
Enantioselective synthesis and absolute configuration determination of hydroxywilfordic acid in sesquiterpene pyridine alkaloids†
Yue
Yuan
a,
Jong-Wha
Jung
 b and
Seung-Yong
Seo
*a
b and
Seung-Yong
Seo
*a
aCollege of Pharmacy and Gachon Institute of Pharmaceutical Sciences, Gachon University, Incheon 21936, Republic of Korea. E-mail: syseo@gachon.ac.kr
bCollege of Pharmacy, Research Institute of Pharmaceutical Sciences, Kyungpook National University, Daegu 41566, Republic of Korea
First published on 3rd December 2018
Abstract
An enantioselective synthetic route to hydroxywilfordic acid, a key subunit of sesquiterpene pyridine alkaloids such as wilfortrine, was developed. Asymmetric cyanation using Jacobsen's (R,R)-amino-thiourea and hydrolysis were performed to afford chiral α-hydroxy-α-methyl acid as the (S)-isomer. Naturally derived hydroxywilfordate prepared by methanolysis of wilfortrine was found to be the (R)-isomer upon comparison with the synthetic compound.
Sesquiterpene pyridine alkaloids are mainly isolated from the Celastraceae family of plants and exhibit immunosuppressive, insecticidal, anti-human immunodeficiency virus (HIV) activity, cytotoxicity, and anti-cancer activity against multi-drug resistant cancer cells.1 Among the anti-HIV sesquiterpene pyridine alkaloids, wilfordate- and hydroxywilfordate-containing compounds such as wilforine, wilforgine, wilfordine and wilfortrine exhibit interesting biological properties2 (Fig. 1). Those containing a tertiary hydroxy group have potent anti-HIV activity (EC50 of >0.01 μM). However, other natural products that contain the wilfordate moiety do not suppress HIV cell replication. Thus, it is reasonable to assume that the tertiary alcohol plays an important role in the anti-HIV activity of sesquiterpene pyridine alkaloids.
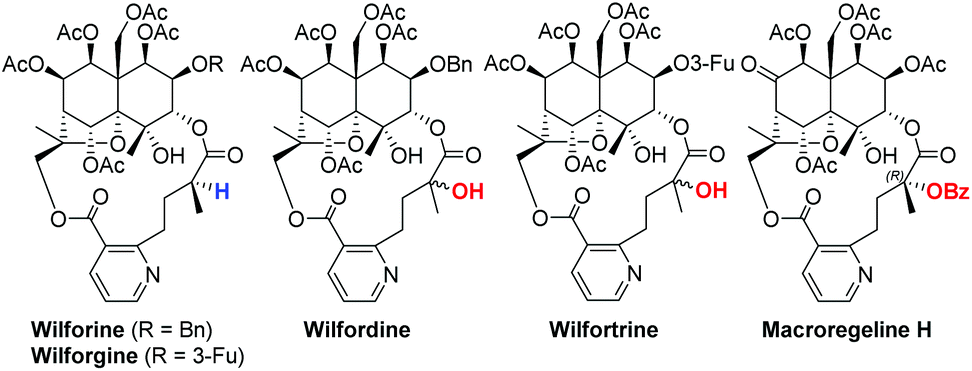 | ||
| Fig. 1 Representative sesquiterpene pyridine alkaloids. | ||
Enormous progress in the development of anti-HIV drugs has been made since the virus was discovered approximately 40 years ago.3 The major classes of anti-HIV agents are reverse transcriptase and protease inhibitors. Recently, therapies involving the combination of different classes have emerged as highly effective drug regimens. However, there is a continuing need for the development of new agents for the treatment of HIV as more easily tolerated, more convenient and less expensive treatments. Furthermore, resistance to existing drugs is also increasing.
The anti-HIV activity of sesquiterpene pyridine alkaloids such as wilfortrine and wilfordine motivated us to investigate the asymmetric synthesis of hydroxywilfordic acid to elucidate the absolute configuration of the quaternary carbon center. The structures of several esterifying substituents in sesquiterpene pyridine alkaloids have already been determined by X-ray crystallography. However, the stereochemistry of the tertiary alcohol in hydroxywilfordic acid has not yet been assigned.4 Recently, a new hydroxywilfordate-type sesquiterpene pyridine alkaloid macroregeline H was isolated and the stereochemistry of the tertiary alcohol was assigned as the (R)-isomer by electron circular dichroism (ECD) even though it has a C9′-benzoyl group on the tertiary alcohol.5
We attempted the asymmetric synthesis of hydroxywilfordic acid, the esterifying subunit of the natural products, to assign the stereochemistry of the α-hydroxy-α-methyl acid by Kusumi's NMR method using amides of the (R)- and (S)-phenylglycine methyl esters.6 Thus, it is necessary to find highly enantioselective synthetic approaches to hydroxywilfordic acid.
Herein, the enantioselective synthesis of the hydroxywilfordic acid fragment of anti-HIV sesquiterpene pyridine alkaloids is described for the first time. We envisioned that our synthesis would be useful for elucidating the absolute configuration of the tertiary alcohol. Hydroxywilfordate-bearing sesquiterpene pyridine alkaloids such as wilfortine can be hydrolyzed into two key fragments, euonyminol and hydroxywilfordic acid, the chirality of which can be compared with that of synthetic hydroxywilfordic acid to determine the stereochemistry of the tertiary alcohol in the natural products (Fig. 2).
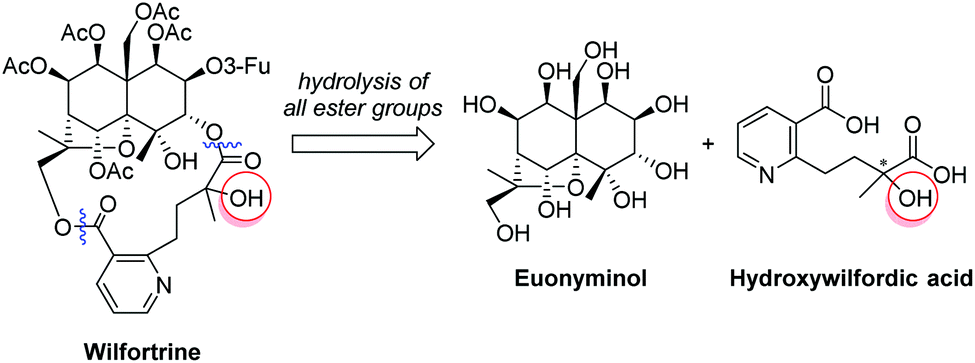 | ||
| Fig. 2 Hydroxywilfordic acid, an esterifying fragment of wilfortrine. | ||
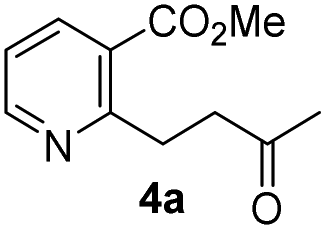
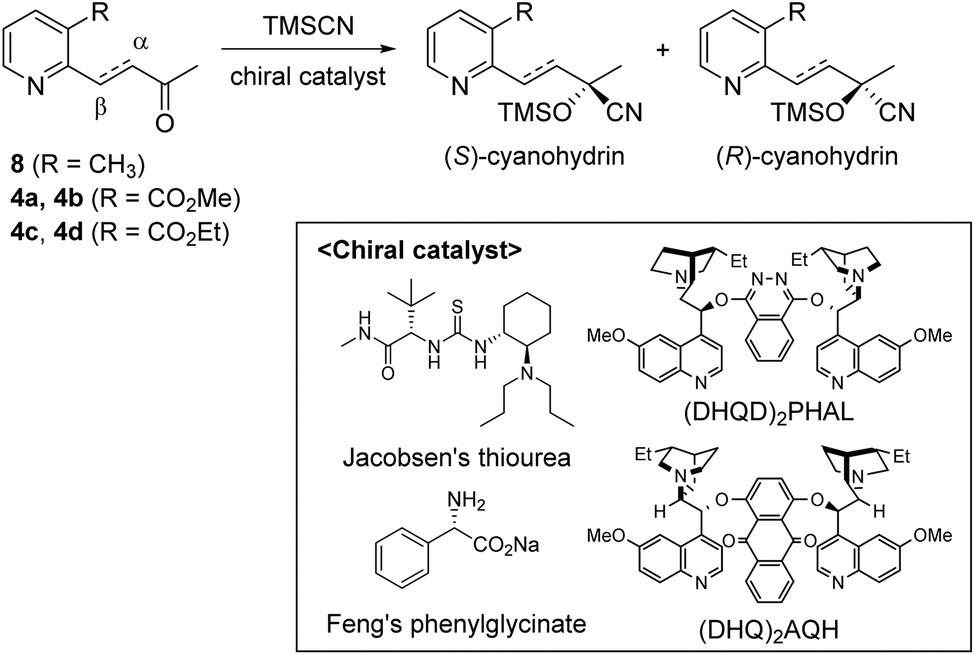
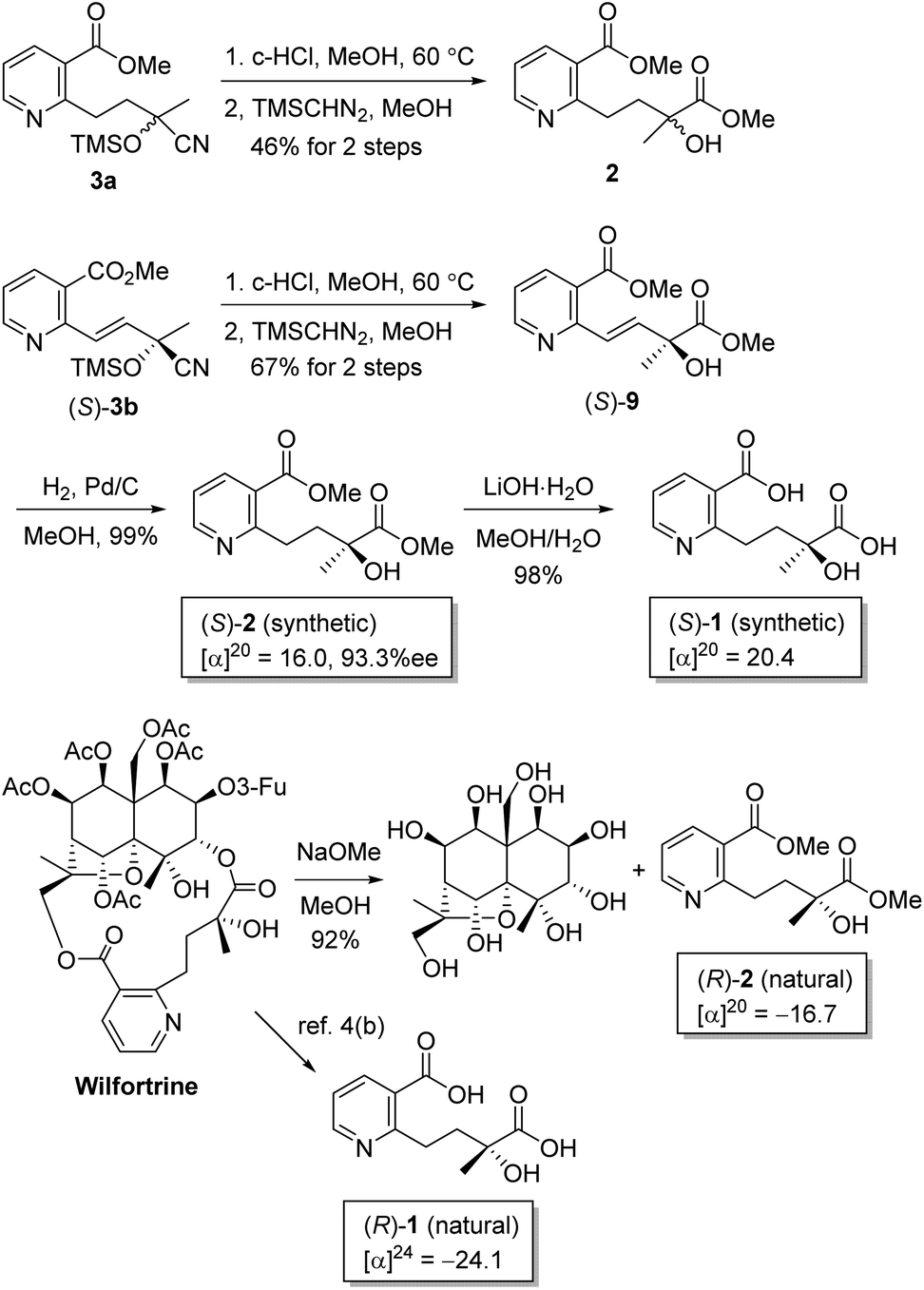
Our strategy for the preparation of chiral hydroxylwilfordic acid 1 and its dimethyl ester 2 involved the asymmetric cyanation of the ketone and careful hydrolysis of the cyanohydrin to generate α-hydroxy-α-methyl acid (Scheme 1).7 Based on our previous work,6 the Jacobsen's (R,R)-amino-thiourea catalyst for asymmetric cyanosilylation of the ketone was utilized.8 To obtain the α-hydroxy-α-methyl acid, we performed the hydrolysis of chiral cyanohydrin 3, derived from the asymmetric cyanosilylation of methyl ketone 4. Palladium-catalyzed cross coupling such as the Stille reaction was considered for constructing methyl ketone 4. To this end, methyl 2-chloronicotinate 5 and 4-(tributylstannanyl)but-3-en-2-ol were used as the starting materials.
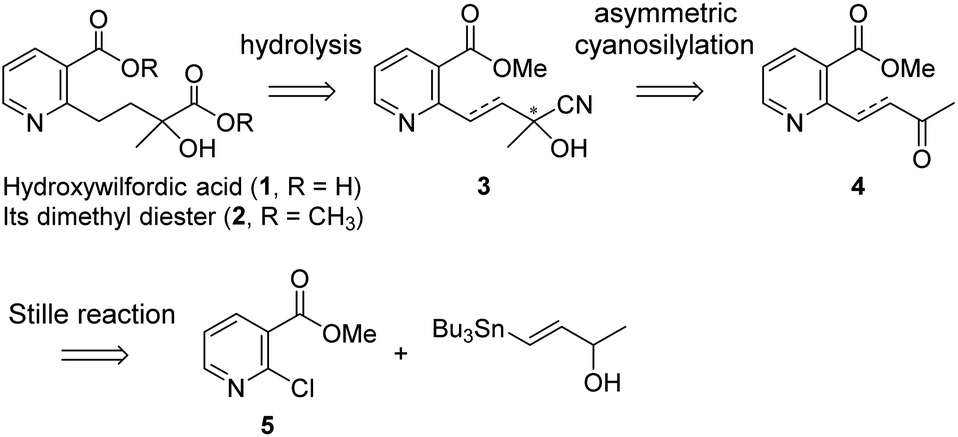 | ||
| Scheme 1 Retrosynthetic analysis of hydroxywilfordic acid and its dimethyl diester. | ||
As shown in Scheme 2, our initial approach toward the synthesis of saturated ketone 4a and α,β-unsaturated ketone 4b began with the Sonogashira reaction of methyl 2-chloronicotinate 5 with 3-butyn-2-ol to afford alkyne 6 in 63% yield.9 Catalytic hydrogenation of alkyne 6 and TPAP-mediated alcohol oxidation afforded the saturated methyl ketone 4a. We attempted the tandem cross coupling-isomerization under microwave-accelerated Sonogashira reaction conditions as developed by the Müller group,10 but the desired ketone 4b was obtained in 15% yield along with 39% yield of 6. Thus, another synthetic route, employing a Stille coupling with (E)-4-(tributylstannyl)but-3-en-2-ol and subsequent MnO2-mediated allylic oxidation was exploited for the synthesis of α,β-unsaturated ketone 4b. Methyl ester 5 was treated with catalytic Pd(PPh3)4 and (E)-4-(tributylstannyl)but-3-en-2-ol, prepared from trans-hydrostannylation, as investigated by the Kim group to afford allylic alcohol 7 in 93% yield.11 With 7 in hand, MnO2 oxidation afforded the α,β-unsaturated ketone 4b in 91% yield.
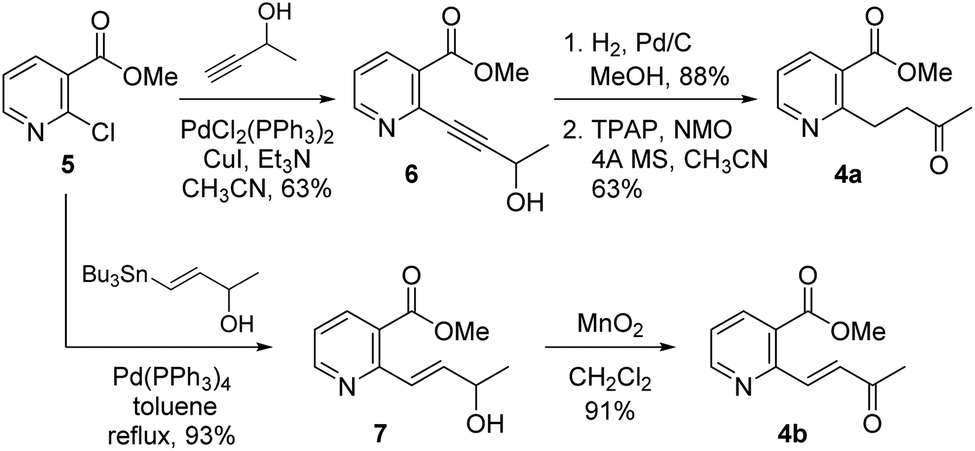 | ||
| Scheme 2 Synthesis of the ketones 4a and 4b. | ||
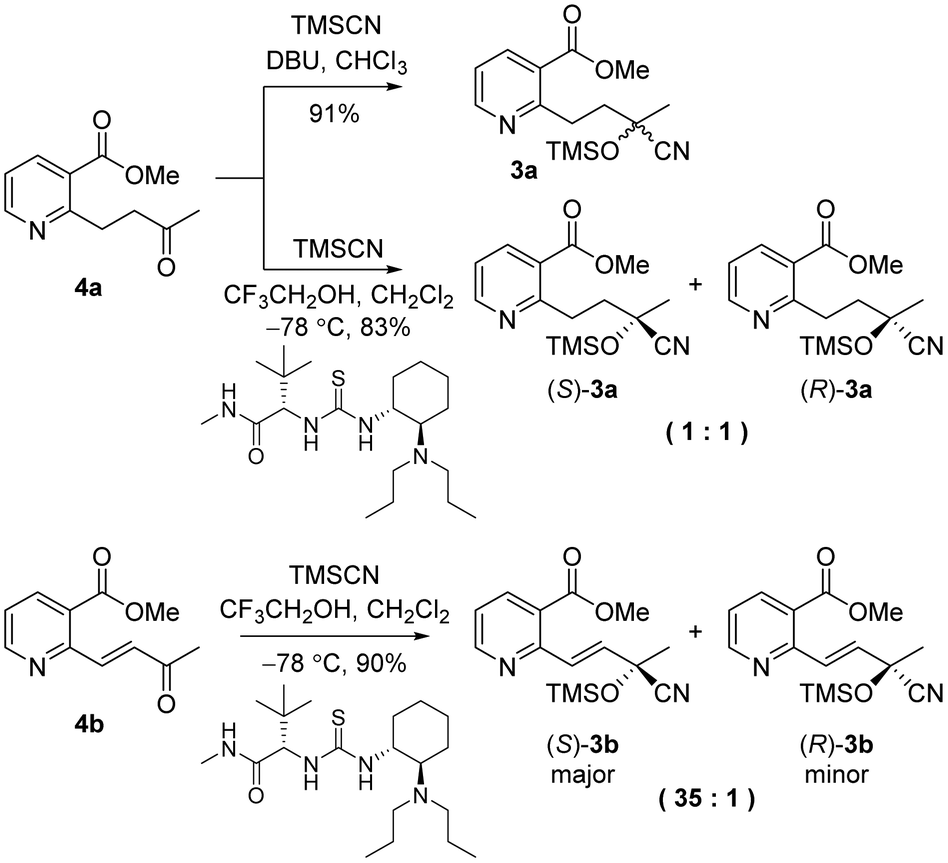
To investigate the asymmetric cyanosilylation of the ketones 4a and 4b, racemic cyanohydrins were prepared in good yields by the treatment of 4a and 4b with TMSCN and DBU (Scheme 3). Previously, we chose two chiral catalysts, Feng's sodium phenylglycinate12 and Jacobsen's amino-thiourea8 for cyanation of the similar ketone 8 having a methyl group on the C3 position of the pyridine ring.6 By Kusumi's NMR method, the (S)-isomer was assigned as a major cyanohydrin by asymmetric cynanation using Jacobsen's catalyst, while the (R)-isomer as a major one using Feng's catalyst. However, the enantioselective cyanations using these reagents were insufficient, providing the (R)-isomer with 39% ee and the (S)-isomer with 50% ee, respectively. With 4a and 4b in hand, we investigated asymmetric cyanosilylation using the commercially available Jacobsen's amino-thiourea catalyst. Regrettably, 4a upon treatment with TMSCN, 2,2,2-trifluoroethanol, and Jacobsen's (R,R)-amino-thiourea afforded cyanohydrin 3a as a racemic mixture. Conversely, the cyanosilylation of α,β-unsaturated ketone 4b using the same catalytic system afforded the desired cyanohydrin 3b in good yield with an enantiomeric ratio of 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
 | ||
| Scheme 3 Asymmetric syntheses of dimethyl hydroxywilfordate. | ||
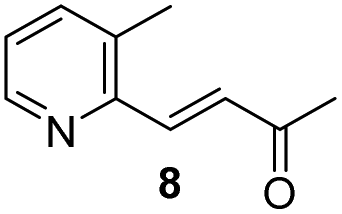
The enantioselectivity of cyanation was proposed to be dependent on the planarity of the ketone substrate. Among ketones 8, 4a, and 4b, 4a reacts with free bond rotation, whereas 4b, which has the largest conjugated system interacts with the amino-thiourea catalyst and the stereospecific reaction proceeds well through the enantiotopic attack of cyanide. As reported by Jacobsen's group, the cyanation of aryl ketones and vinyl ketones is more stereoselective than that of aliphatic ketones.8 The two different approaches for cyanation to 4b employed in the current study are depicted in Fig. 3.
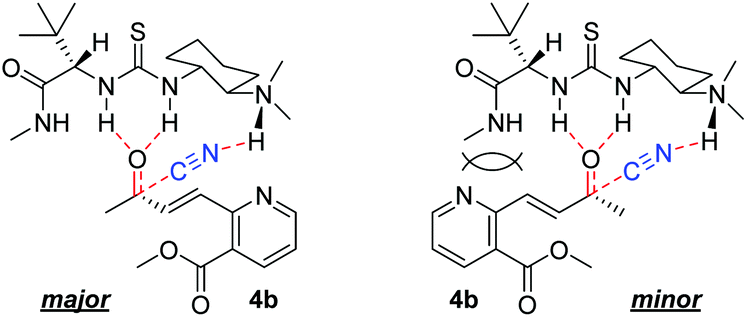 | ||
| Fig. 3 Possible mechanism of asymmetric cyanosilylation to ketone 4b. | ||
We examined the enantioselectivity of cyanosilylation using different catalysts including Jacobsen's thiourea as shown in Table 1. As mentioned above, the Jacobsen's thiourea-catalyzed cyanation to ketone 8 led to the product with an enantiomeric ratio of 4:1. The saturated ketones 4a and 4c were converted into the racemic mixtures of cyanohydrins under the same conditions, whereas the cyanation of the α,β-unsaturated ketones 4b and 4d using Jacobsen's amino-thiourea gave excellent selectivities with ratios of 35:1 and 21:1, respectively. Furthermore, the yields and stereoselectivities for methyl esters 4a and 4b were superior to those for ethyl esters 4c and 4d. The enantioselectivities achieved with sodium phenylglycinate, (DHQD)2PHAL and (DHQ)2AQN,13 were unsatisfactory, although the cyanation gave rise to the (R)-isomer slightly more favorably.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Conditions | Yielda | Ratiob (S:R) |
| a Isolated yield based on the recovered starting material in parentheses. b Determined by chiral HPLC. c See ref. 5. | |||||
| 1c |
|
Jacobsen's thiourea | TMSCN, 2,2,2-trifluoroethanol, CH2Cl2, −78 °C | 82 | 4:1 |
| 2 |
|
85 | 1:1.2 |
||
| 3 |
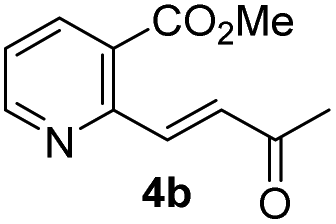
|
90 | 35:1 |
||
| 4 |
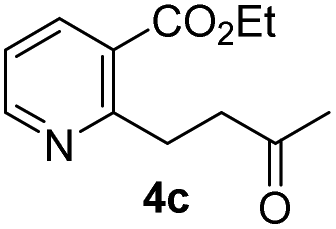
|
29 (91) | 1:1.1 |
||
| 5 |
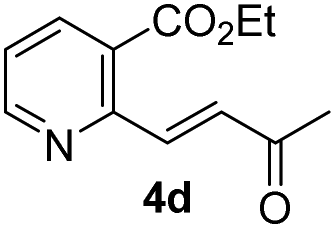
|
79 (99) | 21:1 |
||
| 6c | 8 | Feng's phenylglycinate | TMSCN, iPrOH, THF, −45 °C | 79 | 1:2.3 |
| 7 | 4b | 70 | 1:1.2 |
||
| 8 | 4b | (DHQD)2PHAL | TMSCN, CHCl3, −24 °C | 80 | 1.9:1 |
| 9 | 4b | (DHQ)2AQN | TMSCN, CHCl3, −24 °C | 80 | 1:2.9 |
We then turned to the hydrolysis of cyanohydrins 3a and 3b into the corresponding α-hydroxy-α-methyl acids. However, it is difficult to isolate the diacids containing pyridine, and mixtures of a diacid and a monoacid were obtained upon hydrolysis or methanolysis. Thus, the esters are easier to separate than the acids, allowing increased isolated yields. Cyanohydrin 3a was hydrolyzed using HCl in MeOH to afford a mixture of hydroxywilfordate isomers, which were then treated with (trimethylsilyl)diazomethane in MeOH to afford racemic diester 2. Similarly, (S)-3b was converted into the diester, followed by catalytic hydrogenation to afford the final hydroxywilfordic acid dimethyl diester (S)-2 in good yield with the minimization of racemization and β-elimination. The optical rotation of the final compound (S)-2 with 93.3%ee was determined by chiral HPLC. Finally, the hydroxywilfordic acid (S)-1 was obtained by LiOH-mediated hydrolysis in good yield. The selective/partial hydrolysis of 2 was conducted to afford a monoacid by controlling the reaction time and conditions but the treatment of LiOH over 2 h led to two regioisomers with a ratio of 1.9:1.14
As a means to determine the absolute configurations of the tertiary alcohol groups in the synthetic dimethyl ester and the naturally derived one, methanolysis of the commercially available wilfortrine using sodium methoxide afforded the naturally derived dimethyl diester 2. The 1H- and 13C-NMR spectra of naturally derived 2 were identical to those of synthetic 2. Interestingly, the stereochemistry of synthetic 2 is determined to be opposite to naturally derived 2 by chiral HPLC and optical rotation. The optical rotation of the naturally derived 2 was found to be −16.7; thus, its absolute configuration was observed to be the (R)-isomer by comparison with the synthetic (S)-isomer, the optical rotation of which was +16.0. For synthetic 1, an [α]D value of +20.4 was measured, while for natural compound 1, [α]D = −24.1 was reported.4b As the stereochemistry of macroregeline H was determined by the experimental and calculated ECD spectra,5 the ECD spectra of synthetic 2 and naturally derived 2 were measured to define the absolute configuration although the tertiary alcohol of synthetic 2 was regarded to have the (S)-configuration following Jacobsen group's report and our previous work. Not surprisingly, the experimental ECD spectra confirmed that two compounds have the opposite configuration as enantiomers and the theoretical calculations of the ECD spectra were conducted for a pair of enantiomers of hydroxywilfordic acid dimethyl diester 2 using time-dependent DFT at the B3LYP/6-311+G(d,p) level of theory in MeOH. The calculated ECD spectra of the (S)- and (R)-isomers showed agreement with the experimental curves of synthetic and naturally derived 2, respectively, suggesting that the absolute configuration of the tertiary alcohol in naturally occurring hydroxywilfordic acid turns out to be the (R)-form. Thus, the full stereochemistry of wilfortrine, as shown in Scheme 4, has been determined for the first time.
 | ||
| Scheme 4 Stereochemistry comparison of hydroxywilfordic acid (1) and its dimethyl ester (2) derived from chiral cyanohydrin (S)-3b and by methanolysis of naturally occurring wilfortrine. | ||
Conclusions
In summary, a novel enantioselective strategy for the synthesis of the diester derivative of hydroxywilfordic acid, a key fragment of anti-HIV sesquiterpene pyridine alkaloids, was developed. Asymmetric cyanosilylation of the α,β-unsaturated ketone using Jacobsen's (R,R)-amino-thiourea was performed to synthesize the (S)-isomer. The stereochemistry of the tertiary alcohol in naturally occurring hydroxywilfordic acid could be assigned as the (R)-configuration. Currently, synthetic strategies for regioselective/chemoselective esters or amide formation from hydroxywilfordic acid are under investigation as a means to obtaining simplified analogs of sesquiterpene pyridine alkaloids and evaluating the biological activities of their derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2014M3C1A3054139 and NRF-2016R1D1A1A09917300).Notes and references
- (a) J.-M. Gao, W.-J. Wu, J.-W. Zhang and Y. Konishi, Nat. Prod. Rep., 2007, 24, 1153 RSC; (b) A. C. Spivey, M. Weston and S. Woodhead, Chem. Soc. Rev., 2002, 31, 43 RSC; (c) A. M. Brinker, J. Ma, P. E. Lipsky and I. Raskin, Phytochemistry, 2007, 68, 732 CrossRef CAS PubMed.
- M. Horiuch, C. Murakami, N. Fukamiya, D. Yu, T.-H. Chen, K. F. Bastow, D.-C. Zhang, Y. Takaishi, Y. Imakura and K.-H. Lee, J. Nat. Prod., 2006, 69, 1271 CrossRef CAS PubMed.
- (a) A. S. Fauci, Cell, 2006, 131, 429 CrossRef PubMed; (b) C. Flexner, Nat. Rev. Drug Discovery, 2007, 6, 959 CrossRef CAS PubMed.
- (a) H. Itokawa, O. Shirota, H. Morita, K. Takeyaa and Y. Iitakab, J. Chem. Soc., Perkin Trans. 1, 1993, 1247 RSC; (b) M. Beroza, J. Org. Chem., 1963, 28, 3562 CrossRef CAS.
- D. Fan, T. Li, Z. Zheng, G.-Y. Zhu, X. Yao, Z.-H. Jiang and L.-P. Bai, J. Nat. Med., 2018 DOI:10.1007/s11418-018-1232-8.
- J. S. Eun and S.-Y. Seo, Arch. Pharm. Res., 2009, 32, 1673 CrossRef CAS PubMed.
- N. Kurono and T. Ohkuma, ACS Catal., 2016, 6, 989 CrossRef CAS.
- (a) D. E. Fuerst and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 8964 CrossRef CAS PubMed; (b) S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 15872 CrossRef CAS PubMed.
- Methyl 2-chloronicotinate 5 is commercially available and was easily prepared by the treatment of 2-hydroxynicotinic acid with thionyl chloride and catalytic DMF, followed by the addition of methanol.
- O. G. Schramm and T. J. J. Müller, Adv. Synth. Catal., 2006, 348, 2565 CrossRef CAS.
- T. Lee and S. Kim, Tetrahedron: Asymmetry, 2003, 14, 1951 CrossRef CAS.
- X. Liu, B. Qin, X. Zhou, B. He and X. Feng, J. Am. Chem. Soc., 2005, 127, 12224 CrossRef CAS PubMed.
- S.-K. Tian, R. Hong and L. Deng, J. Am. Chem. Soc., 2003, 125, 9900 CrossRef CAS PubMed.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02364f |
| This journal is © The Royal Society of Chemistry 2019 |