
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(triazolyl methacrylate) glycopolymers as potential targeted unimolecular nanocarriers†
J.
Madeira do O‡
a,
R.
Foralosso‡
a,
G.
Yilmaz
 a,
F.
Mastrotto
b,
P. J. S.
King
c,
R. M.
Xerri
a,
Y.
He
a,
C. F.
van der Walle
d,
F.
Fernandez-Trillo
e,
C. A.
Laughton
a,
I.
Styliari
*f,
S.
Stolnik
*a and
G.
Mantovani
*a
a,
F.
Mastrotto
b,
P. J. S.
King
c,
R. M.
Xerri
a,
Y.
He
a,
C. F.
van der Walle
d,
F.
Fernandez-Trillo
e,
C. A.
Laughton
a,
I.
Styliari
*f,
S.
Stolnik
*a and
G.
Mantovani
*a
aMolecular Therapeutics and Formulation Division, School of Pharmacy, University of Nottingham, NG7 2RD, Nottingham, UK. E-mail: snow.stolnik@nottingham.ac.uk; Giuseppe.mantovani@nottingham.ac.uk
bDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Via F. Marzolo 5, 35131 Padova, Italy
cMalvern Panalytical Ltd, Malvern, WR14 1XZ, UK
dAstraZeneca Ltd, Cambridge, CB21 6GH, UK
eSchool of Chemistry, University of Birmingham, B15 2TT, UK
fUniversity of Hertfordshire, Hatfield, Hertfordshire, UK. E-mail: i.d.styliari@herts.ac.uk
First published on 29th October 2019
Abstract
Synthetic glycopolymers are increasingly investigated as multivalent ligands for a range of biological and biomedical applications. This study indicates that glycopolymers with a fine-tuned balance between hydrophilic sugar pendant units and relatively hydrophobic polymer backbones can act as single-chain targeted nanocarriers for low molecular weight hydrophobic molecules. Non-covalent complexes formed from poly(triazolyl methacrylate) glycopolymers and low molecular weight hydrophobic guest molecules were characterised through a range of analytical techniques – DLS, SLS, TDA, fluorescence spectroscopy, surface tension analysis – and molecular dynamics (MD) modelling simulations provided further information on the macromolecular characteristics of these single chain complexes. Finally, we show that these nanocarriers can be utilised to deliver a hydrophobic guest molecule, Nile red, to both soluble and surface-immobilised concanavalin A (Con A) and peanut agglutinin (PNA) model lectins with high specificity, showing the potential of non-covalent complexation with specific glycopolymers in targeted guest-molecule delivery.
Introduction
The development of synthetic single-chain macromolecular devices has been the subject of considerable research effort in recent years. One notable example of this is single-chain polymeric nanoparticles (SCPNs), where polymer chains are designed to fold at predefined points of their repeating unit sequence,1 mimicking in concept the formation of folded structures in natural proteins. SCPNs can be synthesised following the formation of covalent or non-covalent intramolecular interactions.2–5 Concerted effort between macromolecular chemists and biophysicists has provided access to increasingly sophisticated single-chain molecular devices with potential applications in catalysis, sensors, nanoreactors, and nanomedicine.4–6 For example, Lemcoff,7–9 Pomposo10 and Zimmerman's11 groups engineered single-chain metal–organic nanocatalysts sharing key structural and functional similarity with metalloenzymes.In biomedical settings, the size of delivery carriers affects their ability to cross biological barriers, and distribute in biological tissues.12 Whilst sub-100 nm nano-structures are able to permeate hypervascular solid tumours, only those in the sub-50 nm size range can extravasate and penetrate poorly permeable hypovascular cancer tissues.13,14 Thus, the possibility of scaling the size of specific nanocarriers down to that of individual polymer chains could potentially result in tangible clinical benefits. Outside of the biomedical applications, ultra-small polymer nanoparticles enabled the tuning of the macroscopic properties of polymer nanocomposites (PNCs) over a range inaccessible to conventional PNCs,15 and access to conjugated polymer nanoparticles (Pdots) with unique fluorescence brightness and intraparticle energy migration efficiencies.16
Unimolecular micelles are molecular devices where polymer chains are designed to possess a core–shell morphology mimicking that of the self-assembled micelles.17,18 Based on the individual polymer chains, unimolecular micelles possess the advantage of not unfolding or disassembling at low concentrations, such as those encountered in clinical settings in vivo. This is a key advantage for their potential applications in drug delivery and (bio)imaging, relative to conventional micelles.12,19–24 Unimolecular micelles frequently include, in a single molecule, a hydrophobic core with a hyperbranched or dendrimeric structure which can non-covalently complex hydrophobic drugs, and a hydrophilic periphery, thus, resembling micelles formed by conventional surfactants.19,21,25,26
Alternative unimolecular structures have also been described, such as the ‘polysoaps’ assembled by McCormick and co-workers by statistical RAFT copolymerization of 2-acrylamido-2-methylpropane sulfonic acid (AMPS) and n-dodecyl acrylamide (DDAM) at specific molar ratios.27 Recently, Stenzel and Barner-Kowollik's groups described the synthesis of crosslinked sub-10 nm-sized single chain nanoparticles based on glucose-containing glycopolymers,28 while Stenzel, Paulusse and co-workers utilised analogous nanomaterials to target GLUT1 and GLUT3 glucose transporters in HeLa cells.29
Inspired by these examples, here we explored a minimalistic approach to single-chain vectors, where guest molecules are reversibly incorporated within the individual linear and 4-arm polymer chains – as opposed to self-assembled micelles and/or hyperbranched-like structures – and pendant functionalities are used to provide recognition to specific biological targets.
Synthetic glycopolymers are a class of macromolecules which are ideally suited for this aim, as (i) by varying the nature of pendant sugars and polymer backbone, their physico-chemical properties can be precisely controlled, (ii) they can be designed to possess functional groups which enable selective interaction with low molecular weight payloads, (iii) as biologically-inspired macromolecules they can target a variety of biologically relevant carbohydrate-binding proteins – lectins.30–32
Accordingly, in this work we investigate the ability of a range of glycopolymers to reversibly interact with model hydrophobic guest molecules, and evaluate the physico-chemical characteristics of the resulting nanocomplexes. Furthermore, we provide a prediction of the atomistic structure of these single-chain nanocomplexes through molecular dynamics simulations, and an initial evaluation of their targeting properties, particularly their interactions with carbohydrate-binding proteins.
Results and discussion
Poly(triazolyl methacrylate) glycopolymers: interaction with hydrophobic fluorescent probes
The glycopolymers investigated in this work were prepared by copper-catalysed alkyne–azide cycloaddition (CuAAC) functionalisation of preformed poly(propargyl methacrylate)s with appropriate sugar azides, as first reported by Haddleton and us.33,34 Through this approach, libraries of glycopolymers were prepared via post-polymerization modification of the same parent polymer, leading to families of glycopolymers which only differ in the nature of the pendant carbohydrate units, and share all the other macromolecular characteristics – i.e. degree of polymerisation (DP), macromolecular topology (e.g. linear vs. branched), and molecular weight dispersity (Đ). Importantly for this study, this strategy leads to glycopolymers with a relatively hydrophobic polymethacrylate backbone and triazolyl linkers, which could potentially allow reversible binding with small hydrophobic guest molecules.This work focusses primarily on mannosylated linear and 4-arm star glycopolymers (1)MAN and (2)MAN, respectively (Chart 1), although analogous materials bearing different pendant carbohydrate units – galactose, lactose and trehalose – were also investigated to identify the general structure–activity relationships of these materials (vide infra). Linear (1)MAN and star (2)MAN glycopolymers were made with similar degrees of polymerisation – DP 66 and 77, respectively – to identify the effect of the macromolecular topology on their physico-chemical properties, and on their ability to interact with guest molecules.
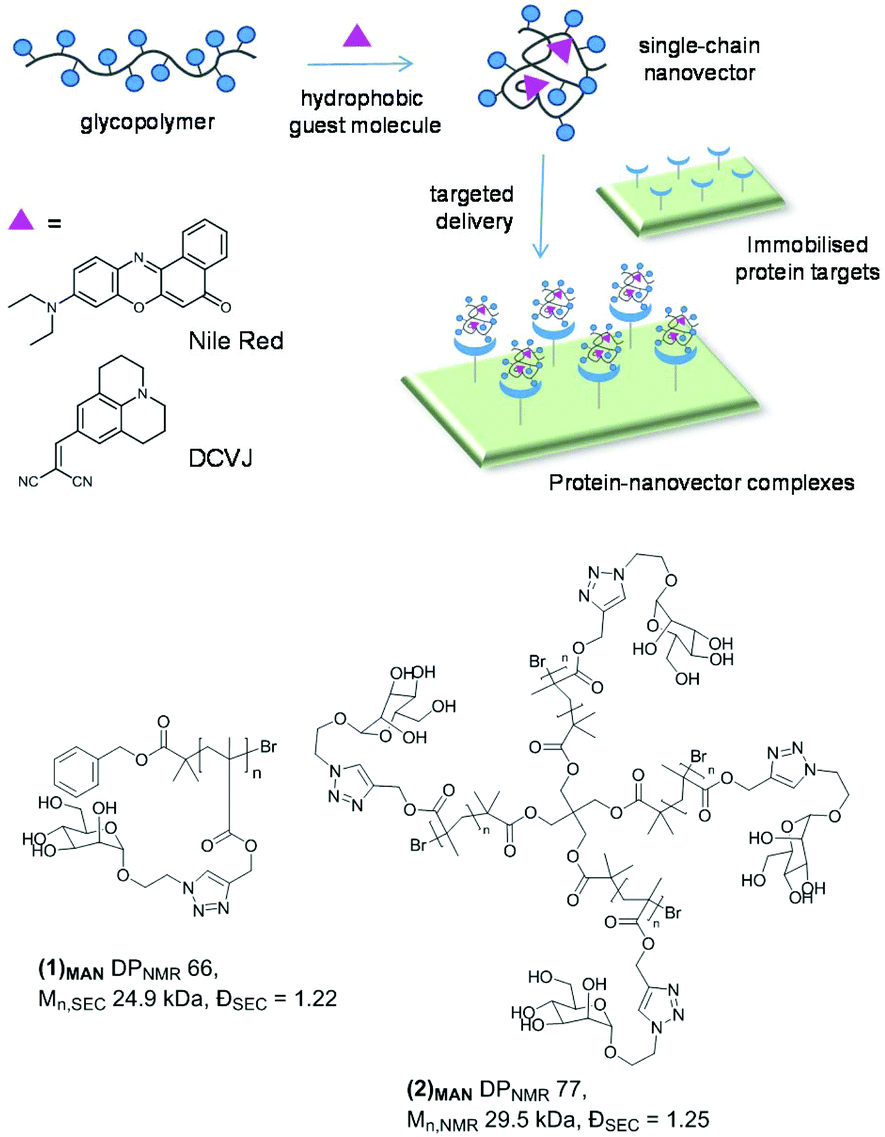 | ||
| Chart 1 Top: Graphical illustration of glycopolymer single chain-guest molecule interaction, and targeted delivery to protein targets on functional surfaces. Hydrophobic fluorescent probes utilised in this study are Nile red and DCVJ. Bottom: Mannosylated linear (1)MAN and 4-arm star (2)MAN glycopolymers investigated in this study. | ||
In a previous study,35 we observed that polarity-responsive extrinsic fluorescent dyes, Nile red and SYPRO-orange, produced highly fluorescent solutions in aqueous media containing triazolyl methacrylate glycopolymers. Both fluorophores possess low quantum yields in aqueous media, resulting in a low fluorescence intensity, which increases in less polar environments.36,37 Triazolyl methacrylate glycopolymers have been extensively investigated.33,34,38–47 Yet, to the best of our knowledge, this behaviour, suggestive of their surfactant-like properties and of their ability to non-covalently bind hydrophobic probes under aqueous conditions, has not been described. Thus, the present study investigates the mechanism and modalities of host–guest interactions between glycopolymers and low molecular weight guest-molecules. We further explore the potential of these materials as nanocarriers for receptor specific, targeted delivery.
Here, Nile red was chosen as the hydrophobic guest molecule because, although a possible structure has been suggested,37 the exact molecular identity of SYPRO-orange has not yet been disclosed. In initial experiments, a solution of Nile red in THF was added to deionised water to achieve a theoretical final concentration of 20 μg mL−1. In the absence of glycopolymers (1)MAN and (2)MAN, after evaporation of the co-solvent, THF, Nile red was found to form a red-pink precipitate in an otherwise colourless or very lightly pink aqueous phase (Fig. 1A). This is consistent with the very low solubility reported for Nile red in water (<1 μg mL−1).48 In contrast, in the presence of triazolyl glycopolymers (1)MAN and (2)MAN (0, 50, 100, 1000 μg mL−1) a visible pink coloration, with intensity increasing as the concentration of glycopolymers increased, was observed.
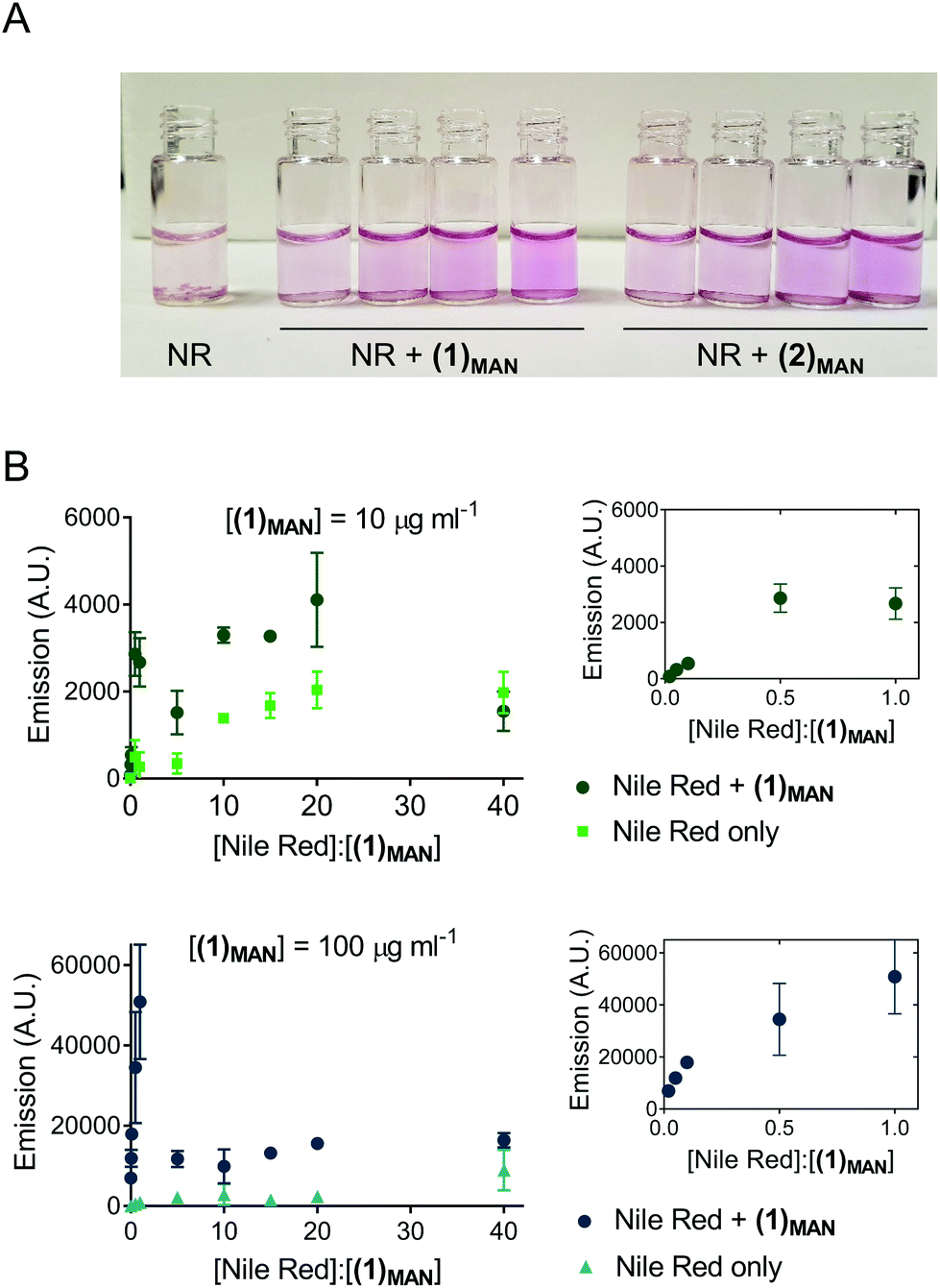 | ||
Fig. 1 Interaction of triazolyl methacrylate glycopolymers with hydrophobic fluorescent probes: (A) Left: Nile red (NR), 20 μg mL−1 in DI H2O. Centre and right: 20 μg mL−1 NR + linear (1)MAN (centre) and 4-arm star (2)MAN (right) at increasing concentration of glycopolymers (0, 50, 100, 1000 μg mL−1). (B) Fluorescence readings (λex 550 nm, λem 630 nm) of Nile red – (1)MAN samples at variable [Nile red]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[polymer chains] molar ratios, in DI H2O. Concentration of (1)MAN is kept constant at either 10 or 100 μg mL−1, while [Nile red] is gradually increased. [Nile red]:[(1)MAN] refers to the molar ratio between Nile red and polymer (1)MAN used in each sample. ‘Nile red only’ refers to control samples identical to ones described above, except that no glycopolymer (1)MAN was added. Insets: Magnification of the 0–1.0 [Nile red]:[(1)MAN] region. :[polymer chains] molar ratios, in DI H2O. Concentration of (1)MAN is kept constant at either 10 or 100 μg mL−1, while [Nile red] is gradually increased. [Nile red]:[(1)MAN] refers to the molar ratio between Nile red and polymer (1)MAN used in each sample. ‘Nile red only’ refers to control samples identical to ones described above, except that no glycopolymer (1)MAN was added. Insets: Magnification of the 0–1.0 [Nile red]:[(1)MAN] region. | ||
Following these initial observations, increasing concentrations of Nile red (50–1000 μg mL−1) were added to solutions of (1)MAN in deionised water (10 and 100 μg mL−1). The measured fluorescence was found to reach its maximum at approximately 0.5–1 molecules of Nile red per polymer chain (Fig. 1B). Interestingly, in the samples with a higher concentration of glycopolymer (1)MAN (100 μg mL−1) a lower fluorescence was observed at high [Nile red]:[(1)MAN] ratios. This could be explained with the fact that in aqueous media Nile red can form non-emissive H-type dimers and larger aggregates through π–π stacking interactions,49,50 and this phenomenon would become predominant at high concentrations of the dye. This effect was more evident in the set of samples with a higher concentration of glycopolymer (1)MAN (100 μg mL−1), which requires larger amounts of Nile red to achieve the same [Nile red]:[(1)MAN] ratios. Analogous fluorescence vs. concentration profiles were observed when a more biologically relevant medium, PBS, was employed as the dispersing medium (Fig. S2†).
Sugar poly(triazolyl methacrylates): unimolecular micelles vs. higher order assemblies
Our initial experiments suggested that the poly(triazolyl methacrylate) glycopolymer could, rather unexpectedly, solubilise in water the otherwise poorly soluble hydrophobic probe Nile red. To gain mechanistic insight into this phenomenon, the propensity of polymers (1)MAN and (2)MAN to self-assemble under aqueous conditions was initially assessed by surface tension analysis.The surface tension (γ) of aqueous solutions is reduced in the presence of chemical species which adsorb at the air/water interface, i.e. surfactants.51 Typical γ vs. [surfactant] plots are sigmoidal curves with three distinct zones. At low concentrations, the air/water interface is scarcely populated by surfactant molecules, which in turn induces only relatively small changes of surface tension.51,52 As the concentration of the surfactant increases, cooperativity among the adsorbed surfactant molecules at the interface induces a sharp decrease of γ, until saturation (or quasi-saturation) of the interface is reached.51,52 After this point – the onset of aggregation (critical micellar concentration, CMC, if micelles are formed) – subsequent addition of the surfactant results in the formation of supramolecular assemblies, and will not induce any further significant decrease of surface tension. Thus, surface tension analysis is routinely used to determine the CMC of surfactants.27,52–54
In this study, for glycopolymers (1)MAN and (2)MAN no CMC could be observed, as the surface tension of their aqueous solutions steadily decreased as the polymer concentration increased throughout the range of concentrations tested (0.020–5.0 mg mL−1), without reaching a plateau (Fig. 2A). This suggested that, under these experimental conditions, glycopolymers (1)MAN and (2)MAN exist in aqueous solution as individual polymeric chains.
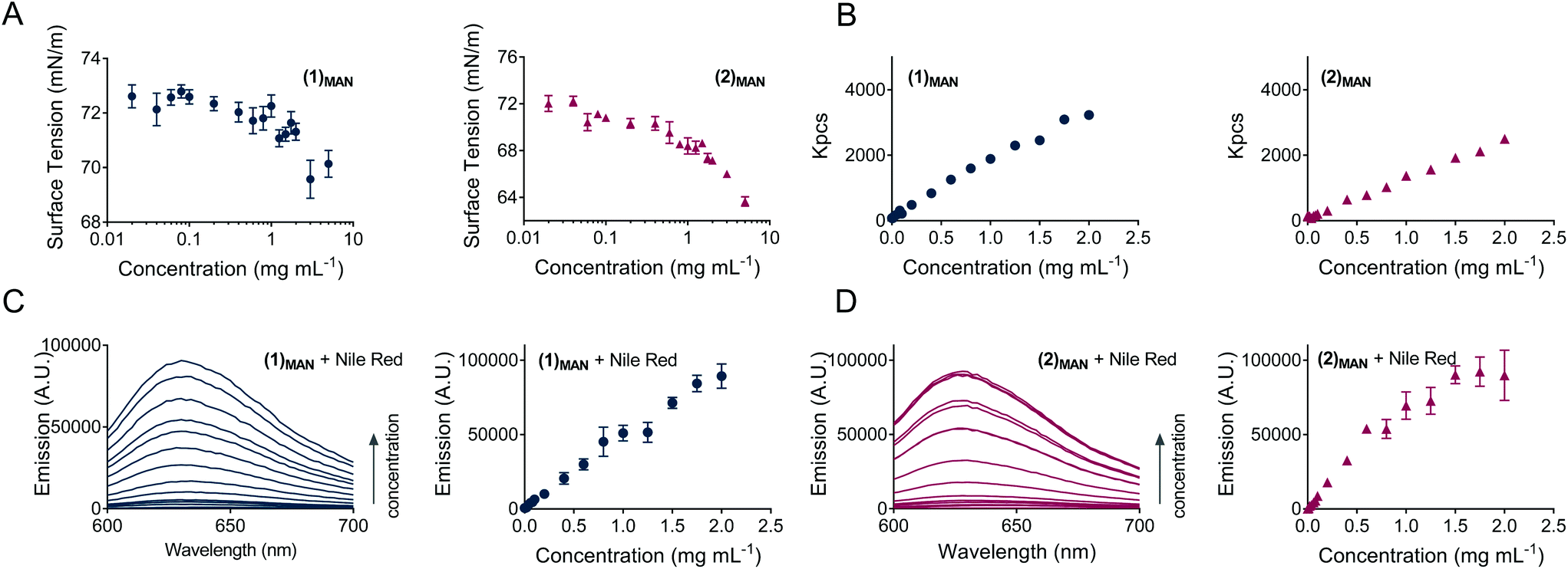 | ||
| Fig. 2 A: Surface tension of solutions of linear (1)MAN and 4-arm star (2)MAN glycopolymers in DI H2O at increasing concentrations (n = 2, triplicate). B: Scattered light intensity as a function of glycopolymer concentration, for (1)MAN and (2)MAN. C: Left: Emission spectra of samples containing Nile red (equivalent to a theoretical final dye concentration of 2.0 μM, 0.64 μg mL−1) and solutions of (1)MAN at increasing polymer concentrations, 0.0050–2.0 mg mL−1 – corresponding to a [polymer]:[dye] = 0.10–40 for (1)MAN. Right: Emission (λem = 630 nm) vs. concentration profiles for these samples. D: Left: Emission spectra of samples containing Nile red (equivalent to a theoretical final dye concentration of 2.0 μM) and solutions of (2)MAN at increasing polymer concentrations, 0.0050–2.0 mg mL−1 – corresponding to 0.084–34 for (2)MAN. Right: Emission (λem = 630 nm) vs. concentration profiles for these samples. | ||
An analogous behaviour – that is, γ vs. concentration plots lacking the final plateau, has been previously described for polymers with topologies which structurally resembled the micellar structure. For example, Uhrich and co-workers utilised surface tension measurements to prove that the encapsulation of hydrophobic lidocaine molecules into mucic acid–fatty acids–PEG branched polymers was caused by individual unimolecular micelles.54 McCormick's group showed that random copolymers with selected molar ratios of 2-acrylamido-2-methylpropane sulfonic acid (AMPS) and n-dodecyl acrylamide (DDAM) possessed unimolecular micellar features, and, similar to what was observed in our work, induced a continuous decrease of surface tension in the range of concentrations investigated (up to 10 mg mL−1).27
In terms of structure–function relationships, 4-arm star (2)MAN induced a larger decrease in surface tension compared to its linear counterpart (1)MAN, 66.5 vs. 70.2 mN m−1 at the highest concentration tested (5.0 mg mL−1), respectively (Fig. 2A).
The observed effect of poly(triazolyl methacrylate)s (1)MAN and (2)MAN on surface tension suggested a partial orientation of their hydrophobic polymer backbone towards air, at the water–gas interphase.
To ascertain whether the balance between the hydrophilic sugar pendant units and the more hydrophobic polymer backbone affected the surface tension of the aqueous solution of these materials, these experiments were repeated using analogous linear and 4-arm star glycopolymers bearing larger sugar units, namely D-lactose and α,α-trehalose disaccharides, (1)LAC and (2)LAC, and (1)TRE and (2)TRE, respectively (Chart S1†). As these were prepared via post-polymerization modification of the same parent polymer precursor,35 they possessed the same macromolecular features – average number of repeating units (DP), dispersity (Đ), and topology (linear and star) – as (1)MAN and (2)MAN, and thus allowed the dissection of the contribution of the nature of the carbohydrate pendant units on the ability of the corresponding glycopolymers to lower the surface tension of water.
The results (Fig. S4†) indicated that: (i) again, no CMC could be observed; (ii) glycopolymers with larger, more hydrophilic55 sugar pendant units induced a lower overall decrease in surface tension, compared to (1)MAN and (2)MAN, showing that the hydrophobic/hydrophilic balance between the polymer backbone and the carbohydrate pendant units does affect the surfactant-like properties of these materials; (iii) 4-arm star polymers still induced a larger decrease in γ, which is in line with what was already observed for (1)MAN and (2)MAN; (iv) when used at the same concentrations utilised for the corresponding glycopolymers, individual D-mannose, D-lactose and α,α-trehalose mono- and disaccharides did not affect γ, showing that the physico-chemical nature of each glycopolymer as a whole, rather than the intrinsic properties of the pendant carbohydrates, is responsible for their effect on the surface tension.
Taken together, these experiments suggested that, in the range of concentrations and conditions investigated, these glycopolymers exist in water solution as individual polymer chains.
The propensity of poly(triazolyl methacrylates) (1)MAN and (2)MAN to aggregate was further probed by following the profile of the intensity of scattering of solutions with increasing glycopolymer concentration (Fig. 2B). The data showed a linear increase of scattering (Kcps) for (1)MAN and (2)MAN in the 0.0010–2.0 mg mL−1 range of concentrations, as previously observed for unimolecular micelles, e.g. by McCormick and co-workers,27 and in agreement with the results from the surface tension experiments.
Whilst surface tension and light scattering experiments suggested that glycopolymers (1)MAN and (2)MAN exist as individual chains in aqueous solution, in principle the presence of a hydrophobic probe may result in guest molecule-induced self-assembly, with the formation of larger aggregates containing multiple polymer chains and guest-molecules. For example, Mohr and co-workers showed that the hydrophobic probe Nile red promotes the formation of dye-loaded SDS, CTAB and Triton X micelles at sub-micellar surfactant concentrations.50
To address this point, here Nile red:glycopolymer interactions were investigated by fluorescence spectroscopy, under the hypothesis that if above a certain polymer concentration micelles or other large assemblies with a hydrophobic core were formed, a discontinuity in the emission intensity vs. polymer concentration plot would have been observed.56 This would be suggestive of the formation of higher-order structures with a hydrophobic core able to enhance Nile red incorporation. Accordingly, a constant amount of Nile red, equivalent to a theoretical dye concentration of 2.0 μM, was added to solutions of glycopolymers (1)MAN and (2)MAN in deionised water at increasing polymer concentrations (Fig. 2C and D). Sample fluorescence increased almost linearly as the concentration of glycopolymers increased, with a tailing at higher concentrations for star polymer (2)MAN (Fig. 2D). The amount of the added dye was the same for all samples, thus the observed increase in fluorescence could be explained with the ability of increasing number of glycopolymer chains to complex a larger number of Nile red molecules. Additional contributing mechanisms may include the disruption of non-emissive dye H-dimers/aggregates which Nile red can form both in aqueous solution or within the hydrophobic domains of polymer carriers.50
Nile red is known to undergo a blue shift of the maximum of emission as the polarity of the surrounding (micro)environment decreases.48,57,58 For example, Rimmer and co-workers utilised Nile red as a polarity-sensitive probe to show that specific highly-branched poly(N-isopropyl acrylamide)s possessed core–shell morphologies.59 In our experiments we observed a shift from ∼650 nm, close to that of Nile red in water, at the lowest [polymer]:[dye] ratios (0.020 and 0.084 for linear (1)MAN and star (2)MAN, respectively), to 632–634 nm, starting from [polymer]:[dye] ∼0.5–1 (Fig. S3†), indicative of Nile red existence in a more hydrophobic environment. This agrees with the studies of Alexiev and co-workers58 and Mohr's group50 where a blue shift of fluorescence emission was ascribed to the incorporation of the dye within the hydrophobic domains of their systems.
The potential self-assembly of (1)MAN was then further investigated by DLS (Fig. 3). The results showed a hydrodynamic diameter of 3–5 nm for linear (1)MAN at 2.0 mg mL−1, with and without added Nile red, at a 1:10 [Nile red]:[(1)MAN] molar ratio (Fig. 3A), consistent with the size expected for individual glycopolymer chains in this molecular weight range in solution.60,61 As the hydrodynamic diameters observed for (1)MAN with and without Nile red were close to the lower limit of detection for DLS, Taylor dispersion analysis (TDA) of (1)MAN was then carried out to obtain a more accurate estimation of its hydrodynamic size. TDA is an absolute method to determine the diffusion coefficients (D), and thus the hydrodynamic radii (Rh), of molecules in solution,62,63 based on the dispersion of a solute plug through a uniform cylindrical tube under laminar Poiseuille flow.64 Following injection, the UV absorbance (λ = 214 nm) of the species in solution is recorded at two detection windows positioned along the capillary (windows 1 and 2, Fig. 3B). The diffusion coefficient of the injected solute(s) can be derived by fitting Taylor's solution to the concentration profile (taylorgram) of the solute(s).62 In turn, this can be used to assess the size of molecules or particles with hydrodynamic diameter (2Rh) from angstroms to submicron size,65,66 typically in the 0.4–100 nm range. Hawe et al. showed that for peptide hormones and neuropeptide oxytocin, with a hydrodynamic radius Rh = 0.9–1.1 nm, not too dissimilar from that expected for our polymers (1)MAN and (2)MAN, TDA was superior to DLS to estimate the peptide size.64
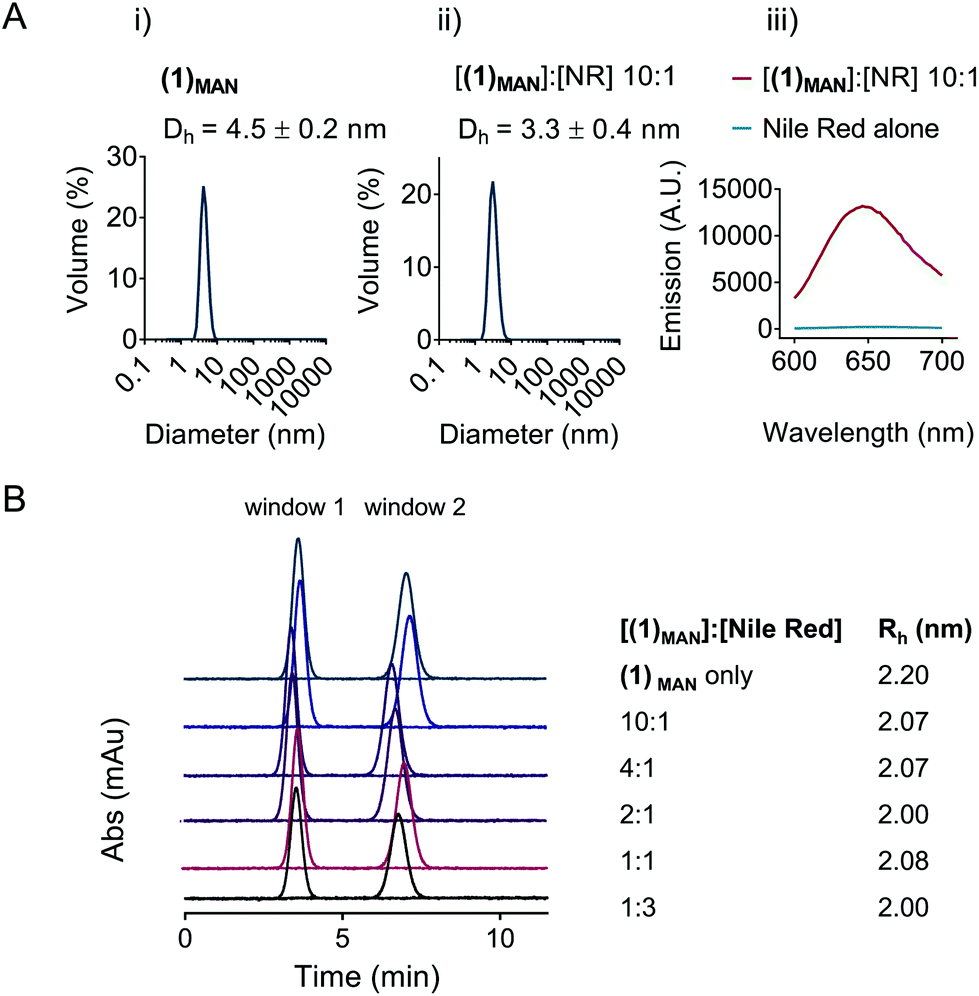 | ||
| Fig. 3 A. DLS size distributions of (i) linear mannose poly(triazolyl methacrylate) (1)MAN alone (top) and (ii) 10:1 mol:mol (1)MAN-Nile red (NR) complex at a [(1)MAN] = 10 mg mL−1 in DI water. (iii) Fluorescence emission spectra of (1)MAN-Nile red complexes, and Nile red alone in water, at λex = 550 nm. B. Taylor dispersion analysis (TDA) of (1)MAN alone and (1)MAN-Nile red complexes. Broadening of the concentration profile due to axial spreading of the solute pulse observed between two detection windows (windows 1 and 2 in the figure) is used to estimate the hydrodynamic radii. All samples in parts A and B were filtered through a 0.22 μm membrane prior to analysis. | ||
Importantly for our study, the deconvolution of taylorgrams from mixtures of species in solution allows the estimation of the hydrodynamic radii of their individual components and their relative proportions,62,67 and has been used to investigate the aggregation of macromolecules in solution.63,64 For example, Latunde-Dada et al. estimated accurately size and relative proportion of the bovine serum albumin (BSA) monomer and thermally-induced aggregates in BSA:(BSA aggregates) in the 3–100% aggregation range.63 Here, experiments were carried out at a concentration of linear (1)MAN of 2.0 mg mL−1 and at variable [glycopolymer]:[Nile red] molar ratios (Fig. 3B). In all samples only one species in solution could be detected, with hydrodynamic radii (Rh) in the 2.0–2.2 nm range, corresponding to diameters of 4.0–4.4 nm, which again was indicative of the presence of unimolecular polymer chains in solution.
Effect of the nature of polymer backbone on guest molecule incorporation: loading and molecular dynamics (MD) simulations
The surface tension experiments showed that for glycopolymers with the same poly(triazolyl methacrylate) backbone, the nature and size of the pendant carbohydrate units have a direct effect on the surfactant properties of these materials. Here, we aimed at investigating the effect of the nature of the polymer backbone on the ability of these glycopolymers to incorporate hydrophobic fluorescent guest molecules.To achieve this aim, linear poly(N-ethylacrylamidoyl-α-D-mannopyranoside) (3)MAN (Fig. 4), analogous to (1)MAN but with a more hydrophilic polymer backbone, was synthesised by SET LRP (DP 67, Đ 1.22) and tested. In the first set of experiments, a fixed amount of Nile red was added to solutions at increasing concentration of polymers (1)MAN and (3)MAN (0.050–1.0 mg mL−1), and the fluorescence of the resulting mixtures was recorded (Fig. 4). The results clearly showed that whilst poly(mannose triazolyl methacrylate) (1)MAN was able to interact with Nile red and incorporate it already at the lowest concentration tested (50 μg mL−1), linear mannose acrylamide (3)MAN showed no detectable polymer–dye interaction. It should be noted that the increase of fluorescence is used here only for a qualitative assessment of relative amounts of Nile red, as a proportion of molecules of fluorophore incorporated within polymeric chains could still interact with each other and form non-fluorescent dimers and H-type aggregates through π–π stacking interactions.50
 | ||
| Fig. 4 Fluorescence of Nile red- and DCVJ (20 μg mL−1, 63 μM and 80 μM, respectively) in the presence of increasing concentrations of mannose poly(triazolyl methacrylate) (1)MAN and poly(N-ethylacrylamidoyl-α-D-mannopyranoside) (3)MAN. Nile red: λex 550 nm, max emission recorded in the λem = 520–700 nm region; DCVJ: λex 430 nm, max emission recorded in the λem = 400–600 nm region. For some points error bars are smaller than data symbols. | ||
9-(2,2-Dicyanovinyl)julolidine (DCVJ, Fig. 1A) was then tested as the hydrophobic probe. DCVJ belongs to a class of fluorophores known as molecular rotors, whose fluorescence depends on the velocity of rotation of a specific bond, presenting maximal fluorescence when the molecule is in a fully planar conformation. Upon irradiation, fluorescent molecular rotors undergo twisted intramolecular charge transfer (TICT), and relax via the nonradiative torsional relaxation pathway.68 The fluorescence of molecular rotors is therefore more sensitive to changes in microenvironments which restrict rotation around key bonds – e.g. the increase of local viscosity or inclusion within the tightly packed microenvironment – than to the polarity of the medium the fluorophore is dissolved in.69 Thus, DCVJ here served two purposes: (i) expanding the range of hydrophobic molecules which can be complexed by glycopolymers based on poly(triazolyl methacrylates), and (ii) gathering information on the molecular mobility of the entrapped guest molecule within dye:polymer complexes. As for the previous tests with Nile red, with DCVJ only poly(mannose triazolyl methacrylate) (1)MAN showed an increase in fluorescence (Fig. 4), suggesting a certain degree of steric rotational restriction of DCVJ guest molecules within the hydrophobic domain of the polymer.
To investigate the nature of triazolyl methacrylate glycopolymer–Nile red interactions at an atomistic level, Molecular Dynamics (MD) simulations70 were employed. Our initial MD experiments focussed on the self-assembly of Nile red alone, in the absence of added glycopolymers. Accordingly, five Nile red molecules were randomly placed in an aqueous environment and were allowed to interact freely over a 100 ns timescale. In agreement with their known aggregation mechanism,49,50 the dye molecules rapidly aggregated through π–π stacking (Fig. S10†). Next, we explored the average conformation in the water of poly(mannose triazolyl methacrylate) (1)MAN and poly(N-ethylacrylamidoyl-α-D-mannopyranoside) (3)MAN, through 80 ns simulations. The polymers were built using tleap of AMBERtools, with the number of sugar repeating units matching exactly their DP calculated by 1H NMR. Three replica simulations were performed for each polymer, and the evolution over time of the static properties of a single polymer chain, namely the end-to-end distance and the radius of gyration, was monitored. In all replicas, within 40 ns poly(mannose triazolyl methacrylate) (1)MAN adopted a globular conformation with a radius of gyration (Rg) of 1.7 nm with the α-D-mannopyranoside sugar moieties of the repeating units pointing towards the surrounding water-filled space. Combined with the hydrodynamic radius (Rh) measured by TDA (Fig. 3), this gave a shape factor ρ = Rg/Rh of 0.77, which is characteristic of spherical conformations.71 In contrast, most likely due to its more hydrophilic poly(N-ethylacrylamidoyl) backbone, within the timescale of the simulation experiments glycopolymer (3)MAN remained in a more solvated, chain-extended conformation, with radius of gyration of ∼3.5–4.0 nm (Fig. 5B).
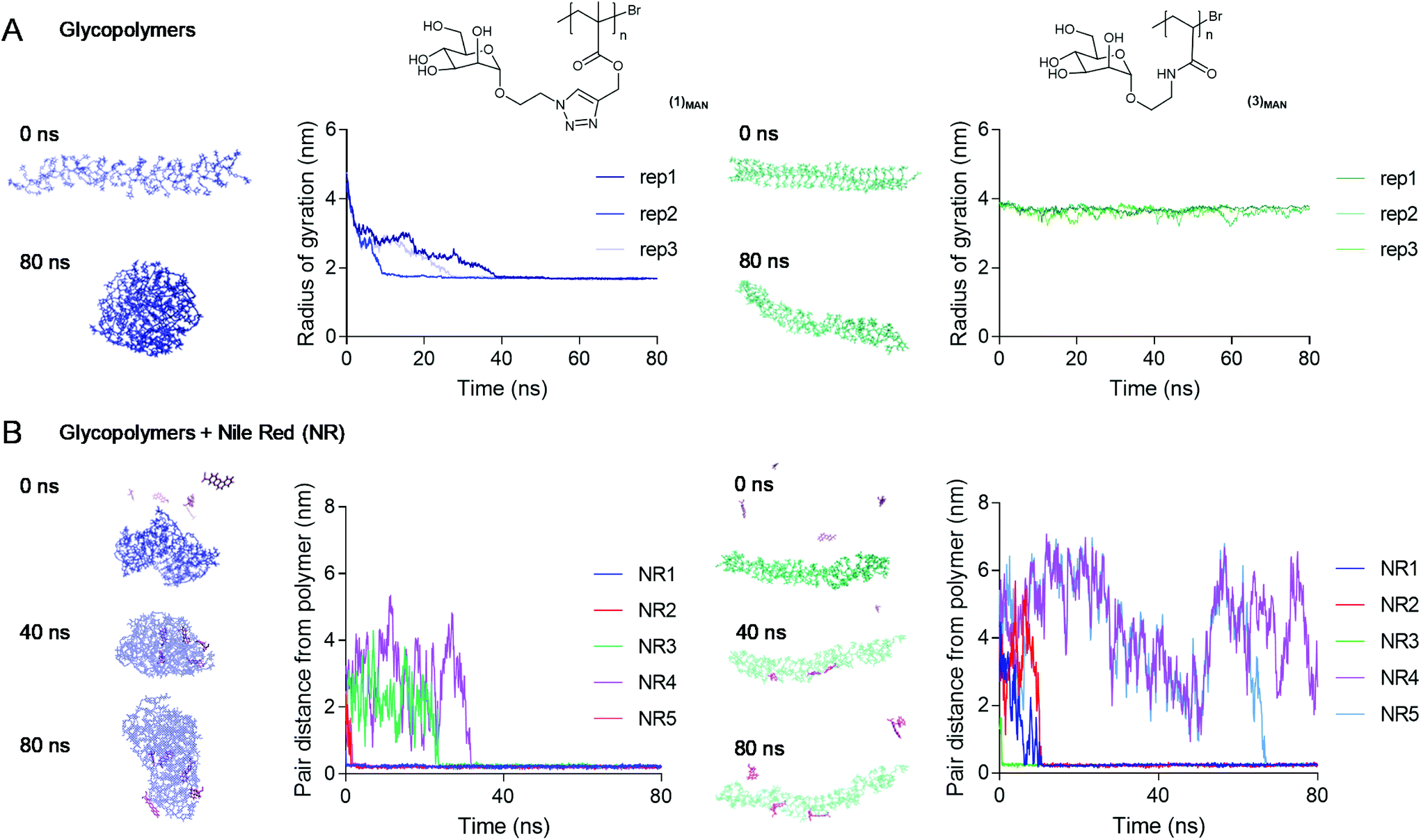 | ||
| Fig. 5 A: Metrics obtained from the 80 ns simulations with poly(mannose triazolyl methacrylates) (1)MAN and poly(N-ethylacrylamidoyl-α-D-mannopyranoside) (3)MAN alone, in the absence of Nile red: evolution of radius of gyration of the glycopolymers in water during the 0–80 ns simulations (3 replicas – rep1, rep2, and rep3 – per polymer). B: Five Nile red (NR1 to NR5) molecules were randomly placed in the space surrounding glycopolymers (1)MAN (left) and (3)MAN (right) (t = 0 ns). The graphs show the evolution of the pair-distances between the centre of mass of the polymer chains and each Nile red molecule for one of the replica simulations. | ||
To assess the propensity of the glycopolymers to complex Nile Red (NR), five Nile red molecules were randomly placed around a single pre-equilibrated polymer chain, and the evolution of the pair-distances between the centre of mass of the glycopolymers and each Nile red molecule was monitored over an 80 ns period. Poly(mannose triazolyl methacrylate) (1)MAN was found to bind all five Nile red molecules within the first 60 ns, as shown by the distances between the centre of mass of the glycopolymers and Nile red molecules decreasing to values close to zero (Fig. 5B, left). In contrast, with glycopolymer (3)MAN at the end of some of the replica simulations, a few NR molecules were found to be still unbound (Fig. 5B, right and S12†). Rapid dye incorporation in poly(mannose triazolyl methacrylate) (1)MAN appeared to prevent Nile red self-aggregation, which was instead observed in the presence of glycopolymer (3)MAN (NR4 and NR5 in Fig. 5, showing identical distance from the polymer centre of mass for a proportion of the simulation experiment).
Interestingly, in these simulations all five Nile red molecules were rapidly incorporated within (1)MAN single-chain nanocarriers, while in our initial complexation studies a maximum of fluorescence at [Nile red]:[polymer] ratios in the 0.5–1 range was observed. While it may be difficult to rationalise this partial discrepancy at this stage, possible explanations may include an overestimation in the simulations of the ability of the glycopolymers to disrupt dye–dye aggregates and incorporate individual Nile red molecules, or the formation of non-emissive H-type π–π stacked Nile red aggregates within each complex, favoured at higher Nile red concentrations.
In a final experiment, we found that two water-equilibrated (1)MAN polymer chains existed as individual coiled chains for the duration of the experiment (80 ns), showing that, under these simulation conditions, glycopolymer (1)MAN did not form larger aggregates (Fig. S14†).
Taken together, these simulations provided an atomistic insight on the Nile red–glycopolymer interactions, and supported the structure–function observations derived from our Nile red–glycopolymer complexation experiments and the literature, that is: (i) under aqueous conditions poly(mannose triazolyl methacrylate) (1)MAN appears to adopt a coiled conformation able to rapidly incorporate Nile red molecules, (ii) more hydrophilic poly(N-ethylacrylamidoyl-α-D-mannopyranoside) (3)MAN is comparatively less prone to incorporate Nile red and exists in solution in a more chain extended conformation; (iii) in the absence of glycopolymers, Nile red rapidly associated into aggregates through π–π stacking interactions; (iv) under the conditions investigated, (1)MAN was not prone to self-assemble into larger aggregates, in agreement with what was observed in our light scattering and TDA experiments.
Delivery of guest molecules to lectin targets
This part of our study assessed the potential of the synthesized glycopolymers to target carbohydrate-binding proteins (lectins). In nature carbohydrates are involved in the regulation of a plethora of biological processes such as cell differentiation, proliferation and adhesion, inflammation, and immunological response,72 thus targeted drug delivery through sugar-binding receptors to modulate these key processes is particularly attractive.73 Initially, an agglutination assay using mannose-binding concanavalin A model lectin74,75 was developed, based on a protocol described by Haddleton and co-workers.76In the first part of the assay, increasing amounts of Con A were added to solutions of glycopolymer–Nile red complexes (obtained from 1.00 mg mL−1 of (1)MAN or (2)MAN and 20 μg mL−1 (63 μM) of Nile red), resulting in the formation of insoluble Con A-complex clusters. The highest Con A concentration, 1.00 μg mL−1, corresponds to one molecule of the Con A monomer per two polymer chains. The fluorescence of (1)MAN-Nile red samples was found to decrease steadily as the concentration of added Con A was increased from 0 to 1.00 mg mL−1 (Fig. 6A). This can be explained in terms of (i) Con A-induced aggregation and precipitation of fluorescent glycopolymer–Nile red complexes, (ii) sequestration of glycopolymers by the added Con A, followed by precipitation of Nile red due to the lack of polymer chains able to solubilise it, or (iii) a combination of these two effects.
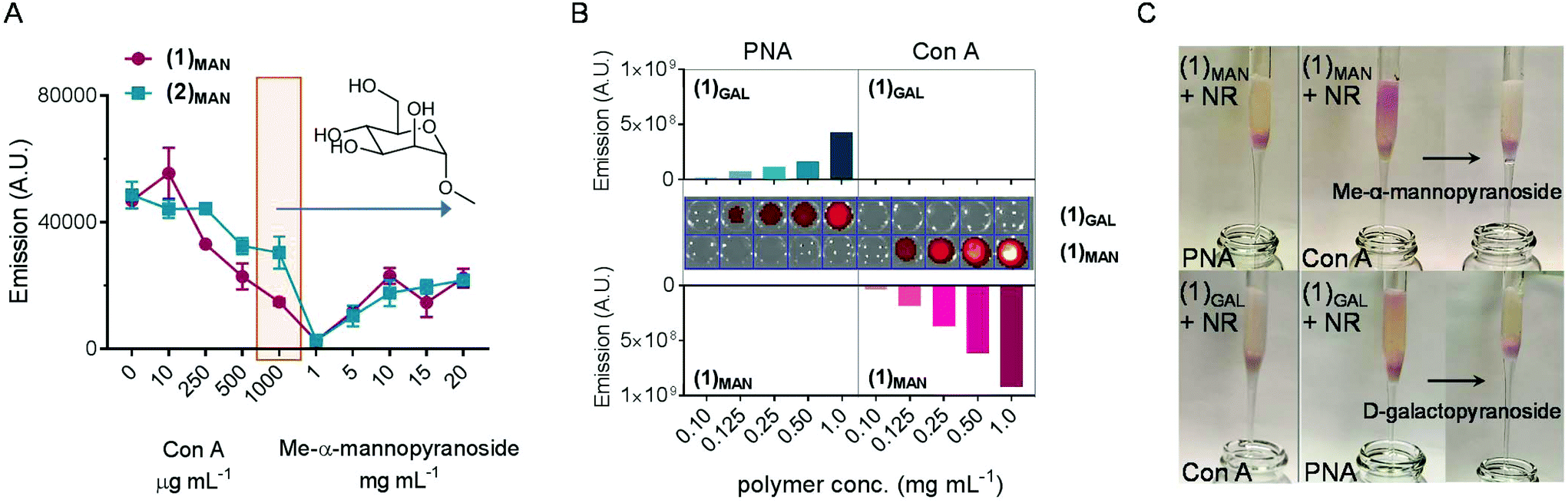 | ||
| Fig. 6 Binding of (1)MAN-Nile red non-covalent conjugates to mannose-specific model lectins. A: Con A agglutination assay. To a polymer solution (1.0 mg mL−1 in 20 mM HEPES buffer (pH 7.4 with 50 mM NaCl, 5 mM CaCl2 and 5 mM MnCl2), Nile red was added (1:1 mol:mol glycopolymer:dye), and the fluorescence at λex = 630 nm (λex = 550 nm) was recorded. Upon addition of Con A, the decrease of fluorescence due to the formation of insoluble Con A-((1)MAN-Nile red) was recorded. Reversible agglutination: the solid precipitate obtained from the 1000 μg mL−1 Con A experiment (highlighted) was treated with different concentrations of Me-α-mannopyranoside competitive ligand, and recovery of fluorescence due to re-dissolution (or re-formation) of (1)MAN-Nile red complexes was measured. B: Agarose-bound lectin beads were treated with different amounts of (1)MAN- and (1)GAL-Nile red complexes (1:1 polymer:dye molar ratio). After extensive rinsing with 20 mM HEPES buffer, the samples were seeded on a 96-well plate and the fluorescence was recorded. C: Affinity chromatography assay. Con A- and PNA-immobilised beads were utilised as stationary phases for affinity chromatography on (1)MAN- and (1)GAL-Nile red complexes (1:1 polymer:dye molar ratio), eluting with HEPES buffer. (1)MAN-Nile red was found to selectively bind to Con A, and (1)GAL-Nile red to PNA. These complexes could be eluted from the lectin stationary phase by the addition of a mobile phase containing an excess of monovalent competitive ligands, Me-α-mannopyranoside, and D-galactopyranoside, respectively. | ||
In the second part of the assay, the reversibility of this process was evaluated. The release of glycopolymers from the clusters formed at 1.00 mg mL−1 Con A concentration was tested by adding increasing concentrations of methyl-α-mannopyranoside (1–20 mg mL−1), which acts as a competitive monovalent ligand for Con A. An increase in fluorescence, proportional to the amount of the monovalent mannose ligand added, was observed, indicating that Nile red was retained within the mannose glycopolymers, or re-complexed, according to the mechanism (ii) above, following decomplexation from Con A lectin.
Next, the ability of these glycopolymers to mediate selective targeting to lectin-functionalised surfaces was investigated. To achieve this aim, commercially available 45–165 μm agarose beads functionalised with mannose-binding Con A and galactose-binding peanut agglutinin (PNA) were utilised, along with a linear galactose glycopolymer (1)GAL analogous to mannosylated (1)MAN. Lectin-decorated beads were treated with (1)MAN- and (1)GAL-Nile red complexes (1:1 glycopolymer:dye ratio) in the 0.10–1.0 mg mL−1 range of polymer concentrations. Upon centrifugation and removal of the supernatant, followed by several washing cycles, the beads were suspended in HEPES buffer and analysed by fluorescence microscopy (Fig. 6B). Selective recognition of (1)MAN- and (1)GAL-Nile red complexes to Con A and PNA, respectively, was observed. No binding to ‘mismatched’ lectins – that is, (1)MAN to PNA and (1)GAL to Con A – was detected, thus ruling out any significant binding to the lectin surfaces due to non-specific adsorption or other non-specific binding mechanisms.
Utilising Con A and PNA agarose beads as stationary phases for affinity chromatography (Fig. 6C, S7 and S8†) showed a retention by Con A of (1)MAN-Nile red samples, while these were readily eluted from the PNA column. This demonstrated that poly(mannose triazolyl methacrylate) (1)MAN was able to efficiently transport Nile red through the PNA stationary phase. Treatment with a 10 mg mL−1 aqueous solution of methyl-α-mannopyranoside monovalent competitive ligand resulted in the complete elution of (1)MAN-Nile red samples from the Con A column, confirming that (1)MAN here acted as a targeted nanocarrier for Nile red. On repeating this experiment using (1)GAL, a linear glycopolymer identical to (1)MAN except for the presence of galactose instead of mannose binding units yielded analogous results, with selective binding of (1)GAL-Nile red complexes to PNA and not to Con A.
Guest probe (Nile red) release profiles were then assessed using Nile red-(1)MAN complexes in deionised water ([(1)MAN]: 1.00 mg mL−1, [Nile red]: 20 μg mL−1 (63 μM)) through a 5 kDa MWCO membrane under sink conditions. To evaluate the effect of polymer topology on Nile red release, 4-arm star (2)MAN was also tested. Consistent with a reversible polymer–probe interaction, near complete Nile red release was observed already after 4 hours for both linear and star glycopolymers (Fig. S6†).
Attempts to quantify the amount of Nile red retained in the polymers following filtration through a 0.22 μm membrane resulted in a significant amount of Nile red being retained in the filter membrane (see the ESI†), possibly suggesting a relatively low stability of the dye:polymer complexes under shear stress and/or the presence of relatively large free Nile red aggregates in equilibrium with complexed dye. Importantly, no loss of glycopolymers following filtration could be detected by 1NMR analysis (Fig. S5†), again confirming that under the conditions utilised to form the glycopolymer–Nile red complexes, only a very small or no appreciable amount of glycopolymers were involved in the formation of large aggregates with Nile red. The stability of the complexes under specific stress conditions will be the focus of future studies. Taken together, this part of our work showed that upon complexation of Nile red, glycopolymers conserved their ability to bind model lectins Con A and PNA, whilst at the same time retained their Nile red payload.
Finally, whilst not within the scope of this present work, initial experiments aiming at using these single-chain nanocarriers for targeted delivery to cells presenting endocytic lectin receptors were carried out. Chinese Hamster Ovary (CHO) cells were selected due to the availability of a mutant cell line expressing Mannose Receptor (MR, CD206),77,78 an endocytic receptor expressed in subpopulations of dendritic cells (DCs), macrophages, and selected endothelial cells, and which plays a role in both innate and adaptive immunity.79,80 Here, MR+-CHO cells were incubated for 30 minutes with glycopolymer:Nile red-containing solutions at constant Nile red theoretical concentration (10 μM if all of it were solubilised) and increasing polymer concentrations (50–1000 μg mL−1). Nile red cell uptake was monitored by flow cytometry (FACS), and was found to be significantly increased in the presence of (1)MAN and (2)MAN, relative to free dye control and to be dependent on the concentration of the added glycopolymers (Fig. 7).
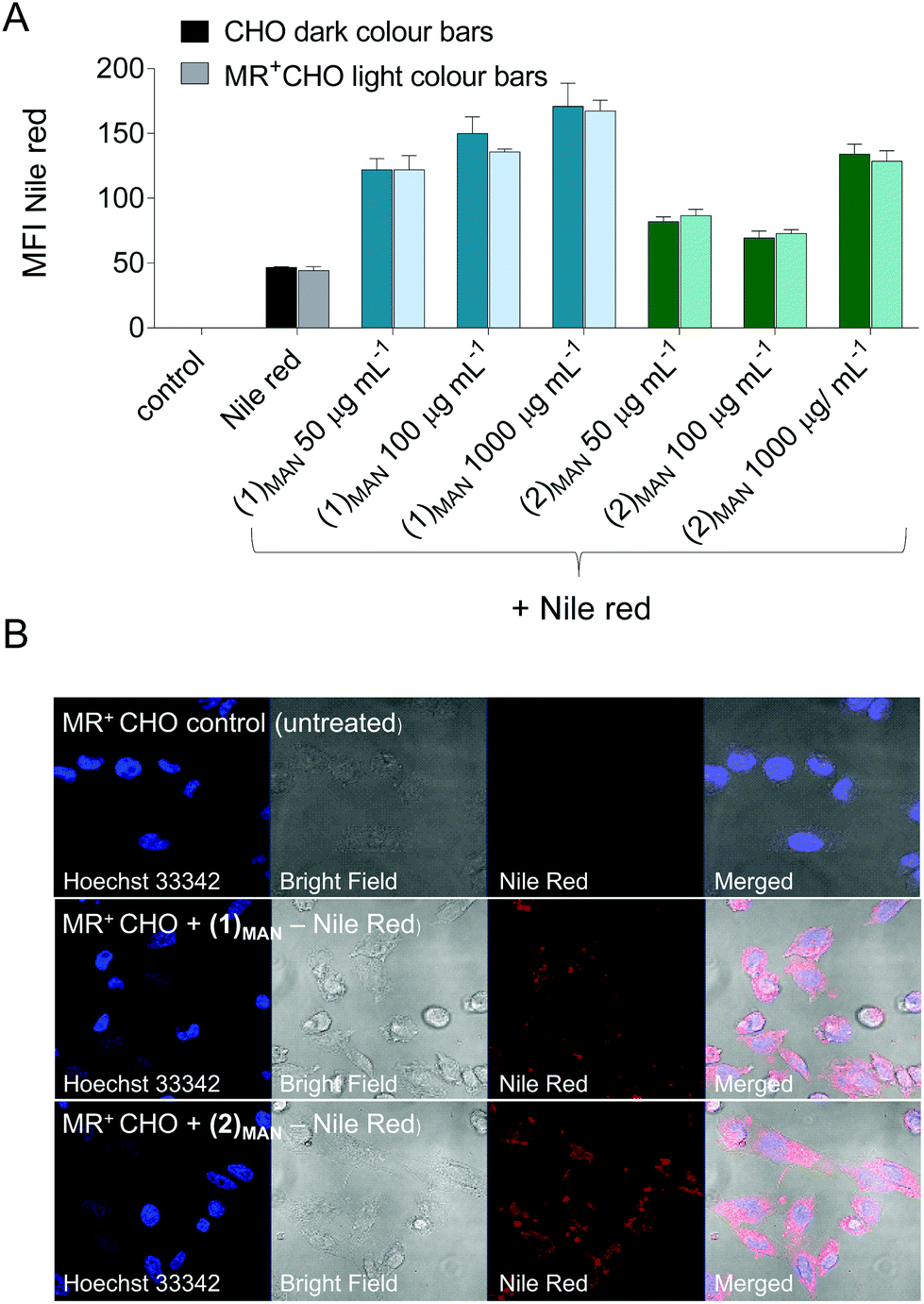 | ||
| Fig. 7 A: Flow cytometry quantification of the uptake of Nile red by CHO (MR negative), and MR+CHO cells incubated with (1)MAN- and (2)MAN-Nile red complexes at a constant concentration of Nile red (10 μM if all of it were solubilised), and increasing concentrations of glycopolymers. ‘Nile red’ columns are relative to cells incubated with Nile red at the same theoretical concentration, in the absence of glycopolymers. ‘Control’ refers to untreated cells. B: Confocal microscopy analysis of these samples: Top. MR+CHO untreated cells (negative control). Middle: MR+CHO cells incubated with 1.00 mg mL−1(1)MAN + 10 μM Nile red, and Bottom: MR+CHO cells treated with 1.00 mg mL−1(2)MAN + 10 μM Nile red, for 30 min. | ||
Interestingly, the uptake was found to be not significantly different in mannose receptor positive and negative CHO cells (Fig. 7A). This was observed even for systems where dye uptake was only marginally higher than that of Nile red alone ((2)MAN-Nile red at 50 and 100 μg mL−1, Fig. 7A), suggesting that this effect may not be due to receptor saturation.
Several studies have described Nile red transfer from nanoparticle systems to hydrophobic acceptors, such as cell membranes,81,82 lipid nanocapsules and triglyceride oil,83 and emulsion droplets.84 Thus, our results could be explained in terms of an equilibrium between Nile red reversibly complexed to the glycopolymers, and the unbound fraction being able to enter CHO cells by diffusion through the plasma membrane, or other mechanisms. Confocal analysis of the cells following treatment with Nile red and (1)MAN or (2)MAN showed diffuse intracellular cytosolic fluorescence, with a few punctuated spots (Fig. 6). This is in line with the analogous fluorophore distribution patterns described by Snipstad et al. who showed that on using Nile red-loaded poly(butylcyanoacrylate) nanoparticles, dye uptake by human prostate adenocarcinoma cells (PC3) did not occur by endocytosis, but rather by nanoparticle–cell contact-mediated transfer directly to the cytosol and, to a lesser extent, release of payload into the medium, followed by diffusion into the cells.82
Taken together our in vitro experiments showed that the targeted delivery of host molecules to soluble and surface-immobilised lectins can be successfully achieved, and preliminary cell experiments revealed that, whilst not directly suitable to probe MR-mediated targeted cell delivery at this stage, poly(triazolyl methacrylate)s (1)MAN and (2)MAN can act as carriers for Nile red and enhance its cellular uptake.
Conclusions
Poly(triazolyl methacrylate) glycopolymers are a class of synthetic carbohydrate-based materials investigated for a range of applications as diverse as inhibition of human lectin interactions with HIV viral envelope, modulation of antibody aggregation and as ligands for specific cancer cells. The work presented here suggests that, intriguingly, in addition to their known modalities of action, under specific conditions they can also function as unimolecular targeted carriers for hydrophobic guest molecules.In this study, these guest molecule–glycopolymer complexes were characterised through a range of techniques – DLS, TDA, fluorescence spectroscopy, surface tension analysis – and an initial structure–function relationship, i.e. how the nature of both the sugar repeating units and polymer backbone affects probe complexation was identified. Molecular dynamics (MD) simulations supported the experimental results and provided an insight into the mechanism of formation of these nanocomplexes. While at this stage the structure of the glycopolymers was not optimised to enhance guest molecule incorporation and delivery, this work provides an ab initio investigation on the factors that control non-covalent complexation of small hydrophobic probes with poly(triazolyl methacrylate) glycopolymers.
Glycopolymers are often utilised as tools for selective targeting of carbohydrate-binding proteins (lectins). While a limited number of studies have looked at the effect of different spacers linking carbohydrate pendant units to the polymer backbone on lectin binding affinity and specificity,85–87 from a synthetic design standpoint focus is typically placed on the nature, number, and orientation of the sugar repeating units. A broader implication of our study is that it suggests that the physico-chemical nature of both spacers and macromolecular backbone can affect the conformation of glycopolymers in solution – thus potentially affecting their ability to span over multiple lectin receptors and the orientation of their sugar binding units – and their ability to reversibly interact with small molecules in solution. The latter phenomenon may need to be considered when glycopolymers are utilised within multicomponent systems such as those encountered in complex biological systems, and could be exploited for the reversible binding and delivery of specific guest molecules within biomedical settings.
Conflicts of interest
CvdW is an employee of AstraZeneca Ltd and PJSK is an employee of Malvern Panalytical Ltd.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/L01646X, and EP/N024818/1] (RF and RMX, and YH); AstraZeneca (JMdO); and the University of Nottingham. We thank Dr Luisa Martinez-Pomares (University of Nottingham) for supplying MR+CHO cells, and Dr Annela Seddon (University of Bristol) for useful discussion. All MD simulations were run in GROMACS 5.1.05 using the High Performance Computing (HPC) cluster of the University of Nottingham or ARCHER, UK's National Supercomputer.Notes and references
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- M. Gonzalez-Burgos, A. Latorre-Sanchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef CAS.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef CAS PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS PubMed.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- I. Berkovich, S. Mavila, O. Iliashevsky, S. Kozuch and N. G. Lemcoff, Chem. Sci., 2016, 7, 1773–1778 RSC.
- S. Mavila, C. E. Diesendruck, S. Linde, L. Amir, R. Shikler and N. G. Lemcoff, Angew. Chem., Int. Ed., 2013, 52, 5767–5770 CrossRef CAS PubMed.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196–4203 RSC.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- A. P. P. Kröger and J. M. J. Paulusse, J. Controlled Release, 2018, 286, 326–347 CrossRef PubMed.
- S. Quader and K. Kataoka, Mol. Ther., 2017, 25, 1501–1513 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815 CrossRef CAS PubMed.
- S. Cheng, S.-J. Xie, J.-M. Y. Carrillo, B. Carroll, H. Martin, P.-F. Cao, M. D. Dadmun, B. G. Sumpter, V. N. Novikov, K. S. Schweizer and A. P. Sokolov, ACS Nano, 2017, 11, 752–759 CrossRef CAS PubMed.
- H. Piwoński, T. Michinobu and S. Habuchi, Nat. Commun., 2017, 8, 15256 CrossRef PubMed.
- X. Fan, Z. Li and X. J. Loh, Polym. Chem., 2016, 7, 5898–5919 RSC.
- G. Chen, Y. Wang, R. Xie and S. Gong, Adv. Drug Delivery Rev., 2018, 130, 58–72 CrossRef CAS PubMed.
- Y. Wang, G. Qi and J. He, ACS Macro Lett., 2016, 5, 547–551 CrossRef CAS.
- G. Liu, H. Gao, Y. Zuo, X. Zeng, W. Tao, H.-i. Tsai and L. Mei, ACS Appl. Mater. Interfaces, 2017, 9, 112–119 CrossRef CAS PubMed.
- X. Fan, Z. Hu and G. Wang, RSC Adv., 2015, 5, 100816–100823 RSC.
- G. Chen, L. Wang, T. Cordie, C. Vokoun, K. W. Eliceiri and S. Gong, Biomaterials, 2015, 41–50 CrossRef CAS PubMed.
- Y. Xiao, H. Hong, A. Javadi, J. W. Engle, W. Xu, Y. Yang, Y. Zhang, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2012, 33, 3071–3082 CrossRef CAS PubMed.
- C. S. Popeney, M. C. Lukowiak, C. Böttcher, B. Schade, P. Welker, D. Mangoldt, G. Gunkel, Z. Guan and R. Haag, ACS Macro Lett., 2012, 1, 564–567 CrossRef CAS.
- C. Porsch, Y. Zhang, C. Ducani, F. Vilaplana, L. Nordstierna, A. M. Nyström and E. Malmström, Biomacromolecules, 2014, 15, 2235–2245 CrossRef CAS PubMed.
- J. Xu, S. Luo, W. Shi and S. Liu, Langmuir, 2006, 22, 989–997 CrossRef CAS PubMed.
- W.-M. Wan, P. D. Pickett, D. A. Savin and C. L. McCormick, Polym. Chem., 2014, 5, 819–827 RSC.
- K. N. R. Wuest, H. Lu, D. S. Thomas, A. S. Goldmann, M. H. Stenzel and C. Barner-Kowollik, ACS Macro Lett., 2017, 6, 1168–1174 CrossRef CAS.
- A. P. P. Kröger, M. I. Komil, N. M. Hamelmann, A. Juan, M. H. Stenzel and J. M. J. Paulusse, ACS Macro Lett., 2019, 8, 95–101 CrossRef PubMed.
- L. L. Kiessling and J. C. Grim, Chem. Soc. Rev., 2013, 42, 4476–4491 RSC.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS PubMed.
- C. R. Becer, Macromol. Rapid Commun., 2012, 33, 742–752 CrossRef CAS PubMed.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS PubMed.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS PubMed.
- J. Madeira do O, F. Mastrotto, N. Francini, S. Allen, C. F. van der Waalse, S. Stolnik and G. Mantovani, J. Mater. Chem. B, 2018, 6, 1044–1054 RSC.
- J. Jose and K. Burgess, J. Org. Chem., 2006, 71, 7835–7839 CrossRef CAS PubMed.
- T. Kroeger, B. Frieg, T. Zhang, F. K. Hansen, A. Marmann, P. Proksch, L. Nagel-Steger, G. Groth, S. H. J. Smits and H. Gohlke, PLoS One, 2017, 12, e0177024 CrossRef PubMed.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130–15132 CrossRef CAS PubMed.
- J. Chen, C. Travelet, R. Borsali and S. Halila, Biomacromolecules, 2017, 18, 3410–3417 CrossRef CAS PubMed.
- Y. Gou, J. Geng, S.-J. Richards, J. Burns, C. Remzi Becer and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2588–2597 CrossRef CAS PubMed.
- Y. Gou, S. Slavin, J. Geng, L. Voorhaar, D. M. Haddleton and C. R. Becer, ACS Macro Lett., 2012, 1, 180–183 CrossRef CAS.
- K. Jono, M. Nagao, T. Oh, S. Sonoda, Y. Hoshino and Y. Miura, Chem. Commun., 2018, 54, 82–85 RSC.
- J. Tanaka, A. S. Gleinich, Q. Zhang, R. Whitfield, K. Kempe, D. M. Haddleton, T. P. Davis, S. Perrier, D. A. Mitchell and P. Wilson, Biomacromolecules, 2017, 18, 1624–1633 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhang, C. Wang, G. Chen and M. Jiang, Biomacromolecules, 2017, 18, 568–575 CrossRef CAS PubMed.
- K. Babiuch, A. Dag, J. Zhao, H. Lu and M. H. Stenzel, Biomacromolecules, 2015, 16, 1948–1957 CrossRef CAS PubMed.
- J. Geng, J. Lindqvist, G. Mantovani, G. Chen, C. T. Sayers, G. J. Clarkson and D. M. Haddleton, QSAR Comb. Sci., 2007, 26, 1220–1228 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- P. Greenspan and S. D. Fowler, J. Lipid Res., 1985, 26, 781–789 CAS.
- A. Eisfeld and J. S. Briggs, Chem. Phys., 2006, 324, 376–384 CrossRef CAS.
- I. N. Kurniasih, H. Liang, P. C. Mohr, G. Khot, J. P. Rabe and A. Mohr, Langmuir, 2015, 31, 2639–2648 CrossRef CAS PubMed.
- I. Mukherjee, S. P. Moulik and A. K. Rakshit, J. Colloid Interface Sci., 2013, 394, 329–336 CrossRef CAS PubMed.
- A. Pan, A. K. Rakshit and S. P. Moulik, Colloids Surf., A, 2015, 464, 8–16 CrossRef CAS.
- G. Kasza, G. Gyulai, A. Abraham, G. Szarka, B. Ivan and E. Kiss, RSC Adv., 2017, 7, 4348–4352 RSC.
- H. Liu, A. Jiang, J. Guo and K. E. Uhrich, J. Polym. Sci., Part A: Polym. Chem., 2000, 37, 703–711 CrossRef.
- M. F. Mazzobre, M. V. Román, A. F. Mourelle and H. R. Corti, Carbohydr. Res., 2005, 340, 1207–1211 CrossRef CAS PubMed.
- We were aware that dye H-dimerization/aggregation within these polymer assemblies could have potentially affected the final fluorescence readings.
- A. Rei, M. I. C. Ferreira and G. Hungerford, J. Fluoresc., 2008, 18, 1083–1091 CrossRef CAS PubMed.
- A. Boreham, M. Pfaff, E. Fleige, R. Haag and U. Alexiev, Langmuir, 2014, 30, 1686–1695 CrossRef CAS PubMed.
- R. Plenderleith, T. Swift and S. Rimmer, RSC Adv., 2014, 4, 50932–50937 RSC.
- A. Muñoz-Bonilla, O. León, M. L. Cerrada, J. Rodríguez-Hernández, M. Sánchez-Chaves and M. Fernández-García, Eur. Polym. J., 2015, 62, 167–178 CrossRef.
- A. Muñoz-Bonilla, O. León, V. Bordegé, M. Sánchez-Chaves and M. Fernández-García, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1337–1347 CrossRef.
- S. Latunde-Dada, R. Bott, K. Hampton, J. Patel and O. I. Leszczyszyn, Anal. Methods, 2015, 7, 10312–10321 RSC.
- S. Latunde-Dada, R. Bott, D. Barker and O. I. Leszczyszyn, Anal. Methods, 2016, 8, 386–392 RSC.
- A. Hawe, W. L. Hulse, W. Jiskoot and R. T. Forbes, Pharm. Res., 2011, 28, 2302–2310 CrossRef CAS PubMed.
- H. Cottet, J.-P. Biron and M. Martin, Analyst, 2014, 139, 3552–3562 RSC.
- L. Cipelletti, J.-P. Biron, M. Martin and H. Cottet, Anal. Chem., 2015, 87, 8489–8496 CrossRef CAS PubMed.
- H. Cottet, J.-P. Biron and M. Martin, Anal. Chem., 2007, 79, 9066–9073 CrossRef CAS PubMed.
- W. L. Goh, M. Y. Lee, T. L. Joseph, S. T. Quah, C. J. Brown, C. Verma, S. Brenner, F. J. Ghadessy and Y. N. Teo, J. Am. Chem. Soc., 2014, 136, 6159–6162 CrossRef CAS PubMed.
- A. Hawe, V. Filipe and W. Jiskoot, Pharm. Res., 2010, 27, 314–326 CrossRef CAS PubMed.
- R. O. Dror, R. M. Dirks, J. P. Grossman, H. Xu and D. E. Shaw, Annu. Rev. Biophys., 2012, 41, 429–452 CrossRef CAS PubMed.
- B. M. Tande, N. J. Wagner, M. E. Mackay, C. J. Hawker and M. Jeong, Macromolecules, 2001, 34, 8580–8585 CrossRef CAS.
- M. Delbianco, P. Bharate, S. Varela-Aramburu and P. Seeberger, Chem. Rev., 2016, 116, 1693–1752 CrossRef CAS PubMed.
- K. Liu, X. Jiang and P. Hunziker, Nanoscale, 2016, 8, 16091–16156 RSC.
- A. Surolia, S. Bishayee, A. Ahmad, K. Balasubramanian, D. Thambi-Dorai, S. Podder and B. Bachhawat, Adv. Exp. Med. Biol., 1975, 55, 95–115 CrossRef CAS PubMed.
- I. Papp, J. Dernedde, S. Enders, S. Riese, T. Shiao, R. Roy and R. Haag, ChemBioChem, 2011, 12, 1075–1083 CrossRef CAS PubMed.
- Y. Gou, J. Geng, S.-j. Richards, J. Burns, C. R. Becer and D. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2588–2597 CrossRef CAS PubMed.
- L. Martinez-Pomares, D. M. Reid, G. D. Brown, P. R. Taylor, R. J. Stillion, S. A. Linehan, S. Zamze, S. Gordon and S. Y. C. Wong, J. Leukocyte Biol., 2003, 73, 604–613 CrossRef CAS PubMed.
- Y. Su, T. Bakker, J. Harris, C. Tsang, G. D. Brown, M. R. Wormald, S. Gordon, R. A. Dwek, P. M. Rudd and L. Martinez-Pomares, J. Biol. Chem., 2005, 280, 32811–32820 CrossRef CAS PubMed.
- P. R. Taylor, S. Gordon and L. Martinez-Pomares, Trends Immunol., 2005, 26, 104–110 CrossRef CAS PubMed.
- L. Martinez-Pomares, J. Leukocyte Biol., 2012, 92, 1177–1186 CrossRef CAS PubMed.
- S. Snipstad, S. Hak, H. Baghirov, E. Sulheim, Ý. Mørch, S. Lélu, E. von Haartman, M. Bäck, K. P. R. Nilsson, S. Klymchenko Andrey, C. de Lange Davies and K. O. Åslund Andreas, Cytometry, Part A, 2016, 91, 760–766 CrossRef PubMed.
- S. Snipstad, S. Westrøm, Y. Mørch, M. Afadzi, A. K. O. Åslund and C. de Lange Davies, Cancer Nanotechnol., 2014, 5, 8 CrossRef PubMed.
- G. Bastiat, C. O. Pritz, C. Roider, F. Fouchet, E. Lignières, A. Jesacher, R. Glueckert, M. Ritsch-Marte, A. Schrott-Fischer, P. Saulnier and J.-P. Benoit, J. Controlled Release, 2013, 170, 334–342 CrossRef CAS PubMed.
- S. Petersen, A. Fahr and H. Bunjes, Mol. Pharm., 2010, 7, 350–363 CrossRef CAS PubMed.
- B. D. Polizzotti and K. L. Kiick, Biomacromolecules, 2006, 7, 483–490 CrossRef CAS PubMed.
- S.-J. Richards, M. W. Jones, M. Hunaban, D. M. Haddleton and M. I. Gibson, Angew. Chem., Int. Ed., 2012, 51, 7812–7816 CrossRef CAS PubMed.
- M. W. Jones, L. Otten, S. J. Richards, R. Lowery, D. J. Phillips, D. M. Haddleton and M. I. Gibson, Chem. Sci., 2014, 5, 1611–1616 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General, experimental procedures, and characterization of all new compounds. See DOI: 10.1039/c9nr05836b |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |