
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis of N-doped porous carbon at room temperature†
Mirian Elizabeth
Casco
 *a,
Sebastian
Kirchhoff
a,
Desirée
Leistenschneider
a,
Marcus
Rauche
b,
Eike
Brunner
b and
Lars
Borchardt
*a
*a,
Sebastian
Kirchhoff
a,
Desirée
Leistenschneider
a,
Marcus
Rauche
b,
Eike
Brunner
b and
Lars
Borchardt
*a
aDepartment of Inorganic Chemistry, Technische Universität Dresden, Bergstrasse 66, 01069, Dresden, Germany. E-mail: lars.borchardt@tu-dresden.de; miriancasco@gmail.com
bDepartment of Bioanalytical Chemistry, Technische Universität Dresden, Bergstrasse 66, 01069, Dresden, Germany
First published on 25th February 2019
Abstract
We report the one-pot mechanochemical synthesis of N-doped porous carbons at room temperature using a planetary ball mill. The fast reaction (5 minutes) between calcium carbide and cyanuric chloride proceeds in absence of any solvent and displays a facile bottom-up strategy that completely avoids typical thermal carbonization steps and directly yields a N-doped porous carbon containing 16 wt% of nitrogen and exhibiting a surface area of 1080 m2 g−1.
Introduction
Tailoring the microstructure and the electrical properties of carbon-based nanomaterials by molecular design is crucial for many advancing applications1,2 in fields such as sensors,3 batteries,4,5 supercapacitors,6 electrocatalysis7,8 or optoelectronics.9 Doping the π-system of an sp2-hybridized carbon framework with nitrogen is a straight-forward strategy to increase wettability10,11 and to enhance catalytic activity (for instance in ORR).12–14 The comparable size of C and N atoms makes their substitution feasible while the stronger electronegativity of the N changes the electronic properties of the carbon network. Pyridinic nitrogen, for instance, induces Lewis basicity at the adjacent carbon atoms13 whereas graphitic nitrogen increases the electron density within the delocalized π-system,15 consequently affecting the electrical conductivity of the graphene network.16,17A growing number of works report N-doping on carbon-based materials by either direct synthesis (e.g. chemical vapour deposition,18,19 solvothermal,20 arc-discharge,21 segregation,22 condensation reaction,2 thermal decomposition,23–25 porous polymer precursor approach26 or post-synthesis approaches (e.g. thermal27,28 or plasma3 treatment). However, most of these syntheses suffer from a high energy penalty, multiple-step protocols and waste accumulation, which increase production costs considerably. A facile synthesis approach towards N-doped carbon-based materials at industrial quantity, while preserving the environment, is fundamental to guarantee the future commercial application of emerging technologies.
In this context, mechanochemical syntheses have attracted much attention as sustainable and cost-effective alternatives to conventional syntheses for a vast number of products ranging from discrete molecules in organic29,30 and inorganic31 chemistry to polymers32,33 and porous materials.34,35 These syntheses rely on mechanical energy, which is provided by the collision of milling balls in ball mills, and which is transferred to the chemical educts. These reactions are fast, scalable and can be conducted in a solvent-free environment, reducing waste accumulation to a minimum.
Porous carbons have already been synthesized by mechanochemistry, among them ordered mesoporous carbons,25,36 and nitrogen-doped carbons.4,37,38 However, a crucial step in all of these syntheses is a subsequent heat temperature treatment to facilitate carbonization and to derive a graphitic conductive structure. The heat treatment renders not only an additional step but also one of the most energetic steps in the synthesis, probably limiting its applicability.
In order to tackle this drawback, self-sustaining, high-temperature synthesis (SHS) has been investigated to instantaneously form carbon materials from highly exothermic solid state reactions by local heating of the mixture (with a hot wire, electric spark, laser, thermal radiation pulse or ion beam).39,40 When the reaction is induced by ball milling rather than a pre-heated solid state mixture, the process is called mechanically-induced self-sustaining reaction (MSR) and is typically performed at room temperature.41–46 Takacs47 has nicely reviewed MSR for several systems. Basically, the process consists on a primary activation period during which size reduction; mixing and inclusion of defect take place. After this time the mixture ignites and the reaction propagates to form the product in a matter of seconds.
Here we report on the synthesis of a nitrogen-doped carbon via MSR at room temperature. We thereby adapt and develop further a recent synthesis concept of our group, the mechanochemical reaction of CaC2 with halogenated hydrocarbons.43 In particular, we use the reaction between CaC2 with cyanuric chloride (C3Cl3N3) (Fig. 1). We show that changing the mass ratio of reactants and the milling time allows us to tailor the textural properties and the nitrogen content of the carbonaceous product, reaching values of surface area as high as 1080 m2 g−1 accompanied with a nitrogen content of 16 wt%.
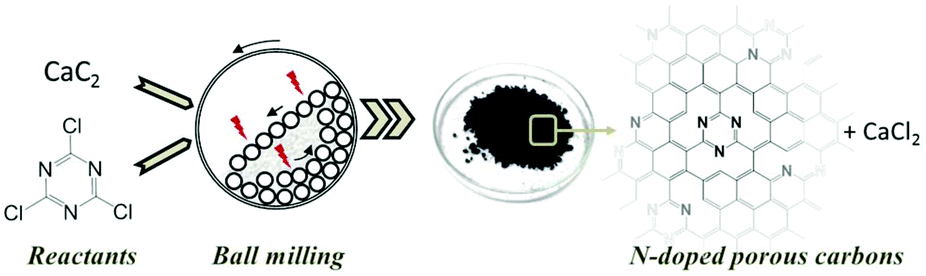 | ||
| Fig. 1 Concept scheme for the mechanochemical reaction between calcium carbide and cyanuric chloride inside a planetary ball mill at room temperature, resulting in N-doped porous carbons. | ||
Materials and methods
Sample preparation
Granular calcium carbide (CaC2 ≤ 75%) was purchased from Sigma Aldrich and ground for 2 h at 500 rpm in a planetary mono mill Pulverisette 6 classic line from Fritsch GmbH. The main impurities of CaC2 are CaO and Ca(OH)2, although traces of S can also be detected (ESI Fig. 1 and 2†). 99% pure cyanuric chloride (C3Cl3N3) was purchased from Sigma Aldrich. The synthesis of the carbonaceous materials was performed by mixing both reactants in a 45 mL zirconium oxide grinding bowl with 22 zirconium oxide balls (d = 10 mm, ∼75.7 g) using a planetary micro mill Pulverisette 7 premium line from Fritsch GmbH at the rotational speed of 500 rpm. The system was prepared under argon atmosphere inside a glove box. We tested four different CaC2/C3Cl3N3 mass ratios, namely 0.5, 0.7, 2.3, 4.6. The evolution of pressure and temperature inside the grinding bowl for all samples was monitored by gas pressure and temperature measurement (GTM) automatic system (45 mL zirconium oxide bowl and 22 zirconium oxide balls), see ESI Fig. 3.† After the ignition was detected with the GTM system, the product was milled for 5 min or 120 min, giving 8 samples in total (see ESI Table 1†). The product was washed first with a HCl solution of 10% v/v and then with hot water overnight using soxhlet system until pH = 7 was reached and impurities were removed.Sample characterization
The nitrogen physisorption isotherms were measured at 77 K using a Quadrasorb EVO/SI apparatus from Quantachrome Instruments. Prior to all measurements, the samples were degassed under vacuum at 150 °C for 12 h. The specific surface area (SSABET) of the N-doped carbons was calculated using the equation from Brunauer–Emmett–Teller (BET) in the range that fits to the consistency criteria proposed by Rouquerol and Llewellyn.48 The total pore volume (Vt) was calculated at a relative pressure of 0.95. The sample composition was measured in the CHNS analyser (Euro EA HEKAtech GmbH). Ash content was determined by thermogravimetric analysis (TGA) on a NETZSCH STA 409 C/CD system using alumina crucibles under air stream with the heating rate of 5 °C min−1. Transmission electron microscopy (TEM) image was observed in a JEOL JEM 1400plus operated at an acceleration voltage of 120 kV. The sample were collected by soaking the TEM copper grid in the already with ethanol well-dispersed carbon suspension. Raman spectra were obtained using a Raman-spectrometer DXR SmartRaman from the company Thermo Scientific. The wavelength for the measurement was 532 nm (100 scans). X-ray photoelectron spectroscopy (XPS, K-alpha, Thermo Scientific) was used to analyse the C, N and O species. All spectra were collected using Al-K radiation (1486.6 eV), monochromatized by a twin crystal monochromator, yielding a focused X-ray spot (elliptical in shape with a major axis length of 400 µm) at 3 mA × 12 kV. The alpha hemispherical analyser was operated in the constant energy mode with survey scan pass energies of 200 eV to measure the whole energy band and 50 eV in a narrow scan to selectively measure the particular elements. XPS data were analysed with Avantage software. Electric powder conductivities were measured with an Agilent 34420A combined with a self-constructed cell with a diameter of 1 cm. The powder was pressed with a mass of 2 t.Results and discussion
Mechanochemical synthesis
In a typical synthesis, the powders of calcium carbide (CaC2) and cyanuric chloride (C3Cl3N3) are placed into a 45 mL zirconia grinding bowl with 22 zirconia milling balls (d = 10 mm, ∼75.7 g). After milling at 500 rpm for a certain time and a consecutive washing step, the final N-doped carbon product is obtained. They were labelled as ‘N-Carb’ following by the CaC2/C3Cl3N3 mass ratio (0.5, 0.7, 2.3 and 4.6). To track the proceeding reaction, we used a vessel with integrated temperature- and pressure-sensors. The evolution of temperature and pressure was recorded for all conditions (Fig. 2) and is characteristic for mechanically-induced self-propagating or self-sustaining reactions.43–47 At the beginning of the experiment, the pressure remains at atmospheric value and the temperature increases gradually only due to the balls friction. During this time (mechanical induction period), the number of structural defects and the particle surface increase. As a result, the reactivity of the system rises significantly. After the induction time, a sudden rise in the pressure and temperature are detected, which is associated with the instantaneous formation of carbon. Macroscopically, a black powder is obtained opening the bowl immediately after ignition.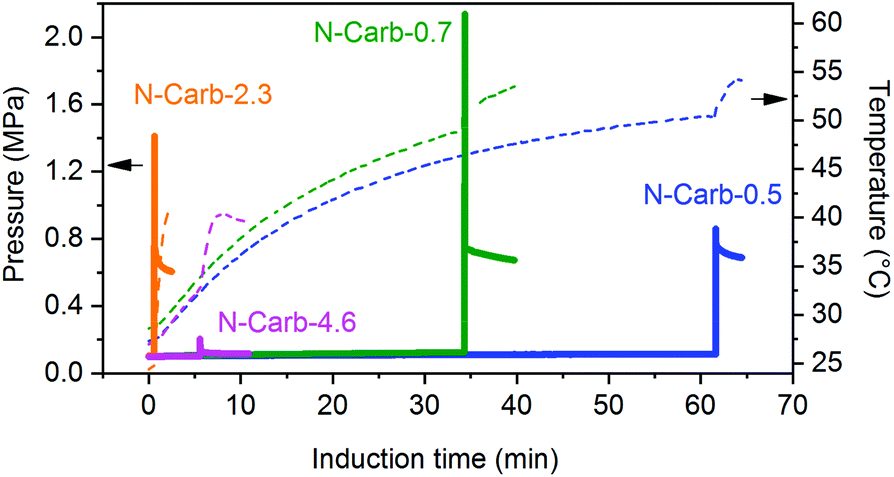 | ||
| Fig. 2 Temperature (dashed line) and pressure (solid line) profiles for the mechanochemical synthesis of N-doped carbons. Reactions were followed using an easy GTM system (Fritsch GmbH, detailed description ESI Fig. 3†). | ||
The reactants quantities are selected according to the following hypothetical eqn (1) (symbol for mechanochemical reactivity is adapted from ref. 49). Please, note that this is not a defined stoichiometry. The experimentally obtained composition can be derived from elemental analysis, which will be discussed later.
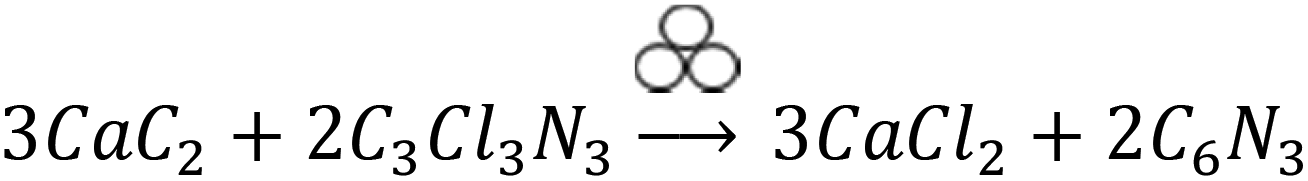 | (1) |
A CaC2/C3Cl3N3 mass ratio of 0.7 is calculated to be the stoichiometric quantity, whereas 0.5 corresponds to an excess of C3Cl3N3 and 2.3 to an excess of CaC2 (the purity of CaC2 75% was taking into account for the calculation). Additionally, we investigated the minimum amount of cyanuric chloride needed to promote the reaction within 1 h, which resulted in a mass ratio of 4.6. Therefore, we can draw two conclusions from comparing the profiles in Fig. 2. Firstly, an excess of CaC2 promotes the mechanochemical reaction, leading to shorter induction times. This is 1 and 5 min for N-Carb-2.3 and N-Carb-4.6, in contrast to 30 and 60 min for sample N-Carb-0.7 and N-Carb-0.5, respectively. In any of these cases, the necessary times are extremely short compared to conventional carbonization methods to prepare activated carbon materials, which usually require around 2 h plus a cooling period. Note that the registered global temperature rises gradually due to the milling process itself (attrition of milling material) and suddenly shows a small spike (not higher than 50 °C) due to the exothermic nature of the reaction (∼−600 kcal). These results demonstrate that, even though the reaction is highly exothermic and provokes a local over-heating, the product can be synthesized at room temperature and in a time scale of minutes. The second conclusion is related to the maximal pressure reached by the system during the ignition. The readers have to be aware that this type of reaction can result in an uncontrolled explosion when large quantities of reactants are used in stoichiometric ratios. The pressure rises abruptly due to the expansion wave followed by the propagation reaction front. The remaining pressure might due to the gases formation during the ignition (e.g. light hydrocarbons, CO, CO2), although the currently system does not allow their extraction for further analysis. Oxygen and hydrogen might come from the impurities of calcium carbide (see ESI, S1† for detailed information). In our experiments, the sample N-Carb-0.7 generated a maximal pressure of 2.3 MPa. However, when using an excess of either calcium carbide or cyanuric chloride, the reaction can be controlled and carried out very safely. We observed maximal pressures of 0.1 MPa, 1.4 MPa and 0.9 MPa for samples N-Carb-4.6, N-Carb-2.3 and N-Carb-0.5, respectively. All samples were milled for 5 and 120 min after the detection of the reaction (the milling time was added to the sample's name in subscript, see Table 1). Longer milling time exerts rather low influence on the yield of the reaction (Table 1), which indicates that most of the product is formed during the ignition. In general, the yield increases when increasing the calcium carbide content in the initial mixture. However, according to elemental analysis (Table 1), this increment is accompanied with a decrease of the nitrogen content. These opposing observations suggest that CaC2 can also react with itself to form carbon. Cyanuric chloride, however, is necessary, since milling of pure CaC2 does not yield any product at all. A proposed mechanism involves the nucleophilic substitution of the anion chloride by the acetylide anion on the triazine ring with the corresponding formation of CaCl2 as a side product. Since triple bond was not found by further structural characterization techniques on the carbon products, the presence of the suggested polymer in eqn (1) can be ruled out. The obtained product is a more stable nitrogen-doped carbon, which structural and textural characteristics will be analyzed along the manuscript. Thermogravimetric analysis shows that the product is stable up to 400 °C in air, with a maximal decomposition rate at around 500–600 °C (see ESI Fig. 4†). The ash content (mainly CaO coming from the calcium carbide impurities) decreases with the milling time (Table 1), since the continuous particle size reduction of the formed carbon allows for a more effective washing process. Traces of S can also be detected in the final carbon by elemental analysis (Table 1).
| Materials | Yielda (%) | SSABETb (m2 g−1) |
V
Total![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (cm3 g−1) c (cm3 g−1) |
Ashd (wt%) | I D/IGe (nm) | Elemental analysisf | XPSg | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | C | H | S | C/N | N | C | O | C/N | ||||||
| (wt%) | (at%) | |||||||||||||
| a Yield (%) = (mfinal × X × MWlimiting reactant × 100)/(mlimiting reactant × 2 × MWC6N3), where X is 3 (or 2) when CaC2 (or C3Cl3N3) is the limiting reactant. MWCaC2 = 64.099 g mol−1. MWC3Cl3N3 = 184.41 g mol−1. MWC6N3 = 114 g mol−1. The yield is calculated according to the hypothetical eqn (1), therefore yields higher than 100% indicates that more acetylene units than expected are reacting. b Multi-point BET-method for 0.05 ≤ P/P0 ≤ 0.3. c Total pore volume at P/P0 = 0.95 (N2 adsorption). d Ash content is the remaining mass of thermogravimetric analysis under air. e I D/IG is the intensities ratio of the D and G peaks. f The remaining elemental composition (wt%) is supposed to be oxygen and Ca. g The remaining composition (at%) is sulfur coming from the CaC2. | ||||||||||||||
| N-Carb-0.55 min | 31 | 410 | 0.55 | 7.2 | 0.94 | 12.0 | 74.7 | 0.2 | 0.3 | 6.2 | — | — | — | — |
| N-Carb-0.5120 min | 41 | 370 | 0.30 | 4.8 | 0.93 | 18.0 | 66.7 | 0.5 | 0.8 | 3.7 | 18.8 | 74.8 | 6.1 | 4.0 |
| N-Carb-0.75 min | 41 | 170 | 0.32 | 7.8 | 0.66 | 4.9 | 83.6 | 0.1 | — | 17 | — | — | — | — |
| N-Carb-0.7120 min | 43 | 280 | 0.20 | 3.8 | 1.01 | 7.0 | 80.3 | — | — | 11.5 | 7.1 | 88.9 | 3.8 | 12.5 |
| N-Carb-2.35 min | 110 | 230 | 0.60 | 6.7 | 0.92 | 1.7 | 81.9 | — | — | 48.2 | — | — | — | — |
| N-Carb-2.3120 min | 98 | 450 | 0.36 | 3.7 | 1.47 | 2.0 | 85.2 | 0.1 | 0.8 | 42.6 | 1.6 | 95.2 | 3.0 | 53.3 |
| N-Carb-4.65 min | 92 | 1080 | 1.00 | 9.5 | 0.75 | 2.2 | 82.7 | 0.2 | 0.7 | 37.6 | — | — | — | — |
| N-Carb-4.6120 min | 110 | 1080 | 1.25 | 2.7 | 1.13 | 16.1 | 60.3 | 2.1 | 2.1 | 3.7 | — | — | — | — |
N-Doped porous carbon
Elemental analysis confirms that nitrogen atoms are incorporated into the carbon network (Table 1). According to the eqn (1), the reaction between calcium carbide and cyanuric chloride might yield a triazine polymer bridged by acetylene units with a theoretical C/N atomic ratio of 2, and a C/N mass ratio of 1.7. However, the experimental results reveal C/N mass ratios between 3.7 and 48, which confirm that the acetylene units react with themselves and the cyanuric chloride is fundamental to start the reaction giving N-doped carbons and CaCl2 as products. A closer look at these values suggests that the nitrogen incorporation process could be divided into two steps: (1) in situ doping during the reaction and, (2) ex situ doping during the subsequent grinding. The in situ doping majorly determines the degree of nitrogen incorporation, which, in principle, is more effective when increasing the amount of cyanuric chloride in the initial mixture. For instance, the nitrogen content is 1.7 wt%, 4.9 wt% and 12 wt% for samples N-Carb-2.35 min, N-Carb-0.75 min and N-Carb-0.55 min, respectively. The ex situ doping, although less effective, still provides further heteroatoms into the carbon network, e.g. the nitrogen content for sample N-Carb-0.55 min is enhanced from 12 wt% up to 18 wt% after 120 min of milling. In the particular case of N-Carb-4.6, in situ doping led to a nitrogen content of 2.2 wt%, that is enhanced up to the value of 16 wt% with the subsequent 120 min of grinding. Please note, that we performed these syntheses several times in order to test the reproducibility of this mechanochemical reaction. As a result, we replicated the product and the nitrogen content, thus confirming the success of the ex situ doping for sample N-Carb-4.6.Structural information about the nitrogen species in the product are obtained by means of X-ray photoelectron spectroscopy (XPS). The survey spectra for samples N-Carb-0.5120 min, N-Carb-0.7120 min and N-Carb-2.3120 min are selected and analyzed (Fig. 3a–d). The intensities of the N 1s spectra decrease with the nitrogen content and the C/N ratios are in close agreement with elemental analysis results (Table 1). The fitting of the N 1s spectrum of N-Carb-0.5120 min reveals three major bands that correspond to pyridinic N (398.8 eV), pyrrolic N (400 eV) and graphitic N (401.1 eV).50 In the cases of N-Carb-0.7120 min and N-Carb-2.3120 min the pyridinic and pyrrolic functionalities dominate in the fitting (Fig. 3c and d). The fitting of the C 1s spectra (see ESI Fig. 5a–c†) reveals two main bands attributed to sp2 graphitic structure at around 284.6 eV and sp3 carbon bond at around 285.5 eV; and two minor bands at around 286 eV and 288.5 eV which can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond and the C–N single bond of the triazine unit, respectively.51,52 Therefore, the XPS results indicate that triazine units are incorporated into the carbon network, and the doping mainly led to pyridinic species (for more detail about N, C and O species percentages refer to ESI Table 2†). TEM image in Fig. 3e shows a flakes-like microstructure for nitrogen-doped carbon obtained in the ball mill, and a proposed scheme for the N-doped graphene layer with integrated triazine units.
N double bond and the C–N single bond of the triazine unit, respectively.51,52 Therefore, the XPS results indicate that triazine units are incorporated into the carbon network, and the doping mainly led to pyridinic species (for more detail about N, C and O species percentages refer to ESI Table 2†). TEM image in Fig. 3e shows a flakes-like microstructure for nitrogen-doped carbon obtained in the ball mill, and a proposed scheme for the N-doped graphene layer with integrated triazine units.
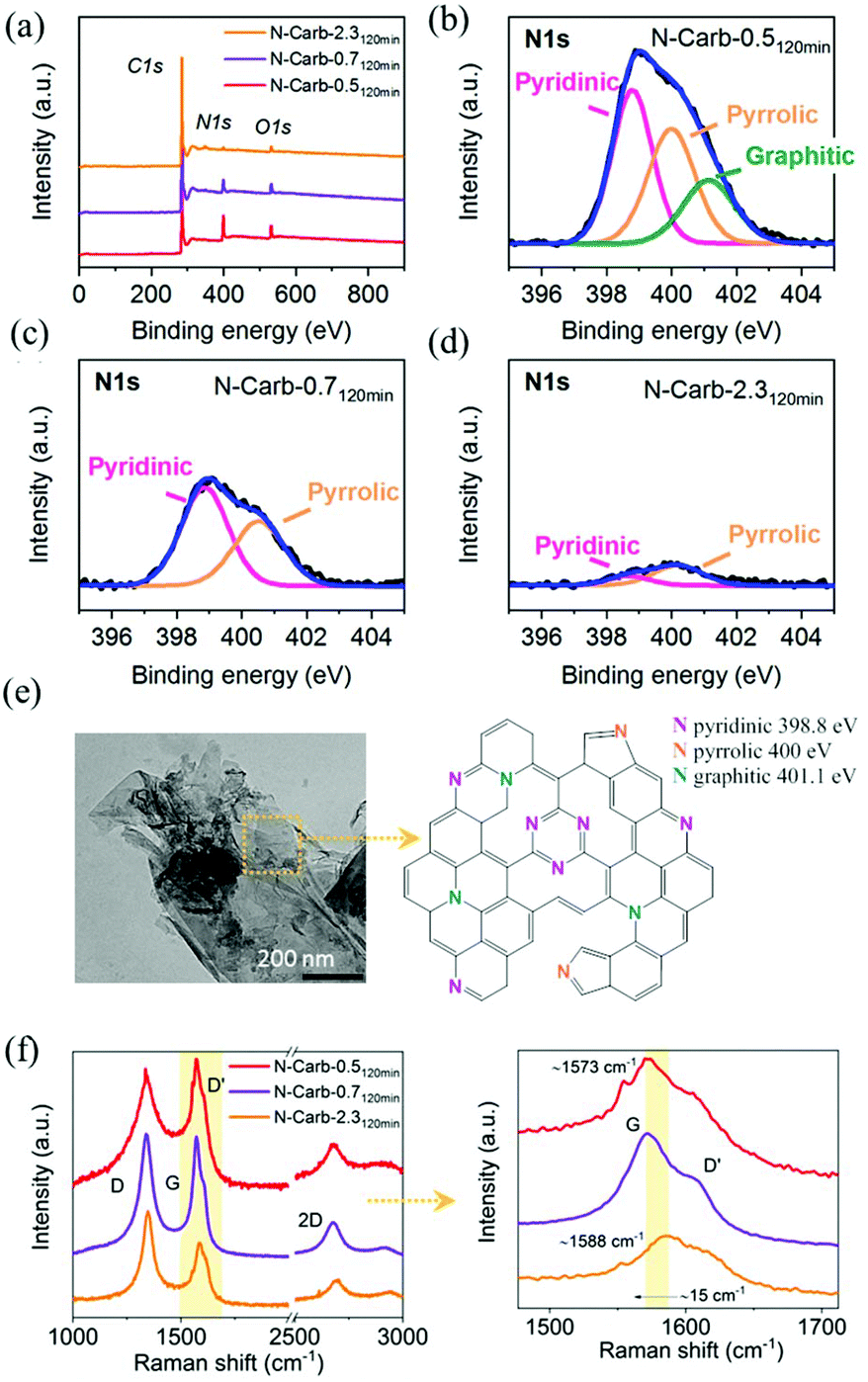 | ||
| Fig. 3 (a) Survey X-ray photoelectron spectra for samples N-Carb-X120 min (X = 0.5, 0.7 and 2.3) obtained in the ball mill. N 1s deconvoluted spectra of (b) N-Carb-0.5120 min, (c) N-Carb-0.7120 min and (d) N-Carb-2.3120 min. (e) Transmission electron microscopy image for sample N-Carb-0.5 and a proposed scheme of a nitrogen-doped graphene layer. (f) Raman spectra for samples N-Carb-X120 min (X = 0.5, 0.7 and 2.3) obtained in the ball mill and a zoom-in on the G band. | ||
Raman spectra were measured to complement the characterization of the carbon structure (Fig. 3f and ESI Fig. 6†). There was no indication of sp hybridized carbon in the Raman spectra and in turn the existence of the polymer proposed in eqn (1). Three peaks characteristic of carbonaceous materials arise at around 1570–1585 cm−1 (G band), 1348 cm−1 (D band), and 2688 cm−1 (2D). The G band corresponds to the optical mode vibration of two neighbouring carbon atoms on a graphene layer. The D band corresponds to the breathing mode of sp2 hybridized carbon in the presence of a defect in the aromatic system. In addition, this defect is also responsible for the small shoulder observed on the G peaks at ∼1610 cm−1 (D′ band).53 The 2D band, which involves a two-phonon process, is a signature for layer thickness and is related to the number of graphene layers.54 This peak is sharper for the carbonaceous materials obtained immediately after the ignition (5 min milling time) and becomes smaller after longer milling times due to the inclusions of defects. In addition, a higher ID/IG intensities ratio is found when comparing same reactant ratio but higher milling time in parallel with a higher degree of disordering in the sp2 hybridized carbon network. For N-doped carbon materials, the substitution of C by N atoms must be accompanied by the introduction of defects into the graphene layer.55 Previous studies from Ferrari and Robertson55 revealed a down-shift of the G peak position with the nitrogen content. Herein, a maximum shift of ∼15 cm−1 is observed in the G band position when comparing N-Carb-0.5120 min (18 wt%N), and N-Carb-2.3120 min (2 wt%N). This observation supports the XPS results that nitrogen is successfully incorporated into the graphene structure.
The textural properties of the N-doped carbons obtained in the ball mill were investigated by means of nitrogen physisorption technique (see ESI Fig. 7† for N2 adsorption isotherms and ESI Fig. 8† for pore size distribution). The surface area is between 170 and 1080 m2 g−1 depending on the mass ratio (Table 1). These surface areas are relatively high considering that neither thermal carbonization nor activation processes were applied. From this, we conclude that mainly the CaC2 and the graphitization of the acetylene units are responsible for the porosity, while the cyanuric chloride, apart from being C and N source, it is necessary to start the reaction. In addition, we observed that further milling does not affect the textural properties of the materials, but increase considerably the nitrogen content, especially for sample N-Carb-4.6. In consequence, all these results strongly point towards a nitrogen-doped carbon rather than a polymer framework. Moreover, X-ray diffraction reveals the typical pattern for turbostratic carbon (Fig. 4).56
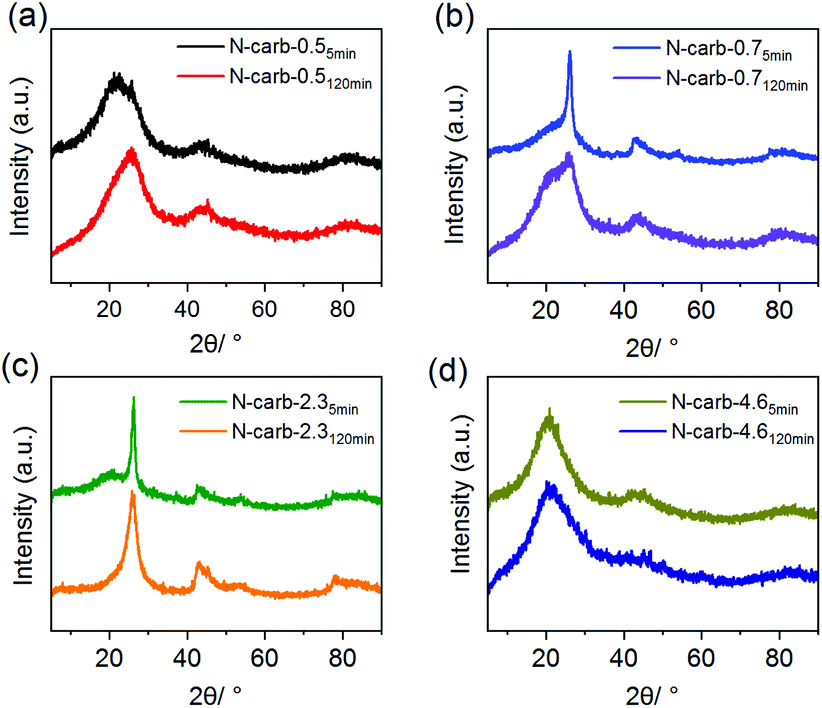 | ||
| Fig. 4 X-ray diffractrograms for the eight N-doped porous carbon materials produced in the ball mill. | ||
We measure the electrical conductivity of the eight samples obtained in the ball mill (Fig. 5). Results reveal a decrease in the conductivity with the milling time. Two factors may explain this trend: the nitrogen content and the surface area. Increasing milling time enhances, at different extent, both of them (Table 1). The high nitrogen content of 16 wt% combined with a surface area of 1080 m2 g−1 give the lowest electrical conductivity for sample N-Carb-4.6120 min (0.5 × 10−4 S cm−1). The increasing trend in conductivity of samples N-Carb-0.5, N-Carb-0.7 and N-Carb-2.3, either comparing 5 or 120 min, is in line with the decrease in the nitrogen content. It was reported in the literature the effect of the nitrogen content on the electrical conductivity is determined by a competition between electron doping and structure disorder.16,57 XPS results demonstrated that mainly pyridinic and pyrrolic species are present in the carbon network, which induce a more defective structure (confirmed by Raman). Nonetheless, the obtained values (Fig. 5) are comparable with that of commercial activated carbons, carbon fibers and carbonized anthracites reported on literature, usually <20 S cm−1.58 These electrical conductivities are quite good considering that mechanical forces are the only external source of energy.
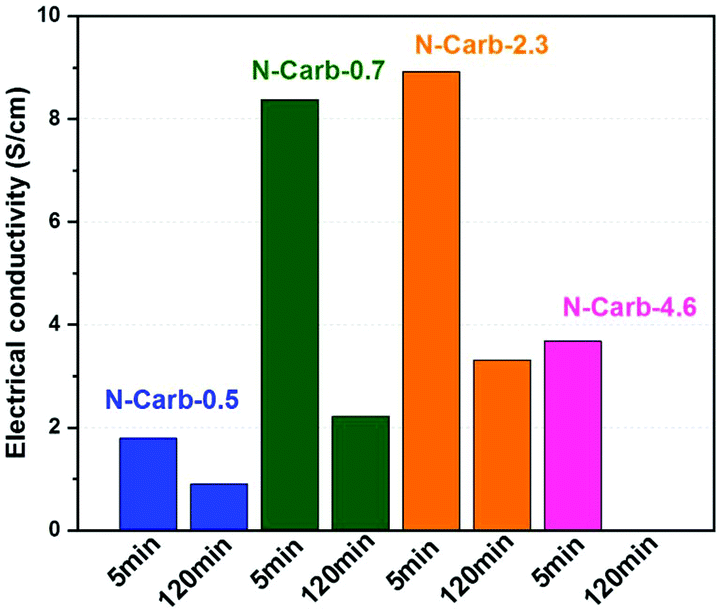 | ||
| Fig. 5 Electrical conductivities for the eight samples obtained in the ball mill. | ||
Conclusions
We demonstrated that N-doped porous carbons can be synthesized via the simple reaction between calcium carbide and cyanuric chloride at room temperature inside a ball mill. We reported further insight into the mechanochemical mechanism. On one side, we found that the cyanuric chloride is critical to start the reaction while in situ incorporates nitrogen species, mainly pyridinic. On the other side, we found that the reaction evolves generating the acetylene units to react to each other, and consequently more carbon is produced. Furthermore, the influence of the nitrogen content on the electrical conductivity was investigated. Results show an increment on the electrical conductivity when decreasing the nitrogen content and the surface area. The reported electrical conductivities are quite good considering that any thermal step was avoided; being the mechanical forces the only external source of energy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MEC thanks the Alexander von Humboldt Foundation for financial support. We thank the German Federal Ministry of Education and Research (BMBF) for funding of the “mechanocarb” project (FKZ 03SF0498). Thanks are further due to the Deutsche Forschungsgemeinschaft (grant no. Br 1278/29-1). We thank Christian Kensy (Fraunhofer-IWS) for electrical conductivity measurements, and Prof. Iniesta (University of Alicante) for XPS measurements.Notes and references
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- L. Borchardt, Q.-L. Zhu, M. E. Casco, R. Berger, X. Zhuang, S. Kaskel, X. Feng and Q. Xu, Mater. Today, 2017, 20(10), 592 CrossRef CAS.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- C. Schneidermann, C. Kensy, P. Otto, S. Oswald, L. Giebeler, D. Leistenschneider, S. Grätz, S. Doerfler, S. Kaskel and L. Borchardt, ChemSusChem, 2019, 12, 310 CrossRef CAS PubMed.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- X. Chen, J. Zhang, B. Zhang, S. Dong, X. Guo, X. Mu and B. Fei, Sci. Rep., 2017, 7, 7362 CrossRef PubMed.
- M. K. Rybarczyk, E. Gontarek, M. Lieder and M.-M. Titirici, Appl. Surf. Sci., 2018, 435, 543 CrossRef CAS.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal., 2017, 7, 1520 CrossRef CAS.
- T. J. Bandosz and C. O. Ania, Adv. Sci., 2018, 5, 1800293 CrossRef PubMed.
- W. Tian, H. Li, B. Qin, Y. Xu, Y. Hao, Y. Li, G. Zhang, J. Liu, X. Sun and X. Duan, J. Mater. Chem. A, 2017, 5, 7103 RSC.
- G. P. Hao, G. Mondin, Z. Zheng, T. Biemelt, S. Klosz, R. Schubel, A. Eychmüller and S. Kaskel, Angew. Chem., Int. Ed., 2015, 54, 1941 CrossRef CAS PubMed.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038 RSC.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamur, Science, 2016, 351, 361 CrossRef CAS PubMed.
- E. Antolini, Renewable Sustainable Energy Rev., 2016, 58, 34 CrossRef CAS.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085 RSC.
- Z. R. Ismagilov, A. E. Shalagina, O. Y. Podyacheva, A. V. Ischenko, L. S. Kibis, A. I. Boronin, Y. A. Chesalov, D. I. Kochubey, A. I. Romanenko, O. B. Anikeeva, T. I. Buryakov and E. N. Tkachev, Carbon, 2009, 47, 1922 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641 CrossRef CAS.
- T.-Y. Kim, K.-R. Lee, K. Y. Eun and K.-H. Oh, Chem. Phys. Lett., 2003, 372, 603 CrossRef CAS.
- X. Yongde and R. Mokaya, Chem. Mater., 2005, 17(6), 1553 CrossRef.
- D. Deng, X. Pan, L. Yu, Y. Cui, Y. Jiang, J. Qi, W.-X. Li, Q. Fu, X. Ma, Q. Xue, G. Sun and X. Bao, Chem. Mater., 2011, 23, 1188 CrossRef CAS.
- N. Li, Z. Wang, K. Zhao, Z. Shi, Z. Gu and S. Xu, Carbon, 2010, 48, 255 CrossRef CAS.
- C. Zhang, L. Fu, N. Liu, M. Liu, Y. Wang and Z. Liu, Adv. Mater., 2011, 23, 1020 CrossRef CAS PubMed.
- R. J. White, M. Antonietti and M.-M. Titirici, J. Mater. Chem., 2009, 19, 8645 RSC.
- S. J. Yang, R. Rothe, S. Kirchhecker, D. Esposito, M. Antonietti, H. Gojzewski and N. Fechler, Carbon, 2015, 94, 641 CrossRef CAS.
- E. Zhang, G.-P. Hao, M. E. Casco, V. Bon, S. Grätz and L. Borchardt, J. Mater. Chem. A, 2018, 6, 859 RSC.
- J. Zhu, C. Yang, C. Lu, F. Zhang, Z. Yuan and X. Zhuang, Acc. Chem. Res., 2018, 51(12), 3191 CrossRef CAS PubMed.
- L.-S. Zhang, X.-Q. Liang, W.-G. Song and Z.-Y. Wu, Phys. Chem. Chem. Phys., 2010, 12, 12055 RSC.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350 CrossRef CAS PubMed.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007 CrossRef PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080 RSC.
- J. Puszkiel, F. C. Gennari, P. Arneodo Larochette, H. E. Troiani, F. Karimi, C. Pistidda, R. Gosalawit-Utke, J. Jepsen, T. R. Jensen, C. Gundlach, M. Tolkiehn, J. Bellosta Von Colbe, T. Klassen and M. Dornheim, J. Power Sources, 2014, 267, 799 CrossRef CAS.
- E. Troschke, S. Grätz, T. Lübken and L. Borchardt, Angew. Chem., Int. Ed., 2017, 56, 6859 CrossRef CAS PubMed.
- S. Grätz and L. Borchardt, RSC Adv., 2016, 6, 64799 RSC.
- P. A. Julien, K. Užarević, A. D. Katsenis, S. A. J. Kimber, T. Wang, O. K. Farha, Y. Zhang, J. Casaban, L. S. Germann, M. Etter, R. E. Dinnebier, S. L. James, I. Halasz and T. Friščić, J. Am. Chem. Soc., 2016, 138, 2929 CrossRef CAS PubMed.
- K. Užarević, T. C. Wang, S.-Y. Moon, A. M. Fidelli, J. T. Hupp, O. K. Farha and T. Friščić, Chem. Commun., 2016, 52, 2133 RSC.
- D. Leistenschneider, N. Jäckel, F. Hippauf, V. Presser and L. Borchardt, Beilstein J. Org. Chem., 2017, 13, 1332 CrossRef CAS PubMed.
- C. Schneidermann, N. Jäckel, S. Oswald, L. Giebeler, V. Presser and L. Borchardt, ChemSusChem, 2017, 10, 2416 CrossRef CAS PubMed.
- O. S. G. P. Soares, R. P. Rocha, A. G. Gonçalves, J. L. Figueiredo, J. J. M. Órfão and M. F. R. Pereira, Carbon, 2015, 91, 114 CrossRef CAS.
- S. Dyjak, W. Kiciński, M. Norek, A. Huczko, O. Łabędź, B. Budner and M. Polański, Carbon, 2016, 96, 937 CrossRef CAS.
- M. Szala, Fullerenes, Nanotubes, Carbon Nanostruct., 2013, 21(10), 879 CrossRef CAS.
- G. Cao, S. Doppiu, M. Monagheddu, R. Orru, M. Sannia and G. Cocco, Ind. Eng. Chem. Res., 1999, 38, 3218 CrossRef CAS.
- Q. Li, C. Yang, L. Wu, H. Wang and X. Cui, J. Mater. Chem. A, 2019 10.1039/C8TA10317H.
- M. E. Casco, F. Badaczewski, S. Grätz, A. Tolosa, V. Presser, B. M. Smarsly and L. Borchardt, Carbon, 2018, 139, 325 CrossRef CAS.
- A. Popovich, V. P. Reva, V. N. Vasilenko and O. A. Belous, Mater. Sci. Forum, 1992, 88–90, 737 CAS.
- C. Deidda, F. Delogu and G. Cocco, J. Mater. Sci., 2004, 39, 5315 CrossRef CAS.
- G. Mulas, S. Loiselle, L. Schiffini and G. Cocco, J. Solid State Chem., 1997, 129, 263 CrossRef CAS.
- L. Takacs, Prog. Mater. Sci., 2002, 47, 355 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49 CrossRef CAS.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352 RSC.
- P. Hammer and F. Alvarez, Thin Solid Films, 2001, 398–399, 116 CrossRef CAS.
- U. Gelius, C. J. Allan, G. Johansson, H. Siegbahn, D. A. Allison and K. Siegbahn, Phys. Scr., 1971, 3, 237 CrossRef CAS.
- A. Snis, S. F. Matar, O. Plashkevych and H. Ågren, J. Chem. Phys., 1999, 111, 9678 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013,(8), 235 CrossRef CAS PubMed.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc., A, 2004, 362, 2477 CrossRef CAS PubMed.
- J. Biscoe and B. E. Warren, J. Appl. Phys., 1942, 13, 364 CrossRef CAS.
- M. Einert, C. Wessel, F. Badaczewski, T. Leichtweiß, C. Eufinger, J. Janek, J. Yuan, M. Antonietti and B. M. Smarsly, Macromol. Chem. Phys., 2015, 216, 1930 CrossRef CAS.
- N. Rey-Raap, E. G. Calvo, J. M. Bermúdez, I. Cameán, A. B. García, J. A. Menéndez and A. Arenillas, Measurement, 2014, 56, 215 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr01019j |
| This journal is © The Royal Society of Chemistry 2019 |