
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the growth mechanism of the co-precipitation of iron oxide nanoparticles with the aid of synchrotron X-Ray diffraction in solution†
Alec P.
LaGrow‡
 a,
Maximilian O.
Besenhard‡
b,
Aden
Hodzic
c,
Andreas
Sergides
a,
Lara K.
Bogart
d,
Asterios
Gavriilidis
*b and
Nguyen Thi Kim
Thanh
*a
a,
Maximilian O.
Besenhard‡
b,
Aden
Hodzic
c,
Andreas
Sergides
a,
Lara K.
Bogart
d,
Asterios
Gavriilidis
*b and
Nguyen Thi Kim
Thanh
*a
aBiophysics Group, Department of Physics and Astronomy, University College London, Gower Street, London, WC1E 6BT, UK. E-mail: ntk.thanh@ucl.ac.uk
bDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: a.gavriilidis@ucl.ac.uk
cCentral European Research Infrastructure Consortium, CERIC-ERIC, Trieste, Italy
dUCL Healthcare Biomagnetics Laboratories, University College London, 21 Albemarle Street, London, W1S 4BS, UK
First published on 8th March 2019
Abstract
Co-precipitation is the most ubiquitous method for forming iron oxide nanoparticles. For a typical co-precipitation synthesis, the pH of a ferrous and/or ferric ion solution is increased via the addition of a base. The latter can be added either slowly (a steady addition over either minutes or hours) or fast (a one-time addition) resulting in an abrupt increase in the pH. However, understanding the mechanism of particle formation is still lacking, which limits the reproducibility of the co-precipitation reaction due to intermediate phases still being present in the final product. In this work, we study in detail a co-precipitation synthesis with an abrupt increase in pH via the addition of sodium carbonate. Fast and reproducible mixing at defined precursor and base solution temperatures was achieved utilising a flow reactor. Transmission electron microscopy, electron diffraction and room temperature 57Fe Mössbauer spectroscopy showed a distinct transition from an amorphous ferrihydrite phase to a mixture of magnetite-maghemite (Fe3O4/γ-Fe2O3). Synchrotron X-ray diffraction revealed the initial formation of crystalline iron hydroxide carbonate (green rust) plates occurring before the Fe3O4/γ-Fe2O3 appeared. The ferrihydrite particles increase in size over time as the proportion of iron hydroxide carbonate plates are re-dissolved into solution, until the ferrihydrite particles crystallise into Fe3O4/γ-Fe2O3.
Introduction
Iron oxide nanoparticles (IONPs) are used extensively in applications in biomedicine, magnetic storage media, water treatment and catalysis.1–7 The most ubiquitous synthetic method used to date to form IONPs is the co-precipitation method, which is carried out in aqueous solutions containing ferric (Fe3+) and ferrous (Fe2+) salts as precursors,8 to which a base is added at moderate temperatures (<100 °C). While IONPs mostly contain magnetite and maghemite phases, there are many other iron phases that can form during synthesis; each of them has very different magnetic properties. Even though the co-precipitation reaction is widely used, the mechanism of the reaction is still poorly understood, due to a severe lack of information on how intermediates are formed. This often leads to difficulties in obtaining reproducible syntheses, as the intermediates can remain present to a varying degree products.Proposed mechanisms for IONP formation originate from studies involving a slow transition in pH (from ∼2 to above 8) and investigating the phases formed during the transition through multiple pH regimes.9,10 These studies show intermediate formation of iron oxyhydroxide species9–15 and iron hydroxide2,10,12,16,17 species, all of which transition to magnetite or maghemite (Fe3O4, and γ-Fe2O3 respectively) over time. From the iron oxyhydroxide intermediates the most commonly reported are goethite,9–11 and a poorly crystalline or amorphous ferrihydrite.14,15 For example, Baumgartner et al. showed with cryo-transmission electron microscopy (TEM) the formation of amorphous ferrihydrite which attaches to the surface of the growing IONP.15
However, most co-precipitation syntheses proceed via a single and abrupt increase in pH, which is also the most common approach for large-scale and continuous reactor systems14,18–21 due to the shorter process times. Hence, the understanding of the abrupt pH transitions is extremely important. Ahn et al. showed that abrupt pH changes result in different particle formation routes than slow pH additions, showing the precipitation of ferrous hydroxide [Fe(OH)2] and its transition to several intermediate phases (also due to pH inhomogeneities in the solution after mixing) including lepidocrocite.10 Milosevic et al. continuous flow reactors to study more abrupt pH transitions and in situ magnetic characterisation to study the growth mechanism. From the magnetic measurements they hypothesised that the particles originated as a matrix of primary ferrihydrite of around 3 nm in size, and grew over time to form 7 nm iron oxide particles.14
In this work, we have used a millifluidic flow reactor to facilitate the fast mixing of the base and the precursor solution at a well-controlled reaction temperature to study the growth mechanism of Fe3O4/γ-Fe2O3 NPs. The flow reactor allowed for an abrupt transition of the pH with minimal inhomogeneities of either the pH or temperature of the solution. Characterisation was performed with transmission electron microscopy (TEM), synchrotron X-ray diffraction (XRD) in solution, and room temperature 57Fe Mössbauer spectroscopy. The abrupt pH transition caused the formation of two intermediate phases, i.e., amorphous ferrihydrite and iron hydroxide carbonate (green rust). The ferrihydrite grew at the expense of the iron hydroxide carbonate and crystallised into magnetite/maghemite.
Experimental procedure
Chemicals
Iron(III) chloride hexahydrate (96%) was obtained from Chem Cruz, USA. Iron(II) chloride tetrahydrate (99.0%) and sodium carbonate decahydrate (99.0%) were from Honeywell, Fluka, USA. All chemicals were used as received. For all experiments deionised water was used.Synthesis of iron oxide nanoparticles
In a typical experiment a 0.033 M FeCl2·4H2O and 0.066 M FeCl3·6H2O solution was made up in deionised water. In a separate flask a 0.34 M solution of Na2CO3·10H2O was prepared in deionised water. These two solutions were then loaded into 25 mL glass syringes (Hamilton 1000 series Gastight; Hamilton, Switzerland) and pumped with a dual syringe pump (Legato210; KDScientific, USA) at 5 ml min−1 for both syringes through 6 m of PTFE tubing with an 1/16 inch (= 1.58 mm) outer diameter, and an inner diameter of 1 mm. This tubing was submerged in a water bath heated to 60 °C. Tubing of 6 m was sufficient to preheat the reagents to the final reaction temperature of 60 °C before mixing at the used flow rates.22 Mixing happened in a T-mixer made of ETFE (P-632; IDEX Health & Science, USA) with a channel diameter of 0.5 mm which was immersed in the same water bath. Due to the relatively high flow rates (10 ml min−1 in total) and the small channel size of the T-mixer, Reynolds numbers of ∼900 were achieved, resulting in mixing times of <50 ms as quantified using the Villermaux–Dushman protocol,23,24 see section 1 of the ESI.† After mixing, the solution passed 30 cm of insulated PTFE tubing (inner diameter 1 mm, hence, residence time <1.5 s) to be collected in a vial, i.e., the aging vessel, where it was kept at 60 °C under stirring. The solution was collected for 30 s (after steady-state operation of the flow reactor was ensured), to fill the aging vessel with 5 ml solution. Subsequently, flow to the aging vessel was stopped and reaction was allowed to continue for up to 10 min. The procedure described above for the synthesis of iron oxide NPs is shown in Fig. 1A.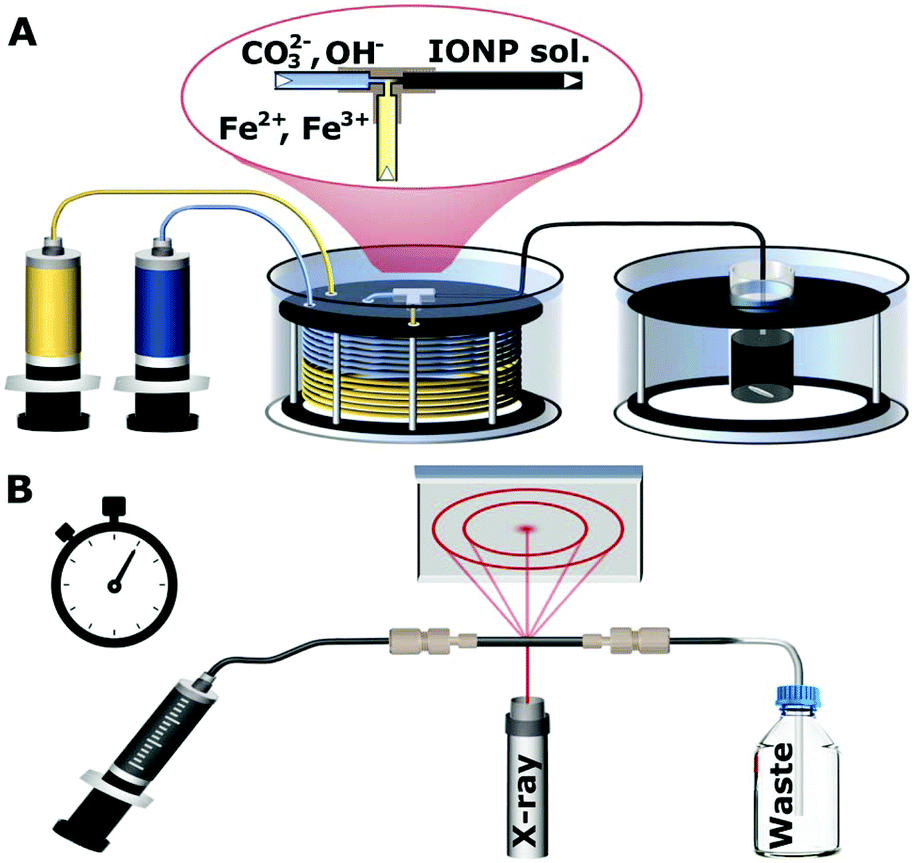 | ||
| Fig. 1 (A) Schematic of the set-up for nanoparticle synthesis showing the syringe pumps used to feed reactants to a T-mixer, pre-heated water bath, and aging bath. (B) Schematic of the synchrotron X-ray diffraction set-up showing the solution injected into a quartz capillary for analysis. | ||
The experiments with only Fe2+ or Fe3+ were carried out identically to the procedure above, except they were either carried out with 0.1 M FeCl2·4H2O only or 0.1 M FeCl3·6H2O only.
Transmission electron microscopy
The transmission electron microscopy (TEM) images and electron diffraction were captured using a JEOL 1200 EX microscope with a tungsten filament operating at a 120 kV acceleration voltage. The high-resolution TEM (HRTEM) imaging was carried out on a FEI Titan G2 60–300 equipped with a Cs image corrector operating at 80 kV. The TEM samples were prepared by withdrawing 20 μL of solution from the stirred reaction flask every min (with the first sample withdrawn immediately after sampling at 30 s) and dropping it onto a carbon coated 200 mesh copper grid (EM Resolutions Ltd) sitting on a filter paper. The filter paper absorbed the excess liquid causing the solution to dry immediately and effectively quenching the reaction.Synchrotron X-ray diffraction
The synchrotron X-ray diffraction was carried out at Elettra Synchrotron in Trieste, Italy using a wavelength of 1 Å for all measurements. Diffraction patterns were collected for 1 min in transmission mode at room temperature (∼24 °C) through the quartz capillary (∼1 mm inner diameter and <0.2 mm wall thickness) of a flow cell mounted in the beamline. This made it possible to characterise the NPs in solution via injection into the quartz capillary after the specified amount of reaction time (1, 2, 3, 4 or 5 min). After these times, a volume of the nanoparticle solution was withdrawn from the aging vessel with a syringe through a thin PTFE tubing (inner diameter 0.5 mm, for rapid cooling to room temperature), and then fed into the quartz capillary, as shown in Fig. 1B. The process of filling the capillary typically took 25 s, and the measurement was started within the next minute. The detector position and angle was calibrated with a boron lanthanide (LaB6) reference powder in the same capillary setup. The resulting pattern was radially averaged25 and the background of the capillary and solution were subtracted as described in section 2 of the ESI (Fig. S1 and S2†). It should be noted that the phases formed are stable upon cooling in the capillary for at least 10 min (Fig. S3†).57Fe Mössbauer spectroscopy
Samples for room temperature 57Fe Mössbauer spectroscopy were withdrawn from the flask at the specified reaction time (1, 3 and 10 min), and pumped through 2 m of 1/16 inch tubing in an ice bath to quench the reaction. The samples were frozen with liquid nitrogen and then freeze dried to remove the water, before storage in a glovebox prior to measurements. Samples were further processed for the measurement by mixing the dried powder with sucrose to form a solid dispersion and mounted in a 2.1 cm coin shaped absorber.A SeeCo W302 spectrometer (SeeCo Inc., UCA) was operated in transmission geometry and in constant acceleration mode. A 57Co in Rh foil was the source of the 14.4 keV γ-rays, with velocity calibration performed by recording a reference spectrum from a 10 μm thick foil of α-Fe at room temperature. All spectra were folded and baseline corrected using a cubic spline correction derived from the α-Fe calibration spectrum and following a protocol implemented in the Recoil analysis program.26 All spectra obtained for the 1 min samples were fitted using pairs of Lorentzian doublets, with the isomer shift and quadrupole splitting allowed to vary as free parameters; the best fit to the spectrum was determined by the lowest value of χ2. The 3 min and 10 min samples were fitted using the ‘center of gravity’ method.27 This method allows for the area weighted mean isomer shift of a magnetically split 57Fe Mössbauer spectrum to be determined without the need to assume any specific underlying model for the microenvironment of the Fe atoms, provided that the data is obtained from thin, texture free absorbers and possesses a flat, well defined background and that the sample comprises magnetite/maghemite only. It is for this reason we were unable to deduce an α-value of the t = 3 min sample, since it appeared to contain an appreciable amount of ferrihydrite (ca. 20%).
Results and discussion
The co-precipitation synthesis was carried out with sodium carbonate, which has been previously reported to result in slower growth kinetics than the strong bases,9 and thus would allow the kinetics to be readily studied. The described semi-flow procedure for the synthesis of iron oxide NPs, facilitated fast mixing (mixing times <50 ms) to rapidly obtain the reaction pH, hence minimise local pH variations, and a controlled reaction temperature of 60 °C throughout the synthesis (including the mixing step). Furthermore, it allowed for the co-precipitation synthesis to be studied multiple times under identical processing conditions, and thus guaranteeing reproducibility needed for a detailed study into the kinetics of nanoparticle synthesis. The samples synthesised were characterised by TEM, synchrotron XRD and room temperature 57Fe Mössbauer spectroscopy, carried out at the reaction times described above.TEM characterisation
The initial particles formed immediately after mixing, as apparent from the change in colour to a dark black-reddish solution when exiting the T-Mixer (Fig. S4†). The first TEM sample was taken immediately after transfer to the aging vessel (i.e., 30 s) and showed particles of 2.2 ± 0.5 nm in size which formed aggregates (Fig. 2A). By 1 min the particles increased to 3.0 ± 0.7 nm. The particles continued to increase approximately linearly in size as the size distribution broadened at the same time (Fig. 2B). This continued until the particles reached a maximum size after 5 min of ∼7 nm. With longer times (up to 10 min) the average particle size stayed the same, although the polydispersity increased (Fig. 2C).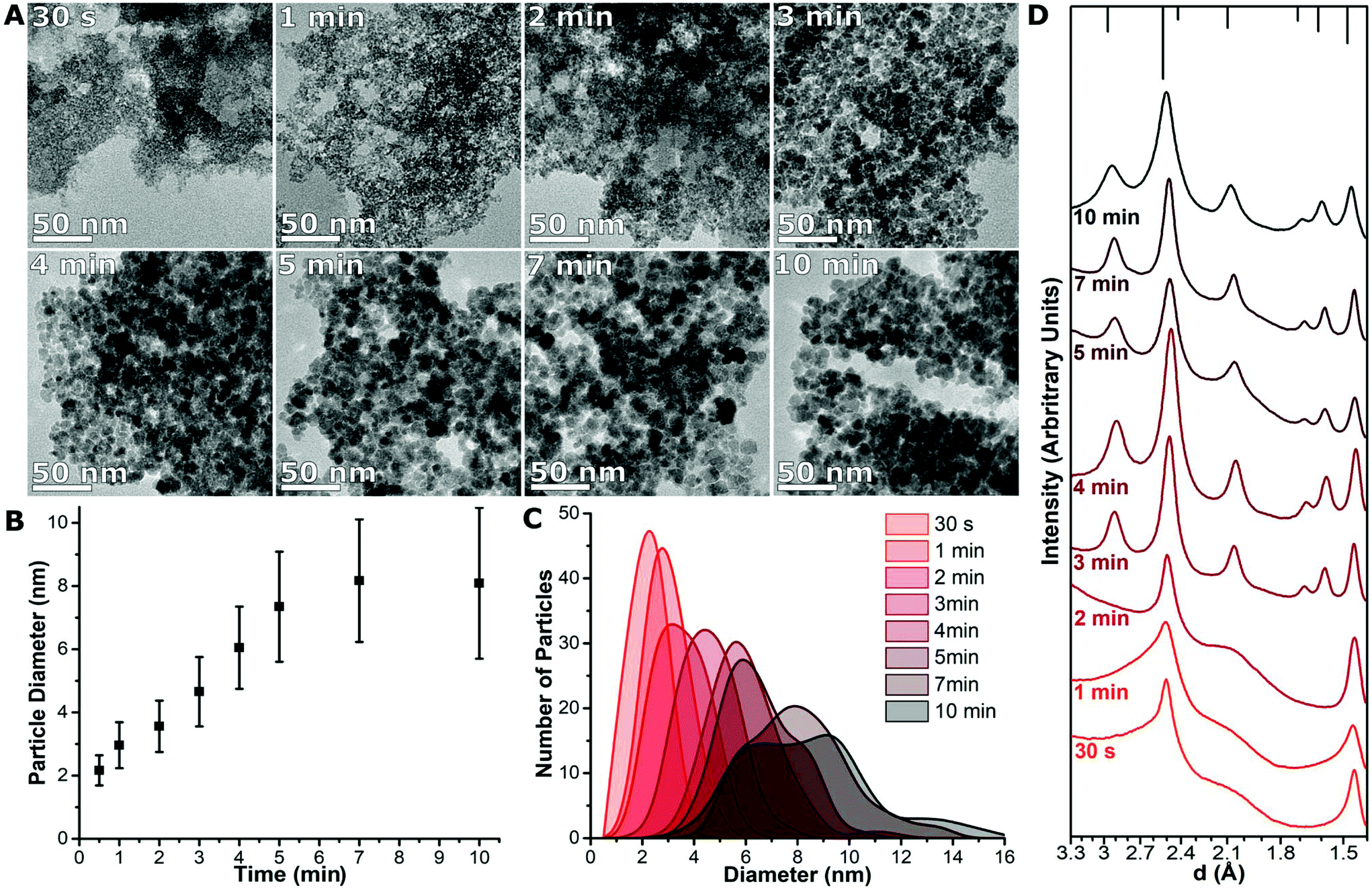 | ||
| Fig. 2 (A) TEM images of the nanoparticles formed after 30 s, 1, 2, 3, 4, 5, 7 and 10 min of reaction. (B) The particle diameter plotted against time with the standard deviation of the particle size distribution plotted as the error bars. (C) Size distributions of each sample were taken from TEM measurements of ca. 100 particles. (D) Radially averaged electron diffraction pattern after 30 s, 1, 2, 3, 4, 5, 7 and 10 min of reaction at 60 °C, with the characteristic peak locations for magnetite indicated by bars at the top of the figure. | ||
The crystal structure of the TEM samples was studied with electron diffraction (Fig. S5†). Each diffraction pattern was radially averaged to allow it to be studied as a two-dimensional plot,13 and normalised (Fig. 2D). The initial sample-formed after 30 s – had two broad peaks in the electron diffraction pattern at ∼2.5 Å and ∼1.5 Å. At 3 min the particles are crystalline; the pattern for iron oxide is clearly observed and stays as the only component observed till the end of the reaction. Electron diffraction on the TEM samples indicate that a transition between the initial phase to crystalline magnetite or maghemite (Fe3O4/γ-Fe2O3) occurred between 2 to 3 min, which corresponds to a particle size of 3.6 ± 0.8 nm to 4.7 ± 1.1 nm.
Aberration corrected TEM was carried out at 80 kV to further study the pre-transition sample after 2 min of reaction. Two distinct structures are seen in TEM showing a smaller proportion of faceted plates of a roughly hexagonal shape and smaller nanoparticles 2–4 nm in size (Fig. 3A). The high-resolution imaging of the 2–4 nm nanoparticles did not show distinct crystallinity, with a few diffraction spots in the Fourier transform of the image at ∼2.4 Å (Fig. 3B) and the high-resolution images showing a partially crystalline structure (Fig. 3D). High resolution imaging of part of the plate and the Fourier transform of the image showed hexagonal symmetry and spacings of ∼2.5 Å and ∼1.5 Å (Fig. 3C and E), indicating that the plate is highly crystalline. The spacings and symmetry seen in the Fourier transform is almost identical to characterisation of iron hydroxide carbonate (green rust) plates reported by McGill et al.28 The spacings of ∼2.5 Å and ∼1.5 Å are in the same position as the peaks in the electron diffraction of Fig. 2D at 30 s, 1 min and 2 min, and thus would originate from these plates. Green rust structures have been shown to adopt hexagonal plate-like structures with the CO32− or SO42− counter-ions.
 | ||
| Fig. 3 (A) Aberration corrected TEM image of the sample taken at 2 min of an area with particles and plates present. High resolution images taken of (B) the particles from the red box, and (C) the plate from the green box. Both images have the Fourier transforms of the image as an inset with the spacing of the spots/rings indicated on the inset. (D) and (E) are zoomed in on the areas in the black boxes of (B) and (C) showing the crystallinity of the structures. | ||
Control experiments were carried out using either solely FeCl3·6H2O or FeCl2·4H2O. For the reaction with FeCl3·6H2O alone, only small poorly crystalline particles were formed (Fig. S6†), which did not grow further when the reaction was continued for up to 10 min. For FeCl2·4H2O alone, plates were formed which had the same electron diffraction patterns as observed in Fig. 2D at 30 s, 1 min and 2 min (Fig. S6†). The FeCl2·4H2O alone sample oxidised over time to form a brown solution. This indicates that the electron diffraction patterns from the mixed samples shown in Fig. 2D originated from the plate structures with the small spherical particles being amorphous, and moreover that the spherical particles formed would not grow further without Fe2+ present in the reaction mixture.
Synchrotron X-Ray diffraction studies
Metal hydroxides can be unstable and the crystal structure decomposes when dried from solution or studied with an electron beam.15,29,30 To avoid drying of sample, XRD was carried out through a capillary in which the nanoparticle solution was injected and studied with a synchrotron light source to achieve prompt measurements during the synthesis. The synchrotron XRD measurements were taken at set time points to retain the benefits of the controlled mixing times and temperature homogeneity of the flow reactor. In contrast, dedicated in situ cells normally have much smaller reaction volumes without the possibility for mixing.31,32 The XRD patterns of samples are shown in Fig. 4A. The presence of Fe3O4/γ-Fe2O3 was first observed at 3 min as shown by XRD, and in agreement with the electron diffraction data (Fig. 2D). At 1 min and 2 min the XRD shows the formation of a crystalline phase which corresponds to iron hydroxide carbonate [Fe6(OH)12CO3], which is formed of 4 Fe2+ centres and 2 Fe3+ centres. At 3 min, the peaks for the iron hydroxide carbonate and iron oxide co-exist, with the latter having a Scherrer size of 7 nm at 3 min, which increases to ∼9 nm at 5 min. The iron hydroxide carbonate makes up a small proportion of the volume of diffracting objects that are eventually seen, surmised by comparing the XRD intensities after 5 min (Fig. 4B). The peak area of the iron hydroxide carbonate linearly decreases until peaks of magnetite (and/or maghemite) are observed, and these increase also linearly in peak area after the iron hydroxide carbonate peaks disappear (Fig. 4B).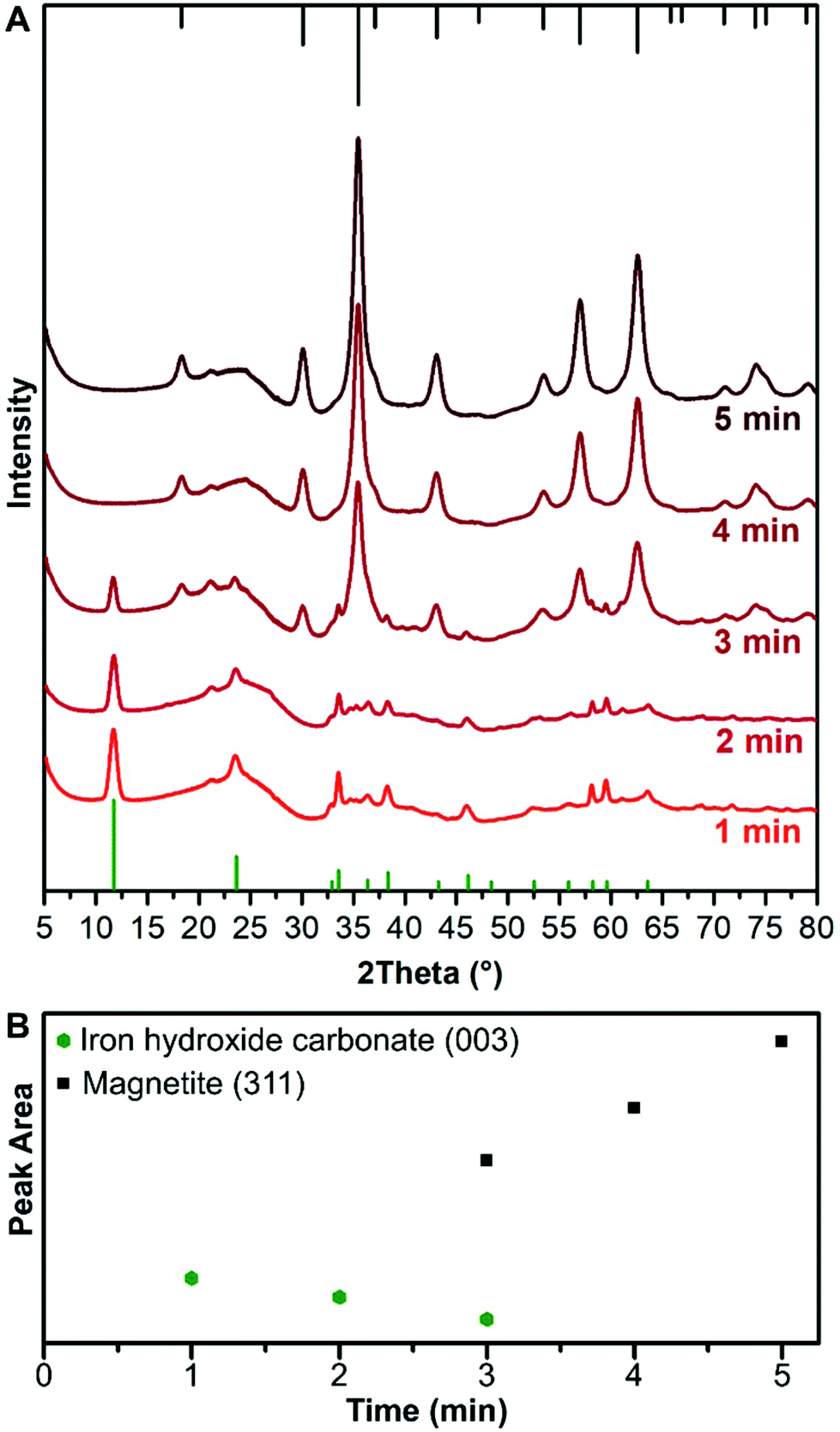 | ||
| Fig. 4 (A) Radially averaged and background subtracted synchrotron X-ray diffraction patterns taken in solution through a capillary with an X-ray wavelength of 1 Å transformed to have a 2Theta from Cu Kα (1.541 Å) after 1, 2, 3, 4, and 5 min of reaction at 60 °C. The green lines at the bottom are the reference pattern for iron hydroxide carbonate (PDF ref. 00-046-0098), the initial phase. The black lines at the top are the reference pattern for magnetite (PDF ref. 03-065-3107), the final phase. (B) Area under the most intense peak for the iron hydroxide carbonate and the magnetite as a function of aging time. Both the intensity and peak area are in arbitrary units. | ||
57Fe Mössbauer spectroscopy studies
Room temperature 57Fe Mössbauer spectroscopy measurements were performed on the 1 min sample (i.e., before transition), the 3 min sample (at the transition) as well as the final product at 10 min. For comparison we also measured the 1 min sample of the Fe3+ sample alone, prepared in the absence of Fe2+. All mixtures were freeze dried to give a fine powder.The spectrum of the product formed at 1 min (Fig. 5) corresponds to that of a quadrupole ferric doublet, with the best fit obtained using two Lorentzian doublets (each with an isomer shift ca. 0.35 mm s−1) and which is reminiscent of ferrihydrite.33 There are very small differences between the samples prepared with and without Fe2+, namely a small increase in the sub-component with higher quadrupole splitting, which could be due to the increase of amorphous material formed when Fe2+ is omitted from the reaction (Fig. S7†). There is no sign of any Fe2+ from the iron hydroxide carbonate plates in either of these samples, which is likely to be due to their oxidation during isolation and the freeze-drying process.
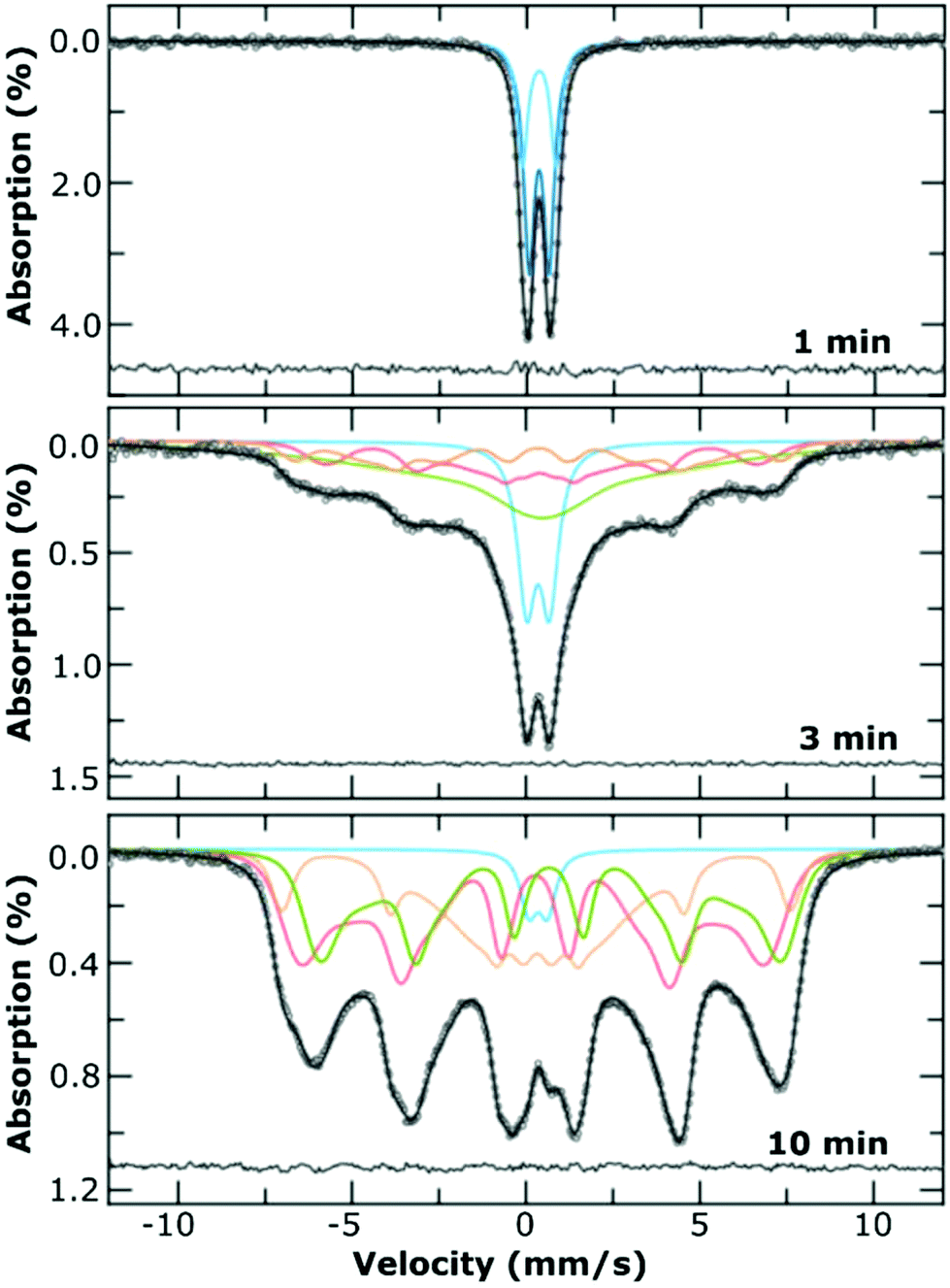 | ||
| Fig. 5 Room temperature 57Fe Mössbauer spectra of the freeze dried samples mixed with sucrose, at the indicated time points. The best fit to the data was achieved using a combination of Lorentzian line shapes (upper panel) or Voigtian line shapes (Gaussian distributions of Lorentzian lines; middle, bottom panel). The sub-component spectra are indicated using coloured lines, which are for presentation purposes only, with the residual of the fit to the measured data is shown as the line at the bottom of each panel. | ||
The room temperature spectrum of the product at 3 min (Fig. 5) is a superposition of a doublet and broad sextet, indicative of a range of Fe environments within the sample. Best fitting of the spectrum was performed using Voigtian lines (to represent a Gaussian distribution of Lorentzian lines) and indicates that ca. 20% of the sample is the original ferrihydrite, with the remainder of the sample comprising of a poorly magnetic iron oxide. The presence of very broad absorption lines, which were best fit using a very broad distribution of hyperfine fields are likely to arise from a non-stoichiometric solid solution of magnetite/maghemite. The spectrum of the final product (10 min), comprises a partially collapsed magnetically split sextet pattern with very broad lines, and is indicative of magnetically blocked particles on the Mössbauer timescale, with a broad size distribution. The spectrum was fitted using the model independent ‘centre of gravity’ method27 from which we surmise that the numerical proportion of Fe atoms in a magnetite environment, α, was 0.40 ± 0.17 i.e., corresponding to a mixture of magnetite and maghemite.27 We did not observe any oxidation ageing of the material during the acquisition of any of the spectra, which took place over at least 3 d for each sample.
Proposed mechanism of iron oxide nanoparticle formation
Ferrihydrite is a poorly crystalline iron oxyhydroxide material with a general formula of Fe10O14(OH)2,34 formed from Fe13 clusters of Fe3+ known as Keggin clusters.35,36 The formation of the initial ferrihydrite, occurs by olation of the hydroxylated iron and oxolation.37 The ferrihydrite structure is formed of a central Keggin cluster with an iron depleted shell.36 These Keggin clusters that are central to ferrihydrite formation also have a similar crystal structure to the central magnetite unit.35 The formation of amorphous ferrihydrite primary particles or pre-nucleation clusters have been reported in numerous previous reports as an intermediate phase in iron oxide formation.10,14,15,36,37Iron hydroxide carbonate [Fe6(OH)12CO3] is a type of green rust. Green rust is a mixed valence 2-dimensional structure which is structurally similar to the amorphous Fe(OH)2 sheets,11 and can be stabilised by a range of counter ions such as CO32−, SO42−, Cl−, Br− in either a hexagonal or rhombohedral crystal structure.17,28 The structure is formed via the olation of Fe2+ hydroxide tetrahedra11 and some incorporation of Fe3+ hydroxide tetrahedral centres, either by incorporation or oxidation.
From our data we observe a linear increase in particle size of the ferrihydrite particles via TEM and room temperature 57Fe Mössbauer spectroscopy with time, starting from initially formed 2.2 ± 0.5 nm particles. We observe a size dependent transition between the ferrihydrite and the magnetite/maghemite which occurs when the particles reach ∼4 nm. This transition corresponds to the disappearance of peaks from the iron hydroxide carbonate phase in the X-ray diffraction pattern. The intensity of the iron hydroxide carbonate pattern decreases as the reaction progresses before the phase disappears at 4 min, which corresponds to the time when the ferrihydrite phase is no longer observed by TEM. The proportion of the iron hydroxide carbonate decreases over time before and after the formation of iron oxide occurs. This indicates that as the reaction progresses the iron hydroxide carbonate structure is re-dissolved and the ions are incorporated into the ferrihydrite, providing a source of Fe2+/3+, causing it to grow and transition into magnetite/maghemite. The iron hydroxide carbonate acts as a reservoir of Fe2+ and Fe3+ ions leading to slow continuous growth first of the ferrihydrite and then subsequently of the magnetite/maghemite. A schematic of this growth mechanism, which differs from classical nucleation and growth concepts, is shown in Fig. 6. In reaction systems without carbonate ions present, a reservoir containing Fe2+ ions could be formed as a green rust structure with another counter ion (SO42−, Cl− or Br−),17,28 or as amorphous Fe(OH)2 sheets.11
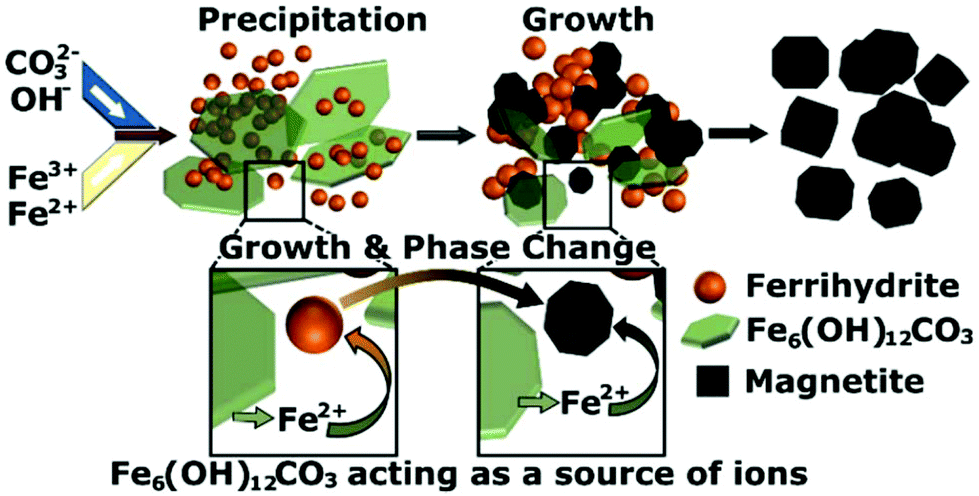 | ||
| Fig. 6 Schematic of growth mechanism showing the initial precipitation of iron hydroxide carbonate [Fe6(OH)12CO3] and ferrihydrite and the growth of ferrihydrite via the re-dissolution of iron hydroxide carbonate to form the magnetite phase. | ||
The incorporation of Fe2+ into the structure of magnetite has been previously predicted to be a rate limiting step.38–41 As Fe2+ has a higher solubility in water,42 it becomes the primary additive causing the primarily Fe3+ ferrihydrite particles to grow. Without the Fe2+ present, the ferrihydrite would not grow to magnetite (as in the control experiment, see Fig. S6†). Therefore, in a common co-precipitation synthesis, if there was an oxidation of the Fe2+, this would lead to its deficiency and hinder the conversion from ferrihydrite to Fe3O4/γ-Fe2O3. This potentially leads to considerable problems with reproducibility of the synthesis of IONPs due to the presence of residual ferrihydrite particles, which are weakly magnetic and amorphous. The amount of residual ferrihydrite in the final product can therefore be reduced by protecting the Fe2+ ions from oxidation or increasing the amount of Fe2+ in the reaction.
Summary and conclusions
For this work a flow reactor was utilised to facilitate fast mixing (<50 ms) and well-controlled, hence reproducible, process conditions during a co-precipitation synthesis. Synchrotron X-ray diffraction (in solution, using a flow cell mounted in the beamline), as well as TEM and room temperature 57Fe Mössbauer spectroscopy studies were performed to unravel the particle formation mechanism.These studies revealed that two initial phases were formed upon the direct mixing of the iron chloride precursor solution with the pH 12 solution of sodium carbonate. The phases were a poorly crystalline ferrihydrite phase formed primarily of Fe3+ and crystalline iron hydroxide carbonate plates formed primarily of Fe2+. The ferrihydrite was characterised by means of room temperature 57Fe Mössbauer spectroscopy and TEM, while the dynamic changes of the (metastable) iron hydroxide carbonate plates could only be monitored via synchrotron X-ray diffraction in solution. The iron hydroxide carbonate concentration decreased over time until it disappeared after 4 min of the reaction. The ferrihydrite phase grew with time from 2 nm particles until it transitioned to Fe3O4/γ-Fe2O3 when the particles reached ∼4 nm in size, which happened after 3–4 min of the reaction. The ferrihydrite phase acted as the seeds that grew into the eventual Fe3O4/γ-Fe2O3 particles, while the iron carbonate plates acted as a feedstock to supply the iron ions for the particles to grow and the phase change to occur. Without the presence of Fe2+ in solution, the formation and growth of the Fe3O4/γ-Fe2O3 NPs did not occur.
A new particle formation mechanism was detailed, differing drastically from the most commonly discussed classical nucleation and growth process. Such comprehensive understanding of the mechanism is essential not only for robust syntheses, but also for the tuning of particle properties for the targeted applications. In particular, the growth mechanism shows a separation of the nucleation and growth stages in the reaction which will allow chemists greater control over the size of the nanoparticles that can be readily synthesised via co-precipitation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
APL, MOB, AG, NTKT would like to thank EPSRC for funding (grant EP/M015157/1). AS thanks the EPSRC CDT for the Advanced Characterisation of Materials (grant EP/L015277/1) for his studentship. NTKT thanks AOARD (FA2386-17-1-4042 award). AH thanks the CERIC ERIC internal research project: Nano Analytics in Pharmaceutics for support. We would like to thank Giorgio Bais and Maurizio Polentarutti for support with the synchrotron XRD studies.References
- A. Hervault and N. T. K. Thanh, Magnetic nanoparticle-based therapeutic agents for thermo-chemotherapy treatment of cancer, Nanoscale, 2014, 6, 11553 RSC.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Use of iron oxide nanomaterials in wastewater treatment: A review, Sci. Total Environ., 2012, 424, 1 CrossRef CAS PubMed.
- N. A. Frey, S. Peng, K. Cheng and S. H. Sun, Magnetic nanoparticles: synthesis, functionalization, and applications in bioimaging and magnetic energy storage, Chem. Soc. Rev., 2009, 38, 2532 RSC.
- A. S. Teja and P. Y. Koh, Synthesis, properties, and applications of magnetic iron oxide nanoparticles, Prog. Cryst. Growth Charact. Mater., 2009, 55, 22 CrossRef CAS.
- Q. A. Pankhurst, N. T. K. Thanh, S. K. Jones and J. Dobson, Progress in applications of magnetic nanoparticles in biomedicine, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- N. T. K. Thanh, Clinical Applications of Magnetic Nanoparticle, CRC Press, Taylor & Francis, Boca Raton, London, New York, 2018 Search PubMed.
- N. T. K. Thanh, Magnetic Nanoparticles: From Fabrication to Clinical Applications, CRC Press, Taylor & Francis, Boca Raton, London, New York, 2012 Search PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications, Chem. Rev., 2008, 108, 2064 CrossRef CAS PubMed.
- C. Blanco-Andujar, D. Ortega, Q. A. Pankhurst and N. T. K. Thanh, Elucidating the morphological and structural evolution of iron oxide nanoparticles formed by sodium carbonate in aqueous medium, J. Mater. Chem., 2012, 22, 12498 RSC.
- T. Ahn, J. H. Kim, H.-M. Yang, J. W. Lee and J.-D. Kim, Formation pathways of magnetite nanoparticles by coprecipitation method, J. Phys. Chem. C, 2012, 116, 6069 CrossRef CAS.
- J.-P. Jolivet, C. Chaneac and E. Tronc, Iron oxide chemistry. From molecular clusters to extended solid networks, Chem. Commun., 2004, 481 CAS.
- T. Sugimoto and E. Matijevic, Formation of uniform spherical magnetite particles by crystallization from ferrous hydroxide gels, J. Colloid Interface Sci., 1980, 74, 227 CrossRef CAS.
- C. Gammer, C. Mangler, C. Rentenberger and H. P. Karnthaler, Quantitative local profile analysis of nanomaterials by electron diffraction, Scr. Mater., 2010, 63, 312 CrossRef CAS.
- I. Milosevic, F. Warmont, Y. Lalatonne and L. Motte, Magnetic metrology for iron oxide nanoparticle scaled-up synthesis, RSC Adv., 2014, 4, 49086 RSC.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nucleation and growth of magnetite from solution, Nat. Mater., 2013, 12, 310 CrossRef CAS PubMed.
- P. Belleville, J.-P. Jolivet, E. Tronc and J. Livage, Crystallization of ferric hydroxide into spinel by adsorption on colloidal magnetite, J. Colloid Interface Sci., 1992, 150, 453 CrossRef CAS.
- C. Ruby, A. Géhin, M. Abdelmoula, J.-M. R. Génin and J.-P. Jolivet, Coprecipitation of Fe(II) and Fe(III) cations in sulphated aqueous medium and formation of hydroxysulphate green rust, Solid State Sci., 2003, 5, 1055 CrossRef CAS.
- K. Kumar, A. M. Nightingale, S. H. Krishnadasan, N. Kamaly, M. Wylenzinska-Arridge, K. Zeissler, W. R. Branford, E. Ware, A. J. deMello and J. C. deMello, Direct synthesis of dextran-coated superparamagnetic iron oxide nanoparticles in a capillary-based droplet reactor, J. Mater. Chem., 2012, 22, 4704 RSC.
- M. Simmons, C. Wiles, V. Rocher, M. G. Francesconi and P. Watts, The preparation of magnetic iron oxide nanoparticles in microreactors, J. Flow Chem., 2013, 3, 7 CrossRef CAS.
- F. Haseidl, B. Müller and O. Hinrichsen, Continuous-Flow Synthesis and Functionalization of Magnetite: Intensified Process for Tailored Nanoparticles, Chem. Eng. Technol., 2016, 39, 2051 CrossRef CAS.
- A. LaGrow, M. Besenhard, R. Hachani and N. T. K. Thanh, Experimental considerations for magnetic nanoparticle synthesis and surface functionalization for clinical applications, in Clinical Applications of Magnetic Nanoparticles, ed. N. T. K. Thanh, CRC Press, Taylor & Francis, Boca Raton, London, New York, 2018 Search PubMed.
- M. O. Besenhard, P. Neugebauer, C.-D. Ho and J. G. Khinast, Crystal Size Control in a Continuous Tubular Crystallizer, Cryst. Growth Des., 2015, 15, 1683 CrossRef CAS.
- J.-M. Commenge and L. Falk, Villermaux–Dushman protocol for experimental characterization of micromixers, Chem. Eng. Process., 2011, 50, 979 CrossRef CAS.
- L. Falk and J. M. Commenge, Performance comparison of micromixers, Chem. Eng. Sci., 2010, 65, 405 CrossRef CAS.
- A. Hammersley, FIT2D: a multi-purpose data reduction, analysis and visualization program, J. Appl. Crystallogr., 2016, 49, 646 CrossRef CAS.
- K. Lagarec and D. G. Rancourt, RECOIL, Mössbauer spectral analysis software for windows (version 1.0), Department of Physics, University of Ottawa, Canada, 1998 Search PubMed.
- J. Fock, L. K. Bogart, D. Gonzalez-Alonso, J. I. Espeso, M. F. Hansen, M. Varon, C. Frandsen and Q. A. Pankhurst, On the ‘centre of gravity’ method for measuring the composition of magnetite/maghemite mixtures, or the stoichiometry of magnetite-maghemite solid solutions, via Fe-57 Mossbauer spectroscopy, J. Phys. D: Appl. Phys., 2017, 50, 265005 CrossRef.
- I. R. McGill, B. McEnaney and D. C. Smith, Crystal structure of green rust formed by corrosion of cast iron, Nature, 1976, 259, 200 CrossRef CAS.
- A. P. LaGrow, L. Sinatra, A. Elshewy, K.-W. Huang, K. Katsiev, A. R. Kirmani, A. Amassian, D. H. Anjum and O. M. Bakr, Synthesis of copper hydroxide branched nanocages and their transformation to copper oxide, J. Phys. Chem. C, 2014, 118, 19374 CrossRef CAS.
- Y. Pan, A. Brown, R. Brydson, A. Warley, A. Li and J. Powell, Electron beam damage studies of synthetic 6-line ferrihydrite and ferritin molecule cores within a human liver biopsy, Micron, 2006, 37, 403 CrossRef CAS PubMed.
- A. P. LaGrow, B. Ingham, M. F. Toney and R. D. Tilley, Effect of surfactant concentration and aggregation on the growth kinetics of nickel nanoparticles, J. Phys. Chem. C, 2013, 117, 16709 CrossRef CAS.
- S. Cheong, J. Watt, B. Ingham, M. F. Toney and R. D. Tilley, In situ and ex situ studies of platinum nanocrystals: growth and evolution in solution, J. Am. Chem. Soc., 2009, 131, 14590 CrossRef CAS PubMed.
- E. Murad, The Mössbauer spectrum of “well”-crystallized ferrihydrite, J. Magn. Magn. Mater., 1988, 74, 153 CrossRef CAS.
- F. M. Michel, L. Ehm, S. M. Antao, P. L. Lee, P. J. Chupas, G. Liu, D. R. Strongin, M. A. A. Schoonen, B. L. Phillips and J. B. Parise, The structure of ferrihydrite, a nanocrystalline material, Science, 2007, 316, 1726 CrossRef CAS PubMed.
- O. Sadeghi, L. N. Zakharov and M. Nyman, Aqueous formation and manipulation of the iron-oxo Keggin ion, Science, 2015, 347, 1359 CrossRef CAS PubMed.
- J. S. Weatherill, K. Morris, P. Bots, T. M. Stawski, A. Janssen, L. Abrahamsen, R. Blackham and S. Shaw, Ferrihydrite Formation: The role of Fe13 Keggin clusters, Environ. Sci. Technol., 2016, 50, 9333 CrossRef CAS PubMed.
- J. Scheck, B. Wu, M. Drechsler, R. Rosenberg, A. E. S. Van Driessche, T. M. Stawski and D. Gebauer, The molecular mechanism of iron(III) oxide nucleation, J. Phys. Chem. Lett., 2016, 7, 3123 CrossRef CAS PubMed.
- S. Mann, N. H. C. Sparks, S. B. Couling, M. C. Larcombe and R. B. Frankel, Crystallochemical characterization of magnetic spinels prepared from aqueous-solution, J. Chem. Soc., Faraday Trans., 1989, 85, 3033 RSC.
- D. Faivre, P. Agrinier, N. Menguy, P. Zuddas, K. Pachana, A. Gloter, J. Y. Laval and F. Guyot, Mineralogical and isotopic properties of inorganic nanocrystalline magnetites, Geochim. Cosmochim. Acta, 2004, 68, 4395 CrossRef CAS.
- J. P. Jolivet, P. Belleville, E. Tronc and J. Livage, Influence of Fe(II) on the formation of the spinel iron-oxide in alkaline-medium, Clays Clay Miner., 1992, 40, 531 CrossRef CAS.
- E. Tronc, P. Belleville, J. P. Jolivet and J. Livage, Transformation of ferric hydroxide into spinel by Fe(II) adsorption, Langmuir, 1992, 8, 313–319 CrossRef CAS.
- M. A. Blesa and E. Matijević, Phase transformations of iron oxides, oxohydroxides, and hydrous oxides in aqueous media, Adv. Colloid Interface Sci., 1989, 29, 173 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional experiments, and additional details on the synchrotron X-ray diffraction measurements. See DOI: 10.1039/c9nr00531e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |