
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoreversible magnetic nanochains†
Jiří
Mikšátko
a,
David
Aurélio
b,
Petr
Kovaříček
 a,
Magdalena
Michlová
a,
Miroslav
Veverka
b,
Michaela
Fridrichová
a,
Irena
Matulková
c,
Martin
Žáček
b,
Martin
Kalbáč
*a and
Jana
Vejpravová
*b
a,
Magdalena
Michlová
a,
Miroslav
Veverka
b,
Michaela
Fridrichová
a,
Irena
Matulková
c,
Martin
Žáček
b,
Martin
Kalbáč
*a and
Jana
Vejpravová
*b
aHeyrovsky Institute of Physical Chemistry of the Czech Academy of Sciences, v.v.i., Dolejškova 2155/3, 182 23 Prague 8, Czech Republic. E-mail: martin.kalbac@jh-inst.cas.cz
bDepartment of Condensed Matter Physics, Faculty of Mathematics and Physics, Charles University, Ke Karlovu 5, 121 16 Prague 2, Czech Republic. E-mail: jana@mag.mff.cuni.cz
cDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, Czech Republic
First published on 16th July 2019
Abstract
The reversible organization of nanomagnets into highly anisotropic assemblies is of considerable interest for many applications, including theragnostic strategies in vivo. The current preparation strategies lead to structures that are not stable without the permanent presence of an applied magnetic field (MF); otherwise, irreversible assemblies are produced with moderate shape anisotropy at nanoscales. Here, we present a new approach based on the thermoreversible Diels–Alder reaction in the presence of an external MF that enables the assembly of single-domain nanomagnets into narrow chains with lengths of several micrometers. The MF-assisted click chemistry approach included (i) the synthesis of nanoparticles through a modified hydrothermal method, (ii) their functionalization via ligand exchange, (iii) the MF-assisted formation of chains, and (iv) the linkage of the nanomagnets in the presence of the magnetic field. Moreover, the chains can be again disassembled at elevated temperatures through a retro-Diels–Alder reaction. We thus demonstrated for the first time that MF-assisted click chemistry is a convenient method for large-scale preparation of highly anisotropic assemblies of nanosized magnets that can be reversibly decomposed by thermal treatment.
Introduction
The organization of nanoscopic magnets into anisotropic structures represents a smart, bottom-up approach for creating highly anisotropic magnets with mesoscopic control over the magnetic anisotropy. Such architectures are of interest for many applications, like drug delivery, bioanalysis, data and energy storage, sensors, and catalysis.1–10 Recently, the magnetic field (MF)-assisted11 self-assembly of magnetic colloids into organized nanostructures like fibers, chains, tubes, and circles12–14 has attracted enormous attention. The studies demonstrated that a nanoscopic and mesoscopic orientational order can be achieved by application of an MF; however, the current strategies and the resulting assemblies do have specific stability aspects, which might be not compatible with some coveted applications.15 The structures formed by chemically non-interacting nanomagnets decompose immediately when the MF is removed and tend to form larger aggregates. This drawback can be overcome by fixing the NPs directly in the MF; several strategies have been reported. For example biocompatible silica-coated clusters of iron oxide NPs were successfully fixed into nanochains and nanobundles with the help of sol–gel proces.16 This strategy was later extended to magnetic NPs coated with plasmonic shells yielding magnetically responsive photonic nanochains.17 A mild chemical solution method using a soft template of trioctylphosphine oxide was used for preparation of one-dimensional Ni/Ni3C and Ni/Ni3S2 core–shell peapod-like chains.18,19 Very recently, Co–Fe–B–P nanochains designed as cost-effective bifunctional electrocatalysts were produced by a facile water-bath synthesis.20In many cases, particularly in biomedical applications, the presence of large aggregates and assemblies strongly reduces biocompatibility due to the difficult transportation of the large objects through the circulatory system.21 Therefore, robust control over the initial formation and decomposition of such assemblies is necessary for further development of therapeutic and diagnostic methodologies in vivo. Beyond the biomedical field, controlled organization of magnetically active carriers may open doors for new strategies in the template-assisted synthesis of nanomaterials or catalysis with spatial selectivity at mesoscale.
In general, magnetic nanoparticles (NPs) are attractive22–27 for the above-mentioned applications due to the remarkable magnetic properties they maintain at scales of a few nanometers. Clever functionalization of the NP surface improves their stability;28 magnetic properties;29 or helps them to mediate their mutual supramolecular interactions,30–35 or to react in the sense of dynamic covalent chemistry.36,37 Moreover, their properties, such as their zeta potential, hydrophobic/-philic character, and ability to interact with other particles, can be tuned by surface modification via ligand exchange.25,26,38,39
In order to achieve control over the organization and decomposition of the nanostructures, we propose the use of a thermoreversible reaction between ligands.40 An example of such reactions is the Diels–Alder [4 + 2] cycloaddition.41,42 Such a well-known reaction between a conjugated diene and dienophile for forming a substituted cyclohexene derivative can be reversible under certain conditions,43–46 which can be further used in the development of self-healing materials.38,47–51
In this study, we report a general strategy for MF-assisted organization of individual magnetic NPs into nanochains linked together by thermoreversible covalent bonds (meaning coupling and subsequent thermally induced decoupling). It is a first ‘proof of concept’ of temperature tunable ‘on–off’ anisotropic magnetic assemblies. First, we functionalized the surface of cobalt ferrite (CoFe2O4) NPs with two complementary Diels–Alder ligands (substituted maleimide and furan; Scheme 1). The mixture of the functionalized NPs was reacted in the presence of an MF, and narrow chains with the length of few hundreds of nanometers were obtained. Furthermore, the thermoreversibility of this particular Diels–Alder reaction enabled us to decompose the chains by means of breakdown into individual NPs. The CoFe2O4 NPs were selected as they are known to be stable against oxidation, thus their magnetic properties do not change under treatment as in case of magnetite, which almost immediately undergoes a topotactic oxidation to maghemite. However, the same chemistry can be easily tranferred to iron oxide NPs functionalized with oleic acid revealed by the same method or decomposition of iron oleates in organic solvents. The remote alignment and connection–disconnection abilities are beneficial for plethora of applications, where the functional mesoscopic structures cannot be directly introduced to the system of interest.
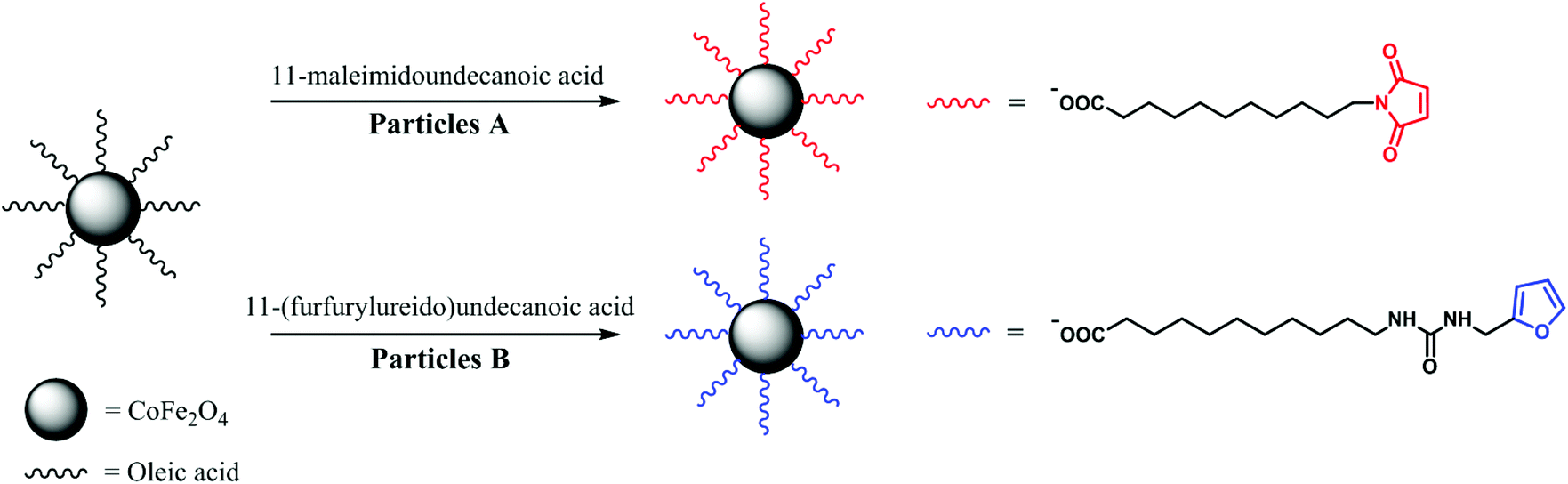 | ||
| Scheme 1 Ligand exchange on the surface of CoFe2O4 oleates. | ||
Results and discussion
Synthesis of oleic acid–coated CoFe2O4
Oleic acid–coated CoFe2O4 NPs were synthesized by hydrothermal decomposition26 of iron(III) nitrate and cobalt(II) nitrate in the presence of sodium oleate directly prepared by the neutralization of oleic acid with NaOH. After a repetitive cleaning procedure consisting of precipitating an n-hexane dispersion of NPs with ethanol, a final centrifugation provided a uniform fraction of CoFe2O4 NPs. The as-prepared NPs formed a stable dispersion in n-hexane or dichloromethane, and a dynamic light scattering (DLS) analysis revealed they had an average hydrodynamic radius of 11.0 ± 0.1 nm. The oleic acid content (21.6 wt%) was determined by a thermogravimetric analysis (TGA), and the mostly sphere-like morphology of the NPs was confirmed by high-resolution transmission electron microscopy (HRTEM; Fig. 1a). The single-phase composition of the samples was confirmed by powder X-ray diffraction (see ESI†). | ||
| Fig. 1 Representative HRTEM images and size histograms of CoFe2O4 NPs functionalized with (a) oleic acid, (b) 11-maleimidoundecanoic acid, and (c) 11-(furfurylureido)undecanoic acid (scale bars = 20 nm). | ||
Ligand exchange on the surface of CoFe2O4
For the Diels–Alder reaction, we chose 11-(furfurylureido)undecanoic acid as a diene and 11-maleimidoundecanoic acid as a dienophile, whose lengths are similar to oleic acid (Scheme 1). While 11-maleimidoundecanoic acid is commercially available, 11-(furfurylureido)undecanoic acid was synthesized by reacting 11-aminoundecanoic acid with an equimolar amount of furfuryl isocyanate in a methanol/acetonitrile mixture and refluxed until the poorly soluble acid dissolved completely. After the isolation, the structure of the product was validated using nuclear magnetic resonance (NMR) spectroscopy, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), and mass spectroscopy (see ESI†).For the ligand exchange, the NPs were dispersed in dichloromethane and separated into two equal fractions (samples A and B). The two ligands, 11-maleimidoundecanoic acid and 11-(furfurylureido)undecanoic acid, were added to samples A and B, respectively, and were allowed to react in a closed vessel for 24 hours. Then, the NPs in both fractions were purified by repeated precipitation with n-hexane followed by magnetic separation. Finally, stable dispersions of both fractions in medium-boiling solvents were prepared. While the A NPs formed a stable dispersion in dichloroethane (DCE), the B NPs were dispersed in a DCE/MeOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixture because of the higher polarity of 11-(furfurylureido)undecanoic acid.
:1, v/v) mixture because of the higher polarity of 11-(furfurylureido)undecanoic acid.
A DLS analysis revealed an average hydrodynamic radii of 12.2 ± 1.8 nm (sample A) and 11.8 ± 0.1 nm (sample B), thus indicating the particles were isolated in both cases. This was further confirmed by the HRTEM analysis (Fig. 1b and c). To demonstrate a successful ligand exchange, a DRIFTS spectroscopy was conducted and evaluated. Typical bands, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1700 cm−1 together with two strong singlets of an out-of-plane bending of a maleimide C–H bond at 825 and 690 cm−1, were found in the spectra of the sample A NPs. Similarly, characteristic N–H stretching around 3300 cm−1, CO stretching bands of the ureido function at 1700 cm−1 and furan's CC ring stretching (1610 and 1590 cm−1) together with three strong singlets between 910 and 700 cm−1 from the C–H out-of-plane deformation of 2-substituted furan are present in the spectra of the sample B NPs. In addition, two typical broad singlets at 600 and 420 cm−1 corresponding to the infrared absorption of spinel-structured CoFe2O4 were present in both cases (see ESI†).52
O stretching at 1700 cm−1 together with two strong singlets of an out-of-plane bending of a maleimide C–H bond at 825 and 690 cm−1, were found in the spectra of the sample A NPs. Similarly, characteristic N–H stretching around 3300 cm−1, CO stretching bands of the ureido function at 1700 cm−1 and furan's CC ring stretching (1610 and 1590 cm−1) together with three strong singlets between 910 and 700 cm−1 from the C–H out-of-plane deformation of 2-substituted furan are present in the spectra of the sample B NPs. In addition, two typical broad singlets at 600 and 420 cm−1 corresponding to the infrared absorption of spinel-structured CoFe2O4 were present in both cases (see ESI†).52
Magnetic properties of the prepared CoFe2O4 nanoparticles
The magnetic properties of the oleic acid-, sample A-, and sample B-functionalized NPs were evaluated. The temperature dependencies of the zero field-cooled (ZFC) and field-cooled (FC) molar susceptibilities are shown in Fig. 2a (more details on its calculation are given in ESI†). All curves are very similar, which suggests negligible disturbance of the magnetic cores by the ligand exchange procedures. The moderate variation of the shape of the ZFC curves, represented as a distribution of the blocking temperature (TB) shown in Fig. 2b can be attributed either to a slight change in the NP size distribution or to a moderate surface anisotropy modification due to the ligand exchange.53 The magnetization isotherms show typical superparamagnetic behavior at 300 K and a loop opening with a coercivity field (Hc) of about 1 T at the blocked state at 10 K (Fig. 2c and d). The results also indicate that the ligand exchange procedure did not significantly affect the magnetic anisotropy, as corroborated by the very similar values of the coercivity field. The slight variation of the absolute magnetization values are more likely attributed to uncertainties in the determination of the organic coating in the NPs. All magnetic parameters are summarized in Table S2.†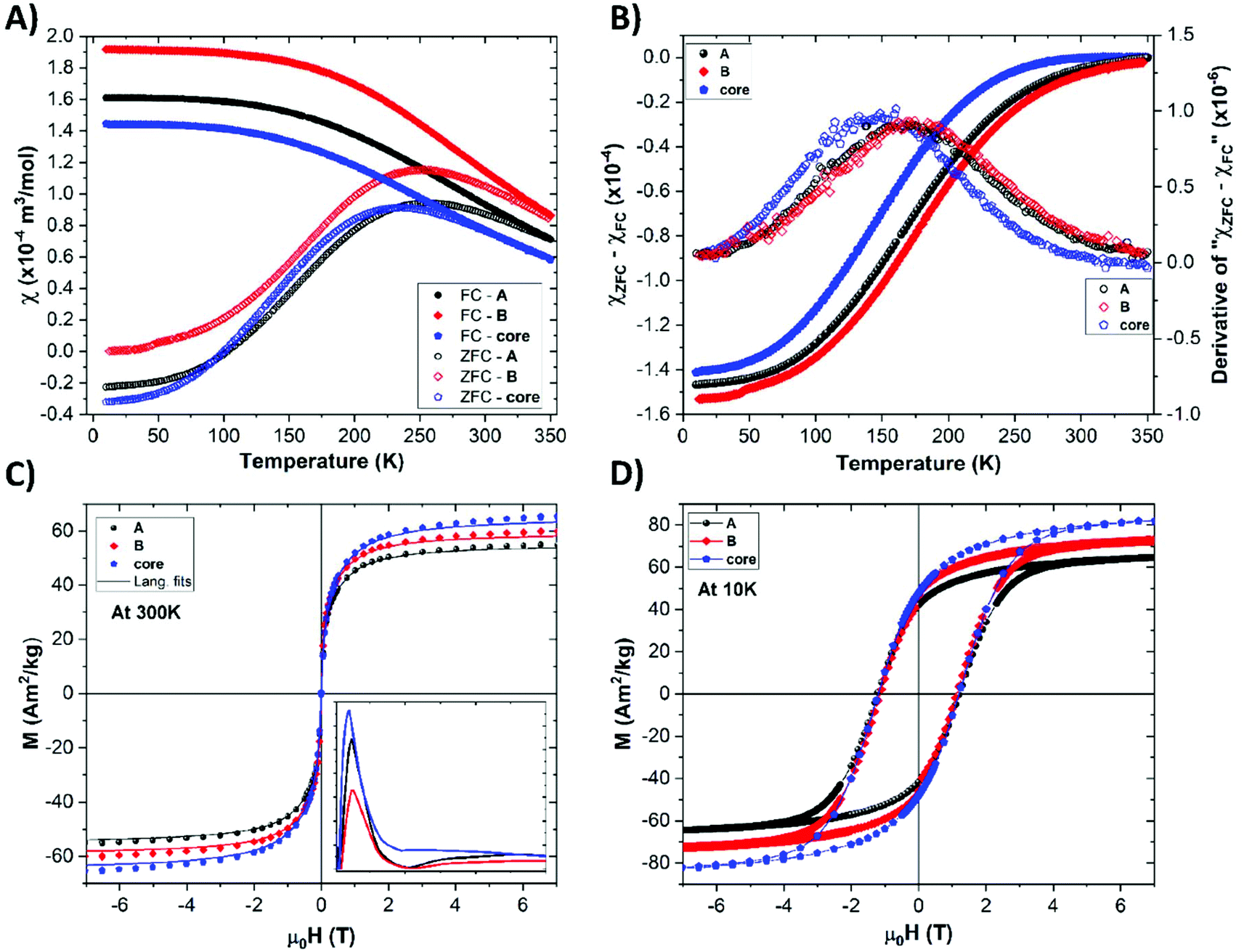 | ||
| Fig. 2 Magnetic measurements of powder samples A and B and their non-functionalized cores. (a) ZFC and FC susceptibility under a field strength of 100 Oe. (b) Determination of the blocking temperature; maximum of the derivative (open scatter lines) of the subtraction of the FC susceptibility (χFC) from the ZFC susceptibility (χZFC), (solid scatter lines). (c) Magnetic isotherm at 300 K showing typical superparamagnetic behavior (magnetic moment distribution in inset). (d) Magnetic isotherm at 10 K showing coercivity fields ≈1.2 T, ≈1.1 T, and ≈1.2 T, for the samples A, B, and oleic acid–only cores, respectively. | ||
The mean magnetic moment per NP (superspin; μm) was evaluated, and the obtained distribution of the magnetic moments extracted from the 300 K isotherms (Fig. 2) is shown in the inset of Fig. 2c. Using the μm values, the so-called magnetic size (dmag) was evaluated. Considering the fact that the cobalt ferrite in the NPs formed a transition between a normal and inverse spinel structure,54 the actual dmag values fall in the interval defined by the normal (∼3.5 nm) and inverse (∼4.7 nm) spinel limits. All values calculated for the two-limit structural arrangements are given in Table S3.† Nevertheless, the magnetic sizes of all types of NPs are very similar, which corroborates the negligible role of ligand exchange in the magnetic response of our nanomagnets.
Diels–Alder reaction without magnetic field
The reversible Diels–Alder reaction between furan and maleimide moieties has been studied thoroughly in the literature.55–58 However, prior to carrying out the click reaction on the A- and B-functionalized NPs, the conversion of this particular Diels–Alder was studied. First, we let the two ligands react together and probed the course of the reaction using 1H NMR spectroscopy. The equimolar ratio of the ligands was dissolved in deuterated chloroform and heated in an NMR tube at 55 °C. The conversion calculated from the time-dependent integral ratios of corresponding signals reached nearly 90% after 7 days of the reaction. The presence of tricyclic product was additionally confirmed by an electrospray ionization mass analysis (ESI) of the reaction mixture, which showed the product signal at m/z 604.5 [M − H]− (ESI, Fig. S18†).Next, we made a click attempt without the presence of an MF. For that purpose, we simply heated an equimolar mixture of the A and B particles (Scheme 2). Because of the multivalency principle59 and the NPs’ enhanced affinity due to the high density of functional groups on their surface, a much shorter reaction time can be expected when assembling these two homomultivalent systems. Therefore, the equimolar mixture was heated at 55 °C for 12 hours. The obtained sample was analyzed by DLS, TEM, and DRIFTS.
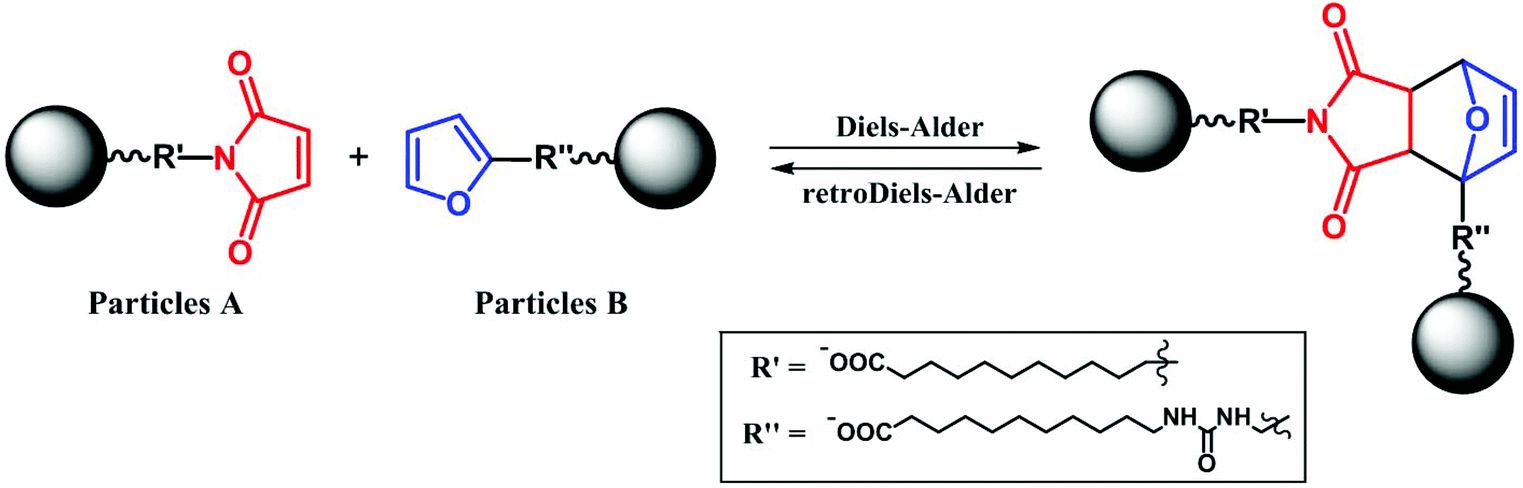 | ||
| Scheme 2 Reaction scheme of Diels–Alder cycloaddition on the surface of CoFe2O4 NPs. | ||
As expected, the first evidence of the formation of larger objects was obtained by measuring the DLS directly after the click reaction. The analyses pointed to the presence of large aggregates with high polydispersity as they did not correspond well within the individual measuring runs. This was supported by an HRTEM analysis, which confirmed the presence of randomly oriented aggregates with an average size of hundreds of nanometers (Fig. 4b). The covalent character of the bonds between the ligands was proved using DRIFTS spectroscopy. The spectra clearly displayed the disappearance of the characteristic bands of the functional groups involved in the reaction. Thus, the three singlets between 910 and 700 cm−1 from the C–H out-of-plane deformation of the furan completely disappeared as well as the singlet at 825 cm−1 from the C–H out-of-plane bending of the maleimide. The bands of the furan's CC ring stretching (1610 and 1590 cm−1) were significantly diminished and slightly shifted as the new double bond was formed. In addition, the presence of hydrogen bonding between the ligands led to the broadening of bands of N–H stretching at around 3300 cm−1 (ESI, Fig. S19†).
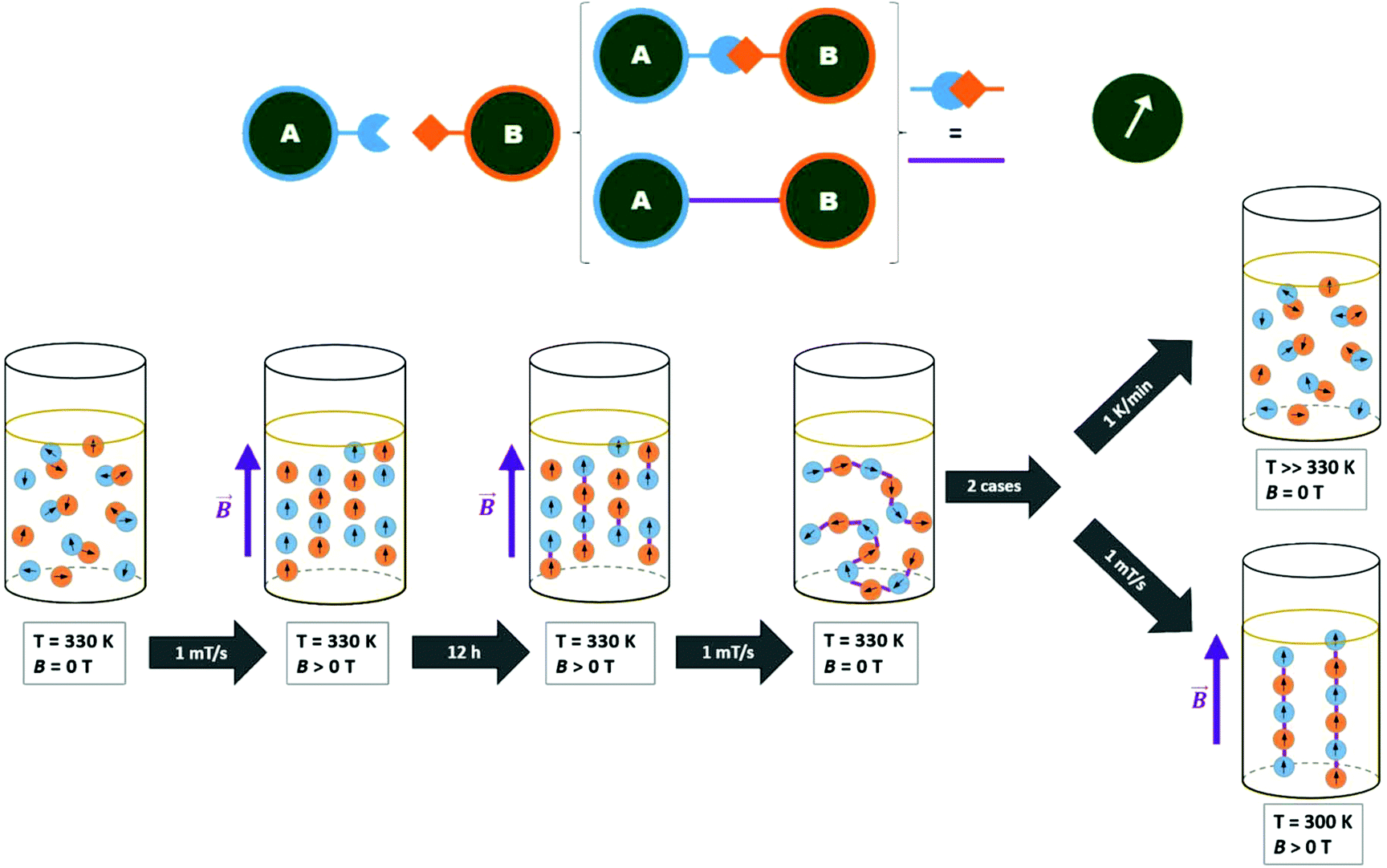 | ||
| Fig. 3 Schematic representation of the magnetic click chemistry procedure: the temperature rises up to the designated value of 330 K with a slow sweep rate of 1 K min−1 and then the external magnetic field is set. The click reactions occur during the wait time under the previous conditions. The chains are stable after bringing the solution back to room temperature and the field switched off. If the chains are heated up the connection is broken, and we return to the initial conditions. Otherwise, when MF is applied, the chains tend to align along the MF lines. | ||
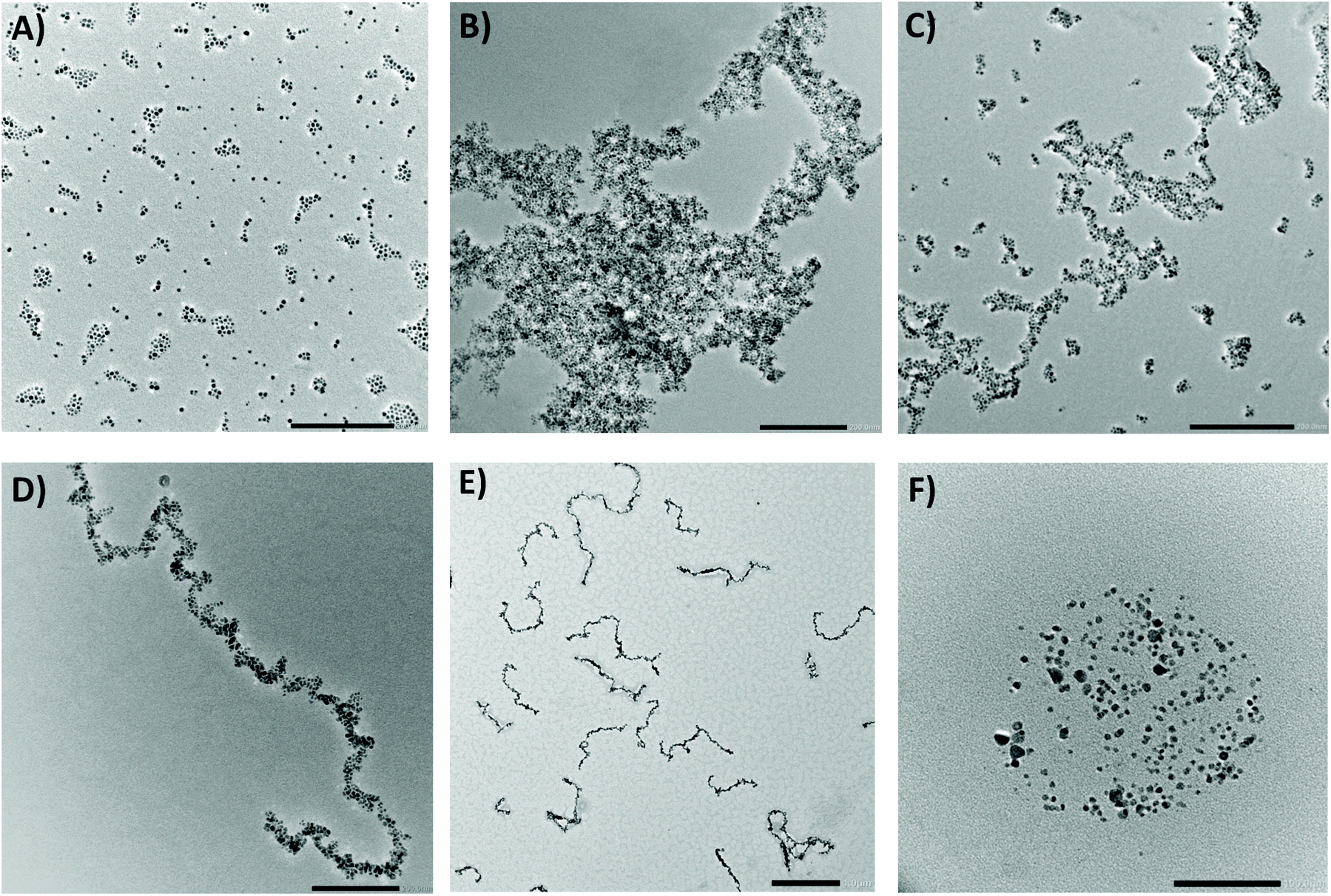 | ||
| Fig. 4 Representative HRTEM images of (a) mixture of samples A and B before the reaction, (b) particles after the reaction without assistance from an MF, (c) mixture of chains and isolated NPs, (d) an organized covalently linked nanochain (all scale bars = 200 nm), (e) large scale image of the narrow chains (scale bar = 1 μm), and (f) isolated NPs after heating the chains at 115 °C (scale bar = 100 nm). | ||
Diels–Alder reaction in the magnetic field
To organize the NPs into anisotropic assemblies, we let a dispersion composed of the A and B samples react inside the cryomagnetic system. An overview of the of the MF-assisted click chemistry process is presented in Fig. 3. The initial reaction sequence was carried out at 1 T at a stable temperature of 60 °C for 12 hours. An HRTEM analysis of the resulting product clearly showed a significant difference compared to the previous case when the NPs reacted without any assistance from an MF (Fig. 4b). Without applying the MF, there was no imprinted shape anisotropy in the formation of covalently bound structures, while under the MF the NPs formed narrow chains. Consequently, heating the sample up to 60 °C caused a Diels–Alder reaction to occur, and thus a covalent linkage between the organized NPs was formed. The resulting chains with lengths from 300 nm to the units of micrometers were then sufficiently stable to withstand the removal of the external MF, allowing us to analyze their morphology (Fig. 4d). The covalent character of the bonds was again confirmed using a DRIFTS analysis that showed the same pattern as in the previous case (ESI, Fig. S20†).We also tried to carry out the reaction at 30 °C. As a result, mostly isolated and thus unreacted NPs were observed in the HRTEM analysis. With a gradual increase of the reaction temperature, we were soon able to observe organized chains, but these were accompanied by a certain number of isolated NPs (Fig. 4c). Therefore, the initial temperature of 60 °C seemed to be appropriate for transforming the isolated NPs into covalently linked chains. A very similar result, as shown in Fig. 4c, was obtained when the reaction time was too short. However, if the strength of the applied MF is 1 T, the reaction time can be shortened to 2 hours. Furthermore, our study showed that the type of the resulting structures is highly concentration-dependent. While at high concentrations (i.e. 0.5 mg mL−1) only large aggregates are formed, the gradual dilution leads to the formation of branched chains (Fig. S22†). Finally, when the nanoparticle concentration is sufficiently low (0.05 mg mL−1), mainly straight chains are present in the solution (Fig. 4d, S23†). A summary of the test preparation protocols for the MF-assisted reaction are given in Table S1 in ESI.†
The chains obtained by the MF-assisted click chemistry are covalently linked; thus, they exhibit very good stability under ambient conditions. An HRTEM analysis of the samples prepared from dispersions stored for several months in tightly closed vials at room temperature provided the same results as the as-prepared samples characterized immediately after the reaction. Finally, the structural and magnetic characterization (Fig. S26, S27 and S31†) did not reveal significant changes in the crystallinity and magnetic size of the NPs in the chains, suggesting a mild nature of the MF-assisted coupling process.60 Inspecting the magnetization isotherm more in detail, a minor modification by means of a paramagnetic-like contribution superimposed on the Langevin curve was identified. This effect is more likely attributed to the internal spin structure reconstruction thanks to the formation of chains. The corrected isotherm was analyzed and increase of the dmag of about 20% was clearly observed (Table S2†). This result corroborates the expectation that formation of chains is associated with moderate change of the internal spin structure and magnetic anisotropy. However, the magnetic properties are still governed by a single-domain response of the individual NPs assembled in the chains. We would like to point out that a robust investigation of anisotropic properties of the chains is impossible without their irreversible fixing in a solid matrix to be able to maintain the chain orientation parallel and perpendicular to the applied MF, respectively. For dense dispersions, the variation will be moderate as the particular geometry of the single-domain NP in oriented dispersion and powders of isolated NPs and in the coupled chains of NPs, respectively, will be very similar (for details, please see Fig. S28–S30†).
In order to decompose the system into individual NPs, the dispersion of covalently linked chains was exposed to an elevated temperature. If we heated the DCE dispersions at 115 °C for 12 hours, the tricyclic product of the Diels–Alder reaction started to decompose, ultimately leading to the recovery of the starting maleimide and furfuryl functions. We were thus able to disconnect the chains and regain the individual NPs (Fig. 4f; for additional HRTEM images, see ESI†).
Conclusion
In conclusion, we have demonstrated for the first time the fully reversible organization of nanomagnets (cobalt ferrite NPs) into narrow chains with lengths up to several micrometers using a thermoreversible Diels–Alder reaction carried out under an MF. The cobalt ferrite NPs were prepared via hydrothermal synthesis, and the oleic acid on their surface was subsequently exchanged with two complementary Diels–Alder ligands. The modified NPs were covalently linked via the Diels–Alder cycloaddition and thus formed stable anisotropic assemblies able to withstand the removal of the external MF. While the MF-unassisted Diels–Alder reaction led to the random formation of large aggregates, the coupling carried out under an MF yielded long nanochains. In addition, the thermoreversibility of the Diels–Alder reaction enabled us to disconnect the nanochains again by heating. The formation of chains is associated with reorganization of the internal spin structure and change of magnetic anisotropy, which brings a possibility of reversible tunning of both parameters in situ directly in the NP dispersions. Our concept can be further generalized to other types of single-domain magnets or quantum magnets. The fully reversible organization of anisotropic nano-objects provides new insight for many applications dealing with magnetically responsive nano- and meso-structures; namely, the revolutionary concept of self-controlled magnetic fluid hyperthermia.Author contributions
J. V., M. K., and P. K. designed the study. M. F. and M. M. prepared and analyzed CoFe2O4 NPs (DLS, TGA, HRTEM). P. K. synthesized the ligands and co-invented the ligand exchange protocol. J. M. co-invented the ligand exchange protocol, characterized the functionalized NPs (DLS, HRTEM), evaluated the DRIFTS spectra, summarized the ESI,† and performed the HRTEM imaging of un-/clicked chains. I. M. measured the DRIFTS spectra. M. V., D. A. measured magnetic properties and performed the D.-A. magnetic click attempts. M. V., D. A. and J. V. analyzed the magnetic measurements. J. V. performed the PXRD and analyzed the data. J. V. and M. K. interpreted the results. J. M. and J. V. co-wrote the paper. M. K., P. K., and D. A. co-edited the manuscript. J. V. attained the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the European Research Council, ERC-Stg-2016 TSuNAMI, project no. 716265. Experiments (magnetic measurements) were performed in MGML (http://www.mgml.eu), which is supported within the program of Czech Research Infrastructures (project no. LM2018096). This work was also supported by the Ministry of Education, Youth and Sports of the Czech Republic and The European Union – European Structural and Investments Funds in the frame of Operational Programme Research Development and Education – project Pro-NanoEnviCz (Project No. CZ.02.1.01/0.0/0.0/16_013/0001821).References
- K. A. López, M. N. Piña, R. Alemany, O. Vögler, F. Barceló and J. Morey, RSC Adv., 2014, 4, 19196–19204 RSC.
- S. Nappini, M. Bonini, F. B. Bombelli, F. Pineider, C. Sangregorio, P. Baglioni and B. Nordèn, Soft Matter, 2011, 7, 1025–1037 RSC.
- N. Wahajuddin and S. Arora, Int. J. Nanomed., 2012, 7, 3445–3471 CrossRef.
- J. Huang, X. Zhong, L. Wang, L. Yang and H. Mao, Theranostics, 2012, 2, 86–102 CrossRef CAS.
- W. Gao and J. Wang, Nanoscale, 2014, 6, 10486–10494 RSC.
- Y. W. Jun, Y. M. Huh, J. S. Choi, J. H. Lee, H. T. Song, S. Kim, S. Yoon, K. S. Kim, J. S. Shin, J. S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- A. Ramazani, M. Almasi-Kashi, E. Golafshan and M. Arefpour, J. Cryst. Growth, 2014, 402, 42–47 CrossRef CAS.
- X. Wang, C. Yan, A. Sumboja and P. S. Lee, Nano Energy, 2014, 3, 119–126 CrossRef CAS.
- S. Senapati, S. K. Srivastava, S. B. Singh and H. N. Mishra, J. Mater. Chem., 2012, 22, 6899–6906 RSC.
- M. Jamal, M. Hasan, A. Mathewson and K. M. Razeeb, Biosens. Bioelectron., 2013, 40, 213–218 CrossRef CAS.
- M. Krajewski, Nanoscale, 2017, 9, 16511–16545 RSC.
- L. Tan, B. Liu, U. Glebe and A. Böker, Langmuir, 2018, 34, 13993–14002 CrossRef CAS.
- L. Ye, T. Pearson, C. Dolbashian, P. Pstrak, A. R. Mohtasebzadeh, B. Fellows, O. T. Mefford and T. M. Crawford, Adv. Funct. Mater., 2016, 26, 3983–3989 CrossRef CAS.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- S. Kralj, T. Potrc, P. Kocbek, S. Marchesan and D. Makovec, Curr. Med. Chem., 2017, 24, 454–469 CrossRef CAS.
- S. Kralj and D. Makovec, ACS Nano, 2015, 9, 9700–9707 CrossRef CAS.
- Y. Hu, L. He and Y. Yin, Angew. Chem., Int. Ed., 2011, 50, 3747–3750 CrossRef CAS.
- W. Zhou, K. Zheng, L. He, R. Wang, L. Guo, C. Chen, X. Han and Z. Zhang, Nano Lett., 2008, 8, 1147–1152 CrossRef CAS.
- W. Zhou, W. Chen, J. Nai, P. Yin, C. Chen and L. Guo, Adv. Funct. Mater., 2010, 20, 3678–3683 CrossRef CAS.
- Z. Wu, D. Nie, M. Song, T. Jiao, G. Fu and X. Liu, Nanoscale, 2019, 11, 7506–7512 RSC.
- A. Hanini, A. Schmitt, K. Kacem, F. Chau, S. Ammar and J. Gavard, Int. J. Nanomed., 2011, 6, 787–794 CAS.
- G. Reiss and A. Hütten, Nat. Mater., 2005, 4, 725–726 CrossRef CAS PubMed.
- B. Zhou, Y. Zhang, C. Liao, C. Yan, L. Chen and S. Wang, Solid State Commun., 2003, 126, 593–596 CrossRef CAS.
- K. Stojak, S. Pal, H. Srikanth, C. Morales, J. Dewdney, T. Weller and J. Wang, Nanotechnology, 2011, 22, 135602 CrossRef CAS.
- S. Nappini, E. Magnano, F. Bondino, I. Píš, A. Barla, E. Fantechi, F. Pineider, C. Sangregorio, L. Vaccari, L. Venturelli and P. Baglioni, J. Phys. Chem. C, 2015, 119, 25529–25541 CrossRef CAS.
- A. Repko, D. Nižňanský and J. Poltierová-Vejpravová, J. Nanopart. Res., 2011, 13, 5021–5031 CrossRef CAS.
- S. Chen, C. Chiang and S. Hsieh, J. Magn. Magn. Mater., 2010, 322, 247–252 CrossRef CAS.
- D. Ling, M. J. Hackett and T. Hyeon, Nano Today, 2014, 9, 457–477 CrossRef CAS.
- I. Bertini, G. Parigi, C. Luchinat and E. Ravera, NMR of Paramagnetic Molecules, Elsevier, 2nd edn, 2017 Search PubMed.
- S. Lin, M. Li, E. Dujardin, C. Girard and S. Mann, Adv. Mater., 2005, 17, 2553–2559 CrossRef CAS.
- S. Si, A. Kotal and T. K. Mandal, J. Phys. Chem. C, 2007, 111, 1248–1255 CrossRef CAS.
- L. Chen, B. Su and L. Jiang, Chem. Soc. Rev., 2019, 48, 8–21 RSC.
- L. Wang, Y. Zhu, L. Xu, W. Chen, H. Kuang, L. Liu, A. Agarwal, C. Xu and N. A. Kotov, Angew. Chem., Int. Ed., 2010, 49, 5472–5475 CrossRef CAS.
- L. Zhu, D. Xue and Z. Wang, Langmuir, 2008, 24, 11385–11389 CrossRef CAS.
- D. Tan, S. Tu, Y. Yang, S. Patskovsky, D. Rioux and M. Meunier, J. Phys. Chem. C, 2016, 120, 21790–21796 CrossRef CAS.
- S. Borsley and E. R. Kay, Chem. Commun., 2016, 52, 9117–9120 RSC.
- E. R. Kay, Chem. – Eur. J., 2016, 22, 10706–10716 CrossRef CAS.
- S. Schäfer and G. Kickelbick, ACS Appl. Nano Mater., 2018, 1, 2640–2652 CrossRef.
- K. Gharbi, F. Salles, P. Mathieu, C. Amiens, V. Collière, Y. Coppel, K. Philippot, L. Fontaine, V. Montembault, L. S. Smiri and D. Ciuculescu-Pradines, New J. Chem., 2017, 41, 11898–11905 RSC.
- J. Zhu, A. J. Kell and M. S. Workentin, Org. Lett., 2006, 8, 4993–4996 CrossRef CAS.
- M. C. Kloetzel, in Organic Reactions, American Cancer Society, 2011, pp. 1–59 Search PubMed.
- H. L. Holmes, in Organic Reactions, American Cancer Society, 2011, pp. 60–173 Search PubMed.
- B. Rickborn, in Organic Reactions, American Cancer Society, 2004, pp. 1–393 Search PubMed.
- B. Rickborn, in Organic Reactions, American Cancer Society, 2004, pp. 223–629 Search PubMed.
- A. Ichihara, Synthesis, 1987, 207–222 CrossRef CAS.
- N. Teramoto, Y. Arai and M. Shibata, Carbohydr. Polym., 2006, 64, 78–84 CrossRef CAS.
- T. T. T. N'Guyen, H. T. T. Duong, J. Basuki, V. Montembault, S. Pascual, C. Guibert, J. Fresnais, C. Boyer, M. R. Whittaker, T. P. Davis and L. Fontaine, Angew. Chem., Int. Ed., 2013, 52, 14152–14156 CrossRef.
- C.-S. Wu, T.-H. Kao, H.-Y. Li and Y.-L. Liu, Compos. Sci. Technol., 2012, 72, 1562–1567 CrossRef CAS.
- T. Engel and G. Kickelbick, Eur. J. Inorg. Chem., 2015, 2015, 1226–1232 CrossRef CAS.
- T. Engel and G. Kickelbick, Chem. Mater., 2013, 25, 149–157 CrossRef CAS.
- J. Li, J. Liang, L. Li, F. Ren, W. Hu, J. Li, S. Qi and Q. Pei, ACS Nano, 2014, 8, 12874–12882 CrossRef CAS.
- T. T. Srinivasan, C. M. Srivastava, N. Venkataramani and M. J. Patni, Bull. Mater. Sci., 1984, 6, 1063–1067 CrossRef CAS.
- B. Pacakova, A. Mantlikova, D. Niznansky, S. Kubickova and J. Vejpravova, J. Phys.: Condens. Matter, 2016, 28, 206004 CrossRef CAS.
- B. Pacakova, S. Kubickova, A. Reznickova, D. Niznansky and J. Vejpravova, Magn. Spinels: Synth., Prop. Appl., 2007 DOI:10.5772/66074.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994–8002 CrossRef CAS.
- R. Göstl and S. Hecht, Angew. Chem., Int. Ed., 2014, 53, 8784–8787 CrossRef.
- V. Froidevaux, M. Borne, E. Laborbe, R. Auvergne, A. Gandini and B. Boutevin, RSC Adv., 2015, 5, 37742–37754 RSC.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS.
- B. Pacakova, S. Kubickova, G. Salas, A. R. Mantlikova, M. Marciello, M. P. Morales, D. Niznansky and J. Vejpravova, Nanoscale, 2017, 9, 5129–5140 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details about the synthesis of ligands and functionalization of NPs; DRIFTS, NMR, DLS, TGA and MS spectra; PXRD of magnetic NPs; magnetic properties of NPs; additional HRTEM images. See DOI: 10.1039/c9nr03531a |
| This journal is © The Royal Society of Chemistry 2019 |