
A comparative study of pomegranate Sb@C yolk–shell microspheres as Li and Na-ion battery anodes†
Junhua
Song
ab,
Dongdong
Xiao
a,
Haiping
Jia
a,
Guomin
Zhu
a,
Mark
Engelhard
 a,
Biwei
Xiao
a,
Shuo
Feng
b,
Dongsheng
Li
a,
David
Reed
a,
Vincent L.
Sprenkle
a,
Yuehe
Lin
*b and
Xiaolin
Li
*a
a,
Biwei
Xiao
a,
Shuo
Feng
b,
Dongsheng
Li
a,
David
Reed
a,
Vincent L.
Sprenkle
a,
Yuehe
Lin
*b and
Xiaolin
Li
*a
aPacific Northwest National Laboratory, 902 Battelle Boulevard Richland, WA 99354, USA. E-mail: xiaolin.Li@pnnl.gov
bSchool of Mechanical and Materials Engineering, Washington State University, Pullman, WA 99164, USA. E-mail: yuehe.lin@wsu.edu
First published on 16th November 2018
Abstract
Alloy-based nanostructure anodes have the privilege of alleviating the challenges of large volume expansion and improving the cycling stability and rate performance for high energy lithium- and sodium-ion batteries (LIBs and SIBs). Yet, they face the dilemma of worsening the parasitic reactions at the electrode–electrolyte interface and low packing density for the fabrication of practical electrodes. Here, pomegranate Sb@C yolk–shell microspheres were developed as a high-performance anode for LIBs and SIBs with controlled interfacial properties and enhanced packing density. Although the same yolk–shell nanostructure (primary particle size, porosity) and three-dimensional architecture alleviated the volume change induced stress and swelling in both batteries, the SIBs show 99% capacity retention over 200 cycles, much better than the 78% capacity retention of the LIBs. The comparative electrochemical study and X-ray photoelectron spectroscopy characterization revealed that the different SEIs, besides the distinct phase transition mechanism, played a critical role in the divergent cycling performance.
1. Introduction
Li- and Na-ion batteries (LIBs and SIBs) are the twin analogies explored in the 1980s for high energy electrochemical storage devices.1,2 LIBs continue their success today by supporting a variety of electrified portable devices and transportation applications.3 Meanwhile, the rapidly expanding integration of renewable energy has stirred up the quest for more economically viable energy storage systems than LIBs. Although falling short of the high energy density, SIBs, accredited to their natural abundance of the precursor, uniform geological distribution and low cost, are still regarded as one of the leading rechargeable energy storage technologies for grid-scale storage systems.4–6 To better serve the purposes in their own respective applications, LIBs and SIBs need to acquire higher energy density and better cyclability than the corresponding state-of-the-art devices, respectively. Using high capacity alloy-type anodes is one of the promising ways to boost the energy density of LIBs and SIBs. Upon full lithiation or sodiation, alloys and their intermetallic compounds, such as FeS2, SnO2, Sn, SnS, Sn4P3, Si, Ge, Sb and NiSb, can deliver 2–10 times higher specific capacity than natural graphite or hard carbon anodes.7–16 Integrated with proper cell design/engineering, the energy density of the full cells with alloy anodes can be greatly improved.Antimony (Sb) stands out with a high specific capacity and reasonable alloying/dealloying potential as an anode for both Li and Na-ion batteries.17 Interestingly, the cycling stability of the Sb anode is different in LIBs and SIBs, despite the many similarities shared between these two neighboring alkali ions.18 Early research on the cyclability of Sb has pointed to the different phase transition pathways between the Na and Li systems, leading to a dissimilar rate of structural breakdown upon volume change.18 Yet, the bulk Sb without any nanostructures induces a large stress variation in the electrode integrity and the unstable solid-electrolyte-interphase (SEI) layer, susceptible to the large volume change and severe interphasial reactions with the electrolyte during cycling. Yolk–shell structured materials can effectively alleviate the volume change of alloy-type anode materials and provide electrode, mechanical and electrical integrity.19,20 Recently, we have developed a Sb@C yolk–shell structure, which demonstrated good cycling stability and rate performance for Na-ion intercalation.21 Here, we further advance the technology to prepare pomegranate Sb@C yolk–shell microspheres (Sb@C MSs), which are not only a high density anode material for LIBs and SIBs, but can also act as a model material to systematically examine the stability difference for alloying/dealloying with Li and Na. The Sb@C MSs have an appropriately controlled porosity to accommodate the volume change and maintain a high packing density of ∼1.1 g cm−3, which is ∼40% of the value of Sb particles of ∼70 micrometers (∼2.63 mg cm−3) and 30% larger than the Sb@C yolk–shell structures. The anode exhibits good electrochemical performance in terms of specific capacity and rate capability for both Li-ion and Na-ion cells. The specific capacities for Li- and Na-ion batteries at a low current density of 50 mA g−1 are close to the theoretical values of 935 mA h g−1 and 637 mA h g−1. It can still deliver 633 mA h g−1 and 441 mA h g−1, respectively, at a current density of 5 A g−1. The SIBs show incredibly good cycling stability with 99% capacity retention over 200 cycles at a current density of 200 mA g−1, while the LIBs demonstrated ∼78% capacity retention under similar conditions. The different electrochemical behaviors in Li and Na cells are systematically investigated to reveal their alloying mechanism, cell impedance evolution, SEI composition and thickness during cycling.
2. Experimental section
2.1 Synthesis of pomegranate Sb@C yolk–shell microspheres (Sb@C MSs)
In a typical synthesis, ∼400 mg of Sb2O3 nanoparticles (Sigma-Aldrich) was dispersed in 200 ml of Tris-buffer solution (10 mM, pH = 8.5, Biotech) and sonicated for 30 minutes. Then 400 mg of dopamine (Sigma-Aldrich) was added into the above solution and mechanically stirred overnight to obtain polydopamine (PDA) coated Sb2O3 nanoparticles (Sb2O3@PDA). The resultant Sb2O3@PDA core–shell structure was then dispersed in 200 ml of polyvinylpyrrolidone (PVP, average molecular weight 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich) solution (10 mg ml−1) and stirred for 6 h. Excess PVP was removed by washing the Sb2O3@PDA with distilled water twice. To coat the Sb2O3@PDA with SiO2, the PVP functionalized Sb2O3@PDA was dispersed in a 400 ml ethanol/water solution (4:1 by volume) with the addition of 4 ml NH4OH (28–30%, Sigma-Aldrich) and 1.2 ml tetraethyl orthosilicate (TEOS, 98%, Sigma-Aldrich) in sequence. The resultant Sb2O3@PDA@SiO2 was washed several times and re-dispersed to form a 40 mg ml−1 aqueous solution.
000, Sigma-Aldrich) solution (10 mg ml−1) and stirred for 6 h. Excess PVP was removed by washing the Sb2O3@PDA with distilled water twice. To coat the Sb2O3@PDA with SiO2, the PVP functionalized Sb2O3@PDA was dispersed in a 400 ml ethanol/water solution (4:1 by volume) with the addition of 4 ml NH4OH (28–30%, Sigma-Aldrich) and 1.2 ml tetraethyl orthosilicate (TEOS, 98%, Sigma-Aldrich) in sequence. The resultant Sb2O3@PDA@SiO2 was washed several times and re-dispersed to form a 40 mg ml−1 aqueous solution.
The emulsion was prepared by adding 1 g of Hypermer emulsifier (Hypermer™ 2524, Croda, USA) into ∼400 ml of 1-octadecene (ODE) and stirring for 20 minutes to form a homogeneous solution. The Sb2O3@PDA@SiO2 precursor solution (∼2 ml) was then mixed with 8 ml of ODE solution and emulsified using a homogenizer for 1 minute. The micron sized Sb2O3@PDA@SiO2 was obtained after water evaporation at 95 °C for 6 h followed by several washing cycles with petroleum ether. To stabilize the structure, the micron sized Sb2O3@PDA@SiO2 was further coated with another layer of PDA. Finally, the Sb@C yolk–shell MSs were obtained by thermal treatment of the sample at 480 °C in Ar/H2 (5%) for 1 h and subsequent HF etching to remove the SiO2 layer and Sb2O3 residual.
2.2 Characterization
Crystal structure characterization of the cycled electrode was carried out on a Rigaku MiniFlex 600 X-ray diffraction (XRD) instrument operating at 40 kV accelerating voltage and a current of 15 mA. The morphological and elemental analyses of the sample were conducted on a field emission scanning electron microscope (SEM) with an energy dispersive spectroscope (EDS) and an electron transmission microscope (Technai G2 F20, FEI Company) equipped with a scanning transmission electron microscopy (STEM) unit. X-ray photoelectron spectroscopy (XPS) was carried out on the cycled electrode in the desodiated state using a Physical Electronics Quantera Scanning X-ray Microprobe. A focused monochromatic Al Kα X-ray (1486.7 eV) source was used for excitation. The packing density analysis was carried out by adding equally weighted Sb@C yolk–shell structure, Sb@C MSs and bulk Sb (200 mesh, Alfa-Aesar) powders into glass tubes and shaking for 30 minutes using an electrical vibrator. The volumetric density was calculated based on the mass and volume of the powders.2.3 Electrochemical testing
The electrode of the pomegranate Sb@C MS yolk–shell material was prepared by slurry casting the mixture of the active material, Super-P and sodium carboxymethyl cellulose (CMC) with a mass ratio of 60:20:20. The slurry was then casted on copper foil and dried under vacuum at 70 °C for 12 h. The half-cell (2032 coin cell from MTI Corp) was assembled in an argon-filled glove box with Celgard 3501 as the separator and sodium foil as the counter electrode. The electrolyte was 1 M NaClO4 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:2 by weight) with 10 wt% fluoroethylene carbonate (FEC) and the amount was maintained at 100 μL unless otherwise mentioned. The average active material loading was ∼1 to 2 mg cm−2. The electrodes were tested on an Arbin BT-2000 battery tester at a charge–discharge potential range of 0.02–2.0 V vs. Na+/Na and vs. Li+/Li for SIBs and LIBs, respectively. The capacity was calculated based on the weight of the Sb@C MS composite. The internal resistance of the cell was measured using the galvanostatic intermittent titration technique (GITT) by applying a pulse current with a duration of 10 minutes, followed by 30 minutes pause time. The electrochemical impedance spectra (EIS) were recorded on an impedance analyzer (Solatron Analytical).
3. Results and discussion
The Sb@C MSs were synthesized through a micro-emulsion method assisted controlled reduction and etching of the Sb2O3@C@SiO2 core–shell structure. As depicted in the schematic illustration in Fig. 1A, the Sb2O3@PDA@SiO2 core–shell structure was prepared as primary particles by coating commercial Sb2O3 nanoparticles with layers of polydopamine and silica in sequence. Then they were assembled into microspheres, partially reduced and etched with HF forming microspheres of the Sb@C yolk–shell structure. The transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) images in Fig. S1† were taken on a typical Sb2O3@PDA@SiO2 particle. The Sb, C, N, and Si elemental mapping (Fig. S1†) clearly revealed the uniform coating layers of polydopamine and SiO2 on the Sb2O3 particles. The Sb@C yolk–shell microspheres were obtained by drying the emulsified micro-droplet of Sb2O3@PDA@SiO2 in a water-in-oil system, carbonization of the polymer, thermal reduction of the Sb2O3 into Sb and subsequent etching of SiO2 and Sb2O3. By controlling the SiO2 layer thickness and reduction temperature, the resultant Sb@C MSs consist of myriads of yolk–shell particles that allow the nano Sb to expand within the electrically conductive carbon shell. The obtained Sb@C MSs possess a similar crystallographic structure and Sb content as in our previous report (Fig. S2†).21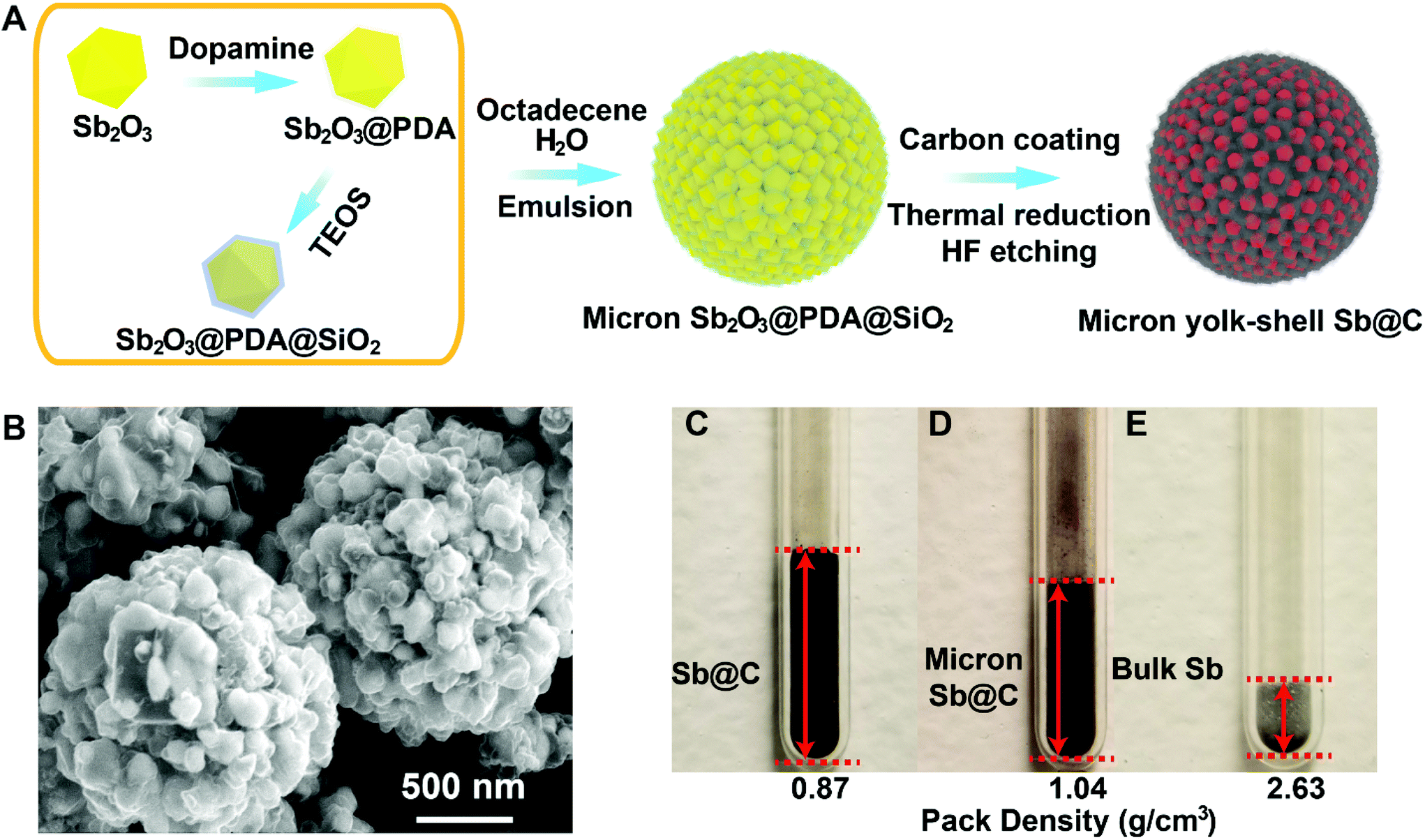 | ||
| Fig. 1 Pomegranate Sb@C yolk–shell microspheres (Sb@C MSs). (A) Schematic illustration of the synthesis of Sb@C MSs. (B) SEM image of typical Sb@C MSs. Packing density comparison of (C) the nano-Sb@C yolk–shell structure, (D) Sb@C MSs and (E) bulk Sb of ∼70 micrometers. | ||
Fig. 1B shows the representative scanning electron microscopy (SEM) image of the Sb@C MSs, in which the nano Sb@C yolk–shell particles were self-assembled and interlocked together in the H2O/octadecene microemulsion system. The average secondary particle sizes of the Sb@C MSs are ∼2 to 10 μm, a good range for the processing of battery materials (Fig. 1B and Fig. S3†).22,23 The Sb particle size is ∼100–200 nm and the gap is ∼50–200 nm from the Sb@C yolk–shell particles at the surface of the microspheres (Fig. 1B) and TEM images of a broken microsphere (Fig. S3†).
The Sb@C MSs are expected to not only have the advantage of controlled porosity in accommodating the volume change of Sb, similar to nano Sb@C yolk–shell particles, but also improved tap density, minimized surface area and mitigated parasitic reactions of electrolyte decomposition. Side-by-side comparison of the packing density of the Sb@C microspheres, nano Sb@C yolk–shell particles, and bulk Sb (74 μm) was scrutinized in Fig. 1C–E. Naturally, bulk Sb showed the highest packing density of 2.63 g cm−3. The Sb@C MSs demonstrated a packing density of ∼1.08 g cm−3, ∼41% of bulk Sb and ∼30% more than the nano Sb@C yolk–shell particle's density (0.87 g cm−3). It has to be noted that for alloy anodes of large swell, the electrode density at the 100% state of lithiation/sodiation is more meaningful in practice than the density after electrode manufacturing. The theoretical volume expansion of the fully lithiated/sodiated Sb is around 135% (Li) and 293% (Na), which will decrease the packing density of bulk Li3Sb/Na3Sb to ∼1.94/0.9 g cm−3, respectively.24 This is close to the density of Sb@C yolk–shell microspheres.21 The density measurement corroborates the pore volume control in our Sb@C yolk–shell MSs to be very close to the optimized value in accommodating the volume expansion of Sb during sodiation or lithiation. TEM and energy dispersive X-ray (EDX) elemental mapping of a broken part of a Sb@C microsphere in Fig. S3D–H† further confirm the uniform coating of the conductive carbon layer and the empty inner space deliberately left after removing the SiO2 layer.
The Sb@C MSs showed good electrochemical performance in both Li- and Na-ion batteries. Fig. 2A shows the specific capacities of the Sb@C MS anodes in LIBs at various charge/discharge current densities. The specific discharge capacities are ∼935 mA h g−1 (2nd cycle), 762 mA h g−1, and 633 mA h g−1 at current densities of 50 mA g−1, 500 mA g−1, and 5 A g−1, respectively. At the same current densities in SIBs, they deliver specific discharge capacities of 637 mA h g−1, 521 mA h g−1, and 441 mA h g−1 (Fig. 2B). Furthermore, the Sb@C MS anodes show good cycling stability. In LIBs, they retained ∼78% of their initial capacity after 200 cycles at 200 mA g−1 (Fig. 2C). The cycling stability is even better for SIBs. In Fig. 2D, the anode showed an amazingly stable cycling with 99% capacity retention over 200 cycles at a similar current density of 200 mA g−1.
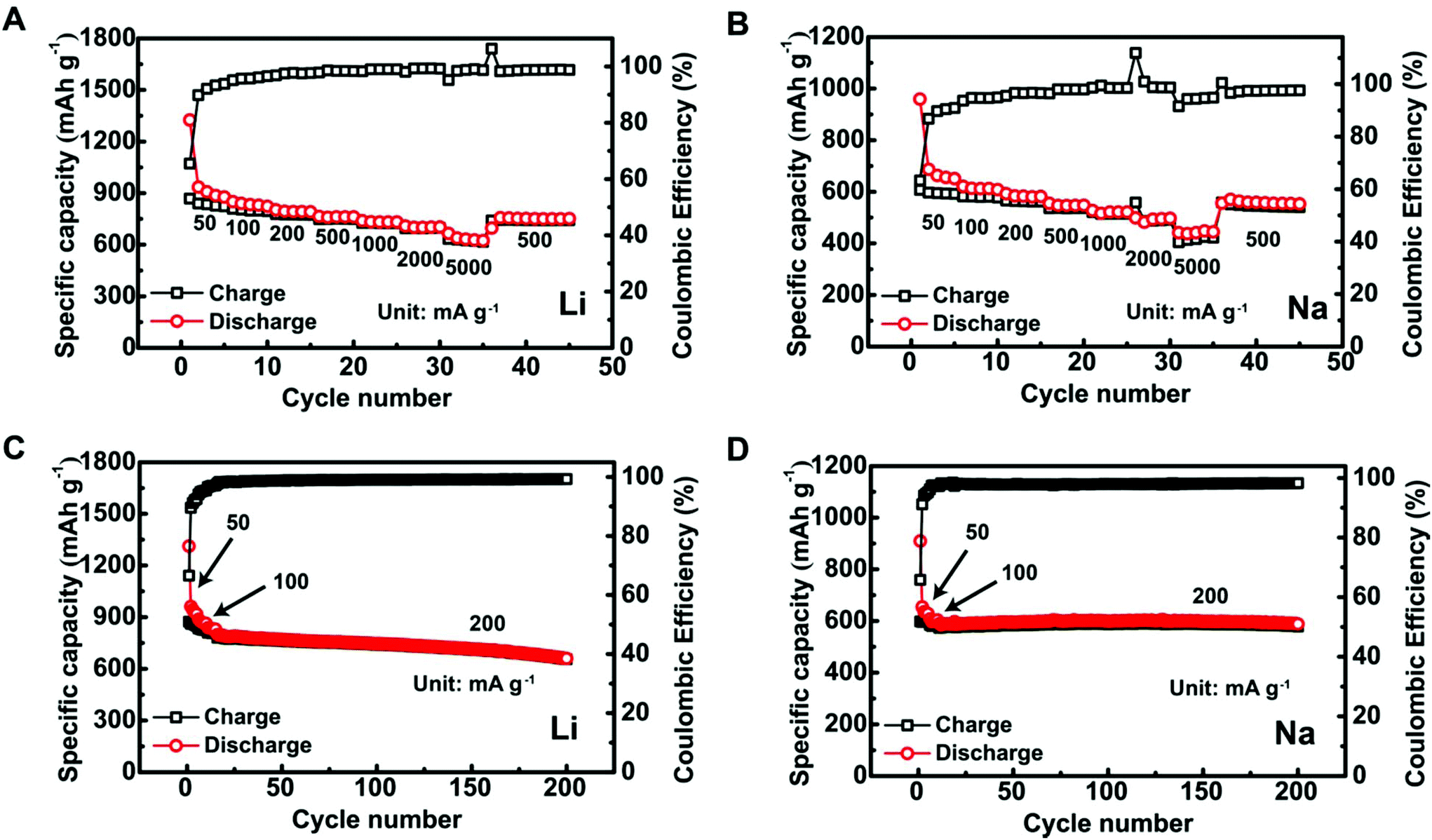 | ||
| Fig. 2 Typical electrochemical performance of the Sb@C MSs in Li- and Na-ion batteries. (A) Li-Ion storage capacity at different current densities. (B) Na-Ion storage capacity at different current densities. (C) Long term cycling of the Sb@C MS anodes against Li. (D) Long term cycling of the Sb@C MS anodes against Na. | ||
The cycling stability difference of Sb in SIBs and LIBs has been observed previously for bulk particles. Bulk Sb showed better cycling stability upon sodiation than lithiation, in spite of the larger volume expansion of Sb at full sodiation (293%) than at full lithiation (135%).24 The distinctive alloying mechanism of Na–Sb with the presence of the amorphous intermediate phase compared to the Li–Sb system was ascribed to be the reason.18,21 However, the detailed understanding of how the different phase transitions and amorphous alloy phase affect the electrochemical performance is missing.
Many factors affect the cycling performance of Sb: (1) Sb expands ∼293% at full sodiation while it only expands ∼135% at full lithiation. The stress from the different volume changes will affect the mechanical and electrical integrity, and hence cycling stability. (2) The specific capacity of Sb in SIBs and LIBs is different. The potentially disparate kinetics of the alloying process of Li and Na may lead to different depths of Sb material utilization at the same current density and hence potentially unalike capacity retention at the same cycling life. (3) The different SEIs in SIBs and LIBs are particularly entangled with different volume changes, degree of electrolyte decomposition and SEI properties. (4) Dissimilar reversible electrochemistry can be introduced through the kinetically controlled method such as the use of nanostructure/amorphous materials to lower the energy/spatial barrier.
The presence of the amorphous intermediate phase in the Na–Sb system may provide a similar function of using the nanostructured material by introducing more reversible electrochemistry because of the fast kinetics and low energy/spatial barrier. However, it does not rule out other factors. Our Sb@C MSs are a good platform for the systematic investigation of the mechanism of cycling stability difference of Sb in SIBs and LIBs: (1) with the appropriate void space in accommodating the Sb volume change, the electrodes can maintain a relatively good mechanical/electrical integrity during cycling. (2) The yolk–shell structure with micron size particles of reduced surface area has controlled parasitic reactions of electrolyte decomposition. (3) Electrochemical measurement showed that Sb@C MSs had similar and very good alloying kinetics with Li and Na. The specific capacities of LIBs and SIBs at a current density of 5 A g−1 retained ∼68% and 69% of their capacities at 50 mA g−1, respectively. (4) The long-term cycling stability for LIBs and SIBs was compared at the same current density of 200 mA g−1.
X-ray photoelectron spectroscopy (XPS) characterization and a comparative study of the electrochemical kinetics revealed that the different SEIs of Sb in LIBs and SIBs were crucial to their long-term cycling performance/failure mechanism besides the contrasting alloying mechanism.
Before investigating the SEI difference, the galvanostatic intermittent titration technique (GITT) was used to study the thermodynamics and kinetics of these two battery systems to understand how the different phase changes and amorphous alloy phases affect the electrochemical performance (Fig. 3). According to the results, Li-ion storage showed lower resistance than the alloying/de-alloying of Na ions even though the Na–Sb system has the amorphous intermediate phase in the alloying process. For the Li and Na cells shown in Fig. 3A and B, the overpotential before and after the current pulse gradually increases during the charge step, and fluctuates during the discharge step, which reflects the changing energy barriers at different alloying/dealloying stages. For a closer look at the reaction kinetics at different lithiation/sodiation degrees, the internal resistances of the cell are derived by dividing the overpotential by the pulse current. Although the resistance of both systems is low, the Na-cell's resistance is ∼1.5 times higher than that of Li-cell's on average (Fig. 3C). The development of internal resistance is closely related to the sodium/lithium diffusion through the SEI layer and alloy with Sb. The GITT results indicate that Na+ ion migration inside Sb faces a larger energy barrier associated with the relatively high internal resistance and sharp increase of the energy barrier (resistance bump) approaching the full sodiation state. The amorphous intermediate phase in SIBs did not help much on the reaction kinetics or the electrochemical reversibility.
 | ||
| Fig. 3 Galvanostatic intermittent titration technique (GITT) study of the alloying/de-alloying process of Sb in Li and Na ion batteries. (A) Typical GITT plot of a Li cell. (B) Typical GITT plot of a Na cell. (C) Comparison of the reaction resistances of the Li (black dots) and Na (red dots) cells in (A) and (B). | ||
The average coulombic efficiency (CE) of the SIBs and LIBs provides indication of the reversibility of the Li/Na ion storage process. At the same current density of 200 mA g−1, the average coulombic efficiency (CE) for the Sb@C MS-Na cell is ∼97.73%, lower than the value of a typical Sb@C MS-Li cell, ∼98.54%. In fact, the higher CE of Li cells holds true at different current densities in the rate test. This indicates more parasitic reactions and SEI formation in the SIBs, while more reversible electrochemical processes in the LIBs. Parasitic reactions at the metal counter electrode also have an effect on the CE and they will be discussed in the electrochemical impedance spectroscopy (EIS) study (vide infra).
More SEI on the Sb@C MS electrode surface in SIBs than in LIBs was corroborated by XPS characterization. Fig. 4 shows the C, Cl and N XPS spectra of the electrodes after cycling for 200 times in LIBs and SIBs. The C 1s spectra (Fig. 4A) reveal that electrodes from both systems have a very similar surface chemical composition consisting of sp2 carbon, alkoxy and alkyl carbon, fluorine contained groups and inorganic sodium salts.25 The electrode cycled in the SIB has a larger ratio of SEI carbon, i.e., sp2 carbon, than that in the LIB. The Cl signal is from the salt decomposition. The Cl 2p spectrum from SIB presents a noticeable difference compared to its Li counterpart, with stronger peak intensity in both ∼200 eV (Cl−) and 210 eV (ClO4−) regions (Fig. 4B).26 This indicates that the decomposition of the Na salt is more dramatic than the Li salt on the electrode surface forming the SEI. The accelerated salt decomposition might be the trigger point of the swift thickening of the SEI in the Na cell, which is evidenced by the N 1s spectra (Fig. 4C). Fig. 4C shows that the nitrogen signal (∼399.6 eV) from the carbonized polydopamine is completely absent in the electrode from SIBs while it is sharp and clear in the electrode from LIBs. Considering the XPS only reflected the surface information, the absence of a nitrogen signal can be ascribed to the thick SEI that covered up the carbon surface of Sb@C MSs cycled in SIBs. In agreement with the XPS analysis, the SEM of the cycled Na electrode (Fig. S4A and B†) shows less recognizable Sb@C MSs than the cycled Li electrode (Fig. S4C and D†), as they are buried in a thick SEI layer.
 | ||
| Fig. 4 XPS spectra of Sb@C MS electrodes after 50 cycles in LIBs and SIBs. (A) C 1s spectra. (B) Cl 2p spectra. (C) N 1s spectra. | ||
The growth of the SEI usually leads to the rise in cell impedance and subsequent cell failure or capacity fade. Hence, electrochemical impedance spectroscopy (EIS) was used to study the SEI layer impedance evolution in the LIBs and SIBs. Fig. 5 shows the representative Nyquist plot of Li and Na cells at charged states before and after cycling. As we can see in Fig. 5A and B, the SIB has larger impedance than the LIB before cycling.27 Although the impedance decreases after the 1st cycle, the SIB impedance is still larger than the LIB impedance (Fig. 5C and D). A detailed comparison of the impedance evolution with cycling in both systems showed gradual resistance increment in the charge-transfer (Rct) region due to the development of the SEI layer. The impedance of the SIBs increased by ∼400% after 50 cycles while the impedance of the LIBs increased slightly by ∼12% (Table S1†).
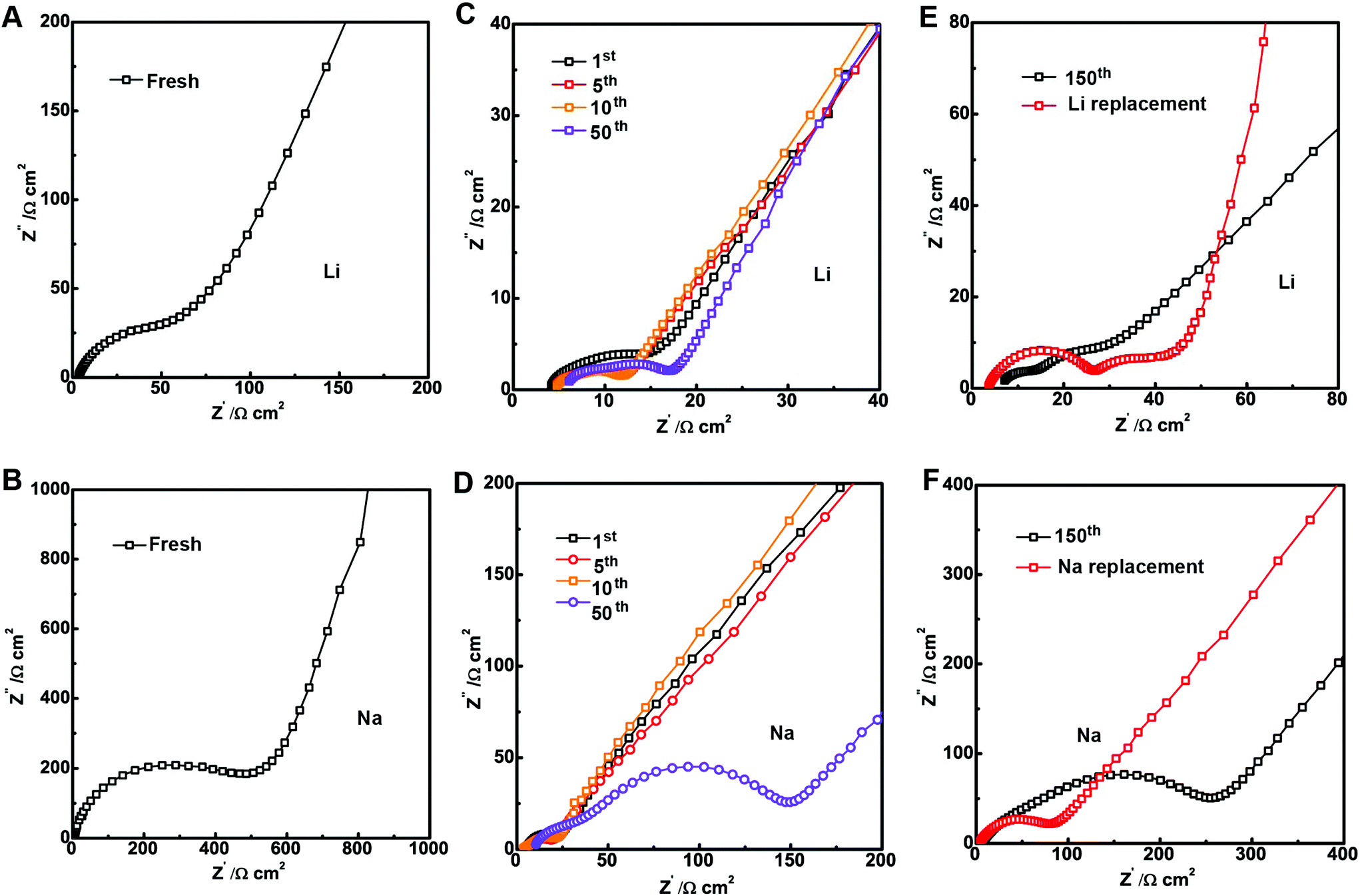 | ||
| Fig. 5 Impedance evolution of Sb@C MSs in LIBs and SIBs. (A) EIS of the LIB before cycling. (B) EIS of the SIB before cycling. (C) EIS comparison of the LIB after the 1st, 5th, 10th and 50th cycles. (D) EIS comparison of the SIB after the 1st, 5th, 10th and 50th cycles. (E) EIS of the LIB after 150 cycles before and after switching to a fresh Li metal. (F) EIS of the SIB after 150 cycles before and after switching to a fresh Na metal. | ||
The SEI impedance increment in the half cell against Li or Na metals includes not only the structural degradation and SEI thickening on the surface of the Sb@C MSs but also the metal surface. To avoid the effect from the counter metal anodes, the anodes after cycling in SIBs and LIBs for 150 cycles were disassembled and then re-assembled with a fresh Li/Na metal anode. Fig. 5E and F show that the impedance of both the SIBs and LIBs decreased. The Rct impedance of the LIBs only decreased slightly by 4 Ω, while the Rct of the SIBs dropped to188 Ω after switching to a fresh Na metal (Table S1†). Even so, the SIB impedance is still much larger than the impedance of the LIBs indicating more SEI on the surface of the Sb@C MSs in the SIBs. The large increase of the Na–Sb cell impedance did not lead to capacity fade, indicating the SIBs are more tolerant to the negative effect of SEI growth.
On the basis of the above results from battery testing, GITT, EIS and XPS, we can see that (1) although the intermediate phase of the Na–Sb alloy is amorphous, the Na–Sb alloying process is more difficult than that of Li–Sb according to the larger resistance from GITT measurement and larger EIS impedance. The different alloying mechanisms and the presence of an amorphous interphase may help the cycling stability by releasing the stress, but does not help the electrochemical reversibility. Since SIBs have more parasitic reactions than LIBs according to the CE difference, Sb amorphorization may not be the sole/major reason for cycling stability improvement. (2) More parasitic reactions, SEI formation and large impedance increase of SIBs did not lead to capacity fade. The difference in the ion transfer kinetics in SIBs and LIBs did not contribute much to the different cycling stabilities. (3) The SEI layer has a main function of stabilizing the electrode–electrolyte interphase, while it is insulating and slows down the ion transfer when it is too thick.28,29 However, for the Sb@C MSs in SIBs, a thick SEI on the surface did not deter Na-ion transfer and hence did not lead to capacity fade. It is believed that the thick SEI is critical as a mechanically robust protecting layer to improve the long-term cycling stability, by mitigating the challenge of the volume expandable alloy.30 The SEM images of cycled electrodes in SIBs (Fig. S4–S6†) show a better preserved spherical morphology than those in LIBs.
4. Conclusion
In summary, we have prepared pomegranate Sb@C yolk–shell microspheres to improve the long-term cycling stability and packing density. As SIB anodes, they demonstrated an incredible 99.8% capacity retention after 200 cycles and 77% for LIBs. The packing density increased by 30% compared to the nano-sized Sb@C yolk–shell structure. Using these as a material platform, we elucidated the SEI effect on the cycling stability difference in LIBs and SIBs. The thick SEI developed on the electrode surface in SIBs did not deter the Na-ion transfer and was very beneficial to the mechanical stability and hence is considered a main reason for the better cyclability. Although it is not practical to form a thick SEI in practical full-cell batteries, this study revealed the importance of designing a stable SEI for high-density alloy based anodes for LIBs and SIBs. The dissimilar SEI properties between Li and Na chemistries also inspire a quest for customized material and electrolyte design for their own respective electrochemical systems.Author contributions
J.S., Y.L. and X.L. initiated the project. J.S. synthesized the materials. D.X., H.J. and G.Z. conducted the TEM and SEM analyses. J.S. and S.F. performed the electrochemical testing. J.S., Y.L. and X.L. prepared the manuscript with input from all the other co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. L. L. would like to acknowledge the financial support from the U.S. Department of Energy's (DOE's) Office of Electricity Delivery & Energy Reliability (OE) under Contract No. 70247A. Y. H. L. would like to acknowledge the support from the start-up funds from the Washington State University. Some of the characterization studies were performed using the EMSL, a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory.References
- G. H. Newman and L. P. Klemann, J. Electrochem. Soc., 1980, 127, 2097–2099 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- J. Liu, J. G. Zhang, Z. G. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Y. Li, J. Z. Hu, C. M. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. L. Li, Y. Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929–946 CrossRef CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 RSC.
- K. Westman, R. Dugas, P. Jankowski, W. G. Wieczorek, G. Gachot, M. Morcrette, E. Irisarri, A. Ponrouch, M. R. Palacin and J.-M. Tarascon, ACS Appl. Energy Mater., 2018, 1, 2671–2680 CrossRef CAS.
- Z. M. Liu, T. C. Lu, T. Song, X. Y. Yu, X. W. Lou and U. Paik, Energy Environ. Sci., 2017, 10, 1576–1580 RSC.
- J.-I. Lee, J. Song, Y. Cha, S. Fu, C. Zhu, X. Li, Y. Lin and M.-K. Song, Nano Res., 2017, 10, 4398–4414 CrossRef CAS.
- H. L. Zhu, Z. Jia, Y. C. Chen, N. Weadock, J. Y. Wan, O. Vaaland, X. G. Han, T. Li and L. B. Hu, Nano Lett., 2013, 13, 3093–3100 CrossRef CAS PubMed.
- X. H. Xiong, C. H. Yang, G. H. Wang, Y. W. Lin, X. Ou, J. H. Wang, B. T. Zhao, M. L. Liu, Z. Lin and K. Huang, Energy Environ. Sci., 2017, 10, 1757–1763 RSC.
- W. J. Li, S. L. Chou, J. Z. Wang, J. H. Kim, H. K. Liu and S. X. Dou, Adv. Mater., 2014, 26, 4037–4042 CrossRef CAS PubMed.
- N. Liu, Z. D. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. T. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- X. W. Li, Z. B. Yang, Y. J. Fu, L. Qiao, D. Li, H. W. Yue and D. Y. He, ACS Nano, 2015, 9, 1858–1867 CrossRef CAS PubMed.
- Y. Jin, S. Sun, M. Ou, Y. Liu, C. Fan, X. Sun, J. Peng, Y. Li, Y. Qiu and P. Wei, ACS Appl. Energy Mater., 2018, 1, 2295–2305 CrossRef CAS.
- J. Liu, L. T. Yu, C. Wu, Y. R. Wen, K. B. Yin, F. K. Chiang, R. Z. Hu, J. W. Liu, L. T. Sun, L. Gu, J. Maier, Y. Yu and M. Zhu, Nano Lett., 2017, 17, 2034–2042 CrossRef CAS PubMed.
- L. T. Yu, J. Liu, X. J. Xu, L. G. Zhang, R. Z. Hu, J. W. Liu, L. C. Yang and M. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 2516–2525 CrossRef CAS PubMed.
- C. M. Park, J. H. Kim, H. Kim and H. J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2013, 135, 10179–10179 CrossRef CAS.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- M. T. McDowell, S. W. Lee, I. Ryu, H. Wu, W. D. Nix, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 4018–4025 CrossRef CAS PubMed.
- J. H. Song, P. F. Yan, L. L. Luo, X. G. Qi, X. H. Rong, J. M. Zheng, B. W. Xiao, S. Feng, C. M. Wang, Y. S. Hu, Y. H. Lin, V. L. Sprenkle and X. L. Li, Nano Energy, 2017, 40, 504–511 CrossRef CAS.
- M. H. Lee, Y. Kang, S. T. Myung and Y. K. Sun, Electrochim. Acta, 2004, 50, 939–948 CrossRef CAS.
- J. M. Zheng, S. J. Myeong, W. R. Cho, P. F. Yan, J. Xiao, C. M. Wang, J. Cho and J. G. Zhang, Adv. Energy Mater., 2017, 7, 1601284 CrossRef.
- L. Baggetto, P. Ganesh, C. N. Sun, R. A. Meisner, T. A. Zawodzinski and G. M. Veith, J. Mater. Chem. A, 2013, 1, 7985–7994 RSC.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- L. W. Ji, M. Gu, Y. Y. Shao, X. L. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. M. Nie, J. Xiao, C. M. Wang, J. G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- Y. H. Xu, Y. J. Zhu, Y. H. Liu and C. S. Wang, Adv. Energy Mater., 2013, 3, 128–133 CrossRef CAS.
- J. Song, B. Xiao, Y. Lin, K. Xu and X. Li, Adv. Energy Mater., 2018, 8, 1703082 CrossRef.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- A. Darwiche, L. Bodenes, L. Madec, L. Monconduit and H. Martinez, Electrochim. Acta, 2016, 207, 284–292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08461k |
| This journal is © The Royal Society of Chemistry 2019 |