
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A robust, modular approach to produce graphene–MOx multilayer foams as electrodes for Li-ion batteries†
Zhen Yuan
Xia
 ab,
Meganne
Christian
c,
Catia
Arbizzani
d,
Vittorio
Morandi
c,
Massimo
Gazzano
a,
Vanesa
Quintano
a,
Alessandro
Kovtun
a and
Vincenzo
Palermo
*ab
ab,
Meganne
Christian
c,
Catia
Arbizzani
d,
Vittorio
Morandi
c,
Massimo
Gazzano
a,
Vanesa
Quintano
a,
Alessandro
Kovtun
a and
Vincenzo
Palermo
*ab
aIndustrial and Materials Science, Chalmers University of Technology, 41258, Göteborg, Sweden. E-mail: palermo@chalmers.se
bIstituto per la Sintesi Organica e la Fotoreattività, CNR, 40129, Bologna, Italy
cIstituto per la Microelettronica e Microsistemi, CNR, 40129, Bologna, Italy
dDipartimento di Chimica “Giacomo Ciamician”, University of Bologna, 40126, Bologna, Italy
First published on 7th March 2019
Abstract
Major breakthroughs in batteries would require the development of new composite electrode materials, with a precisely controlled nanoscale architecture. However, composites used for energy storage are typically a disordered bulk mixture of different materials, or simple coatings of one material onto another. We demonstrate here a new technique to create complex hierarchical electrodes made of multilayers of vertically aligned nanowalls of hematite (Fe2O3) alternated with horizontal spacers of reduced graphene oxide (RGO), all deposited on a 3D, conductive graphene foam. The RGO nanosheets act as porous spacers, current collectors and protection against delamination of the hematite. The multilayer composite, formed by up to 7 different layers, can be used with no further processing as an anode in Li-ion batteries, with a specific capacity of up to 1175 μA h cm−2 and a capacity retention of 84% after 1000 cycles. Our coating strategy gives improved cyclability and rate capacity compared to conventional bulk materials. Our production method is ideally suited to assemble an arbitrary number of organic–inorganic materials in an arbitrary number of layers.
Introduction
Graphene, a two-dimensional (2D) nanomaterial, is composed of a monoatomic layer of carbon atoms. This 2D material possesses fascinating characteristics such as high surface area, excellent electrical conductivity and extraordinary flexibility. Graphene nanosheets can be shaped in three-dimensional (3D) structures, such as porous foams, either by processing from solution or direct growth on metal templates, using a chemical vapour deposition (CVD) approach.1–5 A 3D graphene foam (GF) inherits most of the attractive physical properties of 2D graphene; the self-supported porous network of GF also avoids aggregation or restacking issue of individual graphene sheets.6,7 More importantly, the strong and conductive GF matrix could incorporate different electroactive materials to boost the electrochemical performance of these nanocomposites.8–12 The unique macro/mesoporous morphology of GF provides large electrode/active material contact area and effective multidimensional electrolyte-accessible channels. Thus, the production of 3D graphene-based nanocomposites represents a promising approach to achieve lightweight, durable and efficient electrode materials in energy storage.13–17For example, hematite (α-Fe2O3) is a promising anode material for lithium ion batteries due to low cost, natural abundance, and high theoretical energy capacity values (ca. 1006 mA h g−1). However, the poor electronic conductivity, large volume changes (ca. 74.7%), and serious aggregation of Fe2O3 particles during lithiation/delithiation cycles limit its practical application.18 To solve this issue, reduced graphene oxide (RGO) can be combined with α-Fe2O3.19–25 The self-assembled RGO/α-Fe2O3 3D hybrids efficiently prevent nanoparticle aggregation, alleviate the stress of volume expansion and maintain the mechanical integrity of the electrode. Although RGO-based 3D foam structure can improve the cycling behaviour of the Fe2O3 anode, the complicated oxidative-exfoliation/post-reduction steps of RGO and the subsequent long-term hydrothermal synthesis process of Fe2O3–RGO composites are still a challenge for real industrial applications.
GF can also be directly grown on metallic templates using CVD growth. This gives foams with superior electrical conductivity and better mechanical stability with respect to RGO. However, the hydrophobic and chemically inert CVD graphene surface renders the deposition of uniform metal oxide films on it extremely difficult. To overcome the surface adhesion issue special techniques, such as oxygen plasma, or supercritical carbon dioxide (scCO2) pre-treatment, are required. For example, mesoporous magnetite nanoparticles (Fe3O4) were successfully grafted onto an O2 plasma pre-treated CVD-GF matrix by a sacrificial ZnO layer using atomic layer deposition (ALD) or by the supercritical CO2 (scCO2) assisted method.10,26 The good adhesion of iron oxide on CVD-GF guarantees the long cycle behaviour with high performance in lithium ion batteries. However, the unavoidable oxidative damage of GF during the O2 plasma treatment, the use of an expensive metal organic precursor (diethyl zinc) with vacuum processing in ALD techniques, and the complex high-pressure system in scCO2 techniques inevitably decrease the GF quality and increase its cost.
With the aim of obtaining better performance and simplifying the production process, we developed here an electrodeposition method to functionalize GF with multiple layers of mesoporous α-Fe2O3 arranged in nanowall structures (Fig. 1).27–30 Electrodeposition is a low-cost and solution-processable technique that can be easily upscaled from the beaker level in lab to the industrial prototype size level. By tuning the anodic deposition time and electrolytes, homogeneous and well-adherent iron oxide thin films could be obtained on various conductive substrates.31
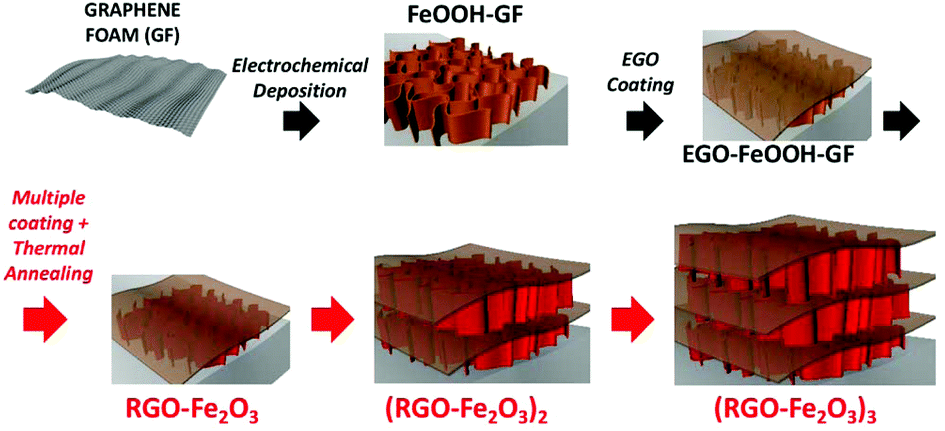 | ||
| Fig. 1 Schematic illustration of the preparation process for multilayer RGO–Fe2O3 graphitic foam architectures. | ||
To further enhance the adhesion of the active Fe2O3 material and increase the loading amount and stability of the coating, we tried to deposit an additional protective layer by solution casting of electrochemically exfoliated graphene oxide (EGO) solution. EGOs are moderately oxidized graphene sheets with less defects and much better electronic conductivity than conventional GO.32,33 The peculiar electronic properties of EGO make it a promising material for energy storage application.25,34 Our synthetic strategy aims at using EGO nanosheets as multifunctional additives in the composite, acting as an electrical conductor, a binder, a spacer and a primer to achieve uniform, multilayer deposition of metal oxides. The modular coating process can be repeated an arbitrary number of times; after alternative coating of FeOOH and EGO layers and subsequent annealing, multilayer (RGO–Fe2O3)x–GF (x = 1–3) composites with hierarchically interconnected meso- and macro-porosity were successfully fabricated, and tested as long life and high capacity anodes in lithium-ion batteries.
Experimental section
Materials
Graphite flakes (+100 mesh series) were purchased from Aldrich. Sodium perchlorate (Sigma-Aldrich, 98%) and sulphuric acid (Sigma-Aldrich, 95–97%) were used as electrolytes; acetonitrile (Sigma-Aldrich, 99%), N,N-dimethylformamide (Sigma, 99%) and deionized (DI) water were used as solvents. A platinum foil (99.99%) with the size of 5 mm × 10 mm × 0.1 mm was used as an anode connector. A platinum wire (99.99%, 0.3 mm) was used as a counter electrode. Nickel foams (380 g m−2 in areal density and 1.5 mm in thickness, compressed to 0.2 mm) were used as 3D templates for graphene coating. FeCl3 (Sigma-Aldrich, 45%) and HCl (Sigma-Aldrich, 37%) were utilized as etching acids for Ni foam. CH3COONa (Sigma-Aldrich, 99%) and Fe(NH4)2(SO4)2·6H2O (Sigma-Aldrich, 99%) were used for the electrochemical deposition of FeOOH on GF foam.Synthesis of graphene-based compounds
A platinum foil was then connected to the graphite on the top of the tube through a slot in the plastic cap, to act as the working electrode. Another platinum wire was used as a counter-electrode. The ionic solution was prepared by dissolving 1.3 g of sodium perchlorate in 10 mL of acetonitrile (1 mol L−1). Then, exfoliation was performed following an approach already described in previous work.34 In brief, uncharged acetonitrile molecules were intercalated in the graphite by the electrochemical treatment, due to the synergistic action of perchlorate ions dissolved in the acetonitrile. Then, the acetonitrile molecules were decomposed with microwaves, causing gas production and rapid graphite exfoliation. This method uses the gas produced by the decomposition of acetonitrile molecules as a powerful blowing agent to promote graphite exfoliation in a few seconds.
The first electrochemical intercalation/exfoliation stage was carried out for 30 min by applying a DC bias on the graphite electrode at a voltage of +5 V. After the reaction, the partially exfoliated graphite samples were washed with acetonitrile several times and blow-dried with dry nitrogen for 2 min.
A commercial microwave oven (Whirlpool JT379) with a rotating tray was used for further expansion. The graphite samples were placed in a porcelain crucible (capacity 50 mL) and heated in the microwave oven for 30 s under 90 W. Then, the expanded foam-like graphite was compressed again to make the whole material coherent. The material was fixed a second time in a porous plastic tube, connected to a Pt foil and further exfoliated in 0.1 M H2SO4 solution as an anode at +10 V for 2 hours. Then, the graphene sheets were collected by vacuum filtration onto a PTFE membrane and cleaned several times by repeated washing with DI water. Afterwards, the exfoliated flakes were re-dispersed in DMF by sonication for 30 min (37 kHz, 100 W effective ultrasonic power, Model ELMA P70H Ultrasonic). The graphene oxide solution was centrifuged at 2000 rpm for 30 min to remove any re-aggregated particles.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) solution containing 0.04 M CH3COONa and 0.01 M Fe(NH4)2(SO4)2 at room temperature. Electrodeposition of FeOOH nanostructures was performed with a three-electrode system, where the 3D graphene foam was used as the working electrode, a platinum wire electrode as the counter electrode, and a Ag/AgCl electrode as the reference electrode. The mass of deposited iron oxide films was controlled by adjusting the deposition time. The electrodeposition times used were 5, 10, 20 and 40 min, corresponding to mass loading amounts of FeOOH of 0.10, 0.15, 0.26 and 0.39 mg cm−2 Electrodeposition was done at a current density of 0.2 mA cm−2 under a constant N2 atmosphere and gentle stirring.
:1 volume ratio) solution containing 0.04 M CH3COONa and 0.01 M Fe(NH4)2(SO4)2 at room temperature. Electrodeposition of FeOOH nanostructures was performed with a three-electrode system, where the 3D graphene foam was used as the working electrode, a platinum wire electrode as the counter electrode, and a Ag/AgCl electrode as the reference electrode. The mass of deposited iron oxide films was controlled by adjusting the deposition time. The electrodeposition times used were 5, 10, 20 and 40 min, corresponding to mass loading amounts of FeOOH of 0.10, 0.15, 0.26 and 0.39 mg cm−2 Electrodeposition was done at a current density of 0.2 mA cm−2 under a constant N2 atmosphere and gentle stirring.
Double and triple layers of EGO–FeOOH coating were obtained by repeating the previous steps two or three times. After that, the samples were annealed at 400 °C in a muffle furnace for 1 hour with N2 protection, to obtain the Fe2O3 nanocrystals and reduced graphene oxide (RGO).
PDI-F solutions with a fixed concentration of 1.125 × 10−6 M in chloroform were exposed to different GF based materials for 7 days, without stirring. The GF based foam matrix acts as a trap for the molecules in solution, thereby capturing the dyes from the solution. The amount of this dye adsorption process was calculated by simply observing the decrease in the optical absorption intensity of the PDI-F in solution. The fraction of molecules adsorbed on graphite was calculated by monitoring the change in the maximum absorption peak (the molar attenuation coefficient of PDI-F is 79489 L mol−1 cm at 527 nm) for each sample.35 The specific surface area (SSA) of the GF related materials was estimated using the formula:
| SSA = Xm × N × A |
UV absorption data were recorded with a LAMBDA 650 UV/Vis spectrophotometer. For spectrophotometric measurements, all the solutions, with and without graphite, were filtered using “CHROMAFIL® XTRA H-PTFE-45/25” filters with a mesh size of 25 μm.
Electrochemical measurements
We used as working electrodes the multilayer GF samples prepared as described above, without any binder or conductive additive. Each sample had a diameter ∅ = 14 mm and was dried under vacuum overnight. Then, the electrodes were pressed at 1 MPa for 5 min, and assembled in an El-Cell® jar, using a two-electrode configuration with lithium foil as the counter electrode. The electrolyte was 1 M LiPF6 (LP30, BASF) in a 1:1 (w/w) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC), and the separator was a glass fibre membrane (Whatman; Grade: GF/A; ∅ 14 mm). Cell assembly was performed under an Ar atmosphere in an MBraun Labmaster SP dry box (H2O and O2 < 0.1 ppm) and all the electrochemical measurements were carried out by using Biologic VSP and Perkin Elmer VMP potentiostats/galvanostats; the electrochemical impedance spectra of the electrode in the charged stage were obtained in the range of 100 kHz–10 mHz with a perturbation amplitude of 5 mV.
Structural and electrical characterization
Scanning Electron Microscopy (SEM) images were obtained with a ZEISS LEO 1530 microscope equipped with a Schottky emitter, operated at 5 kV and secondary electrons were collected by means of an In-Lens detector. X-ray diffraction (XRD) measurements were collected with a PANalytical X'PertPro instrument in Bragg–Brentano reflection mode (λ = 0.1542 nm, X'Celerator detector). The mass of deposited Fe2O3 and EGO composites was measured by using a microbalance (CPA225D, Sartorius). X-ray photoemission spectroscopy (XPS) was performed exploiting an ultrahigh vacuum (UHV) apparatus (base pressure 1 × 10−10 mbar) using a non-monochromatic Mg Kα excitation source (XR-50, Specs) and a hemispherical energy analyser (Phoibos 100, Specs). Zeta potential determination was performed by using a NanoBrook OMNI (Brookhaven Instruments).The electrical resistance of the graphene foams was measured with a four-point probe system (Keithley 2700 multimeter). First, silver contacts were evaporated onto glass slides in a van der Pauw configuration, then the GF was placed in the centre and brought into contact with the geometry using silver paste on the four corners. Metallic probes were placed onto the evaporated silver contacts for the measurements to achieve a reproducible contact.
Electrochemical impedance spectroscopy (EIS) analysis
The EIS spectrum of the as-prepared electrodes was collected from fully charging the cells, after waiting 5 minutes for a stable open circuit voltage (OCV) before the EIS measurements. The EIS spectra were composed of two partially overlapped semicircles at the high and medium frequency region and a straight sloping line at the low frequency region. The pattern of EIS can be fitted by an equivalent circuit as shown in Fig. S11c.† In this case, Re is the internal resistance of the cell, which reflects a combined resistance of the electrolyte, separator, and electrodes. RSEI and CSEI are resistance and capacitance related to the SEI film on the surface of two electrodes from the high frequency region. Rct and Cdl are charge-transfer resistance and the corresponding double-layer capacitance from the medium frequency region. W is the Warburg impedance related to a combined effect of the solid-state diffusion of Li+ ions on the electrolyte–electrolyte interfaces, which is related to the sloping line at the low frequency region. The combination of Re and RSEI is considered as ohmic impedance, which reflects their ohmic characteristic. Meanwhile, the combination of Rct and Cdl is regarded as faradaic impedance.Electrochemical impedance spectroscopy (EIS) measurements were performed from 100 kHz to 10 mHz at the open-circuit potential, to further evaluate the electrochemical behaviours of the RGO–Fe2O3 coated anode materials. It was difficult to monitor the charge-transfer process in these cells due to the mixed state-of-lithiation for both graphite and Fe2O3 under the OCV conditions. Hereby, we only discuss about the ohmic behaviour of the above electrodes. As shown in the Nyquist plots in Fig. S11a,† the values of the SEI film resistance RSEI of the RGO–Fe2O3–GF, (RGO–Fe2O3)2–GF and (RGO–Fe2O3)3–GF were 24.1, 23.2 and 18.5 Ω respectively, which were lower than those of Fe2O3–GF (30.3 Ω) and pure GF (31.3 Ω) electrodes. The results indicate that the unique architecture of the RGO multilayer efficiently improves the electrical conductivity of the whole electrodes. After long-term cycling at 2C, there was a decrease in the electrode resistance on the RGO coated electrodes (Fig. S11b†), which might be due to the formation of better passivation layers on the surfaces of electrodes compared to the bare Fe2O3 electrode.
Results and discussion
Fig. 2a shows a typical SEM image of the macroporous GF networks synthesized by chemical vapour deposition (CVD) using a commercial nickel foam as a scaffold template. After etching away nickel in acidic solution, free-standing ultrathin GF with low mass per unit area (∼0.8 mg cm−2) and high electrical conductivity (10 S cm−1) was obtained. Due to the good mechanical properties of our GF, it was not necessary to apply any polymer protective coating before metal etching, as typically done with GO or RGO foams,25 which greatly simplified the whole fabrication process. The obtained graphitic foam formed a continuous hollow network structure with a layer thickness of around 18 nm (∼50 layers), and no collapsed areas or cracks were observed from the SEM image (Fig. S1†). | ||
| Fig. 2 (a) Representative SEM images of FeOOH–GF foam. (b–d) Zoom in of different FeOOH–GF foams obtained with different ratios of ethanol/water co-solvents. | ||
The multilayer functionalization process of the foam with Fe2O3 and EGO nanostructures was performed as schematised in Fig. 1 using:
(i) electrodeposition of iron oxyhydroxides from water/EtOH co-solvent solution.
(ii) Coating of EGO onto the FeOOH grafted foam.
(iii) Repetition of previous steps to create multilayer 3D structures.
(iv) Thermal annealing at 400 °C to convert FeOOH into Fe2O3 and EGO into reduced graphene oxide (RGO).
The initial FeOOH coating was deposited onto the GF matrix via anodic electrodeposition in 0.04 M CH3COONa and 0.01 M Fe(NH4)2(SO4)2.
A common problem in the functionalization of foams is the poor wetting of the inner pores by the solvent. For this, we used a co-solvent system (water and EtOH), instead of using destructive pre-treatment (e.g. O2 plasma) usually needed to ensure hydrophilicity of GF. Different ratios of ethanol (10 vol%, 50 vol% and 90 vol%) were mixed with water to improve the wettability of the graphitic foam and enhance the adhesion of deposits onto the GF electrode surfaces. The morphology of the nanostructures after deposition with different ethanol/water ratios was monitored by scanning electron microscopy (SEM), as shown in Fig. 2b–d. FeOOH deposited with a low volume of ethanol (10 vol%) showed some coated areas. However, the coating of FeOOH layers was not uniform on the GF network, as the inner branches of the foam were only partially covered with the FeOOH layer (Fig. 2b). On increasing the ethanol content to 50 vol% a homogeneous coating of FeOOH perfectly covered both the outer and the inner part of the GF networks. The FeOOH formed vertical nanowalls with nanometric thickness, roughly oriented perpendicular to the substrate (Fig. 2c). The uniform coating could be attributed to the lower surface tension of the mixture of solvents, which improved the wetting of the GF foam in aqueous solution and helped the diffusion of iron ions into the inner part of the networks. However, a further increase of the ethanol fraction (90 vol%) was not beneficial, giving poor solubility of the inorganic salts and thus poor deposition of FeOOH on the foams (Fig. 2d).
Using the best EtOH concentration (50%), we then optimized the amount of the material deposited on the substrate by varying the electrodeposition time. Only amorphous island-like structures, due to the nucleation of the iron oxyhydroxide crystals, were observed on the GF surface after 5 min deposition (Fig. 3a, loading 0.10 mg cm−2). Increasing the electrodeposition time to 10 min led to a homogeneous coating of FeOOH, assembled in nanoscopic lamellar structures (Fig. 3c and S2a, b,† loading 0.15 mg cm−2). However, the maximum loading achievable was limited; for example long electrodeposition time (40 min) caused cracks in the FeOOH layer (Fig. 3b, loading 0.39 mg cm−2), as we also observed in previous work.25 The FeOOH could then be converted to nano-porous Fe2O3 by heating at 400 °C under an N2 atmosphere.25 The lamellar structure was well preserved upon conversion to Fe2O3 (Fig. S2c†).
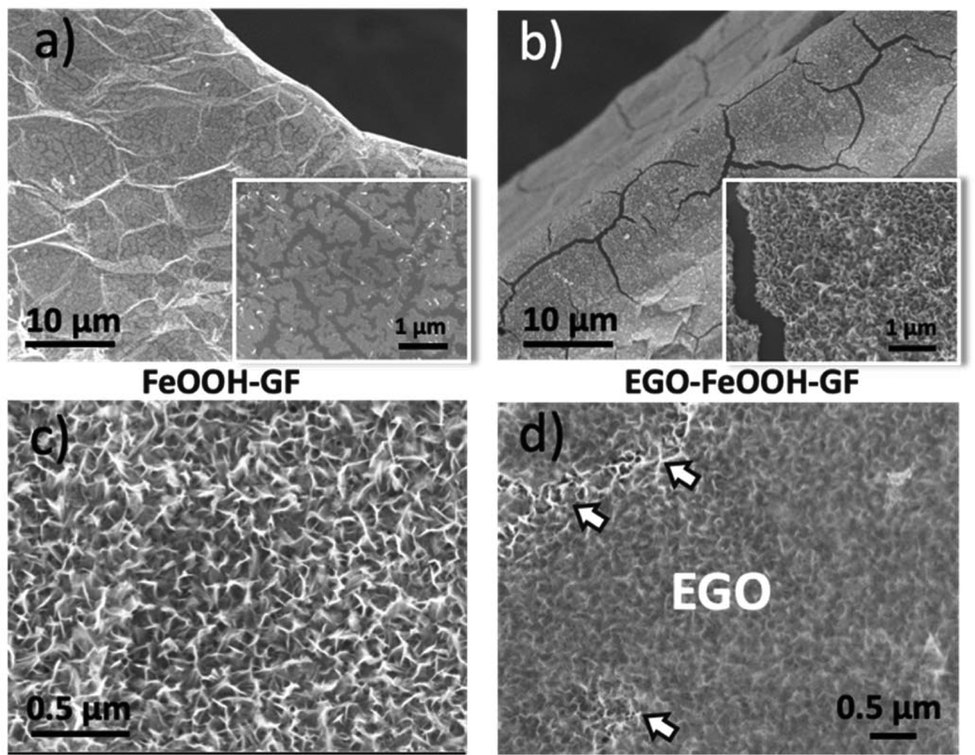 | ||
| Fig. 3 (a, b) SEM image of CVD–GF with low and high loading amounts of FeOOH: (a) 0.10 mg cm−2, (b) 0.39 mg cm−2. The insets show higher magnification of the surface. (c) FeOOH nano-walls grown on GF with a moderate loading (0.15 mg cm−2) showing a uniform nanowall morphology. (d) The same nano-walls of (c), coated with EGO. Brighter areas (shown by white arrows) indicate holes in the EGO coating. | ||
In order to improve the adhesion of Fe2O3 on the GF surface, we coated it with a protective layer of EGO drop-cast from dimethylformamide (DMF) solutions (Fig. 3d). The EGO layer adhered efficiently on the iron hydroxide thanks to the electrostatic attractions between the two materials; measured zeta potential in DMF for EGO was ζ = −30 mV, while for FeOOH it was ζ = +32 mV.
The EGO coating layer had multifunctional roles in the composite structures, acting as an electrically conductive layer, a binder and a spacer, favouring further FeOOH adsorption. During thermal annealing of FeOOH, EGO could be reduced to conductive EGO (RGO).25 Its crumpled structure, analogue to the conventional RGO, can allow unimpeded flow of ions while ensuring mechanical containment and good electrical connection.
Noteworthily, the coating steps could be repeated multiple times on the same substrate. On performing the coating steps x times we could obtain (EGO–FeOOH)x multilayer structures, then transformed into (RGO–Fe2O3)x by annealing. The layered sandwich-structures could be clearly identified by SEM on the cross section of the GF networks (see Fig. 4 and S2d†). Each Fe2O3 layer was ca. ∼100 nm thick, with some variability due to the curved structure of the foam. In comparison, the direct growth of FeOOH by repeated electrodeposition without EGO spacers caused the formation of cracks on the nanowall arrays. Obviously, the introduction of the EGO layer could increase the loading amount of mesoporous iron oxide, and yield a crack-free, homogeneous multilayer architecture.
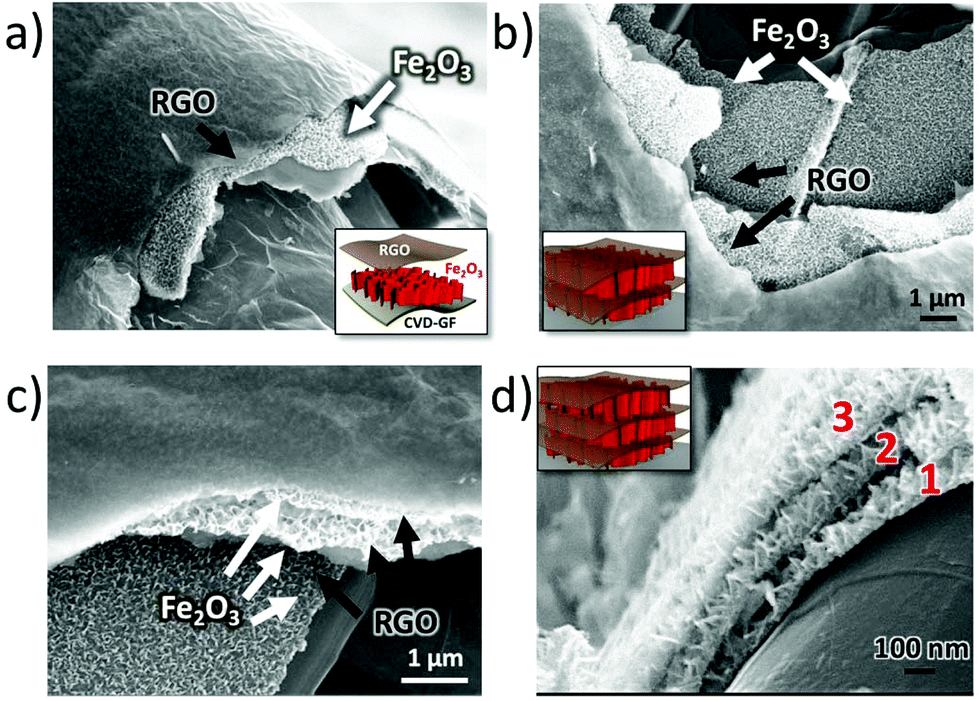 | ||
| Fig. 4 SEM images of GF coated with an increasing number of Fe2O3 and RGO layers; (a) single RGO–Fe2O3 coating; (b) double (RGO–Fe2O3)2 coating; (c, d) triple (RGO–Fe2O3)3 coating. Red numbers in (d) indicate the well-separated different Fe2O3 layers formed by vertical nanowalls. Insets show the schematic representation of the different layers in the sandwich structure. | ||
To better understand the surface morphology changes due to multiple coatings, the specific surface areas (SSA) of the pure GF and the RGO/Fe2O3 functionalized GF were measured. Standard SSA measurements in gas could not be used due to the low SSA of the mesoscopic foams, thus we measured SSA by adsorption of a commercial organic dye, as described in ref. 35, 36 and in exp. details (see Fig. S3 and Table S1†). Pure GF, Fe2O3/GF and single, double, and triple layer RGO coated electrodes gave SSA equal to 2.3, 5.7, 7.3, 10.9 and 11.6 m2 g−1, respectively. The initial SSA value was quite low, as expected due to the mesoscopic structure of the graphitic foam, but the increasing trend of the surface area indicates that the grafting of Fe2O3 nano-wall patterns and further coating of EGO multiple layers could efficiently improve up to 5 times the available surface area on the GF.
The chemical composition of iron oxide and RGO functional layers was confirmed by XPS analysis of the binding energy (B.E.) of C 1s and Fe 2p spectra (see in Fig. 5 and S4a†). The chemical state of Fe 2p was used to detect the presence of iron oxide:37 no FeO (Fe2+) was present (Fe 2p3/2 B.E. = 709.6 eV), while the peak values of Fe2O3 (711.0–711.6 eV) and FeOOH (711.9 eV) were close to the experimental value reported in the literature.37,38 The O 1s peak (see in Fig. S4a†) was fitted by using two main components and a shoulder: FeO (530.1 eV), Fe–OH (531.4 eV) and mixed oxides (532.8 eV).38 After thermal annealing treatment at 400 °C, there was a decrease of the Fe–OH signal from 26% to 12% of the total O 1s signal (Fig. S4a†), indicating a conversion of FeOOH to Fe2O3.
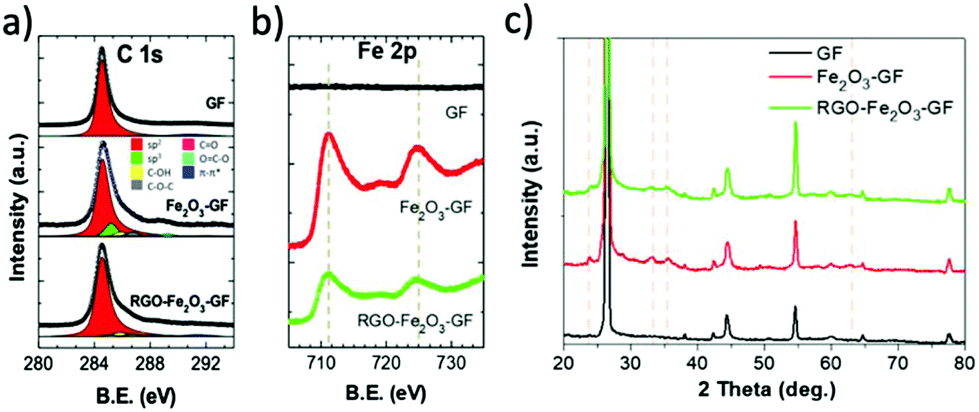 | ||
| Fig. 5 XPS (a) C 1s and (b) Fe 2p spectrum of bare GF, Fe2O3–GF and RGO–Fe2O3–GF composites; (c) XRD patterns of bare GF, Fe2O3–GF and RGO–Fe2O3–GF composites. | ||
The C 1s spectrum was measured on bare GF, Fe2O3–GF and RGO–Fe2O3–GF samples; a sharp peak was observed in all the samples at 284.6 eV (Fig. 5a) due to the sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The O/C ratio was calculated from the fit of the C 1s spectrum with CC sp2 (asymmetric red peak), C–C, C–OH, C–O–C, CO and O–CO components. More details on the C 1s fitting procedure are in ref. 39. After Fe2O3 coating, the overall O/C ratio increased from 0.02 to 0.10, but the intensity of C 1s drastically decreased due to coverage of the iron oxide layer (the oxygen contribution from the substrate or Fe2O3 was removed from the estimate, and only the carbon–oxygen bond was considered). The C 1s spectra of the pristine EGO and the EGO after annealing (RGO) are shown in Fig. S4b.† The O/C ratio decreased significantly from 0.20 ± 0.01 to 0.07 ± 0.01; moreover, the chemical structure of EGO evolved from a C–O defect rich structure to a mainly sp2 aromatic carbon structure with few C–O defects. The C 1s peak of RGO was similar to the one of RGO–Fe2O3–GF.
C bond. The O/C ratio was calculated from the fit of the C 1s spectrum with CC sp2 (asymmetric red peak), C–C, C–OH, C–O–C, CO and O–CO components. More details on the C 1s fitting procedure are in ref. 39. After Fe2O3 coating, the overall O/C ratio increased from 0.02 to 0.10, but the intensity of C 1s drastically decreased due to coverage of the iron oxide layer (the oxygen contribution from the substrate or Fe2O3 was removed from the estimate, and only the carbon–oxygen bond was considered). The C 1s spectra of the pristine EGO and the EGO after annealing (RGO) are shown in Fig. S4b.† The O/C ratio decreased significantly from 0.20 ± 0.01 to 0.07 ± 0.01; moreover, the chemical structure of EGO evolved from a C–O defect rich structure to a mainly sp2 aromatic carbon structure with few C–O defects. The C 1s peak of RGO was similar to the one of RGO–Fe2O3–GF.
Noteworthily, after subsequent coating of EGO and thermal annealing (RGO–Fe2O3–GF), the absolute intensity of carbon increased. The O/C ratio of RGO–Fe2O3–GF was similar to the O/C of pure RGO under the same annealing conditions, suggesting that the thermal treatment efficiently reduced EGO into graphene (RGO). We observed two peaks in the Fe 2p spectra, 724.7 and 711.1 eV, in both Fe2O3–GF and RGO–Fe2O3–GF samples, corresponding to the Fe 2p1/2 and Fe 2p3/2 states (Fig. 5b). Such peaks are characteristic of ferric (Fe3+) compounds, belonging to the Fe2O3 functional nanocomposites, and were not observed on the bare GF samples.
Further characterization of the iron oxide structures was obtained by XRD. The weak and broad peaks of FeOOH (see in Fig. S4c†) could be indexed to the orthorhombic phase of α-FeOOH with low-crystalline structures (goethite, JCPDS 29-0713).29 After the annealing process (Fig. 5c), a rhombohedral phase of α-Fe2O3 (hematite, JCPDS 33-0664) was observed with the characteristic peaks (2θ = 24.1°, 33.2°, 35.6°, 40.9°, 49.5°, 62.4°, and 64.1°). The other diffraction peaks at 26°, 44° and 55° observed in all the samples were due to the graphitic carbon matrix (JCPDS 12-0212). EDS elemental mapping images (Fig. S5 and S6†) also confirmed a homogeneous distribution of carbon, iron and oxygen elements on both Fe2O3 and EGO–Fe2O3 coated foam composites.
The multilayer foams could be compressed into thin films and used as anodes in half-cells with lithium foil as the counter electrode (see SEM images in Fig. S7 and Table S2†). Cyclic voltammograms (CV) of Fe2O3–GF and RGO–Fe2O3–GF electrodes were recorded between 0.05 and 3.2 V (vs. Li/Li+) at a scan rate of 1 mV s−1. The two step reversible Li+ insertion/extraction process with α-Fe2O3 (Fe2O3 + 6Li+ + 6e− ⇄ 2Fe + 3Li2O) was clearly identified during the first two cycles (see Fig. 6) for both electrodes. Two cathodic peaks located at 0.73 and 1.65 V and the corresponding anodic ones at 1.61 and 2.35 V were observed during the first cycle. The cathodic peak at around 0.73 V was due to the two-step reversible lithium insertion/extraction process within Fe2O3 (the reduction of Fe3+ and Fe2+ to Fe0). The cathodic peak at 1.65 V disappeared after the first cycle, likely due to the irreversible formation of LixFe2O3. Another pair of redox peaks located at 0.14 and 0.40 V came from the intercalation/de-intercalation of lithium ions within the graphite foam. The CV curve of the RGO–Fe2O3–GF electrode showed similar results to that of Fe2O3–GF, indicating that the RGO coating did not hinder Li ion transport to/from the electrode surface.
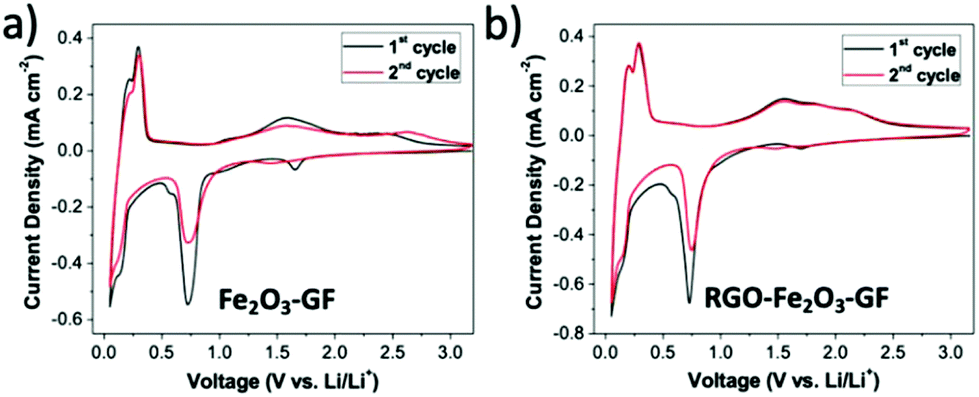 | ||
| Fig. 6 Cyclic voltammetry curves of (a) Fe2O3–GF composites and (b) RGO–Fe2O3–GF composites for the 1st and 2nd cycles at a scan rate of 1 mV s−1. | ||
Galvanostatic charge–discharge cycling performances of the as-prepared Fe2O3–GF, RGO–Fe2O3–GF, (RGO–Fe2O3)2–GF, and (RGO–Fe2O3)3–GF electrodes were performed at various C rates between 0.02 and 3 V (vs. Li/Li+) (Table S3†). The presence of the sandwich layer structures had a significant influence on the electrochemical performance of these anode materials.
First, we observed that the areal capacity of uncoated Fe2O3–GF electrodes at 0.2 C rapidly decreased from 1052 μA h cm−2 (coulombic efficiency: 77.3%) to 473 μA h cm−2 after 5 cycles and to 211 μA h cm−2 after 25 cycles (Fig. 7a and S9a†). Longer cycling of the Fe2O3–GF electrode at 2 C also resulted in serious decay of the specific capacity. This is because of the detachment of Fe2O3 from the conductive GF substrate, due to the large volume changes during the Li+ insertion/extraction process. The capacity of Fe2O3–GF after 1000 cycles was only 27% of the initial value, as bad as the one of bare GF.
 | ||
| Fig. 7 Charge–discharge profiles of (a) Fe2O3–GF and (b) (RGO–Fe2O3)3–GF electrodes at 0.2 C. (c) Cycling performance of different electrodes at a charge rate of 2 C. (d) Discharge rate capability of the different electrodes at different discharge rates, after 1000 cycles at 2 C. | ||
Conversely, the RGO-coated electrodes demonstrated better and more stable electrochemical performance. The initial capacity gradually increased to 1070 μA h cm−2 for the single RGO–Fe2O3 layer (coulombic efficiency 87.0%); to 1182 μA h cm−2 for the bilayer (coulombic efficiency 87.7%); and to an excellent 1403 μA h cm−2 for the tri-layer (coulombic efficiency 93.1%), see Fig. 7b, S8, S9a, and b.†
All the RGO coated electrodes maintained a high capacity after 1000 cycles at 2 C (Fig. 7c). Single, double and triple layers of RGO–Fe2O3 showed capacities of 650 μA h cm−2, 976 μA h cm−2 and 1175 μA h cm−2, with capacity retention of 61%, 83% and 84%, respectively (Fig. 7d). Given that the thickness of these thin films was ≈6.0 μm, the volumetric energy and power density in the half-cell system were significantly high. In particular, with the thicker (RGO–Fe2O3)3–GF electrode, a specific energy density of 176 μW h cm−2 μm−1 at 0.2 C (144 μA cm−2) and a specific power density of 544 μW cm−2 μm−1 at 2 C (1445 μA cm−2) were delivered after long-term cycling. Thus, our multilayer RGO–Fe2O3 based electrode showed an excellent performance in half cell, as compared to previously reported 3D thin film electrodes (10–100 μW h cm−2 μm−1).40–42 Noteworthily, the capacity increased gradually during the cycling test for all the samples (Fig. 7c). This behaviour was observed in other metal oxide/graphene hybrid materials10,43 and might be explained by the gradual activation of the iron oxide nanoparticles in the porous structure during the cycling measurements.14
We also studied by SEM the changes in the material structure after extensive charge/discharge cycling tests (Fig. S10†). Cycling of uncoated Fe2O3–GF composites (1000 cycles) showed an extensive detachment of Fe2O3, causing the capacity to drop close to the one of bare GF. On the other hand, the coated RGO–Fe2O3–GF electrodes showed well preserved mesoporous iron oxide structures. This indicates that the RGO sheets can act as a binder and a flexible spacer to accommodate the volume expansion of Fe2O3 layers without detachment during cycling.
The significantly enhanced cyclability of the RGO–Fe2O3–GF anodes can be attributed to the collective and synergistic interaction between the Fe2O3 layers and RGO sheets at the nanoscale. RGO not only acts as a conductive additive, but works as a binder to promote the adhesion of additional iron oxide nanoflakes on the foam. More importantly, RGO is an excellent flexible spacer to achieve high loading of crack-free Fe2O3 mesoporous structures. The resulting hierarchical sandwich architecture shows uniformly distributed Fe2O3 nanowall patterns and guarantees the extraordinary cycling stability of these Li ion batteries.
Conclusion
In summary, we demonstrated a fast and highly efficient electrochemical approach for the uniform deposition of Fe2O3 nanowall structured layers on graphene foam substrates. The low-cost of these electrodeposition techniques would allow the facile upscaling from the beaker level to large scale industrial reactors. We showed the beneficial role of a protective thin layer of EGO to improve the cycling stability of the Fe2O3–GF composite electrodes. Sandwich structures of multilayer RGO–Fe2O3 could be successfully fabricated with improved areal capacitance and good cycling stability. We envisage that the versatility of such multilayer functionalization on porous substrates could allow the fabrication of other multilayer graphene/metal oxide sandwich structures for energy storage applications; while we used here the same material (Fe2O3) for each layer, more complex structures with different materials on each layer could also be produced, to obtain more tunable or exotic electrochemical properties.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The research leading to these results has received funding from the European Union's Horizon 2020 research and innovation programme under GrapheneCore2 785219 – Graphene Flagship and grant agreement no. 642196 – Marie-Curie ITN- iSwitch.References
- Z. P. Chen, W. C. Ren, L. B. Gao, B. L. Liu, S. F. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- M. Mecklenburg, A. Schuchardt, Y. K. Mishra, S. Kaps, R. Adelung, A. Lotnyk, L. Kienle and K. Schulte, Adv. Mater., 2012, 24, 3486–3490 CrossRef CAS PubMed.
- H. Hu, Z. B. Zhao, W. B. Wan, Y. Gogotsi and J. S. Qiu, Adv. Mater., 2013, 25, 2219–2223 CrossRef CAS PubMed.
- S. Drieschner, M. Weber, J. Wohlketzetter, J. Vieten, E. Makrygiannis, B. M. Blaschke, V. Morandi, L. Colombo, F. Bonaccorso and J. A. Garrido, 2D Mater., 2016, 3, 045013 CrossRef.
- R. Mo, D. Rooney, K. Sun and H. Y. Yang, Nat. Commun., 2017, 8, 13949 CrossRef CAS PubMed.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- B. H. Min, D. W. Kim, K. H. Kim, H. O. Choi, S. W. Jang and H. T. Jung, Carbon, 2014, 80, 446–452 CrossRef CAS.
- M. B. Sassin, A. N. Mansour, K. A. Pettigrew, D. R. Rolison and J. W. Long, ACS Nano, 2010, 4, 4505–4514 CrossRef CAS PubMed.
- K. Parvez, R. J. Li, S. R. Puniredd, Y. Hernandez, F. Hinkel, S. H. Wang, X. L. Feng and K. Mullen, ACS Nano, 2013, 7, 3598–3606 CrossRef CAS PubMed.
- J. S. Luo, J. L. Liu, Z. Y. Zeng, C. F. Ng, L. J. Ma, H. Zhang, J. Y. Lin, Z. X. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136–6143 CrossRef CAS PubMed.
- X. M. Feng, Y. Zhang, J. H. Zhou, Y. Li, S. F. Chen, L. Zhang, Y. W. Ma, L. H. Wang and X. H. Yan, Nanoscale, 2015, 7, 2427–2432 RSC.
- Y. Bai, W. Q. Wang, R. R. Wang, J. Sun and L. Gao, J. Mater. Chem. A, 2015, 3, 12530–12538 RSC.
- Z. S. Wu, Y. Sun, Y. Z. Tan, S. B. Yang, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 19532–19535 CrossRef CAS PubMed.
- J. Y. Ji, L. L. Zhang, H. X. Ji, Y. Li, X. Zhao, X. Bai, X. B. Fan, F. B. Zhang and R. S. Ruoff, ACS Nano, 2013, 7, 6237–6243 CrossRef CAS PubMed.
- T. Q. Lin, I. W. Chen, F. X. Liu, C. Y. Yang, H. Bi, F. F. Xu and F. Q. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS PubMed.
- B. Wang, S. M. Li, X. Y. Wu, J. H. Liu and W. M. Tian, Phys. Chem. Chem. Phys., 2016, 18, 908–915 RSC.
- Z. Y. Xia, C. Arbizzani, L. Ortolani, V. Morandi, V. Bellani, G. Giambastiani, M. Gazzano and V. Palermo, FlatChem, 2017, 3, 8–15 CrossRef CAS.
- Q. M. Su, D. Xie, J. Zhang, G. H. Du and B. S. Xu, ACS Nano, 2013, 7, 9115–9121 CrossRef CAS PubMed.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS PubMed.
- W. F. Chen, S. R. Li, C. H. Chen and L. F. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- S. Guo, G. K. Zhang, Y. D. Guo and J. C. Yu, Carbon, 2013, 60, 437–444 CrossRef CAS.
- D. Z. Chen, G. S. Wang, S. He, J. Liu, L. Guo and M. S. Cao, J. Mater. Chem. A, 2013, 1, 5996–6003 RSC.
- S. K. Liu, Z. X. Chen, K. Xie, Y. J. Li, J. Xu and C. M. Zheng, J. Mater. Chem. A, 2014, 2, 13942–13948 RSC.
- J. S. Cho, Y. J. Hong and Y. C. Kang, ACS Nano, 2015, 9, 4026–4035 CrossRef CAS PubMed.
- Z. Y. Xia, D. Wei, E. Anitowska, V. Bellani, L. Ortolani, V. Morandi, M. Gazzano, A. Zanelli, S. Borini and V. Palermo, Carbon, 2015, 84, 254–262 CrossRef CAS.
- X. Hu, M. Ma, M. Zeng, Y. Sun, L. Chen, Y. Xue, T. Zhang, X. Ai, R. G. Mendes, M. H. Ruemmeli and L. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 22527–22533 CrossRef CAS PubMed.
- M. Cohen and V. Markovac, J. Electrochem. Soc., 1967, 114, 674–678 CrossRef.
- R. L. Spray and K.-S. Choi, Chem. Mater., 2009, 21, 3701–3709 CrossRef CAS.
- S. H. Jiao, L. F. Xu, K. L. Hu, J. J. Li, S. Gao and D. S. Xu, J. Phys. Chem. C, 2010, 114, 269–273 CrossRef CAS.
- O. Zandi, A. R. Schon, H. Hajibabaei and T. W. Hamann, Chem. Mater., 2016, 28, 765–771 CrossRef CAS.
- M.-S. Wu, R.-H. Lee, J.-J. Jow, W.-D. Yang, C.-Y. Hsieh and B.-J. Weng, Electrochem. Solid-State Lett., 2009, 12, A1–A4 CrossRef CAS.
- Z. Y. Xia, S. Pezzini, E. Treossi, G. Giambastiani, F. Corticelli, V. Morandi, A. Zanelli, V. Bellani and V. Palermo, Adv. Funct. Mater., 2013, 23, 4684–4693 CrossRef CAS.
- P. Yu, Z. M. Tian, S. E. Lowe, J. C. Song, Z. R. Ma, X. Wang, Z. J. Han, Q. L. Bao, G. P. Simon, D. Li and Y. L. Zhong, Chem. Mater., 2016, 28, 8429–8438 CrossRef CAS.
- Z. Y. Xia, G. Giambastiani, C. Christodoulou, M. V. Nardi, N. Koch, E. Treossi, V. Bellani, S. Pezzini, F. Corticelli, V. Morandi, A. Zanelli and V. Palermo, ChemPlusChem, 2014, 79, 439–446 CrossRef CAS.
- A. Liscio, K. Kouroupis-Agalou, A. Kovtun, E. Gebremedhn, M. El Garah, W. Rekab, E. Orgiu, L. Giorgini, P. Samorì, D. Beljonne and V. Palermo, ChemPlusChem, 2017, 82, 358–367 CrossRef CAS.
- P. T. Hang and G. W. Brindley, Clays Clay Miner., 1970, 18, 203–212 CrossRef.
- P. Mills and J. L. Sullivan, J. Phys. D: Appl. Phys., 1983, 16, 723–732 CrossRef CAS.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- A. Kovtun, D. Jones, S. Dell'Elce, E. Treossi, A. Liscio and V. Palermo, Carbon, 2019, 143, 68–275 CrossRef.
- Y. Yang, X. J. Fan, G. Casillas, Z. W. Peng, G. D. Ruan, G. Wang, M. J. Yacaman and J. M. Tour, ACS Nano, 2014, 8, 3939–3946 CrossRef CAS PubMed.
- J. H. Pikul, H. G. Zhang, J. Cho, P. V. Braun and W. P. King, Nat. Commun., 2013, 4, 1732 CrossRef PubMed.
- W. H. Lai, Y. Wang, Z. W. Lei, R. H. Wang, Z. Y. Lin, C. P. Wong, F. Y. Kang and C. Yang, J. Mater. Chem. A, 2018, 6, 3933–3940 RSC.
- M. F. Hassan, Z. P. Guo, Z. X. Chen and H. K. Liu, Mater. Res. Bull., 2011, 46, 858–864 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr09195a |
| This journal is © The Royal Society of Chemistry 2019 |