
Highly selective AlCl3 initiated intramolecular α-alkylation of α,β-unsaturated lactams and lactones†
Dawen
Xu
a,
Felix
Kaiser
a,
Han
Li
a,
Robert M.
Reich
 a,
Hao
Guo
*b and
Fritz E.
Kühn
*a
a,
Hao
Guo
*b and
Fritz E.
Kühn
*a
aMolecular Catalysis, Catalysis Research Center and Department of Chemistry, Technische Universität München, Lichtenbergstr. 4, 85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China. E-mail: hao_guo@fudan.edu.cn
First published on 7th December 2018
Abstract
An unprecedented example of AlCl3 initiated intramolecular α-alkylation of α,β-unsaturated lactams and lactones is reported. A variety of substrates containing an intramolecular diene yield exclusively regioselective six-membered ring products. This reaction protocol generates a new stereo centre which may be of high interest for the functionalization of bioactive coumarin and quinolinone derivatives.
α,β-Unsaturated carbonyl compounds are highly valuable building blocks in organic synthesis, as these structures can be utilized for example in [4 + 2] thermocycloadditions1,2 and [2 + 2] photocycloadditions.3,4 α-Functionalization of α,β-unsaturated carbonyl compounds, while preserving the α,β-unsaturation, represents a very useful transformation. In this way, complex products can be produced through subsequent thermocycloaddition, photocycloaddition, or Michael addition of the functionalized carbonyl derivatives. Starting from α,β-unsaturated carbonyl compounds, there are a few methods available that allow α-functionalization. The Baylis–Hillman reaction, typically catalyzed by amines or phosphines, can facilitate α-hydroxyalkylation between activated alkenes and aldehydes at the α-position of Michael acceptors.5,6 In the Lewis base catalyzed Rauhut–Currier reaction, electron deficient olefins such as Michael acceptors can be dimerized.5a,7,8 Pd-Catalyzed cross-coupling reactions between α-iodo or α-triflyloxyenones and alkenylmetal derivatives yield α-alkenylation products.9 Furthermore, α-arylation and α-alkylation can be achieved by a multi-step synthesis via nucleophilic or electrophilic attack after generation of the corresponding α-ketovinyl cation or the α-ketovinyl anion.10 However, until now, the intramolecular α-alkylation of α,β-unsaturated carbonyl compounds always requires activated olefins,11 while for non-activated olefins an analogous reaction has not been reported yet. While investigating the behavior of 4-(pent-4-en-1-yl) quinolinone under Lewis acid catalysis, an interesting result was found. When the substrate was subjected to two equivalents of AlCl3 in anhydrous dichloromethane, a six-membered ring α-alkylation product was observed selectively, even under ambient conditions. To the best of our knowledge, this represents an unprecedented example for AlCl3 initiated α-alkylation of α,β-unsaturated lactams and lactones with non-activated olefins.
Initially, the influence of Lewis acids by using 4-(pent-4-en-1-yl) quinolin-2(1H)-one (1a) as the reaction substrate and CH2Cl2 as a solvent has been explored. Several Lewis acids had been screened, the results in terms of time and yields are listed in Table S1 (ESI, S1†). AlCl3 was chosen as the best available Lewis acid for this reaction, two equivalents of AlCl3 were applied as standard loading and CH2Cl2 as solvent of choice.
With the optimized reaction conditions in hand, attention turned towards the exploration of the substrate scope (Table 1). At first, N-methylated substrate 2a was examined; the reaction gave 98% desired product 2b after 2 h (2b). The exact structure of 2b was determined by single crystal X-ray diffraction analysis.12 Replacement of lactam by lactone resulted in a slight decrease in yield (88%, 3b). The electronic effects on the aromatic ring were examined by the formal addition of various functional groups (4b–8b). An acetoxy moiety was tolerated in this transformation and the respective substrate 4a afforded the desired product in moderate yield 63%. Substrate 5a with an electron withdrawing fluorine substitution also furnished the corresponding product in good yield 78%. Electron-rich substrates, such as methyl, hydroxyl, and dimethoxyl substituted substrates gave the desired products in good to excellent yields (83–95%, 6b–8b). Thus, the reaction was applicable for both electron-poor and electron-rich substrates. Subsequently, substituent effects on the terminal alkene were investigated (9b–11b). Substrate 9a yields the target molecule in 80% yield, while 10a, with an ethyl substituent on the terminal alkene, leads only to 52% yield. When subjecting iso-methylated substrate 11a to the reaction conditions, a significantly decreased yield was observed (24%). This was attributed to the hindrance in the product caused by a quaternary carbon center (11b). Finally, the impact of the alkenyl tether chain length on the α-alkylation results was studied. For substrate 12a with an alkenyl chain that was one carbon shorter than the one in 1a, α-alkylation no longer took place at α-position of the lactam group. Instead, Friedel–Crafts alkylation product 12b was obtained, after a prolonged reaction time of 50 h. However, with the methylated terminal alkene derivative 13a, a six-membered ring product was formed in a yield of 65% (13a → 1b). The results of 12a and 13a indicated that the reaction proceeded through a six-membered ring transition state. To investigate the importance of the presumed six-membered ring transition state for the course of the reaction, substrate 14a with a longer alkenyl tether was synthesized. Upon treatment with AlCl3, six-membered ring product 9b was generated in 49% yield (14a → 9b); no traces of a seven- or eight-membered rings were detected.
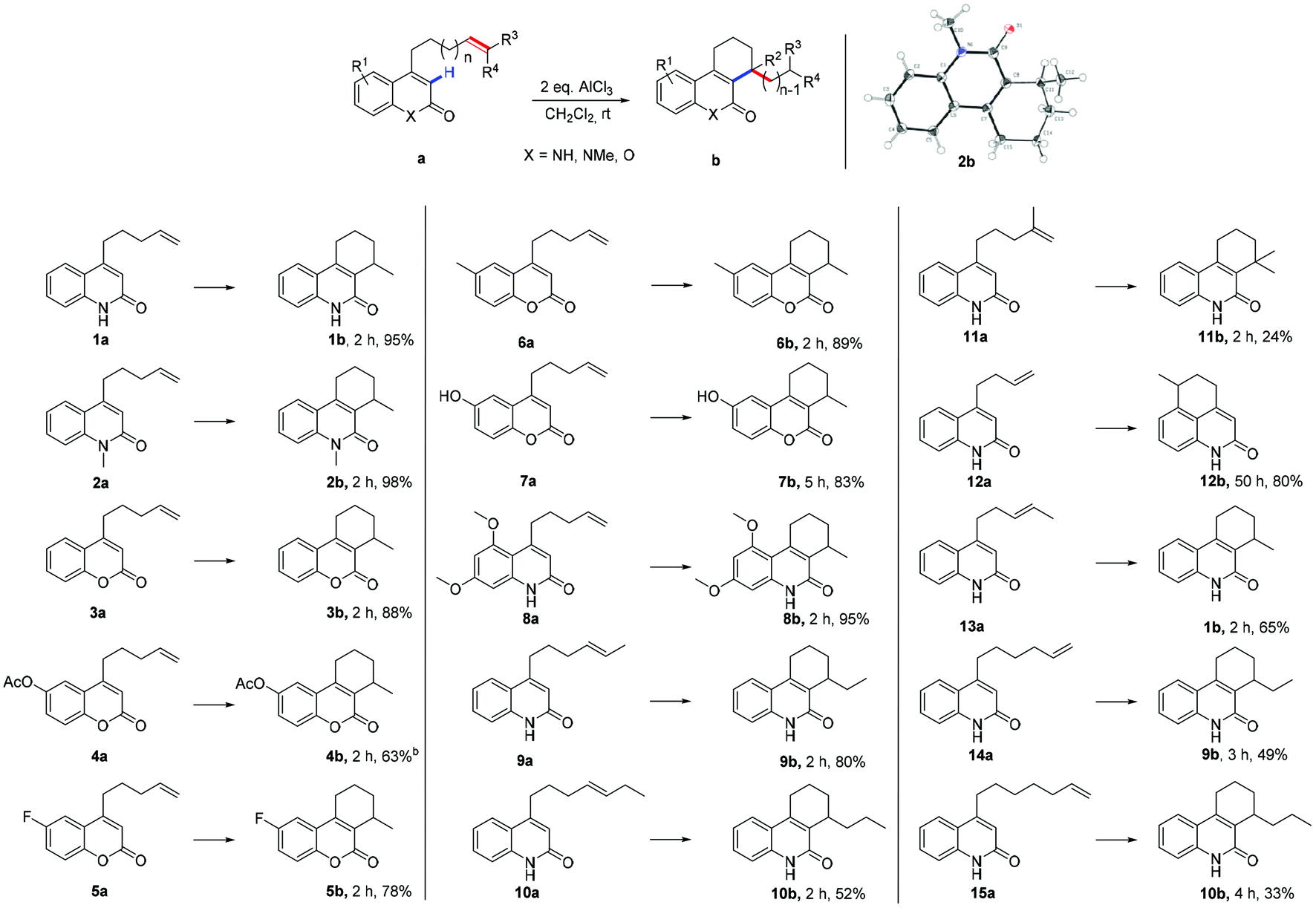
Substrate 15a, bearing two additional CH2– groups within the alkenyl tether, was then as well subjected to the standard reaction conditions. Again, only six-membered ring product 10b was detected, albeit with a lower yield of 33% (15a → 10b). Again, no traces of larger ring products could be observed. These results suggest, that cyclization can only occur via a six-membered ring transition state and therefore, carbocationic rearrangements are involved in the process (vide infra).
In order to gain more information about the reaction mechanism, a set of experiments were carried out (Scheme 1). With finely sublimated AlCl3, the reaction only offered traces of product. When deuterated oxide was added, the cyclized product was then generated with a deuterium content of 7% (Fig. S2†). These experiments indicate that small amounts of water in the medium are required for this transformation and a hydronium ion propagation takes place, since the deuterium content does not match its loading.
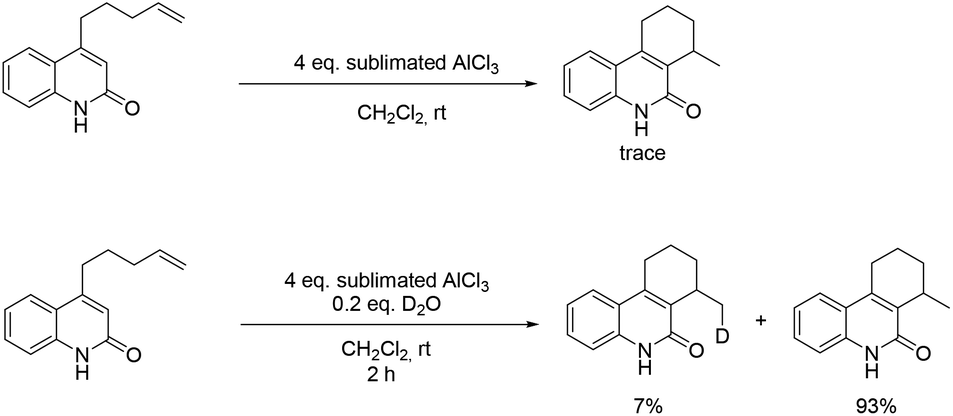 | ||
| Scheme 1 Reaction with (water-free) sublimated AlCl3. | ||
Based on above proofs, a preliminary reaction mechanism for this transformation is proposed (Scheme 2). The general mechanism proceeds through the coordination of the first equivalent of AlCl3 to the lactam carbonyl of starting material A, forming species B.13 Consequently, AlCl3 reacts with trace moisture in the medium to generate hydronium ions, which subsequently protonate the alkene to generate a carbocation in species C. The carbocation generated attacks the α-carbon atom of the lactam ring as in a conventional Friedel–Craft alkylation, yielding the observed six membered ring D.14 Presumably, a deprotonation process occurs to regenerate the hydronium ions to propagate the reaction.
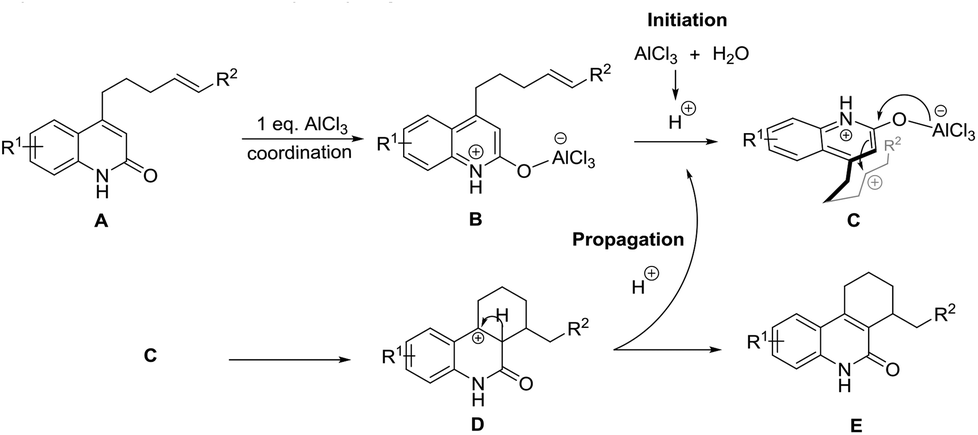 | ||
| Scheme 2 Proposed mechanism of the reaction. | ||
The results for the reaction of 13a, 14a and 15a suggests a preceding carbocationic rearrangements, respectively, between B′ and C′ for different lengths of the alkenyl tether, as depicted in Scheme 3. After coordination of the first equivalent of AlCl3 to the starting material affording B′, this species reacts with a hydronium ion to yield a carbon cation, respectively. The carbocation rearranges via 1,2-hydride shifts,15 which occur reversibly and proceeding in both directions of the alkenyl chain. These shifts are suggested to be reversible and proceeding in both directions of the alkenyl chain. Once the carbon cation is in 4-position of the alkenyl chain (C′), a six-membered ring transition state can be formed. It subsequently follows the general reaction path to yield target molecule E′ (see Scheme 2). Accompanying decomposition processes are assumed during these shifts, explaining the relatively low yields of 13a, 14a and 15a, which decreases with increasing lengths of the alkenyl tether.
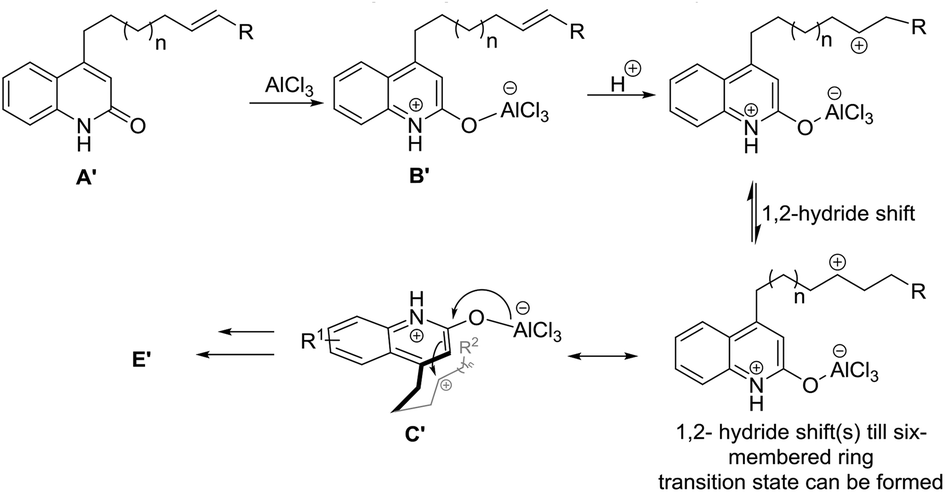 | ||
| Scheme 3 Proposed preceding carbocation rearrangements equilibrium for the reactions of 13a, 14a and 15a. | ||
In summary, the intramolecular α-alkylation of α,β-unsaturated carbonyl derivatives with β-alkenyl tethered olefins has been studied. A variety of functional groups was tolerated and six-membered ring products were selectively formed in this transformation. A preliminary mechanism for the reaction was proposed. A six-membered ring transition state and preceding carbocation rearrangements are proposed key steps for this type of reaction. As this reaction protocol generates tricyclic compounds and allows the introduction of an additional stereo centre, it may pave the way to an easier and more convenient synthesis of coumarine and quinolinone derivatives for the synthesis of natural products and industrially relevant drugs. However, additional mechanistic and computational studies to further increase the mechanistic understanding are still needed.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2018, 118, 2080–2248 CrossRef CAS PubMed; (b) M. Xie, L. Lin and X. Feng, Chem. Rec., 2017, 17, 1184–1202 CrossRef CAS PubMed; (c) K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 3558–3588 CrossRef.
- (a) L. F. Tietze, J. Fennen and E. Anders, Angew. Chem., Int. Ed., 1989, 101, 1420–1422 CrossRef CAS; (b) X. L. Huang, L. He, P. L. Shao and S. Ye, Angew. Chem., Int. Ed., 2009, 48, 192–201 CrossRef CAS PubMed.
- (a) S. Poplata, A. Troster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed; (b) M. Oelgemoller and N. Hoffmann, Org. Biomol. Chem., 2016, 14, 7392–7442 RSC.
- (a) H. Guo, E. Herdtweck and T. Bach, Angew. Chem., Int. Ed., 2010, 49, 7782–7785 CrossRef CAS PubMed; (b) Z. D. Miller, B. J. Lee and T. P. Yoon, Angew. Chem., Int. Ed., 2017, 56, 11891–11895 CrossRef CAS PubMed; (c) F. M. Hormann, T. S. Chung, E. Rodriguez, M. Jakob and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 827–831 CrossRef PubMed; (d) S. Poplata and T. Bach, J. Am. Chem. Soc., 2018, 140, 3228–3231 CrossRef CAS PubMed.
- (a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed; (c) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811–892 CrossRef CAS PubMed.
- (a) M. E. Krafft, K. A. Seibert, T. F. Haxell and C. Hirosawa, Chem. Commun., 2005, 5772–5774 RSC; (b) E. L. Myers, J. G. de Vries and V. K. Aggarwal, Angew. Chem., Int. Ed., 2007, 46, 1893–1896 CrossRef CAS PubMed; (c) S. K. Chittimalla, M. Koodalingam, C. Bandi, S. Putturu and R. Kuppusamy, RSC Adv., 2016, 6, 1460–1465 RSC; (d) V. K. Rai, F. Verma, M. Satnami, M. Singh and A. Rai, Tetrahedron Lett., 2018, 59, 1783–1786 CrossRef CAS; (e) J. Qi, J. Zheng and S. Cui, Org. Lett., 2018, 20, 1355–1358 CrossRef CAS PubMed; (f) C. Peter, P. Geoffroy and M. Miesch, Org. Biomol. Chem., 2018, 16, 1381–1389 RSC; (g) T. Gupta, J. B. Singh, K. Mishra, B. Maiti and R. M. Singh, Eur. J. Org. Chem., 2018, 1130–1135 CrossRef CAS.
- (a) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS; (b) C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069–4084 CrossRef CAS; (c) C. E. Aroyan, A. Dermenci and S. J. Miller, J. Org. Chem., 2010, 75, 5784–5796 CrossRef CAS PubMed; (d) P. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 6213–6226 CrossRef CAS; (e) K. Chandra Bharadwaj, RSC Adv., 2015, 5, 75923–75946 RSC.
- (a) X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621–1624 CrossRef CAS PubMed; (b) M. Frias, R. Mas-Balleste, S. Arias, C. Alvarado and J. Aleman, J. Am. Chem. Soc., 2017, 139, 672–679 CrossRef CAS PubMed; (c) S. Jeon and S. Han, J. Am. Chem. Soc., 2017, 139, 6302–6305 CrossRef CAS PubMed; (d) S. Li, Y. Liu, B. Huang, T. Zhou, H. Tao, Y. Xiao, L. Liu and J. Zhang, ACS Catal., 2017, 7, 2805–2809 CrossRef CAS; (e) H. Wang, K. Wang, Y. Man, X. Gao, L. Yang, Y. Ren, N. Li, B. Tang and G. Zhao, Adv. Synth. Catal., 2017, 359, 3934–3939 CrossRef CAS; (f) S. Maity, S. Sar and P. Ghorai, Org. Lett., 2018, 20, 1707–1711 CrossRef CAS PubMed.
- (a) D. R. Williams and S. A. Bawel, Org. Lett., 2017, 19, 1730–1733 CrossRef CAS; (b) B. M. Trost and X. Luan, Nat. Protoc., 2012, 7, 1497–1501 CrossRef CAS PubMed.
- (a) G. Stork and A. Ponaras, J. Org. Chem., 1976, 41, 2937–2939 CrossRef CAS; (b) E. Wenkert, T. E. Goodwin and B. C. Ranu, J. Org. Chem., 1977, 42, 2137–2141 CrossRef CAS; (c) P. T. Lansbury, R. W. Erwin and D. A. Jeffrey, J. Am. Chem. Soc., 1980, 102, 1602–1608 CrossRef CAS; (d) W. R. Leonard and T. Livinghouse, J. Org. Chem., 1985, 50, 730–732 CrossRef CAS.
- (a) S. A. Frank, D. J. Mergott and W. R. Roush, J. Am. Chem. Soc., 2002, 124, 2404–2405 CrossRef CAS PubMed; (b) L.-C. Wang, A. L. Luis, K. Agapiou, H.-Y. Jang and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 2402–2403 CrossRef CAS; (c) E. Marqués-López, R. P. Herrera, T. Marks, W. C. Jacobs, D. Könning, R. M. de Figueiredo and M. Christmann, Org. Lett., 2009, 11, 4116–4119 CrossRef; (d) S. Takizawa, T. M.-N. Nguyen, A. Grossmann, M. Suzuki, D. Enders and H. Sasai, Tetrahedron, 2013, 69, 1202–1209 CrossRef CAS; (e) H. Jin, Q. Zhang, E. Li, P. Jia, N. Li and Y. Huang, Org. Biomol. Chem., 2017, 15, 7097–7101 RSC; (f) S. Maity, S. Sar and P. Ghorai, Org. Lett., 2018, 20, 1707–1711 CrossRef CAS PubMed.
- For crystal data of 2b, see Experimental section. CCDC 1861781.†.
- S. Shambayati and S. L. Schreiber, Lewis acid carbonyl complexation, Pergamon Press, Oxford, 1992 Search PubMed.
- J. March, March's advanced organic chemistry: reactions, mechanisms, and structure, John Wiley & Sons, 2nd edn, 1977, pp. 485–488 Search PubMed.
- (a) G. J. Karabatsos and C. E. Orzech, J. Am. Chem. Soc., 1962, 84, 2838–2839 CrossRef CAS; (b) M. Saunders and J. J. Stofko, J. Am. Chem. Soc., 1973, 95, 252–253 CrossRef CAS; (c) C. I. F. Watt, in Adv. Phys. Org. Chem, Elsevier, 1988, vol. 24, pp. 57–112 Search PubMed; (d) M. B. Smith and J. March, March's advanced organic chemistry: reactions, mechanisms, and structure, John Wiley & Sons, 2007, pp. 1565–1568 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1861781. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob02961j |
| This journal is © The Royal Society of Chemistry 2019 |