
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmbient synthesis of nanomaterials by in situ heterogeneous metal/ligand reactions†
Boyce S.
Chang
ab,
Brijith
Thomas
bc,
Jiahao
Chen
 a,
Ian D.
Tevis
a,
Paul
Karanja
a,
Simge
Çınar
a,
Amrit
Venkatesh
bc,
Aaron J.
Rossini
*bc and
Martin M.
Thuo
*a
a,
Ian D.
Tevis
a,
Paul
Karanja
a,
Simge
Çınar
a,
Amrit
Venkatesh
bc,
Aaron J.
Rossini
*bc and
Martin M.
Thuo
*a
aDepartment of Materials Science and Engineering, Iowa State University, 2220 Hoover Hall, Ames, IA 50011, USA. E-mail: mthuo@iastate.edu
bUS DOE Ames Laboratory, Ames, Iowa, USA 50011. E-mail: arossini@iastate.edu
cDepartment of Chemistry, Iowa State University, 1605 Gilman Hall, Ames, IA 50011, USA
First published on 9th July 2019
Abstract
Coordination polymers are ideal synthons in creating high aspect ratio nanostructures, however, conventional synthetic methods are often restricted to batch-wise and costly processes. Herein, we demonstrate a non-traditional, frugal approach to synthesize 1D coordination polymers by in situ etching of zerovalent metal particle precursors. This procedure is denoted as the heterogeneous metal/ligand reaction and was demonstrated on Group 13 metals as a proof of concept. Simple carboxylic acids supply the etchant protons and ligands for metal ions (conjugate base) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This scalable reaction produces a 1D polymer that assembles into high-aspect ratio ‘nanobeams’. We demonstrate control over crystal structure and morphology by tuning the: (i) metal center, (ii) stoichiometry and (iii) structure of the ligands. This work presents a general scalable method for continuous, heat free and water-based coordination polymer synthesis.
:1 ratio. This scalable reaction produces a 1D polymer that assembles into high-aspect ratio ‘nanobeams’. We demonstrate control over crystal structure and morphology by tuning the: (i) metal center, (ii) stoichiometry and (iii) structure of the ligands. This work presents a general scalable method for continuous, heat free and water-based coordination polymer synthesis.
Introduction
Nanostructured materials have garnered significant interest over the last few decades due to a myriad of applications in diverse fields such as medicine, catalysis, separation, gas storage, optics and insulation, among others.1–3 In particular, 2D materials such as graphene and other van der Waals hetero-structures have received considerable interest due to their tunable properties, and unparalleled ability to form functional surfaces.4–8 Alternatively, coordination polymers (CP) are known to self-assemble into a variety of structures and offer extensive morphological tunability.9–11 The ability to select a variety of building blocks (metal center and coordinating ligands) widely expands the applications of these hybrid materials whereby tunable behaviors include optics,12 electronic,13 surface chemistry14 and pore structure.15 1D coordination polymers are also potentially useful for the formation of sol-gels, templating nanomaterials,16 and mechanical actuators.17However, significant challenges in fabrication often render the cost of nanostructured materials impractical for widespread applications. For example, coordination polymers are typically produced by solvent induced precipitation and solvothermal methods,18 which are strictly batch processes. Such methods are effective for rapid prototyping but undesirable for large scale production. Thus, the search for green and scalable synthetic routes suitable for industry remains an active endeavor.19 Herein, we demonstrate a soft chemistry, water-based method for producing 1D coordination polymers from metal droplets, known as heterogeneous in situ metal/ligand reaction (HetMet) (Fig. 1a). Our approach exploits: (i) ability of metal droplets to maintain a chemical potential gradient in solution, leading to ad infinitum coordination polymerization, (ii) the ability of a conjugate acid–base pair (H3O+ and RCOO−) to serve as an etchant and a ligand respectively, (iii) kinetically resolved selective oxidation of metal alloy components leading to dominance of the surface by most reactive component,20–22 (iv) control over the dimensions of the structure, thus, potential to tune material properties.23
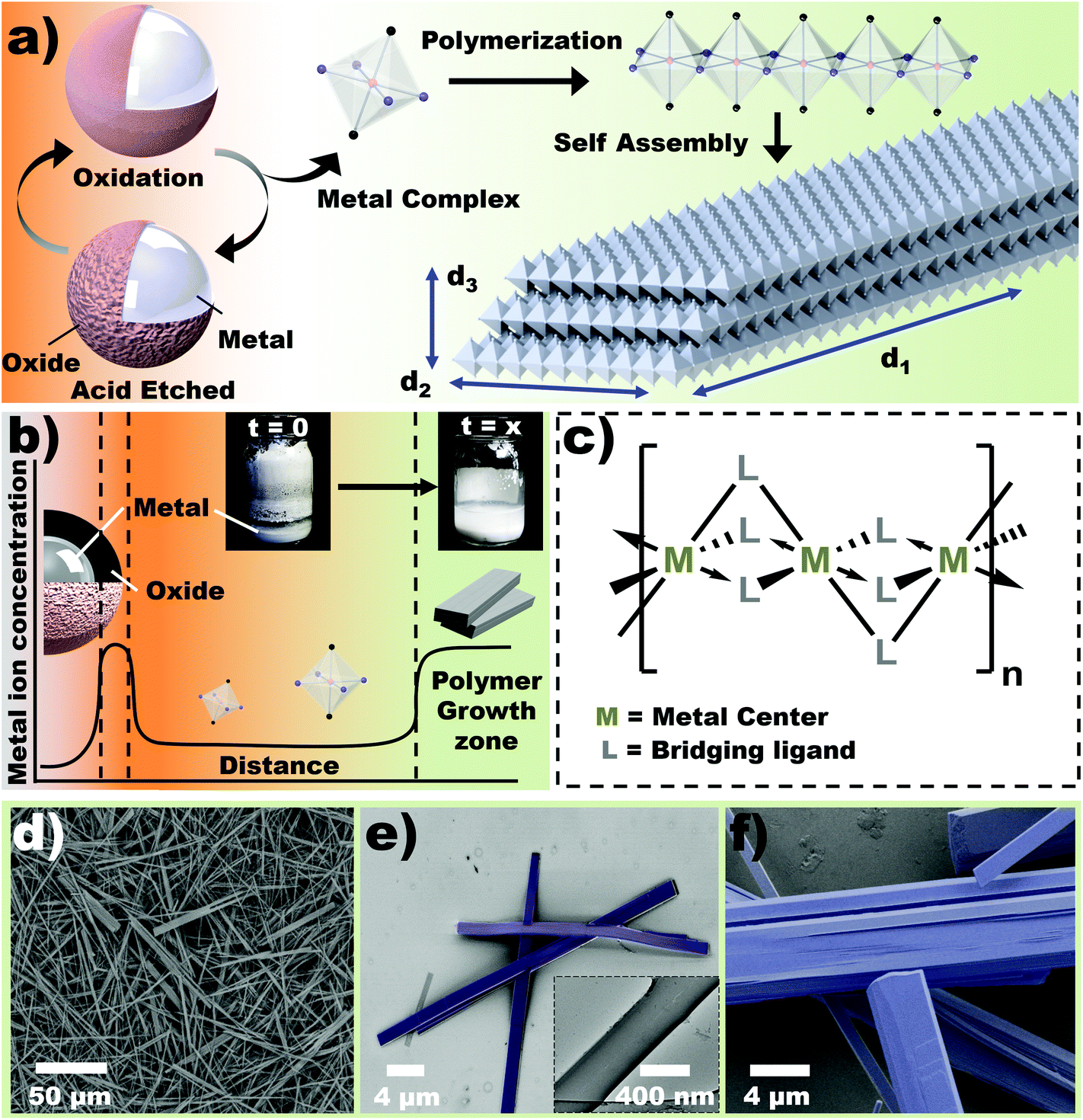 | ||
| Fig. 1 Heterogeneous in situ metal/ligand reaction (HetMet) for coordination polymer (nanobeam) synthesis. (a) Schematic illustration of coordination polymer synthesis with concomitant self-assembly of 1D structures derived from reaction between acid and metal particles. Inter-digitation upon self-assembly gives a rigid planar beam structure. (b) Profile of metal ion concentration showing solubility-controlled concentration of the metal chelate ‘monomer’. Inset demonstrates quantitative reaction. The reaction is mapped relative to the reaction coordinate, where the etching is at the start and the product is at the end – represented via a ‘distance’. (c) Hypothetical chemical structure with a looping/bridging bidentate ligand that drives the reaction towards polymerization. (d) Scanning electron microscopy (SEM) of Ga-based nanobeams. (e–f) Higher magnification SEM images revealing a layered structure. Inset (e) shows a transmission electron micrograph (TEM). | ||
Background
Solvothermal methods of coordination polymer synthesis offers several advantages including control over the crystallite size and high temperature/pressure conditions, which allow utility of otherwise stable reactants.24 This method operates by creating a thermal gradient in a closed reactor. Fundamentally, this creates a chemical potential gradient in solution, which leads to diffusion and precipitation of ions in the growth zone. HetMet utilizes the steep concentration difference between the metal particles and solvent to establish a constant chemical potential gradient (Fig. 1b). This unique concept allows crystal growth at ambient temperatures and pressure. Furthermore, a constant concentration of metal ions—due to limited solubility—in solution allows water-based synthesis, despite growing moisture sensitive crystals. Analogous to a ‘living’ polymerization, crystal growth occurs continuously until all the metal is consumed, thus producing a quantitative reaction (Inset Fig. 1b). The flexibility of using metal particles and reactive ligands should ideally control chain structure as illustrated in Fig. 1c, unlocking new pathways to rationally design nanomaterials.Results and discussion
Inspired by the limitations of batch processes, we envision the use of a high-density monomer reservoir (metals) that would significantly prolong the reaction lifetime. Core–shell Eutectic Gallium Indium (EGaIn) particles were first used as source for gallium ions. The liquid metal allows production of particles by shearing.25 For simplicity, aqueous acetic acid (5%) was used as the etchant, leading to incorporation of acetate ligands into the final coordination polymer. Fig. 1a illustrates the three-step synthesis of self-assembled coordination polymers. Dissolution of the oxide layer from the EGaIn reservoir results in a high concentration of Ga3+ ions26 that chelate with the conjugate base to form a Ga-acetate coordination complex. It has been shown that these particles can be significantly etched when exposed in aqueous acetic acid.25 As the solution reaches a critical concentration of Ga complex, precipitation occurs, hence polymerization of the metal complex. This phenomena establishes a concentration gradient between the EGaIn reservoir and growth (precipitation) zone, which drives further production of Ga complex. Once steady-state conditions between etching of the oxide and precipitation is established, the HetMet reaction continuously generates 1D coordination polymers (Ga-acetate based polymers are known to produce 1D chains)27 whose solubility, akin to the metal ions, decreases with increased growth (Fig. 1b). Polymerization directs growth of the nanobeams in one direction (say X-direction), while the growth in other dimensions are driven by asymmetry in anti-periplanar moieties (i.e. XY vs. YZ Cartesian planes) across the coordination center. Control over the 3D structure of the nanobeams can, therefore, be achieved by changing the structure of the monomer (ligand and metal ion). Furthermore, ad infinitum polymerization maintains chain growth if monomers are present, thus achieving complete conversion analogous to ‘living polymerization’ in organic polymers. Inset Fig. 1b demonstrates that practically all the metal in solution is converted into white precipitates. Growth rate, however, depends on surface area of the metal (particle size) and acid concentration (ca. 95% yield has been observed in a period of 4 days).Physical characteristics of nanobeams
The isolated coordination polymer shows highly regular beam-like structures – so called nanobeams (Fig. 1d). The nanobeams appear as layers stacked in the lateral direction (Fig. S1†), yielding dimensions of approximately 20 μm in length and 2 μm width. The beams bend under low load (Fig. 1e), which indicates that the layered structure is held by weak intermolecular interactions.17 Fractures can be observed to predominantly propagate along the longest (d1) axis (Fig. 1f).Characterization of nanobeams
Collating data from thermogravimetric analysis (TGA) and IR-MS of the evolved gases (Fig. 2a and b, S2†) the molecular formula of Ga-based CP was determined to be Ga(OH)2(O2CCH3). The molecular formula of the acetic acid CP was corroborated by other analytical techniques (vide infra). Intense sharp peaks from WAXS are observed at low 2θ corresponding to large interplanar spacings (∼3 Å, Fig. 2c) while broad distortions in SAXS signify periodic length scales at 3–4 nm (Inset Fig. 2c), which are attributed to layered structure observed in SEM (Fig. 1f). Heating of the CP up to 800 °C in air causes transformation to Ga2O3 as a final product (Fig. S3†).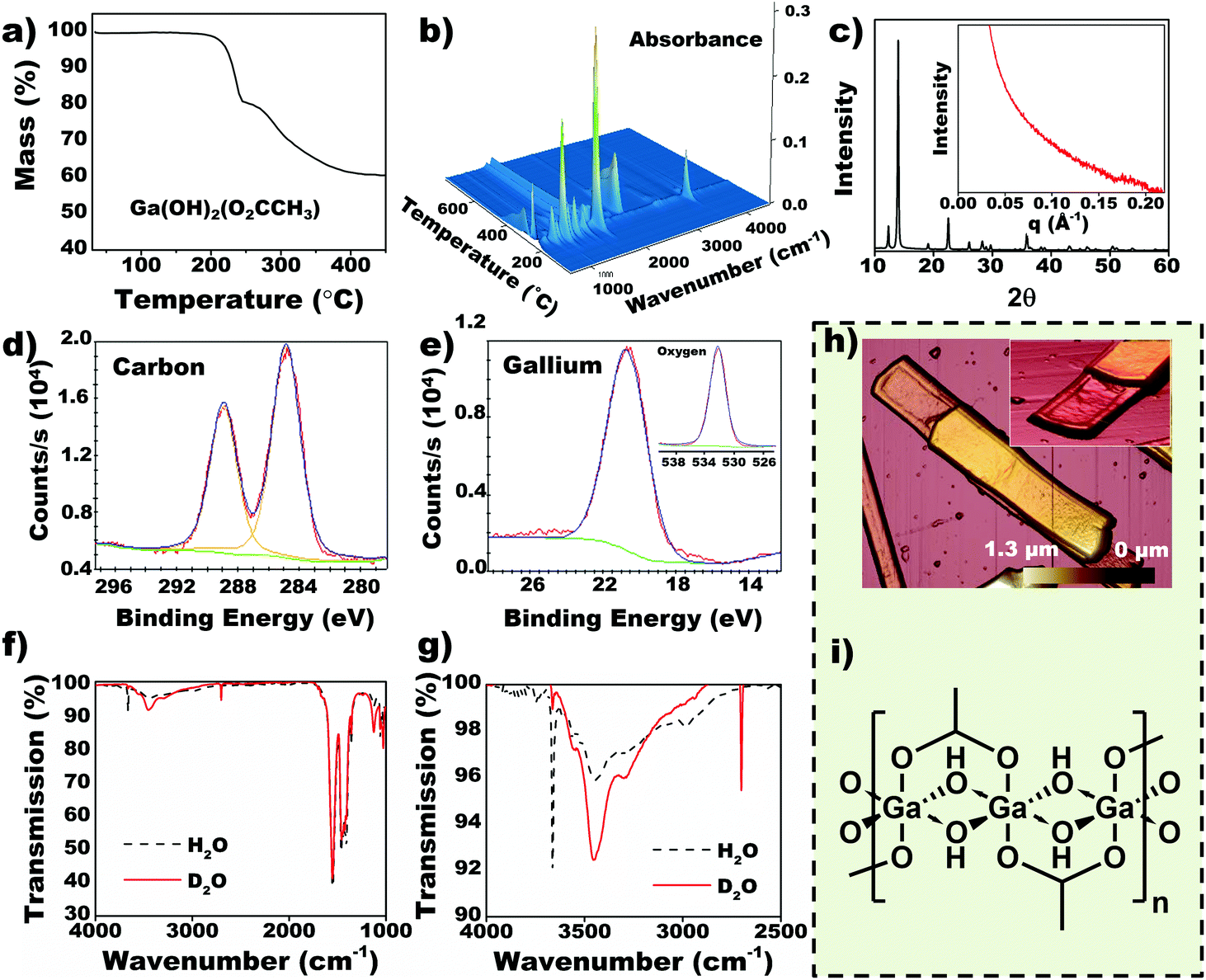 | ||
| Fig. 2 Characterization of Ga-based nanobeams. (a) Thermogravimetric analysis (TGA) used for empirical formula prediction (inset). (b) Coupled IR absorption spectra of gaseous exudates from the TGA. (c) Wide angle X-ray scattering (WAXS) patterns indicating a periodic structure with high intensity peaks at lower angles (large lattice spacing). Inset shows small angle X-ray scattering (SAXS), whereby scattering peaks at 0.15–0.2 Å−1 indicate length scales between 3–4 nm. (d–e) X-ray photoelectron spectroscopy (XPS) showing that the material consists of carbon, gallium and oxygen (inset). (f–g) Infrared (IR) spectroscopy of nanobeams grown in water (dotted) and deuterium oxide (solid) confirms the bridging –OH group. (h) Atomic force microscopy (AFM) showing stacking and capillary warping of the nanobeams upon drying. (i) Molecular formula of the Ga-based nanobeam. | ||
Binding energies obtained from X-ray photoelectron spectroscopy (XPS) (Fig. 2d and e) indicate that the nanobeams contain carbonyl carbon (288.9 eV), Ga3+ cations (20.7 eV) and oxygen (532 eV)—we intentionally disregard the sp3 carbon due to potential presence of adventitious carbon. The composition of the nanobeams corroborate with energy dispersive X-ray spectroscopy (EDS) data (Fig. S4†).20 However, inductively coupled plasma mass spectroscopy (ICP-MS) and atomic emission spectroscopy (ICP-AES) indicates that EGaIn-derived nanobeams contain up to 7% indium atoms. As expected, Ga is preferentially oxidized from EGaIn alloy and polymerized due to its lower reduction potential compared to In.20–22 We infer that the In ions found in the nanobeams correspond to Ga substituted sites, which occur due to stoichiometric balance between the polymer and solvent. This feature could be utilized as a doping strategy through a selection of alloys based on the reduction potential hierarchy. Infrared (IR) spectroscopy (Fig. 2f and g) indicates a sharp peak at 3662 cm−1, which is characteristic of highly ordered O–H groups.28,29 To validate presence of ordered –OH moieties, nanobeams were grown in D2O. The OH signal shifted to 2700 cm−1 on deuteration, corresponding to the expected H2O to D2O spring constant changes (1.347 (ref. 30)), which confirms the coordination of O–H groups with the metal center. A comparison between IR spectrum of nanobeams and sodium acetate is provided in Fig. S5.† The layered structure observed by atomic force microscopy (AFM) (Fig. 2h and Fig. S6–S7†) supports the notion of polymeric structure, thus 1D chains held together by weak secondary interactions. Based on the molecular formula, we determined the most probable structure of the nanobeams to be a 1D chain with its repeat unit being a six coordinate Ga center chelated by acetate and hydroxyl bridging ligands, [Ga(μ-OH)2(μ-O2CCH3)]n (Fig. 2i).
NMR-based molecular structure
To deduce the molecular structure of the nanobeams, we applied solid-state 1H, 2H, 13C and 71/69Ga magic angle spinning (MAS) multi-nuclear magnetic resonance (NMR) experiments.31 The MAS 1H solid-state NMR spectrum of nanobeams (MAS frequency = 50 kHz) is shown in the upper part of Fig. 3a, and depicts a single peak with an isotropic chemical shift of ca. 1.9 ppm, which corresponds to the methyl groups of acetate and hydroxyl groups. The 1H chemical shift of the hydroxyl groups and acetate methyl protons are coincident, and this is demonstrated by 2H solid-state NMR experiments on CP grown from D2O (details provided in the ESI†). A proton detected 1H–13C dipolar HETCOR spectrum (Fig. 3b) confirms presence of acetate in the nanobeams, and the presence of only one set of 13C signal suggest that all acetate ligands within the lattice are equivalent and likely bound to Ga. Fig. 3c shows static (i.e., stationary sample) and MAS 71Ga SSNMR spectra of nanobeams. Paired with static 69Ga SSNMR, accurate electric field gradient (EFG) and chemical shift (CS) tensor parameters from simulations of the experimental spectra (Table S1†) were obtained. These results suggest that all Ga sites within the nanobeam lattice are equivalent and the repeat unit is a highly distorted 6-coordinate coordination environment (additional details and discussion of solid-state NMR experiments in the ESI†). Finally, a 2D 1H–71Ga constant time D-HMQC dipolar correlation spectrum directly confirms spatial proximity of Ga to the acetate and/or hydroxyl ligands (Fig. 3d).31,32 The products of TGA was also confirmed to be Ga2O3 by SSNMR (Fig. S8†). The crystal structure of the Ga-based nanobeam was solved using a combination of PXRD, solved molecular formula and SSNMR (beam-like structures were unsuitable for single crystal diffraction).33 The simulated pattern predicts that 1D chains stack perpendicular to the chain axis, with the acetate ligand interdigitated between chains (Fig. 3e).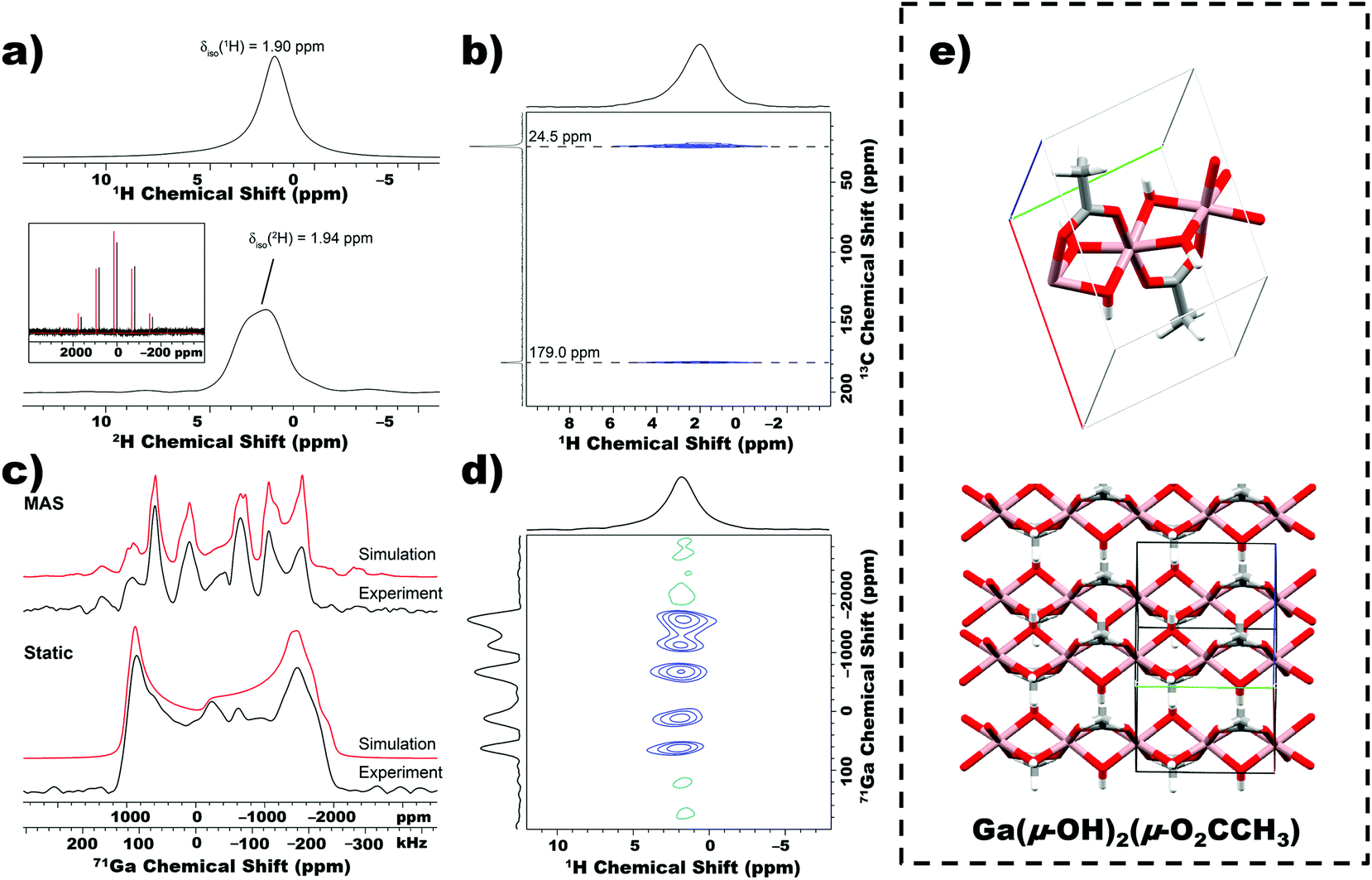 | ||
| Fig. 3 Chemical structure confirmation by solid-state NMR spectroscopy for CP derived from etching of EGaIn in 5% acetic acid solution. (a) MAS 1H spin echo spectrum (upper) and MAS 2H spin echo spectrum of nanobeams. MAS frequency of 50 kHz was used in both cases. The inset shows the whole 2H NMR spectrum and a simulation of the sideband intensities. (b) Proton detected 2D 1H–13C cross-polarization HETCOR spectrum of CP acquired with contact times of 2.5 ms. The 13C chemical shifts are indicated. (c) 71Ga solid-state NMR spectra acquired with a 50 kHz MAS frequency (upper set) and a static sample (lower set). Analytical simulations (red trace) are overlaid on the experimental spectra (black traces). (d) 1H–71Ga D-HMQC correlation spectrum acquired with MAS frequency of 50 kHz and super-cycled R412 dipolar recoupling applied on 1H. (e) Proposed molecular structure of nanobeams and corresponding simulated crystal structure. | ||
Effect of ligand stoichiometry
Control over the chain structure and morphology of the CP would ideally be achieved by changing the coordination environment of the metal. Thus, we performed the etching reaction with different concentrations of acid, which varies the stoichiometry of the available ligands. Products from higher acid concentrations (>50% Ac) appear as smaller analogs of the 5% acid grown nanobeam (5% Ac) (Fig. 4a and b). TGA predicts the molecular formula of the CP obtained with 50% Ac to be Ga(μ-OH)(μ-O2CCH3)2 (Fig. 4c), indicating that a hydroxyl ligand is substituted with an acetate group at higher acetic acid concentration in the solution (Fig. 4d and e). The PXRD pattern confirms that the solid precipitated from >50% acetic acid solution adopts a distinct crystal structure from the solid obtained from the 5% acetic acid solution (Fig. 4f). Mensinger et al. have previously synthesized a gallium coordination polymer with molecular formula Ga(μ-OH)(μ-O2CCH3)2·(HO2CH3)·H2O that incorporates free water and acetic acid molecules in the lattice.27 The PXRD pattern for 50% Ac nanobeam shows low Bragg peaks analogous to those observed in the Mensinger structure. The compounds, however, are likely different given the distinct differences in their empirical formulas. As the 50% Ac nanobeam is exposed to ambient air, we observed that it transitions into the same solid-state structure as the 5% Ac CP (Fig. 4f). The additional acetate ligand in the 50% Ac CP likely exchanges with adventitious water to form a hydroxyl ligand. Furthermore, this transition was not observed when the sample is left in vacuum for a prolonged period. The 1H{13C} HETCOR NMR spectrum shows multiple acetate NMR signals, indicating presence of free and bound acetate ions (Fig. 4g). We hypothesize that adventitious water protonates the acetate groups with concomitant OH-Ac ligand substitution to form the 5% Ac structure, which constitutes the free acetate observed in NMR (Fig. 4g). Due to the metastability of the 50% Ac CP, we exercise caution in assigning a specific structure to the as-synthesized product since we cannot, with certainty, identify amount of occluded H2O or acetic acid. We can, however, confirm that with increased acetic acid concentration, a second, albeit labile, acetate ligand is coordinated to the Ga as confirmed by NMR. The 71Ga static NMR spectrum is clearly distinct from that of the 5% Ac CP, however, the simulated NMR parameters indicate that the Ga coordination environment is also a distorted octahedron (Fig. 4h, Table S2†). Finally, probing the crystal growth over time verifies that both acid concentration and time can be used as parameters to control crystal dimensions (Fig. 4i).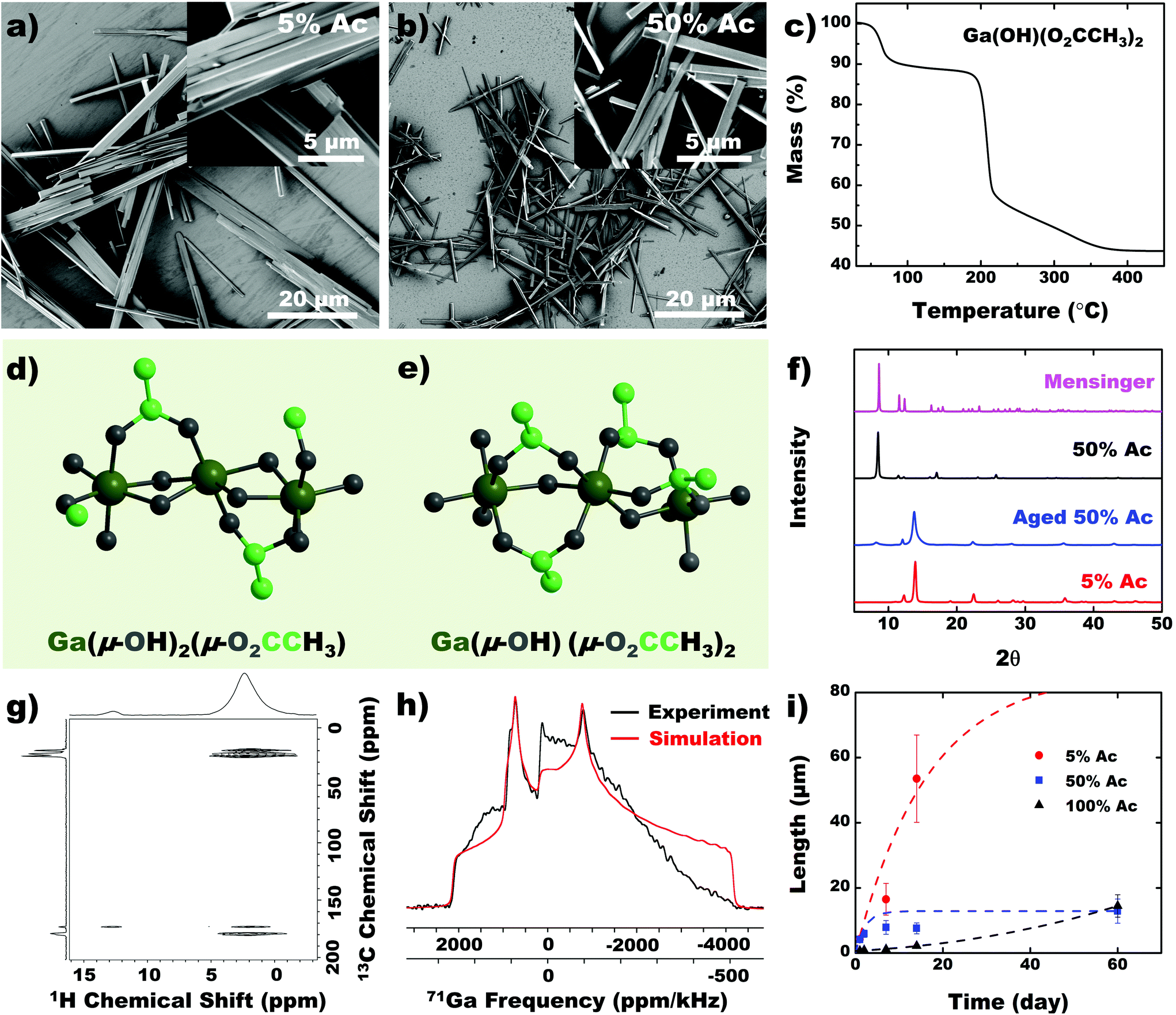 | ||
| Fig. 4 Effect of acid concentration on CP growth. SEM images of CP derived from etching EGaIn with (a) 5% acetic acid and (b) 50% acetic acid solutions. (c) TGA of 50% Ac CP, predicting a new empirical formula (inset). Molecular structure of (d) 5% Ac and (e) 50% Ac CP. (f) PXRD of materials grown in different acid concentrations. Aging refers to exposure of the isolated material to ambient conditions for 12 days. (g) Proton detected 2D 1H{13C} cross-polarization HETCOR and (h) static 71Ga solid-state NMR spectra of 50% Ac. (i) Length of the different crystals over time measured by SEM. | ||
Effect of the metal center
CPs were synthesized using other metals across group 13 in acetic acid to assess the effect of the metal center on crystal structure and morphology. Using pure Ga metal, a similar structure was obtained with EGaIn as expected. Al exhibited growth of ∼1 μm sheet-like material (Fig. 5a) while In preserved the beam-like structure observed in Ga, albeit with larger thickness and width (Fig. 5b and c). In-Based CP also consists of bundles of fibers instead of sheets (Fig. 5c). Crystals formed using alloys of the corresponding metals demonstrate similar morphology (Fig. 5d and f) with their parent metals. In all cases, the most reactive metal is preferentially etched and concomitantly forms CPs. The PXRD of the 3 metals shows similar pattern (Fig. 5g) indicating that they are isostructural. The peaks in In-based CP, however, expressed significant broadening. PXRD of the alloys for NiAl and GaInSn matches that of pure Al and Ga, confirming that the metal with the lowest E° is preferentially etched (Fig. S9†). InBiSn, however, showed a different pattern due to high contents of Sn and Bi incorporated into the CP (Table S3†). We hypothesize that the proximity of their atomic size and E° leads to statistical polymerization of all three species, which could be utilized as a doping strategy. FTIR spectra of the crystals showed both hydroxyl and acetate ligands are present in all cases (Fig. S10 and S11†). Focusing on the hydroxyl groups, we observed a decreasing trend in the peak wavenumber from Al to In (Fig. 5h). Interestingly, force constant, k of the O–H bonds—calculated using the peak wavenumber, ν and Hookes's Law,34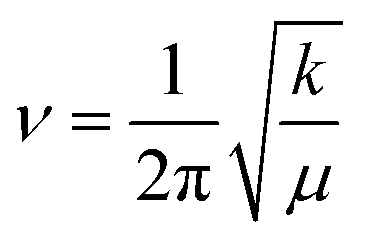 where μ is the reduced mass—was observed to decrease linearly with higher standard reduction potential of the metals (Fig. 5i). The stronger metal–ligand interaction in Al (lowest reduction potential, thus higher force constant) could increase the reorganization energy of the ligands required for self-assembly, which restricts its crystallization to smaller sheets. Conversely, the weaker interactions in In readily reorganizes to form larger precipitates. Weak interactions coupled with higher degrees of freedom, however, promotes the development of defects in the crystal, which explains the broader diffraction peaks observed in In-based CP (Fig. 5g).
where μ is the reduced mass—was observed to decrease linearly with higher standard reduction potential of the metals (Fig. 5i). The stronger metal–ligand interaction in Al (lowest reduction potential, thus higher force constant) could increase the reorganization energy of the ligands required for self-assembly, which restricts its crystallization to smaller sheets. Conversely, the weaker interactions in In readily reorganizes to form larger precipitates. Weak interactions coupled with higher degrees of freedom, however, promotes the development of defects in the crystal, which explains the broader diffraction peaks observed in In-based CP (Fig. 5g).
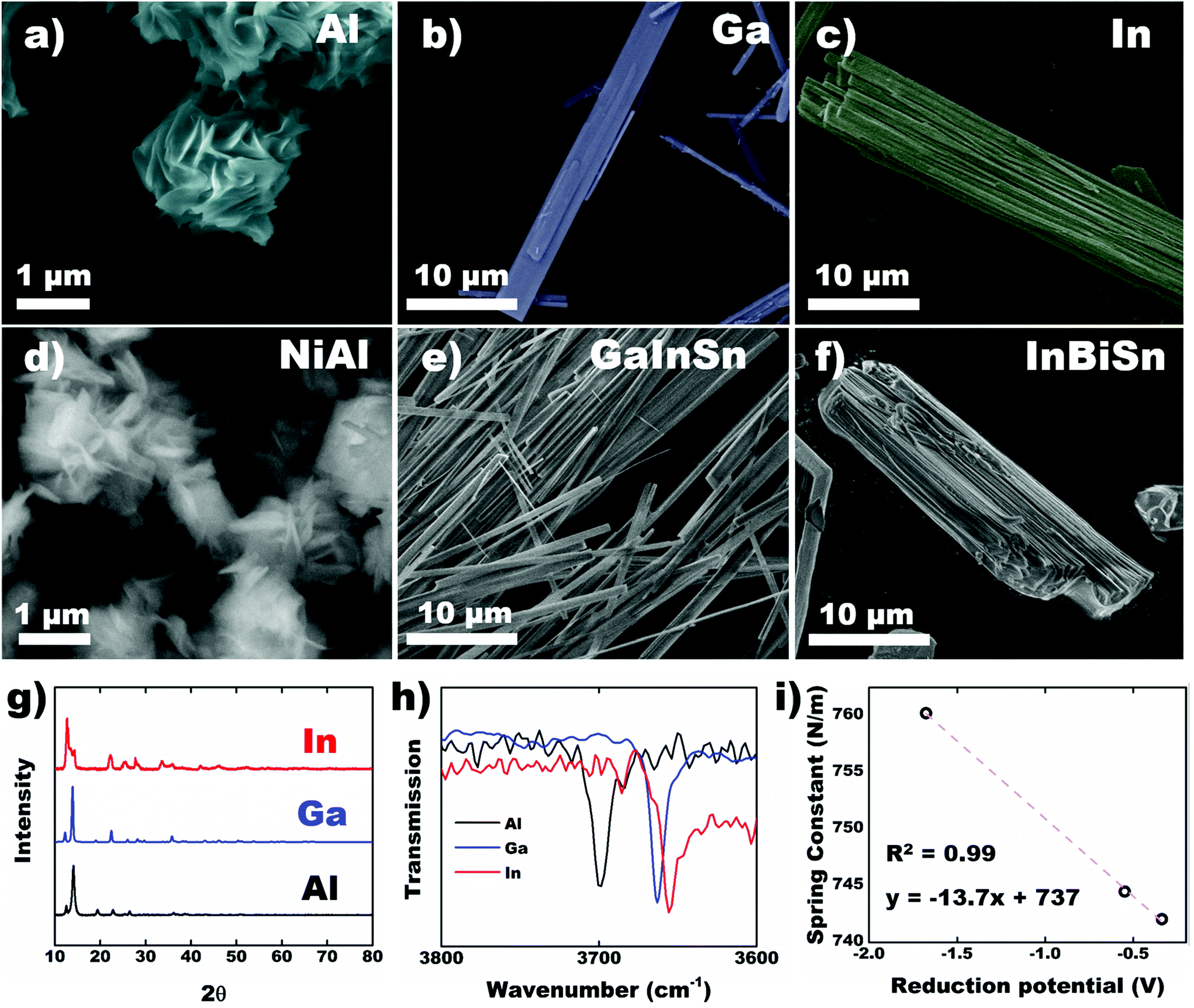 | ||
| Fig. 5 Coordination polymers (CPs) synthesized across (a–c) Group 13 metals and (d–f) their associated alloys. (a–f) SEM images, (g) PXRD patterns and (h) FTIR spectra of the CPs with different metal centers (as shown in a–c), focusing on O–H bond stretch. (i) Calculated force constant of O–H bond based on their peak wavenumbers as a function of reduction potential (Al ≫ Ga > In). | ||
Effect of the ligand structure
Using EGaIn metal (Ga ions), analogs of acetic acid such as formic and propionic acid was used to synthesize CPs. Formic acid demonstrated a variety of structures such as large brick-like blocks, beam-like rods and smaller aggregated particles (Fig. 6a). We attribute the variability of this structure to the highly electron poor carbon of the formate group, which likely interacts with neighboring chains in different directions. Propionic acid forms significantly larger crystals with low aspect ratio (Fig. 6b). Analogous to self-assembled monolayers (SAM), longer chain lengths improve secondary interactions between chains and promote crystal growth.35 The PXRD pattern shows that the dominant diffraction peak shifts to lower angles with increasing chain length (Fig. 6c), which signals an increasing interplanar distance. 71Ga static SSNMR spectra (Fig. 6g and h) verifies that the Ga coordination environment in both coordination polymers is likely distorted 6-coordinate octahedral structure. Analytical simulations of the 71Ga NMR spectrum of the formic acid CP confirm that a small amount of a secondary Ga site is present in the material, possibly due to the presence of a secondary phase or impurity (Fig. 6a). This observation is consistent with the SEM image which shows a minor amount of secondary phase with a distinct crystal morphology. Details of the 71Ga NMR parameters are provided in the ESI (Table S2, Fig. S12†). Collating information from mass balance in TGA (Fig. 6f), SSNMR spectroscopy (Fig. 6g and h) and FTIR (Fig. 6i), CPs precipitated from all three ligands result in a similar chemical environment (with slight deviations in the case of formic acid), giving rise to the general formula Ga(μ-OH)2(μ-O2CX), with X being a proton, methyl or ethyl group for formic acid, acetic acid and propionic acid, respectively (Fig. 6d and e).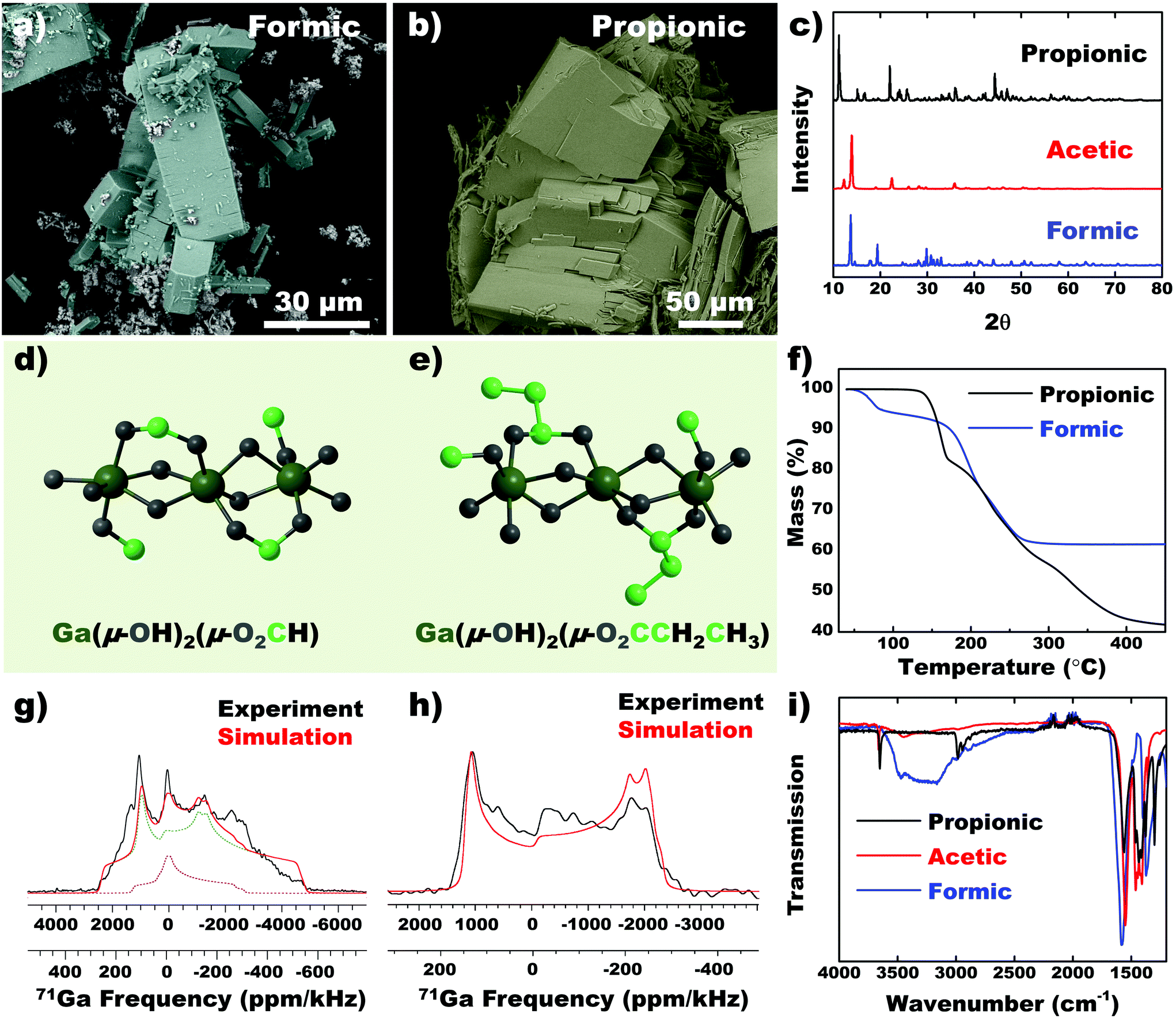 | ||
| Fig. 6 Investigating effect of conjugate acid–base pairs on CP structures. Ga-Based CPs synthesized from formic and propionic acid. (a–b) SEM images, (c) PXRD patterns of CPs derived from different ligands, (d–e) predicted chemical structure of the CPs, and their corresponding (f) TGA. Static 71Ga solid-state NMR spectra with analytical simulations (red trace) overlaid on the experimental spectra (black traces) of CP derived from (g) formic and (h) propionic acid. (i) FTIR of CPs derived from different ligands. | ||
Conclusion
We demonstrate the heterogeneous in situ metal/ligand reaction (HetMet) as a general method for ambient, water-based approach to synthesizing 1D coordination polymer. This process exploits the etching of the passivating oxide layer of metal particles to maintain steady state metal ion concentration in a continuous process. The facile nature and adaptability of the reported method extends paradigms or tools in the synthesis of coordination polymers and nanostructured materials. This work further addresses the following:(i) Quantitative reaction: HetMet achieves complete conversion of the metal precursors to coordination polymers, analogous to ‘living’ polymerization.
(ii) Controlling ligand stoichiometry: The ligand stoichiometry can be tuned by changing acid concentration and/or number of acidic units per molecule (e.g. using dibasic conjugates).
(iii) Controlling metal center: Different metals in group 13 can be used to grow CPs. In general, larger atoms (down the group) result in larger precipitates due to lower reorganization energy.
(iv) Controlling ligand structure: Shorter chain carboxylic acids demonstrate non-uniform crystals, due to absence of preferred growth direction. Longer chain length acids results in larger precipitates due to improved secondary interactions between chains.
Methods
Synthesis of nanobeams
The SLICE method,25 which involves breaking liquid metal droplets into nano and micron size particles under the effect of shear with concomitant formation of gallium oxide-acetate shell,20,36–38 was applied to form EGaIn core shell particles. As a general procedure, 2 g of EGaIn was placed into a 50 ml solution of acetic acid solution (5% v/v, pH = 3.8). The solution was then sheared in a blender (Waring 7010G) for 20 minutes, yielding EGaIn core shell particles. The contents were stored in a glass jar and changes in its appearance was recorded. After several weeks, the precipitates (nanobeams) are purified by ethanol and/or acetone washes through centrifugation and filtration. Similar procedure was used for all other forms of coordination polymers except solid metal particles were used.Instruments
Scanning electron micrographs were obtained using Zeiss Supra55VP field emission SEM and FEI Helios NanoLab G3 UC under immersion mode. X-ray photoelectron spectroscopy was performed using Thermo Scientific K-Alpha XPS. Infrared spectroscopy was performed using PerkinElmer Frontier FT-MIR spectrometer in attenuated total reflectance (ATR) mode. The Rigaku Smartlab X-ray diffractometer was used to perform all powder wide angle X-ray diffraction experiments with Cu K-alpha radiation. Small angle X-ray scattering (SAXS) was performed in vacuo using a Xenocs Xeuss 2.0 set at 2.5 m camera length and Cu K-alpha radiation. Thermogravimetric analysis coupled with differential scanning calorimetry (TGA-DSC) was performed using Netzsch STA449 F1 at 20 °C min−1, using argon as the purge gas. UV-vis spectroscopy was performed using an Agilent 8453 Diode Array UV-Vis, using KCl salt pellets as the substrate.TGA-IR-MS-DSC
Thermogravimetric analysis (TGA) coupled differential scanning calorimetry (DSC) was performed using Netzsch STA449 F1 at 20 °C min−1, using argon as the purge gas. The gases evolved were analyzed by Fourier transform infrared spectroscopy using Bruker Tensor 37 (liquid nitrogen cooled) and mass spectroscopy using Netzsch QMS 403D. Samples were heated from 40 °C to 800 °C, a blank run was performed and subtracted from the sample run to compensate for thermally induced mass drift.Solid-state NMR spectroscopy
All solid-state NMR (SSNMR) experiments were performed on a Bruker Avance III 9.4 T (400 MHz) widebore NMR spectrometer equipped with a broadband fast MAS 1.3 mm double resonance probe or a static 4 mm double resonance probe. Additional details are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Iowa State University through startup funds and through a Black and Veatch faculty fellowship to MT. BT, AV and AJR were supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, Materials Science and Engineering Division. The Ames Laboratory is operated for the U.S. DOE by Iowa State University under contract # DE-AC02-07CH11358. AJR thanks Iowa State University and the Ames Laboratory (Royalty Account) for additional support. JC was supported by Catron Fellowship from the Catron Solar Energy Center. The authors would like to thank Lucas Newcomb for ICP-AAS measurements, Michelle Foster for initial AFM images, Giovanni Rodriguez for technical assistant, R. Sam Houk and Katherine-Jo Galayda for their help with ICP-MS measurements and Eric Cochran for SAXS measurements.References
- M. E. Davis, Ordered porous materials for emerging applications, Nature, 2002, 417, 813–821 CrossRef CAS.
- S. Yan, L. Wan, Z. Li, Y. Zhou and Z. Zou, Synthesis of a mesoporous single crystal Ga2O3 nanoplate with improved photoluminescence and high sensitivity in detecting CO, Chem. Commun., 2010, 46, 6388–6390 RSC.
- B. Chang, L. Zhong and M. Akinc, Low cost composites for vacuum insulation core material, Vacuum, 2016, 131, 120–126 CrossRef CAS.
- A. K. Geim and I. V. Grigorieva, van der Waals heterostructures, Nature, 2013, 499, 419–425 CrossRef CAS.
- J. Deng, D. Deng and X. Bao, Robust Catalysis on 2D Materials Encapsulating Metals: Concept, Application, and Perspective, Adv. Mater., 2017, 29, 1606967 CrossRef.
- T. Low, et al. Polaritons in layered two-dimensional materials, Nat. Mater., 2017, 16, 182–194 CrossRef CAS.
- M. Ebrahimi and F. Rosei, Materials science Organic analogues of graphene, Nature, 2017, 542, 423–424 CrossRef CAS.
- A. Molle, et al. Buckled two-dimensional Xene sheets, Nat. Mater., 2017, 16, 163–169 CrossRef CAS.
- S. C. Junggeburth, et al. Ultrathin 2D Coordination Polymer Nanosheets by Surfactant-Mediated Synthesis, J. Am. Chem. Soc., 2013, 135, 6157–6164 CrossRef CAS.
- H. Maeda, R. Sakamoto and H. Nishihara, Coordination Programming of Two-Dimensional Metal Complex Frameworks, Langmuir, 2016, 32, 2527–2538 CrossRef CAS.
- Y. Peng, Y. Li, Y. Ban and W. Yang, Two-Dimensional Metal-Organic Framework Nanosheets for Membrane-Based Gas Separation, Angew. Chem., Int. Ed., 2017, 56, 9757–9761 CrossRef CAS.
- F. Wang, et al. Facile synthesis of a Ag(i)-doped coordination polymer with enhanced catalytic performance in the photodegradation of azo dyes in water, J. Mater. Chem. A, 2015, 3, 5908–5916 RSC.
- K. Liu, et al. Room-Temperature Synthesis of Multi-Morphological Coordination Polymer and Tunable White-Light Emission, Cryst. Growth Des., 2010, 10, 16–19 CrossRef CAS.
- D. Zhao, et al. Surface Functionalization of Porous Coordination Nanocages Via Click Chemistry and Their Application in Drug Delivery, Adv. Mater., 2010, 23, 90–93 CrossRef PubMed.
- Y. Sun and H.-C. Zhou, Recent Progress in the Synthesis of Metal–Organic Frameworks, Sci. Technol. Adv. Mater., 2015, 16, 054202 CrossRef.
- W. L. Leong and J. J. Vittal, One-Dimensional Coordination Polymers: Complexity and Diversity in Structures, Properties, and Applications, Chem. Rev., 2011, 111, 688–764 CrossRef CAS.
- S. Ghosh, A. Mondal, M. S. R. N. Kiran, U. Ramamurty and C. M. Reddy, The Role of Weak Interactions in the Phase Transition and Distinct Mechanical Behavior of Two Structurally Similar Caffeine Co-crystal Polymorphs Studied by Nanoindentation, Cryst. Growth Des., 2013, 13, 4435–4441 CrossRef CAS.
- A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Infinite coordination polymer nano- and microparticle structures, Chem. Soc. Rev., 2009, 38, 1218–1227 RSC.
- M. Klimakow, P. Klobes, A. F. Thünemann, K. Rademann and F. Emmerling, Mechanochemical Synthesis of Metal–Organic Frameworks: A Fast and Facile Approach toward Quantitative Yields and High Specific Surface Areas, Chem. Mater., 2010, 22, 5216–5221 CrossRef CAS.
- L. Cademartiri, et al. Electrical Resistance of AgTS-S(CH2)n-1CH3//Ga2O3/EGaIn Tunneling Junctions, J. Phys. Chem. C, 2012, 116, 10848–10860 CrossRef CAS.
- R. N. Sodhi, P. Brodersen, L. Cademartiri, M. M. Thuo and C. A. Nijhuis, Surface and Buried Interface Layer Studies on Challenging Structures as Studied by ARXPS, Surf. Interface Anal., 2017, 49, 1309–1315 CrossRef CAS.
- J. Cutinho, et al. Autonomous Thermal-Oxidative Composition Inversion and Texture Tuning of Liquid Metal Surfaces, ACS Nano, 2018, 12, 4744–4753 CrossRef CAS PubMed.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Hybrid nanorod-polymer solar cells, Science, 2002, 295, 2425–2427 CrossRef CAS.
- S. Feng and L. Guanghua, in Modern Inorganic Synthetic Chemistry, ed. R. Xu, W. Pang and Q. Huo, Elsevier, 2011, pp. 63–95 Search PubMed.
- I. D. Tevis, L. B. Newcomb and M. Thuo, Synthesis of Liquid Core-Shell Particles and Solid Patchy Multicomponent Particles by Shearing Liquids Into Complex Particles (SLICE), Langmuir, 2014, 30, 14308–14313 CrossRef CAS.
- C. F. Baes and R. E. Mesmer, Hydrolysis of cations, Wiley, 1976 Search PubMed.
- Z. L. Mensinger, L. N. Zakharov and D. W. Johnson, Synthesis and Crystallization of Infinite Indium and Gallium Acetate 1D Chain Structures and Concomitant Ethyl Acetate Hydrolysis, Inorg. Chem., 2009, 48, 3505–3507 CrossRef CAS PubMed.
- M. Clausen, L. O. Ohman, J. D. Kubicki and P. Persson, Characterisation of gallium(III)-acetate complexes in aqueous solution: A potentiometric, EXAFS, IR and molecular orbital modelling study, J. Chem. Soc., Dalton Trans., 2002, 2559–2564 RSC.
- K. D. Dobson and A. J. McQuillan, In situ infrared spectroscopic analysis of the adsorption of aromatic carboxylic acids to TiO2, ZrO2, Al2O3, and Ta2O5 from aqueous solutions, Spectrochim. Acta, Part A, 2000, 56, 557–565 CrossRef CAS.
- J.-J. Max and C. Chapados, Isotope effects in liquid water by infrared spectroscopy, J. Chem. Phys., 2002, 116, 4626–4642 CrossRef CAS.
- A. J. Rossini, M. P. Hanrahan and M. Thuo, Rapid acquisition of wideline MAS solid-state NMR spectra with fast MAS, proton detection, and dipolar HMQC pulse sequences, Phys. Chem. Chem. Phys., 2016, 18, 25284–25295 RSC.
- Z. Gan, J. P. Amoureux and J. Trébosc, Proton-detected 14N MAS NMR using homonuclear decoupled rotary resonance, Chem. Phys. Lett., 2007, 435, 163–169 CrossRef CAS.
- T. Brijith, B. S. Chang, M. M. Thuo and A. J. Rossini, Solid-State NMR Assisted Structure Determination of Coordination Polymers In preparation, 2019 Search PubMed.
- R. Chang, in General chemistry: the essential concepts, McGraw-Hill, Boston, 5th edn, 2007 Search PubMed.
- J. Chen, B. Chang, S. Oyola-Reynoso, Z. Wang and M. Thuo, Quantifying Gauche Defects and Phase Evolution in Self-Assembled Monolayers through Sessile Drops, ACS Omega, 2017, 2, 2072–2084 CrossRef CAS.
- M. D. Dickey, Emerging Applications of Liquid Metals Featuring Surface Oxides, ACS Appl. Mater. Interfaces, 2014, 6, 18369–18379 CrossRef CAS.
- M. D. Dickey, et al. Eutectic Gallium-Indium (EGaIn): A Liquid Metal Alloy for the Formation of Stable Structures in Microchannels at Room Temperature, Adv. Funct. Mater., 2008, 18, 1097–1104 CrossRef CAS.
- W. F. Reus, M. M. Thuo, N. D. Shapiro, C. A. Nijhuis and G. M. Whitesides, The SAM, Not the Electrodes, Dominates Charge Transport in Metal-Monolayer//Ga2O3/Gallium-Indium Eutectic Junctions, ACS Nano, 2012, 6, 4806–4822 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr05448k |
| This journal is © The Royal Society of Chemistry 2019 |