a (30.0 mg, 28.0 μmol, 70%) as amorphous colourless solid. LC-MS: tR = 13.34 min, m/z = 1079 ([M + Na]+) (Column: Phenomenex Luna C18(2), gradient: acetonitrile/H2O + 0.1% HCOOH, 40% acetonitrile increase to 99% acetonitrile in 7.5 min, a plateau for 15 min at 99% prior to return and reequilibration with the initial conditions, flow rate: 0.6 mL min−1). [α]20D = −11.5 (c = 0.1, CHCl3). 1H NMR (700 MHz, methanol-d4): δ = 0.17 (d, J = 6.7 Hz, 3 H), 0.89 (m, 1 H), 0.95 (d, J = 6.5 Hz, 3 H), 0.97 (d, J = 6.7 Hz, 3 H), 0.99 (d, J = 7.3 Hz, 3 H), 1.03 (d, J = 6.9 Hz, 3 H), 1.05 (d, J = 6.9 Hz, 3 H), 1.12 (d, J = 6.6 Hz, 3 H), 1.13–1.23 (m, 2 H), 1.62–1.66 (m, 2 H), 1.67 (d, J = 1.1 Hz, 3 H), 1.71 (s, 6 H), 1.82 (d, J = 1.1 Hz, 3 H), 2.31 (m, 1 H), 2.43 (m, 1 H), 2.53 (dd, J = 10.6 Hz, J = 7.5 Hz, 1 H), 2.61 (s, 3 H), 2.71–2.78 (m, 2 H), 2.88 (s, 3 H), 3.20 (dd, J = 13.5 Hz, J = 4.5 Hz, 1 H), 3.25 (s, 3 H), 3.28 (m, 1 H), 4.40–4.48 (m, 2 H), 4.54 (d, J = 10.2 Hz, 1 H), 4.65 (dd, J = 10.7 Hz, J = 4.8 Hz, 1 H), 4.82 (dd, J = 12.1 Hz, J = 2.9 Hz, 1 H), 4.91 (m, 1 H), 5.01 (m, 1 H), 5.04 (m, 1 H), 5.17 (d, J = 17.6 Hz, 1 H), 5.22 (d, J = 10.6 Hz, 1 H), 5.29 (dd, J = 11.3 Hz, J = 3.0 Hz, 1 H), 6.12 (dd, J = 17.5 Hz, J = 10.7 Hz, 1 H), 7.01–7.09 (m, 2 H), 7.13 (s, 1 H), 7.50 (d, J = 7.8 Hz, 1 H), 7.54 (d, J = 7.0 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 2 H), 7.65 (d, J = 8.2 Hz, 1 H), 8.19 (d, J = 8.7 Hz, 2 H), 8.44 (d, J = 10.0 Hz, 1 H), 8.48 (d, J = 9.6 Hz, 1 H) ppm. 13C NMR (175 MHz, methanol-d4): δ = 15.8, 19.2, 19.8, 19.9, 20.4, 21.5, 22.6, 24.1, 26.2, 26.6 (d, t), 28.4, 28.5, 29.9 30.3, 31.4, 33.4, 37.0, 40.3, 48.9, 51.6, 52.3, 57.3, 57.5, 58.8, 59.3, 59.4, 60.1, 60.2, 68.3, 84.6, 108.6, 114.2, 115.3, 119.7, 120.4, 122.2, 124.4, 125.5, 126.4, 129.5, 130.6, 135.2, 136.8, 145.5, 145.9, 149.2, 170.1, 170.2, 171.5, 173.3, 173.7, 173.8, 174.0 ppm. HRMS (ESI+) m/z calculated for C56H82N9O11 [M + H]+ 1057.6162; found, 1057.6163.cyclo-(2S,3R)-(2-Amino-3,5-dimethyl-hex-4-enoyl)-(S)-N′-tert-prenyl-tryptophanyl-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-aminophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine (16b)
Heptapeptide 16a (5.0 mg, 4.7 μmol) was dissolved in EtOH (0.5 mL) and tin(II) chloride (9.3 mg, 47.0 μmol) was added at room temperature. After 8 hours at 40 °C (LC-MS) the reaction mixture was cooled to room temperature and 10 mL dichloromethane was added. After washing with brine, drying over Na2SO4 and removing the solvent under reduced pressure, the remaining residue was purified with preparative HPLC. The desired heptapeptide 16b (4.2 mg, 4.1 μmol, 86%) was obtained as an amorphous colourless solid. LC-MS: tR = 12.48 min, m/z = 1049 ([M + Na]+) (Column: Phenomenex Luna C18(2), gradient: acetonitrile/H2O + 0.1% HCOOH, 40% acetonitrile, increase to 99% acetonitrile in 7.5 min, a plateau for 15 min at 99% prior to return and reequilibration with the initial conditions, flow rate: 0.6 mL min−1). [α]20D = −4.9 (c = 0.1, CHCl3). 1H NMR (700 MHz, methanol-d4): δ = 0.17 (d, J = 6.7 Hz, 3 H), 0.89 (m, 1 H), 0.95 (d, J = 6.6 Hz, 3 H), 0.97 (d, J = 6.6 Hz, 3 H), 1.03 (d, J = 6.7 Hz, 3 H), 1.05 (d, J = 6.4 Hz, 3 H), 1.06 (d, J = 7.2 Hz, 3 H), 1.11 (d, J = 6.6 Hz, 3 H), 1.15–1.21 (m, 2 H), 1.60 (d, J = 1.0 Hz, 3 H), 1.62–1.69 (m, 2 H), 1.71 (s, 6 H), 1.81 (d, J = 1.1 Hz, 3 H), 2.29 (m, 1 H), 2.41 (m, 1 H), 2.54 (dd, J = 10.5 Hz, J = 7.4 Hz, 1 H), 2.62 (s, 3 H), 2.67–2.76 (m, 2 H), 2.87 (s, 3 H), 3.18 (s, 3 H), 3.20 (m, 1 H), 3.26 (m, 1 H), 4.40–4.48 (m, 2 H), 4.51 (d, J = 10.3 Hz, 1 H), 4.64 (bs, 2 H), 4.67 (dd, J = 10.5 Hz, J = 4.9 Hz, 1 H), 4.79 (d, J = 3.6 Hz, 1 H), 4.81 (dd, J = 11.9 Hz, J = 2.8 Hz, 1 H), 4.92 (m, 1 H), 4.98 (m, 1 H), 5.18 (dd, J = 17.4 Hz, J = 0.4 Hz, 1 H), 5.23 (dd, J = 10.6 Hz, J = 0.5 Hz, 1 H), 5.28 (dd, J = 11.2 Hz, J = 3.0 Hz, 1 H), 6.13 (dd, J = 17.5 Hz, J = 10.7 Hz, 1 H), 6.62 (d, J = 8.5 Hz, 2 H), 7.02 (d, J = 8.4 Hz, 2 H), 7.03–7.09 (m, 2 H), 7.13 (s, 1 H), 7.51 (d, J = 7.8 Hz, 1 H), 7.54 (d, J = 7.6 Hz, 1 H), 8.42 (d, J = 9.7 Hz, 1 H) ppm. 13C NMR (175 MHz, methanol-d4): δ = 15.8, 19.2, 19.7, 19.9, 20.5, 21.3, 22.6, 24.1, 26.2, 26.6, 28.4, 28.5, 29.9 30.3, 31.3, 33.4, 37.0, 40.4, 48.9, 51.8, 52.2, 57.3, 57.9, 58.0, 59.3, 59.4, 60.1, 60.2, 68.3, 84.6, 108.7, 114.1, 115.3, 116.1, 119.7, 120.4, 122.2, 125.5, 126.2, 126.5, 129.3, 130.6, 135.0, 136.8, 145.5, 149.0, 170.1, 170.2, 172.4, 173.0, 173.5, 173.7, 174.0 ppm. HRMS (ESI+) m/z calculated for C56H84N9O9 [M + H]+ 1026.6387; found, 1026.6388.cyclo-(2S,3R)-(2-Amino-3,5-dimethyl-hex-4-enoyl)-(S)-N′-tert-prenyl-tryptophanyl-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-bromophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine (16c)
According to general procedure H, the linear heptapeptide 15c (10.0 mg, 7.6 μmol) was converted to cyclopeptide 16c. Purification with preparative HPLC resulted in the desired cyclic heptapeptide 16c (4.9 mg, 4.5 μmol, 59%) as amorphous colourless solid. LC-MS: tR = 12.85 min, m/z = 1113 ([M + Na]+) (Column: Phenomenex Luna C18(2), gradient: acetonitrile/H2O + 0.1% HCOOH, 40% acetonitrile, increase to 99% acetonitrile in 7.5 min, a plateau for 15 min at 99% prior to return and reequilibration with the initial conditions, flow rate: 0.6 mL min−1). [α]20D = −8.1 (c = 0.1, CHCl3). 1H NMR (700 MHz, methanol-d4): δ = 0.17 (d, J = 6.7 Hz, 3 H), 0.89 (m, 1 H), 0.96 (d, J = 6.6 Hz, 3 H), 0.97 (d, J = 6.7 Hz, 3 H), 1.01 (d, J = 7.3 Hz, 3 H), 1.03 (d, J = 7.0 Hz, 3 H), 1.05 (d, J = 7.0 Hz, 3 H), 1.11 (d, J = 6.6 Hz, 3 H), 1.13–1.23 (m, 2 H), 1.62 (d, J = 0.9 Hz, 3 H), 1.64–1.69 (m, 2 H), 1.71 (s, 6 H), 1.81 (d, J = 1.0 Hz, 3 H), 2.30 (m, 1 H), 2.43 (m, 1 H), 2.53 (dd, J = 10.4 Hz, J = 7.4 Hz, 1 H), 2.61 (s, 3 H), 2.71–2.78 (m, 2 H), 2.87 (s, 3 H), 3.17 (m, 1 H), 3.21 (s, 3 H), 3.26 (m, 1 H), 4.41–4.48 (m, 2 H), 4.53 (d, J = 10.2 Hz, 1 H), 4.65 (dd, J = 10.7 Hz, J = 4.9 Hz, 1 H), 4.81 (dd, J = 11.8 Hz, J = 2.6 Hz, 1 H), 4.90 (m, 1 H), 4.96–5.01 (m, 2 H), 5.17 (d, J = 17.5 Hz, 1 H), 5.23 (d, J = 10.7 Hz, 1 H), 5.28 (dd, J = 11.3 Hz, J = 2.9 Hz, 1 H), 6.13 (dd, J = 17.5 Hz, J = 10.7 Hz, 1 H), 7.01–7.09 (m, 2 H), 7.13 (s, 1 H), 7.24 (d, J = 8.4 Hz, 2 H), 7.45 (d, J = 8.4 Hz, 2 H), 7.50 (d, J = 7.8 Hz, 1 H), 7.54 (d, J = 7.3 Hz, 1 H), 7.56 (d, J = 7.3 Hz, 1 H), 8.41–8.48 (m, 2 H) ppm. 13C NMR (175 MHz, methanol-d4): δ = 15.8, 19.3, 19.7, 19.9, 20.4, 21.4, 22.6, 24.1, 26.2, 26.6, 28.4, 28.5, 29.9 30.3, 31.3, 33.4, 37.0, 40.4, 48.9, 51.7, 52.3, 57.3, 57.4, 58.5, 59.3, 59.4, 60.1, 60.2, 68.3, 84.4, 108.7, 114.1, 115.3, 119.7, 120.4, 122.2, 123.0, 125.5, 126.4, 130.4, 130.6, 135.0, 136.8, 137.2, 145.5, 170.1, 170.2, 171.8, 173.2, 173.6, 173.7, 174.0 ppm. HRMS (ESI+) m/z calculated for C56H82BrN8O9 [M + H]+ 1091.5362; found, 1091.5386.cyclo-(2S,3R)-(2-Amino-3,5-dimethyl-hex-4-enoyl)-(S)-N′-tert-prenyl-tryptophanyl-N-methyl-(2S,4R)-[5-(tert-butyldimethylsilyl)oxy-leucyl]-(S)-alanyl-(2S,3R)-(3-methoxy-para-azidophenylalanyl)-(S)-valyl-(S)-N-methyl-leucine (16d)
In accordance with general procedure H, linear heptapeptide 15d (10.0 mg, 7.8 μmol) was converted to cyclopeptide 16d. Purification with preparative HPLC resulted in the desired cyclic heptapeptide 16d (3.0 mg, 2.9 μmol, 37%) as amorphous colourless solid. LC-MS: tR = 13.64 min, m/z = 1053 ([M + H]+) (Column: Phenomenex Luna C18(2), gradient: acetonitrile/H2O + 0.1% HCOOH, 40% acetonitrile increase to 99% acetonitrile in 7.5 min, a plateau for 15 min at 99% prior to return and reequilibration with the initial conditions, flow rate: 0.6 mL min−1). [α]20D = −5.4 (c = 0.1, CHCl3). 1H NMR (700 MHz, methanol-d4): δ = 0.17 (d, J = 6.7 Hz, 3 H), 0.88 (m, 1 H), 0.96 (d, J = 6.6 Hz, 3 H), 0.98 (d, J = 6.7 Hz, 3 H), 1.00 (d, J = 7.3 Hz, 3 H), 1.03 (d, J = 6.8 Hz, 3 H), 1.05 (d, J = 6.8 Hz, 3 H), 1.12 (d, J = 6.6 Hz, 3 H), 1.14–1.21 (m, 2 H), 1.61–1.66 (m, 2 H), 1.67 (d, J = 1.1 Hz, 3 H), 1.71 (s, 3 H), 1.72 (s, 3 H), 1.83 (d, J = 1.1 Hz, 3 H), 2.32 (m, 1 H), 2.43 (m, 1 H), 2.53 (dd, J = 10.5 Hz, J = 7.6 Hz, 1 H), 2.61 (s, 3 H), 2.53 (dd, J = 10.5 Hz, J = 6.1 Hz, 1 H), 2.80 (m, 1 H), 2.87 (s, 3 H), 3.20 (dd, J = 13.4 Hz, J = 4.5 Hz, 1 H), 3.24 (s, 3 H), 3.28 (m, 1 H), 4.41 (q, J = 7.2 Hz, 1 H), 4.46 (d, J = 10.7 Hz, 1 H), 4.54 (d, J = 10.2 Hz, 1 H), 4.65 (dd, J = 10.7 Hz, J = 4.8 Hz, 1 H), 4.83 (dd, J = 12.0 Hz, J = 2.6 Hz, 1 H), 4.90 (m, 1 H), 5.00 (m, 1 H), 5.03 (m, 1 H), 5.17 (dd, J = 17.5 Hz, J = 0.5 Hz, 1 H), 5.23 (dd, J = 10.7 Hz, J = 0.6 Hz, 1 H), 5.29 (dd, J = 11.4 Hz, J = 2.9 Hz, 1 H), 6.12 (dd, J = 17.5 Hz, J = 10.8 Hz, 1 H), 7.00–7.09 (m, 2 H), 7.13 (s, 1 H), 7.50 (d, J = 7.8 Hz, 1 H), 7.54 (d, J = 7.4 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 2 H), 8.19 (d, J = 8.7 Hz, 2 H), 8.53 (bs, 1 H) ppm. 13C NMR (175 MHz, methanol-d4): δ = 15.8, 19.2, 19.7, 19.9, 20.4, 21.4, 22.6, 24.1, 26.2, 26.6, 28.4, 28.5, 29.9 30.3, 31.3, 33.4, 37.0, 40.3, 48.9, 51.7, 52.3, 57.4, 57.5, 58.4, 59.3, 59.4, 60.1, 60.2, 68.3, 84.2, 108.7, 114.1, 115.3, 119.7, 119.9, 120.4, 122.2, 125.6, 126.4, 130.1, 130.6, 134.8, 135.1, 136.8, 141.3, 145.5, 170.1, 170.2, 171.9, 173.2, 173.6, 173.7, 174.0 ppm. HRMS (ESI+) m/z calculated for C56H82N11O9 [M + H]+ 1052.6291; found, 1052.6290.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Saarland University is gratefully acknowledged. We also thank Dr med. Dr nat. med. Jan Rybniker (University of Cologne) for determing the biological activities against Mtb. and Chantal Bader (HIPS Saarbrücken) for preparative HPLC measurements and recording high-resolution mass spectra.References
- M. K. Renner, Y.-C. Shen, X.-C. Cheng, P. R. Jensen, W. Frankmoelle, C. A. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Am. Chem. Soc., 1999, 121, 11273–11276 CrossRef CAS.
- N. Bürstner, S. Roggo, N. Ostermann, J. Blank, C. Delmas, F. Freuler, B. Gerhartz, A. Hinniger, D. Hoepfner, B. Liechty, M. Mihalic, J. Murphy, D. Pistorius, M. Rottmann, J. R. Thomas, M. Schirle and E. K. Schmitt, ChemBioChem, 2015, 16, 2433–2436 CrossRef PubMed.
- E. K. Schmitt, M. Riwanto, V. Sambandamurthy, S. Roggo, C. Miault, C. Zwingelstein, P. Krastel, C. Noble, D. Beer, S. P. S. Rao, M. Au, P. Niyomrattanakit, V. Lim, J. Zheng, D. Jeffery, K. Pethe and L. R. Camacho, Angew. Chem., Int. Ed., 2011, 50, 5889–5891 CrossRef CAS PubMed.
- K. Weinhäupl, M. Brennich, U. Kazmaier, J. Lelievre, L. Ballell, A. Goldberg, P. Schanda and H. Fraga, J. Biol. Chem., 2018, 293, 8379–8393 CrossRef PubMed.
- C. Intaraudom, P. Rachtawee, R. Suvannakad and P. Pittayakhajonwut, Tetrahedron, 2011, 67, 7593–7597 CrossRef CAS.
- T. Kumamoto, H. Koshino, D. Watanabe, Y. Matsumoto, K. Aoyama, K. Harada, T. Ishikawa and Y. Mikami, Heterocycles, 2010, 80, 281–288 CrossRef CAS.
- A. W. Schultz, D. C. Oh, J. R. Carney, R. T. Williamson, D. W. Udwary, P. R. Jensen, S. J. Gould, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2008, 130, 4507–4516 CrossRef CAS PubMed.
- A. W. Schultz, C. A. Lewis, M. R. Luzung, P. S. Baran and B. S. Moore, J. Nat. Prod., 2010, 73, 373–377 CrossRef CAS PubMed.
- Q. Qian, A. W. Schultz, B. S. Moore and M. E. Tanner, Biochemistry, 2012, 51, 7733–7739 CrossRef CAS PubMed.
- M. Shibata, E. Higashide and H. Yamamoto, Agric. Biol. Chem., 1962, 26, 228–233 CAS.
- E. Higashide, M. Shibata, H. Yamamoto, K. Nakazawa, H. Iwasaki, J. Ueyanagi and A. Miyake, Agric. Biol. Chem., 1962, 26, 234–237 CrossRef CAS.
- Y. Iitaka, H. Nakamura, K. Takada and T. Takita, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2817–2825 CrossRef.
- L. W. Cary, T. Takita and M. Ohnishi, FEBS Lett., 1971, 17, 145–148 CrossRef CAS PubMed.
- J. Ma, H. Huang, Y. Xie, Z. Liu, J. Zhao, C. Zhang, Y. Jia, Y. Zhang, H. Zhang, T. Zhang and J. Ju, Nat. Commun., 2017, 8, 391 CrossRef PubMed.
- T. Haruhiko, T. Takehiko, T. Yoshihiro and H. Fumiaki, Chem. Lett., 1992, 21, 431–434 CrossRef.
- P. W. Ford, K. R. Gustafson, T. C. McKee, N. Shigematsu, L. K. Maurizi, L. K. Pannell, D. E. Williams, E. D. de Silva, P. Lassota, T. M. Allen, R. Van Soest, R. J. Andersen and M. R. Boyd, J. Am. Chem. Soc., 1999, 121, 5899–5909 CrossRef CAS.
- (a) H. Sugiyama, T. Shioiri and F. Yokokawa, Tetrahedron Lett., 2002, 43, 3489–3492 CrossRef CAS; (b) T. Beulshausen, U. Groth and U. Schöllkopf, Liebigs Ann. Chem., 1991, 1207–1209 CrossRef CAS.
- S. J. Wen and Z. J. Yao, Org. Lett., 2004, 6, 2721–2724 CrossRef CAS PubMed.
- D. B. Hansen and M. M. Joullié, Tetrahedron: Asymmetry, 2005, 16, 3963–3969 CrossRef CAS.
- M. A. Blaskovich and G. A. Lajoie, J. Am. Chem. Soc., 1993, 115, 5021–5030 CrossRef CAS.
- P. Barbie and U. Kazmaier, Org. Biomol. Chem., 2016, 14, 6036–6054 RSC.
- P. Barbie and U. Kazmaier, Org. Lett., 2016, 18, 204–207 CrossRef CAS PubMed.
- P. Barbie and U. Kazmaier, Org. Biomol. Chem., 2016, 14, 6055–6064 RSC.
- A. Ullrich, J. Herrmann, R. Müller and U. Kazmaier, Eur. J. Org. Chem., 2009, 6367–6378 CrossRef CAS.
- S. Kappler, L. Karmann, C. Prudel, J. Herrmann, G. Caddeu, R. Müller, A. Vollmar, S. Zahler and U. Kazmaier, Eur. J. Org. Chem., 2018 DOI:10.1002/ejoc.201801391.
- D. Becker and U. Kazmaier, Eur. J. Org. Chem., 2015, 4198–4213 CrossRef CAS.
- L. Karmann, K. Schultz, J. Herrmann, R. Müller and U. Kazmaier, Angew. Chem., Int. Ed., 2015, 54, 4502–4507 CrossRef CAS PubMed.
- E. A. Krasnokutskaya, N. I. Semenischeva, V. D. Filimonov and P. Knochel, Synthesis, 2007, 81–84 CrossRef CAS.
- M. P. Doyle, B. Siegfried, R. C. Elliott and J. F. Dellaria, J. Org. Chem., 1977, 42, 2431–2436 CrossRef CAS.
- Y. M. Lee, M. E. Moon, V. Vajpayee, V. D. Filimonov and K. W. Chi, Tetrahedron, 2010, 66, 7418–7422 CrossRef CAS.
- K. Barral, A. D. Moorhouse and J. E. Moses, Org. Lett., 2007, 9, 1809–1811 CrossRef CAS PubMed.
- H. Meerwein, H. Allendörfer, P. Beekmann, F. Kunert, H. Morschel, F. Pawellek and K. Wunderlich, Angew. Chem., 1958, 70, 211–215 CrossRef CAS.
- J. I. G. Cadogan and G. A. Molina, J. Chem. Soc., Perkin Trans. 1, 1973, 541–542 RSC.
- K. Burglova, S. Okorochenkov and J. Hlavac, Org. Lett., 2016, 18, 3342–3345 CrossRef CAS PubMed.
- M. P. Doyle, J. F. Dellaria, B. Siegfried and S. W. Bishop, J. Org. Chem., 1977, 42, 3494–3498 CrossRef CAS.
- M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 1999, 64, 2564–2566 CrossRef CAS.
- E. Rozners and Q. Xu, Org. Lett., 2003, 5, 3999–4001 CrossRef CAS PubMed.
- D. M. Shendage, R. Fröhlich and G. Haufe, Org. Lett., 2004, 6, 3675–3678 CrossRef CAS PubMed.
- S. Lemaire-Audoire, M. Savignac, E. Blart, G. Pourcelot, J. P. Genêt and J.-M. Bernard, Tetrahedron Lett., 1994, 35, 8783–8786 CrossRef CAS.
- P. Li, Encycl. Reagents Org. Synth, 2003, pp. 2–3 Search PubMed.
- L. Peng and J. C. Xu, J. Org. Chem., 2000, 65, 2951–2958 CrossRef.
- P. Barbie and U. Kazmaier, Org. Biomol. Chem., 2015, 13, 9267–9275 RSC.
- M. R. Luzung, C. A. Lewis and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 7025–7029 CrossRef CAS PubMed.
- S. J. Wen, T. S. Hu and Z. J. Yao, Tetrahedron, 2005, 61, 4931–4938 CrossRef CAS.
- F. D. Bellamy and K. Ou, Tetrahedron Lett., 1984, 25, 839–842 CrossRef CAS.
- J. Rybniker, A. Vocat, C. Sala, P. Busso, F. Pojer, A. Benjak and S. T. Cole, Nat. Commun., 2015, 6, 7659 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra. See DOI: 10.1039/c8ob02777c |
| This journal is © The Royal Society of Chemistry 2019 |