
Self-assembly dynamics and antimicrobial activity of all L- and D-amino acid enantiomers of a designer peptide†
Zhou
Ye
 a,
Xiao
Zhu
ab,
Sergio
Acosta
c,
Dhiraj
Kumar
a,
Ting
Sang
ad and
Conrado
Aparicio
*a
a,
Xiao
Zhu
ab,
Sergio
Acosta
c,
Dhiraj
Kumar
a,
Ting
Sang
ad and
Conrado
Aparicio
*a
aMDRCBB, Minnesota Dental Research Center for Biomaterials and Biomechanics, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: apari003@umn.edu
bThe State Key Laboratory Breeding Base of Basic Science of Stomatology & Key Laboratory of Oral Biomedicine Ministry of Education, School of Stomatology, Wuhan University, Wuhan 430079, China
cG.I.R. Bioforge, University of Valladolid, CIBER-BBN, Valladolid 47011, Spain
dJiangxi Provincial Key Laboratory of Oral Biomedicine, School of Stomatology, Nanchang University, Nanchang 330006, China
First published on 21st November 2018
Abstract
Recent studies have shown that antimicrobial peptides (AMPs) can self-assemble into supramolecular structures, but this has been overlooked as causative of their antimicrobial activity. Also, the higher antimicrobial potency of D-enantiomers compared to L-enantiomers of AMPs cannot always be attributed to their different resistance to protease degradation. Here, we tested all L- and D-amino acid versions of GL13K, an AMP derived from a human protein, to study structural links between the AMP secondary structure, supramolecular self-assembly dynamics, and antimicrobial activity. pH dependence and the evolution of secondary structures were related to a self-assembly process with differences among these AMPs. The two GL13K enantiomers formed analogous self-assembled twisted nanoribbon structures, but D-GL13K initiated self-assembly faster and had notably higher antimicrobial potency than L-GL13K. A non-antimicrobial scrambled amino acid version of L-GL13K assembled at a much higher pH to form distinctively different self-assembled structures than L-GL13K. Our results support a functional relationship between the AMP self-assembly and their antimicrobial activity.
Introduction
The development of antimicrobial peptides (AMPs), also called host defense peptides, has notably advanced in the past few decades due to increasing prevalence of bacterial resistance to conventional antibiotics. Cell-secreted or synthetic AMPs are typically cationic amphiphilic molecules, which accumulate on bacterial surfaces and disrupt the integrity of the bacterial membrane or attack protoplasmic targets.1 In addition to the direct killing of bacteria, some AMPs show immunomodulatory properties.2 The low toxicity of AMPs against mammalian cells has been attributed to the inherent structural and functional differences in the membranes of mammalian cells and bacterial pathogens. Compared to mammalian cells, bacterial cytoplasmic membranes contain much more anionic components, such as phosphatidylserine and cardiolipin.3 As a result, cationic AMPs are more likely to be attracted to the bacterial surfaces by electrostatic forces and neutralized by negatively charged components. Several models have been proposed to describe membrane permeabilization mechanisms by AMPs, including the barrel-stave model, the carpet model, and the toroidal-pore model.4 All of these models are based on interactions between a group of individual antimicrobial molecules and the bacterial membrane lipids, without considering the self-assembly or oligomerization of AMPs.It is known that a number of amphiphilic peptides can self-assemble into supramolecular nanostructures, which has been mainly exploited to build structures for tissue engineering and drug delivery.5 However, little is known about the functional link of self-assembly of peptides and their antimicrobial activity.6 Instead, peptide secondary structures have been the focus of study and found to be closely related to the bacterial membrane permeabilization. Many AMPs are mainly unordered in solution or in the vicinity of neutral phospholipids, but present molecular structures rich with α-helix or β-sheet components in the vicinity of negatively charged phospholipids.7 The secondary structure change is commonly attributed to reorganization of residues within a single AMP molecule and triggered by interactions of the peptides with the anionic and hydrophobic components of the bacterial membrane. However, the secondary structures are usually not stable but would change with time, suggesting that a process of self-assembly or supramolecular reorganization of the molecules is involved.8
In contrast to AMPs, molecular self-assembly has been widely discussed in the context of amyloid peptides. Amyloid peptides share key structural and functional features with AMPs.9 As it is the case of AMPs, most amyloid peptides contain cationic and hydrophobic residues. Some amyloid peptides also display antimicrobial activity.10–13 Several neurological diseases, including both Alzheimer's disease and Parkinson's disease, are associated with the initial formation of fibrillar amyloids of self-assembled/aggregated peptides and proteins. The amyloids can then disrupt the cell membrane. Similar interaction between self-assembled fibers and lipid vesicles or bacterial membranes has been discovered with surfactant-like peptides14,15 and peptide amphiphiles.16 Different from AMPs, the membrane disrupting mechanisms of amyloids have been commonly related to peptide assembly. This might be related to the fact that amyloid peptide self-assembled nanostructures can be readily found in physiological solutions, whereas AMPs are hardly found as supramolecular assemblies in neutral or bacterial culture media. However, charge density and hydrophobicity near or on bacterial cell membranes are markedly different from the aquatic ionic environment of the bacteria culture and thus, self-assembly of AMPs on the bacterial membrane cannot be disregarded as a potential mechanism for the antimicrobial activity of these peptides. In a recent study, L. Schnaider et al. demonstrated the significance of self-assembled nanostructures of diphenylalanine on membrane permeation and depolarization, upregulation of stress response regulons and damage to bacterial morphology.17
Here, we used a designer AMP, GL13K, its D-enantiomer, D-GL13K and a non-antimicrobial scrambled amino acid sequence of GL13K, L-GL13K-R (Fig. 1a–c) to explore relevant environmental and molecular properties that affect the secondary structure, self-assembly, antimicrobial activity, and associations between these peptide properties. We also aimed to study the effect of time on the structural and self-assembly properties of these peptides.
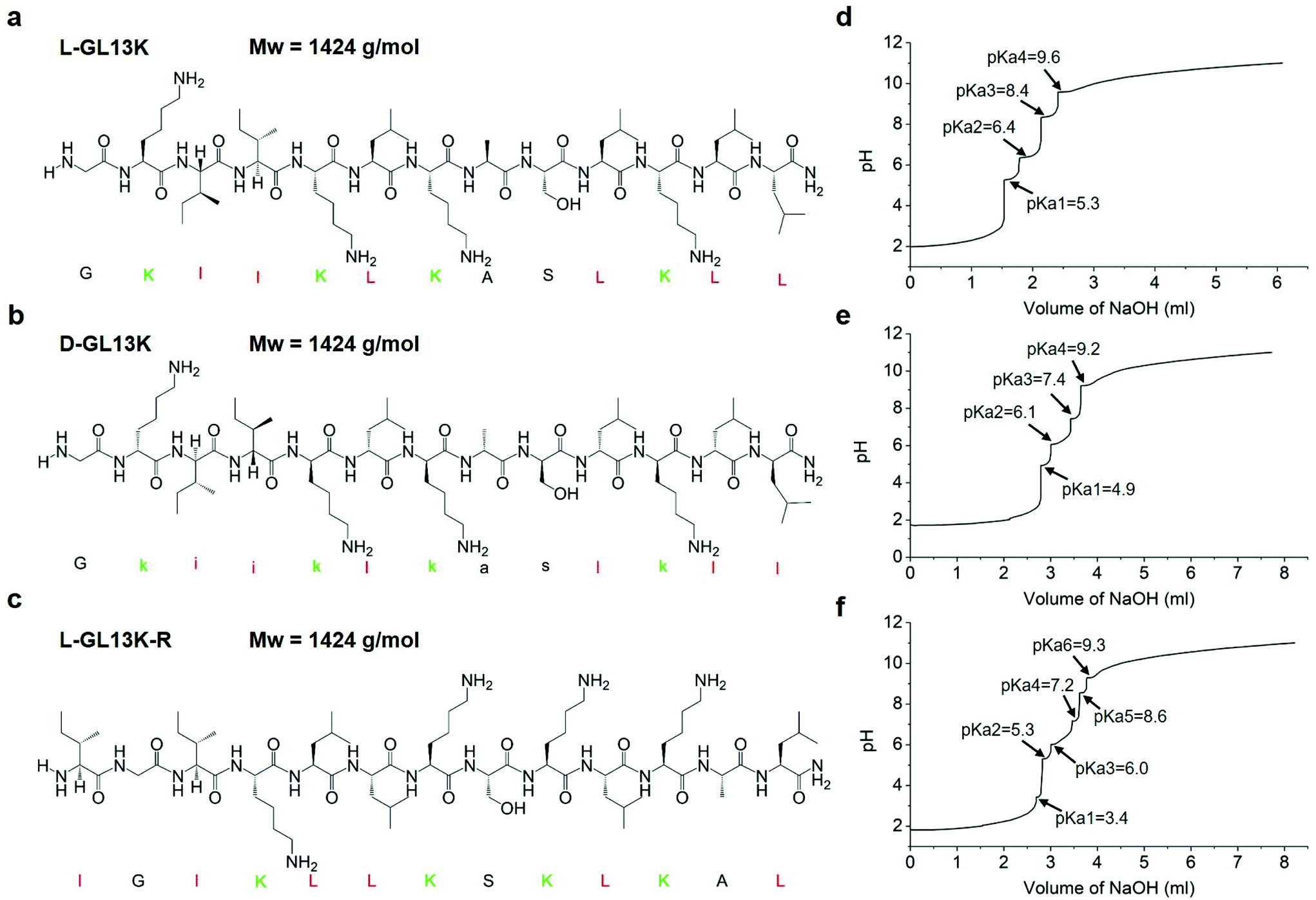 | ||
| Fig. 1 (a–c) Molecular structures and Mw of L-GL13K, D-GL13K, and L-GL13K-R. Cationic residues are marked in green and highly hydrophobic residues are marked in red. (d–f) Titration curves and pKa values of L-GL13K, D-GL13K, and L-GL13K-R. | ||
GL13K was derived from the human salivary protein BPIFA2 (formerly human Parotid Secretory Protein, hPSP).18 The original peptide sequence, GL13NH2 (hPSP 141–153), did not exhibit bactericidal activity, whereas substitution of three of its polar residues for cationic lysines converted it to the AMP, GL13K.19L-GL13K is bactericidal and effective against Gram negative bacteria; e.g. Pseudomonas aeruginosa and Escherichia coli; and Gram-positive bacteria, Streptococcus gordonii.19 It also kills P. aeruginosa biofilms.20L-GL13K immobilized on titanium surfaces kills and reduces biofilm burden of bacterial pathogens, such as Porphyromonas gingivalis21 and primary colonizers, such as S. gordonii.22 Previous studies demonstrated that L-GL13K disrupts anionic bacterial model membranes, but did not affect the integrity of neutral eukaryotic model membranes.23,24 This was consistent with their observation that the secondary structure of L-GL13K selectively changed in solutions with a high concentration of negatively charged phospholipids.
D-GL13K is significantly more active than L-GL13K against Gram positive bacteria, such as Enterococcus faecalis and S. gordonii.25 The higher bactericidal potency of D-amino acid versions of AMPs than their L-amino acid counterparts is often attributed to their increased stability against enzymatic degradation as bacteria secreted proteases are composed of L-amino acids.26 Recent work by H. Hirt et al. suggests that protease-resistance is not the only difference in killing effectiveness of E. faecalis between L- and D-enantiomers of AMPs.25 Structural differences between L- and D-enantiomers of the same peptide have been scarcely explored,27 mainly because identical mirrored structures between the two enantiomers are expected. However, recent work has noted that peptide chirality induces differences in the molecular structure and self-assembly of peptides in solution. S. Marchesan et al. found that the circular dichroism (CD) spectra peak of a tripeptide DPhe-LPhe-LVal was at 228 nm whereas the peak of its enantiomer LPhe-DPhe-DVal shifted to 224 nm, indicating nonidentical secondary structures.28 However, all L-amino acid and all D-amino acid tripeptides did not show similar structural differences. Z. Luo et al. observed that self-assembled structures of the D- and L-enantiomers of the EAK16 peptide had different temperature-dependent responses.29 L-EAK16 formed and maintained a β-sheet structure after heating from 25 °C to 110 °C, whereas D-EAK16 changed from β-sheet to α-helix and remained α-helix after cooling back to 25 °C. These structural and self-assembly differences might result in distinct biological responses. A better understanding of the differences in self-assembly and antimicrobial activity between L- and D-enantiomers could guide the design of AMPs to increase their bactericidal efficiency and broaden their spectrum of activity and applications.
Experimental section
Peptide solutions
GL13K with all L-amino acids, L-GL13K (GKIIKLKASLKLL-NH2), GL13K with all D-amino acids, D-GL13K (Gkiiklkaslkll-NH2), and a scrambled amino acid sequence of GL13K, L-GL13K-R (IGIKLLKSKLKAL-NH2) peptides were purchased from AAPPTec, LLC (Louisville, KY, USA) with purity >98%. Stock solutions of L-GL13K, D-GL13K, and L-GL13K-R peptides were dissolved in deionized (DI) water at a concentration of 10 mM. Borax-NaOH buffer solutions were prepared by mixing a 0.025 M borax solution and a 0.1 M NaOH solution to final pH. A stock solution of 10 μl was added into a 990 μl buffer solution to a final concentration of 0.1 mM. L-GL13K and D-GL13K solutions were prepared at a pH of 9.4, 9.6, 9.8, 10.0, 10.4, and 10.8; L-GL13K-R solutions were prepared at a pH of 9.0, 10.0, 11.0, and 12.0. All peptide solutions were stored at 4 °C before testing.Circular dichroism (CD)
All aqueous peptide solutions were measured right after preparation (t = 0 d) and after 1 d, 2 d, 4 d, and 8 d storage at 4 °C. 200 μL of peptide solution were dispensed into a quartz cuvette with a 1 mm path-length. CD spectra were obtained with a Jasco J-815 CD spectrometer (Easton, MD, USA) at room temperature. The CD signal was monitored over a range of wavelengths from 190 to 260 nm with a data pitch of 1.0 nm, a scanning speed of 50 nm min−1 and a response time of 2 s. All measurements were averaged over 3 scans and were corrected by subtracting their corresponding buffer spectrum. The CD signal for the pH 12.0 L-GL13K-R sample had an excessively high noise to signal ratio for wavelengths below 205 nm.Transmission electron microscopy (TEM)
TEM samples were prepared by dropping 3 μl peptide solution onto negatively charged copper grids with a carbon film, followed by negative staining with 0.75% uranyl formate. TEM visualization of negatively stained samples was performed using a FEI Tecnai G2 F30 (Hillsboro, Oregon, USA) with an accelerating voltage of 300 kV.Cryogenic TEM (cryo-TEM) samples were initially prepared by plunging 2.5 μl peptide solution on lacey carbon/formvar grids for 7 s using an FEI Vitrobot (Hillsboro, Oregon, USA). Cryo-TEM visualization was performed using a FEI Tecnai G2 Spirit BioTWIN (Hillsboro, Oregon, USA) with an accelerating voltage of 120 kV. To improve our ability to find self-assembled nanostructures, we increase peptide concentration to 1 mM to conduct our Cryo-TEM experiments. To validate the existence of peptide self-assembled structures in pH 10.8 solution at a lower concentration of 0.1 mM, the critical aggregation concentration was assessed to be 0.02–0.05 mM by Nile Red assay (Fig. S1†). Thus, solutions used for both the negative-staining TEM (0.1 mM) and cryo-TEM (1 mM) experiments contained self-assembled structures in aqueous environments.
Estimation of secondary structure contents
Estimations for the different secondary structure contents were obtained from further analysis of CD spectra using CDPro software (http://sites.bmb.colostate.edu/sreeram/CDPro/, last accessed September 7, 2018).30 We used a reference set of 48 proteins from four sources (reference set 7 in CDPro) in a wavelength range of 190–240 nm. Before conducting the secondary structure analysis, CD spectra data were converted from ellipticity (θ) to mean residue ellipticity ([θ]) and molar circular dichroism (Δε), where [θ] = millidegrees/(path length in mm × the concentration of peptide in mg ml−1 × the number of residues) and Δε = [θ]/3298. The data were analyzed by three common methods implemented in software packages, SELCON3, CDSSTR, and CONTIN/LL. Data obtained from each of the methods were analyzed and we report average values from the three methods.pH titration
To determine the pKa values of L-GL13K, D-GL13K, and L-GL13K-R, the pH titration curve was assessed between pH 2.0 and 11.0. Each peptide was prepared at 0.1 mM in DI water and 10 ml solution was used for titration with a Zeta potential analyser (Stabino, Particle Metrix, Meerbusch, Germany). 0.1 M HCl solution was added at an increment of 100 μl to reach pH 2.0 as the pre-dosage step. Then 0.1 M NaOH solution was added dynamically to finally reach pH 11.0. At each step, the pH and accumulated NaOH volume were recorded. The dynamic time interval was set at 4–20 s and the dynamic NaOH volume was set at 2–50 μl to keep 0.1 ≤ ΔpH ≤ 0.2. Each pKa value was determined by the half-way point of each buffer region according to the Henderson–Hasselbalch equation.Minimal inhibitory concentration (MIC)
The antimicrobial activity of L-GL13K, D-GL13K, and L-GL13K-R was assessed by MIC assays with Gram-positive S. gordonii M5 and Gram-negative P. aeruginosa Xen41 (a luminescent derivative from the parental strain P. aeruginosa PAO1, PerkinElmer Inc., Waltham, MA, USA) as previously described.31S. gordonii M5 was grown in brain heart infusion (BHI) broth and P. aeruginosa Xen41 was grown in Luria–Bertani (LB) broth at 37 °C overnight. Overnight cultures were diluted to a concentration of 5 × 105 CFU ml−1 in BHI or Mueller-Hinton broth (MHB), respectively. 90 μl of the bacteria solution was incubated with a 10 μl serial peptide dilution from a 10 mg ml−1 stock solution in 96-well polypropylene plates at 37 °C for 20 h. The bacterial growth was evaluated by optical density (OD) at 570 nm. Two control groups were tested, bacterial growth in the absence of peptides and growth medium without bacteria. Each experiment was repeated at least three times.Results and discussion
Effect of pH and time on L-GL13K self-assembly
We analyzed changes of the L-GL13K secondary structure in varying pH solutions (9.4–10.8) for up to 8 days using CD (Fig. 2). In solutions with pH ≥ 9.6, L-GL13K peptides showed a progressive transition over time from mostly unordered to β-sheet structures. The rate of structural transition increased with increasing solution pH. At pH ≥ 10.4, the structural transition occurred almost spontaneously upon the dissolution of the peptides (t = 0 d). At pH 9.4, L-GL13K peptides maintained unordered structures for the whole experiment period.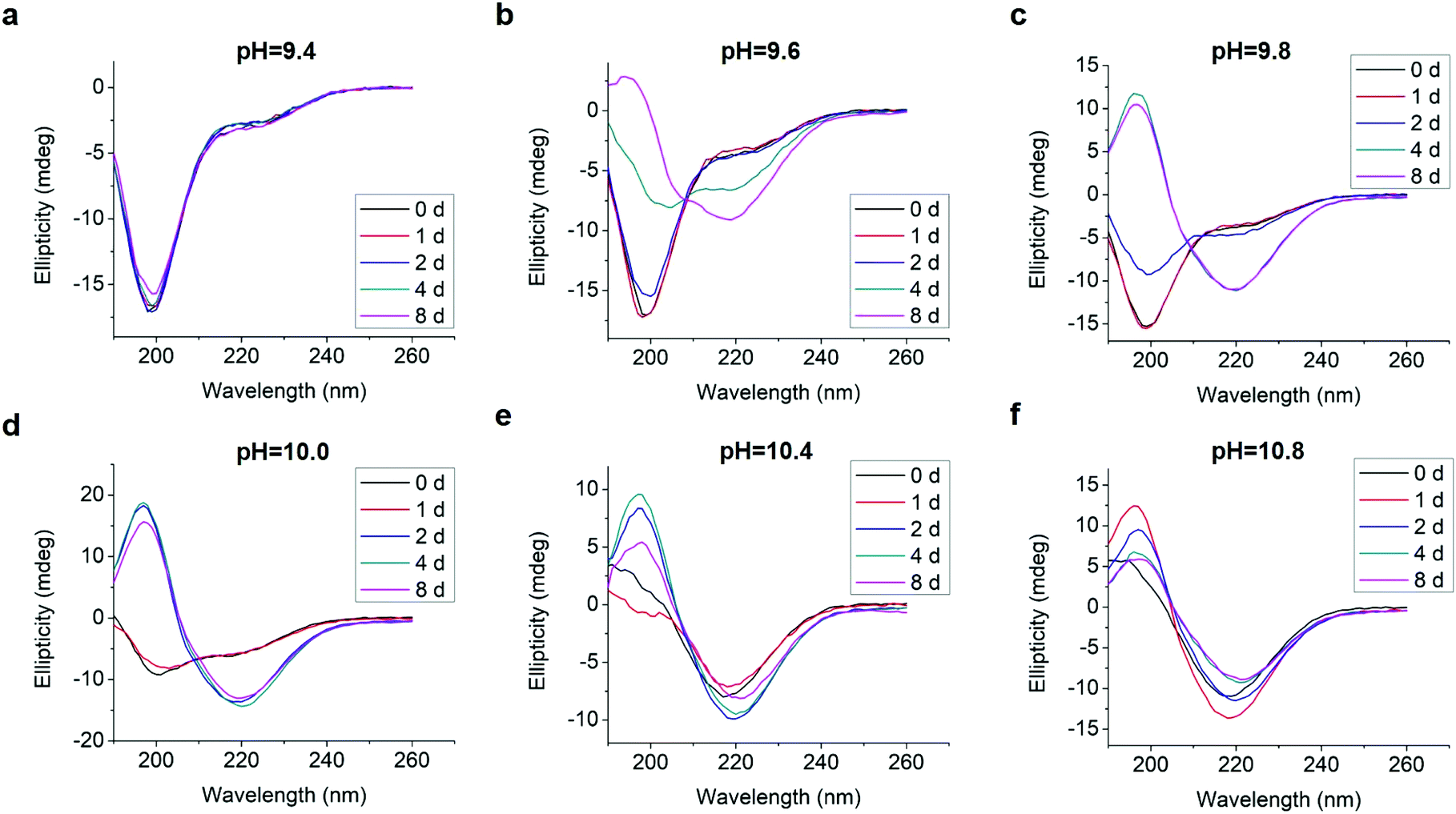 | ||
| Fig. 2 CD spectra of 0.1 mM L-GL13K in borax-NaOH buffer with pH ranging from 9.4 to 10.8 for 0 day, 1 day, 2 days, 4 days, and 8 days. | ||
To explore whether the changes of the secondary structure of L-GL13K peptides were caused by molecular self-assembly, we visualized the formation and evolution of supramolecular structures by both negative-staining TEM (Fig. 3a–e) and cryo-TEM (Fig. 3f). We selected a set of conditions that showed different secondary structures with gradual transition from a few small aggregates at low pH to highly organized and self-assembled nanostructures increasing in number when pH and/or time increased. The self-assembled structures were nanoribbons with varying widths from less than 10 nm to ∼30 nm and a length of as high as a few microns. TEM images at higher magnifications revealed that the nanoribbons were composed of twisted bundles of smaller nanofibrils with a uniform width of ∼3 nm (Fig. S2 and S3†). Notably, the progressive formation of the self-assembled supramolecular structures was in accordance with the progressive transformation of the secondary structure; from unordered to β-sheet signals in the CD spectra (insets in Fig. 3a–e). In solutions with the lowest pH 9.4, the peptides did not self-assemble and the CD spectrum was dominated by the characteristic signal of an unordered structure (Fig. 3a). When the solution pH was 9.8, a portion of the peptide molecules started to form nanofibrils after 2 days in solution, which was recorded in the CD spectra as a small peak at around 220 nm (Fig. 3b). Longer times (Fig. 3c and d) and/or higher pH (Fig. 3e) resulted in larger portions of self-assembled peptides with increasing number of nanofibrils and nanoribbons and increasing dominance of the β-sheet signal. Thus, changes in the secondary structure determined by CD spectra were induced by the self-assembling of L-GL13K peptides. The self-assembly of the peptides was pH reversible. For example, the transformation to a β-sheet rich structure of the peptides by increasing pH to 10.5 was reverted to a mostly unordered structure after titration back to pH 7.4 (Fig. S4†).
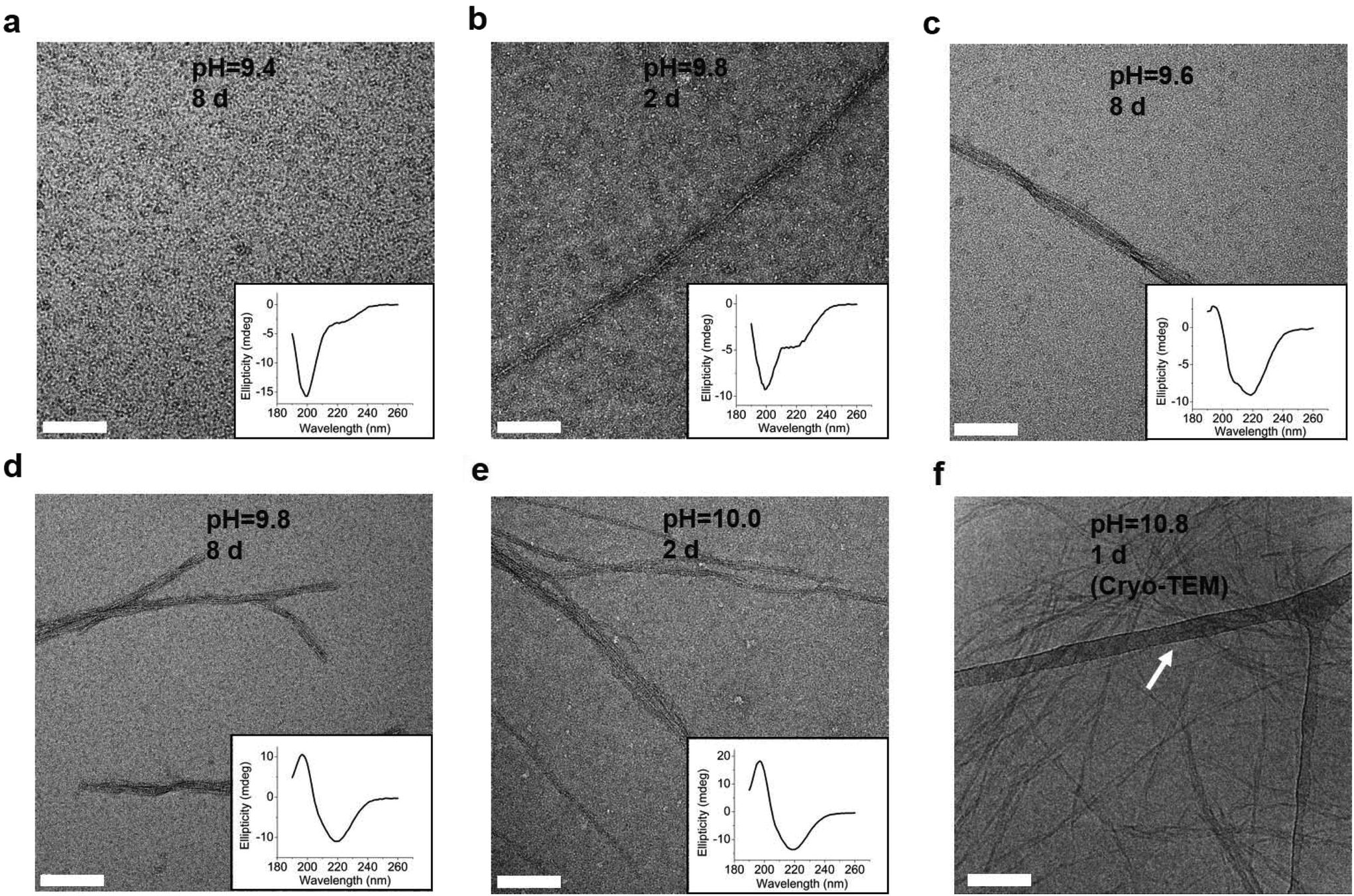 | ||
| Fig. 3 TEM images of negatively stained 0.1 mM L-GL13K agglomerations or self-assembled supramolecular structures (nanofibrils and nanoribbons) in buffer solutions of (a) pH 9.4 after 8 d, (b) pH 9.8 after 2 d, (c) pH 9.6 after 8 d, (d) pH 9.8 after 8 d, (e) pH 10.0 after 2 d. The inset plots are the corresponding CD spectra. (f) Cryo-TEM image of 1 mM L-GL13K nanoribbons in buffer solution at pH 10.8 for 1 d. The nanoribbons were embedded in vitreous ice and the arrow points at the lacey carbon/formvar support grid. All scale bars are 100 nm. | ||
As the structural transformation of the L-GL13K was slowed down; i.e., not immediate in the pH range between 9.6 and 10.0 we were able to investigate in detail specific characteristics of this transformation. Thus, we further used CDPro software to estimate the secondary structure contents from the CD spectra by using three common methods.30,32 We report averages and standard deviations calculated by the three methods against the time the peptides were dissolved in the solutions (Fig. 4). When peptide molecules were not self-assembled, 75% unordered structure was estimated in the peptide solutions. The unordered peptide structures transformed to β-sheet and β-turn before transformed to α-helix. In peptide solutions at pH 9.6 and pH 9.8 (Fig. 4a and b), β-sheet and β-turn contents started to increase after 2 days and 1 day, respectively; whereas α-helix only started to notably increase after 4 days and 2 days, respectively. At pH 10.0 (Fig. 4c), the peptide solutions had notable β-sheet and β-turn contents immediately after dissolution of the molecules (t = 0 day), but the α-helix content increased between 1 day and 2 days. When stable conformations were reached, the final content of each secondary structure was also dependent on solution pH. The higher the pH, the higher the α-helix content (30–34% for pH 10.0 vs. 22–23% for pH 9.8) and the lower the β-sheet content (20% for pH 10.0 vs. 30% for pH 9.8).
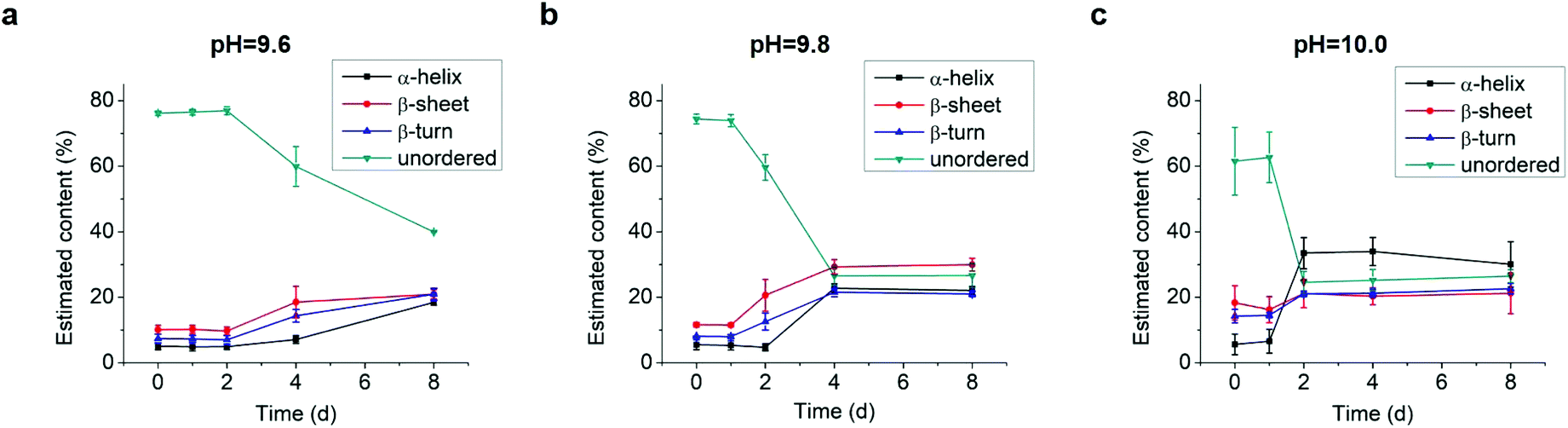 | ||
| Fig. 4 Estimation of secondary structure (α-helix, β-sheet, β-turn, and unordered) contents from CD spectra of 0.1 mM L-GL13K in buffer solutions of pH 9.6, 9.8, and 10.0 for up to 8 days. Content values were averaged from estimations by three different methods, SELCON, CDSSTR, and CONTIN/LL. | ||
Combined results of CD, TEM and estimation of molecular structural contents indicate that the L-GL13K peptides initially self-assembled to form nanofibrils (Fig. 3b, pH 9.8 2 d). The process of self-assembly was triggered by the increasing neutralization of positive charges (increased pH and/or time) and the self-assembled structures were stabilized by the establishment of β-sheet intermolecular structures. Subsequently, twisted nanoribbons formed (Fig. 3d, pH 9.8 8 d) from bundles of parallel nanofibrils and the content of peptides in α-helix conformation increased, which suggest that the transformation to α-helix is triggered by further neutralization of positive charges in the L-GL13K peptides. In summary, L-GL13K peptides completed supramolecular self-assembly and structural changes in 2 to 8 days at solution pH from 10.0 to 9.6, respectively.
Previous work also showed that the secondary structure of L-GL13K peptides was affected by either the pH of the aqueous solution or the concentration of surrounding bacterial model membranes.23,24 These findings support that the detected molecular transformations are triggered by the neutralization of the charged groups in the peptides. Here, we demonstrate that the secondary structure of the peptides in solution not only was affected by the solution pH, but also was not stable over time and changed in both cases as a result of the supramolecular self-assembly of the peptides. Together, these results suggest that the self-assembly of the peptides into supramolecular structures is favored in high pH solutions and/or highly negatively charged interfacial environments. Thus, our findings might contribute to a better understanding of the mechanism of bactericidal activity of these peptides by further investigating the process by which the peptides challenge the integrity of model or real bacterial membranes.
Effect of the chirality and amino acid sequence on peptide self-assembly
We tested an all D-amino acid version of the GL13K peptide to study the effect of chirality on the peptide structural change and self-assembly process. Though D-GL13K and L-GL13K had analogous CD spectra (Fig. 5), substantial differences were detected in solutions of pH 9.8 and pH 10.0 (Fig. S5†). In this pH range, D-GL13K initiated the structural transformation from unordered to ordered structures distinguishably faster (Fig. 5c, d and 6a, b) compared to L-GL13K (Fig. 2c, d and 4b, c). Thus, D-GL13K underwent a faster self-assembly initiation than L-GL13K. Note that after 2 days in solutions with pH 9.8, L-GL13K had only initiated the formation of self-assembled nanofibrils (Fig. 3b), whereas D-GL13K had assembled into a high concentration of long twisted nanoribbons, each formed by a number of bundled nanofibrils (Fig. 6c).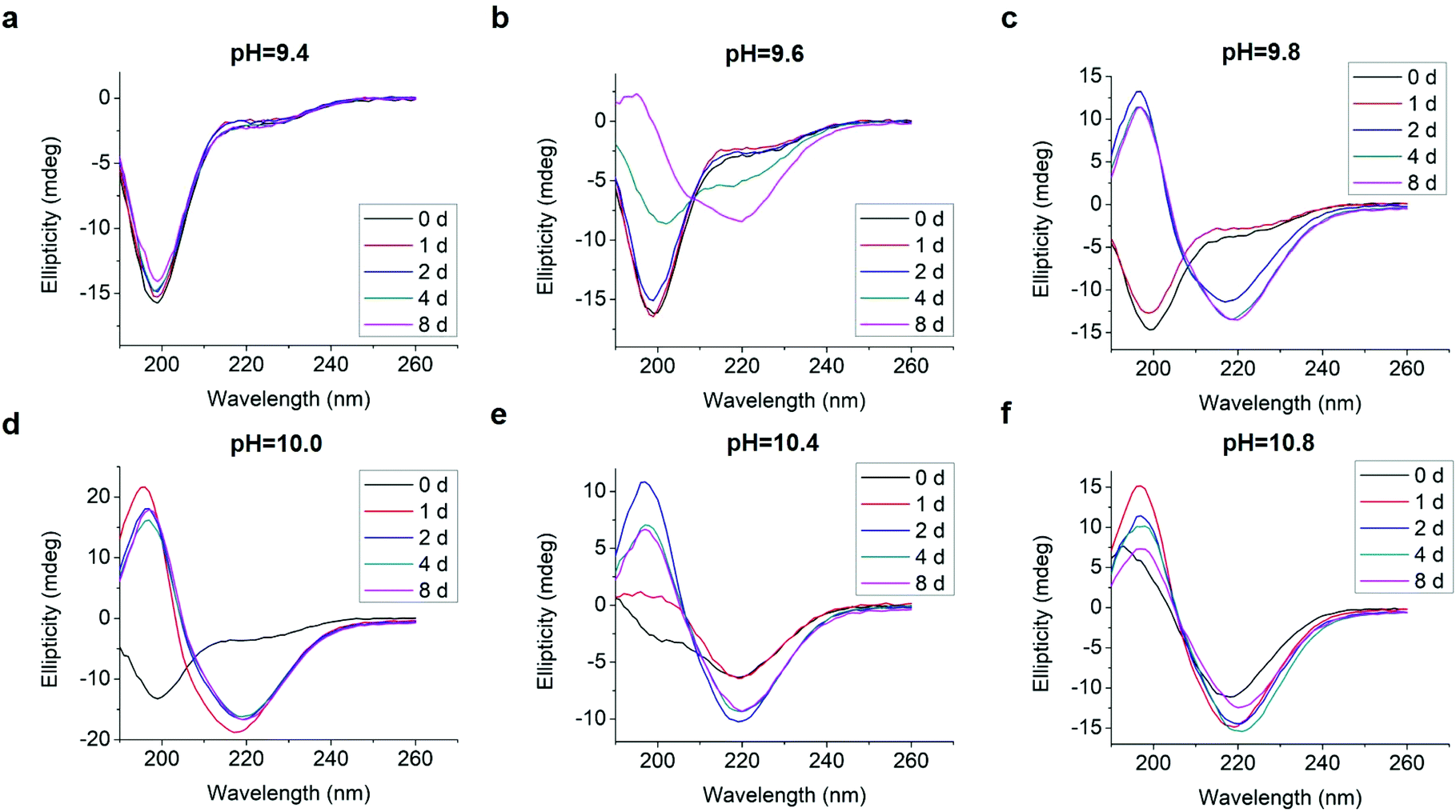 | ||
| Fig. 5 Inverted CD spectra of 0.1 mM D-GL13K in borax-NaOH buffer with pH ranging from 9.4 to 10.8 for 0 day, 1 day, 2 days, 4 days, and 8 days. | ||
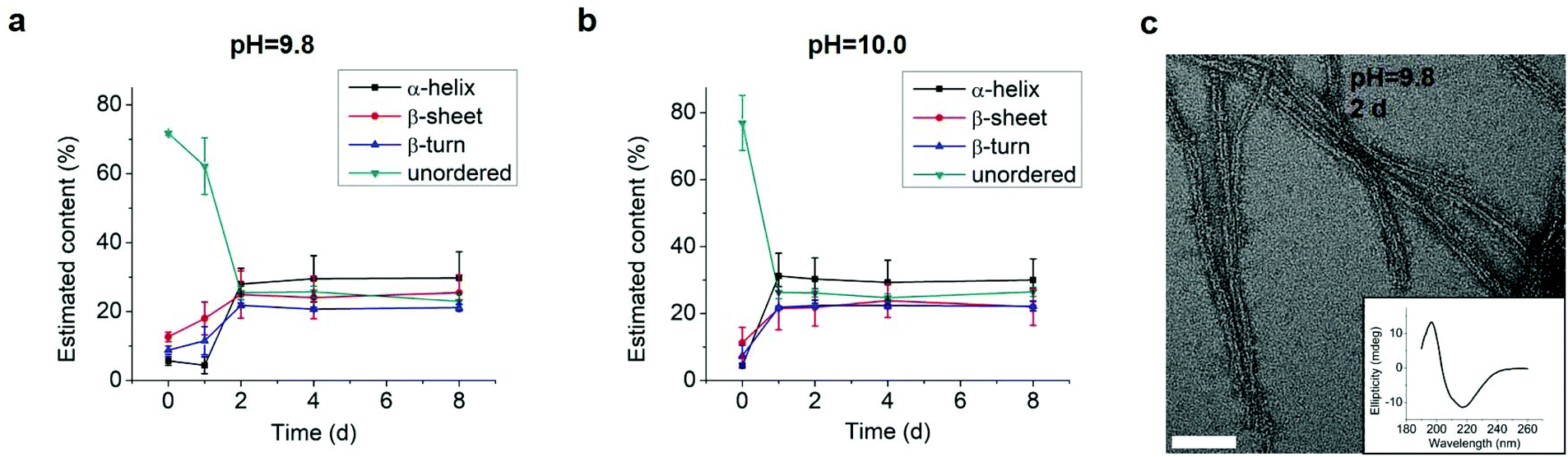 | ||
| Fig. 6 Estimation of secondary structure (α-helix, β-sheet, β-turn, and unordered) contents from inverted CD spectra of 0.1 mM D-GL13K in buffer solutions of (a) pH 9.8 and (b) pH 10.0. Content values were averaged from estimations by three different methods, SELCON, CDSSTR, and CONTIN/LL. (c) TEM image of negatively stained D-GL13K self-assembled nanoribbons in pH 9.8 solution for 2 days. The inset is the corresponding inverted CD spectrum. The scale bar is 50 nm. | ||
At higher solution pH, these time-dependent differences in the formation of the supramolecular assemblies were most likely masked. At pH ≥ 10.4 the driving force for triggering molecular transformation by the two peptides seemed to be high enough that the two peptides immediately started molecular transformations and assembly after dissolution.
The faster assembly initiation of D-GL13K compared to L-GL13K suggests structural differences between the two peptides that result in different effect on molecular transformation and/or intermolecular interactions during charge neutralization. Others have shown that L-peptides with specific amino acids substituted by their D-enantiomers induce changes in amphipathicity of the peptides that favour molecular self-assembly.33 However, the same has not been yet demonstrated for all D-amino acid peptides when compared to their all L-amino acid counterparts. Also, the additional chiral side chain of isoleucine might affect the backbone conformations and contribute to the different self-assembly dynamics of L- and D-GL13K. Molecular dynamics simulation of conformational propensities of the GGXGG host–guest pentapeptide for three isoleucine antipodes (X = L-Ile, D-Ile, and D-allo-Ile) showed that both D-Ile and D-allo-Ile exhibited significant differences in conformational regions compared to L-Ile.34
We also tested L-GL13K-R, a randomized-amino acid sequence of L-GL13K, with the purpose of studying the effects of the arrangement of cationic and hydrophobic peptide residues on the supramolecular assembly and antimicrobial activity. This scrambled peptide was designed to preserve a similar distribution of cationic residues and hydrophobic residues to that of L-GL13K. L-GL13K-R maintained an unordered molecular structure for up to 8 days in buffer solutions with pH 9.0–11.0 (Fig. 7a–c). Only when the solution pH was as high as 12.0 a weak characteristic peak for β-sheet configuration was detected (Fig. 7d). Negative-staining TEM images revealed that L-GL13K-R formed small aggregates of about 10–15 nm at pH ≤ 11.0; i.e., when no obvious β-sheet peak was detected in CD spectra (Fig. 7e). At pH = 12.0, when L-GL13K-R showed partial transformation to the β-sheet, these peptides assembled into sphere-like nanostructures with a diameter of around 40 nm (Fig. 7f); which were distinctively different structures than the nanofibrils and nanoribbons obtained with L- and D-GL13K. Thus, the self-assembly process was not only determined by the total charge and overall hydrophobicity of the molecules, but also significantly affected by the distribution of the cationic and hydrophobic residues of these peptides.
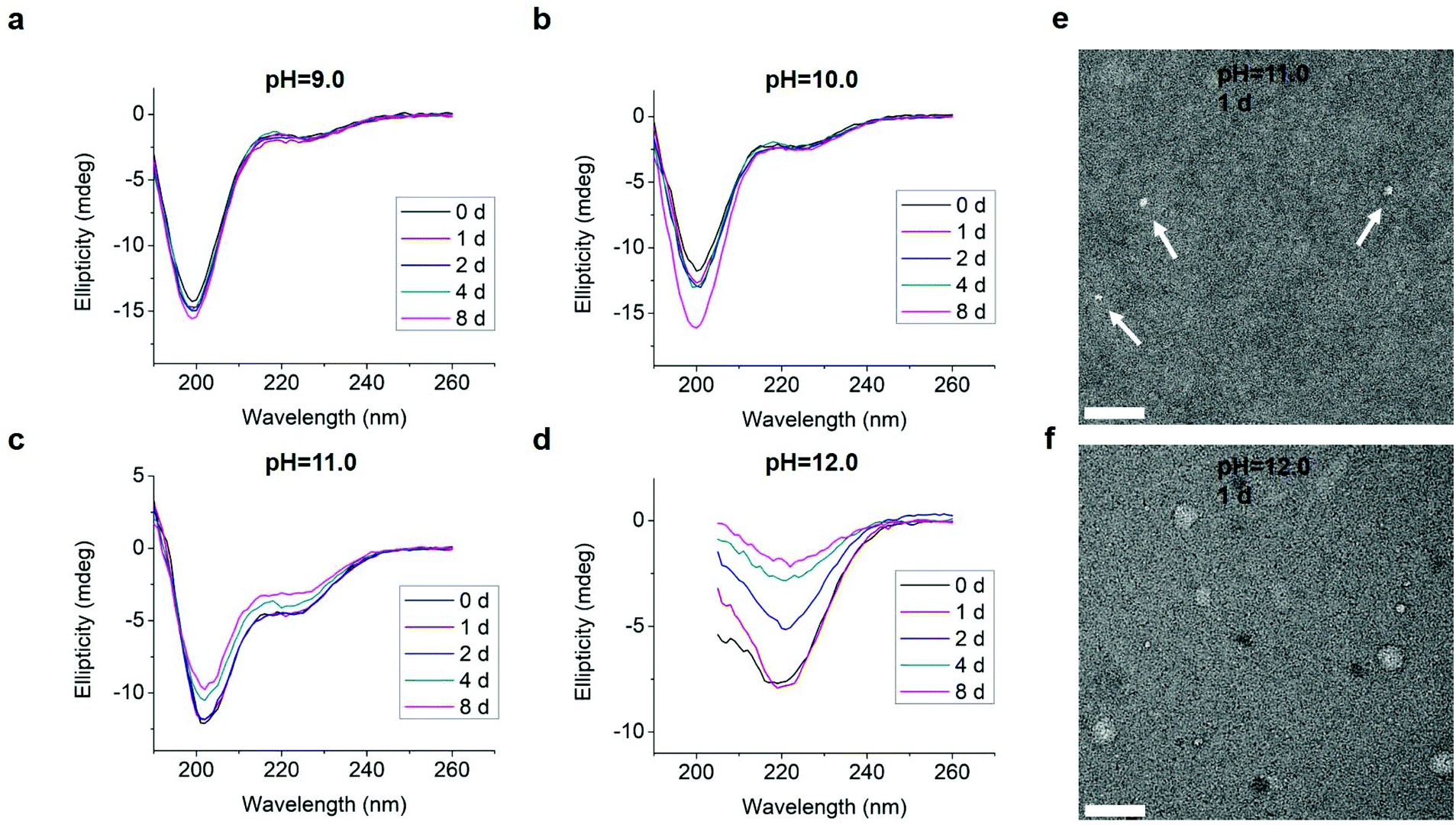 | ||
| Fig. 7 (a–d) CD spectra of 0.1 mM L-GL13K-R in borax-NaOH buffer solutions with pH ranging from 9.0 to 12.0 for 0 day, 1 day, 2 days, 4 days, and 8 days. (e and f) TEM images of negatively stained L-GL13K-R aggregates and nanospheres in pH 11.0 and 12.0 buffer solutions, respectively, for 1 day. Arrows point at small aggregates. All scale bars are 100 nm. | ||
Both L- and D-GL13K had 4 pKa values, but each pKa value of D-GL13K was slightly lower than that of L-GL13K, which might have contributed to the faster initiation of self-assembly of D-GL13K (Fig. 1d–f). In contrast, L-GL13K-R had 6 pKa values and showed a markedly different self-assembly process and structures.
Peptide self-assembly and antimicrobial activity
Chirality and specific amino acid sequence markedly affected the molecular self-assembly process and the supramolecular nanostructures obtained with L-GL13K, D-GL13K, and L-GL13K-R. We then explored potential relationships between the structural differences of the peptides and their biological function; i.e., antimicrobial activity. We determined MICs of the three peptides against both Gram-positive, S. gordonii M5 and Gram-negative, P. aeruginosa Xen41 bacteria (Table 1). MICs for D-GL13K were much smaller than those for L-GL13K against both strains, which demonstrated that the D-enantiomer had a higher antimicrobial potency. L-GL13K-R was not antimicrobial against either of the two bacterium strains in the range of concentrations tested; i.e., up to 512 μg ml−1.| Microbial strain | MIC, μg ml−1 (mM) | ||
|---|---|---|---|
| L-GL13K | D-GL13K | L-GL13K-R | |
| S. gordonii M5 | 128 (0.091) | 8 (0.006) | >512 (>0.366) |
| P. aeruginosa Xen41 | 32 (0.023) | 8 (0.006) | >512 (>0.366) |
When intrinsically structural differences of L- and D-enantiomers of antimicrobial peptides are not considered, the lower antimicrobial potency of the L-peptides in comparison with the D-peptides is commonly attributed to the higher susceptibility of L-peptides to be degraded by bacteria secreted proteases. However, different protease susceptibility did not seem to explain differences in antimicrobial activity between L-GL13K and D-GL13K peptides. H. Hirt et al.25 compared MICs of L-GL13K and D-GL13K against wild-type E. faecalis and a protease deficient mutant of this bacterium strain, TX5128. D-GL13K had low MICs for both wild-type (13 μg ml−1 against) and TX5128 (11 μg ml−1); however, L-GL13K did not show antimicrobial activity against any of the two strains up to concentrations of 200 μg ml−1 or higher. This suggested that the different antimicrobial potency of L-GL13K and D-GL13K against E. faecalis was not only caused by the bacteria secreted proteases. In addition, not all L-enantiomers have lower antimicrobial potency than D-enantiomers. S. Porter et al.35 found that L-diphenylalanine self-assembled nanotubes had increased antimicrobial activity against Gram-positive S. aureus and Gram-negative E. coli and P. aeruginosa compared to D-diphenylalanine.
The MICs for L-GL13K and D-GL13K were close to or lower than the concentration in the buffer solutions (0.1 mM or 140 μg ml−1) used for molecular structural analysis. Also, the MICs for L-GL13K against both strains (0.091 and 0.023 mM, Table 1) were in the range of or higher than the critical aggregation concentration of L-GL13K in pH 10.8 buffer solution (0.02–0.05 mM) (Fig. S1†). Although there are notable differences in the physiochemical conditions on a bacteria membrane than in a buffer solution, V. Balhara et al.24 and N. Harmouche et al.23 demonstrated that the secondary structure of L-GL13K could be similarly controlled either by the buffer pH or by the concentration of bacterial model membranes. Their observations and conclusions were restricted to spectroscopic analysis of the molecular structures and to conditions analogous to the ones that produced the final and stable structures we show here. When comparing D-GL13K and L-GL13K under final stable conditions, we did not find structural differences between the two peptides, including the self-assembled nanostructures. However, we assessed relevant differences in the respective self-assembly processes as D-GL13K peptides initiated assembly faster than L-GL13K peptides. We also assessed that the non-antimicrobial L-GL13K-R peptides self-assembled in a different range of pH and into markedly different self-assembled structures. These findings suggest a link between the process of antimicrobial peptide self-assembly and antimicrobial activity, which has also been recently suggested by others.6,9–13,17
Conclusions
We have demonstrated that all L- and D-amino acid enantiomers of GL13K, a designer AMP derived from a human protein, transformed with time in secondary structures that subsequently evolved into supramolecular self-assemblies. However, differences in the chirality and the arrangement of the cationic and hydrophobic residues led to distinct differences in the dynamics of the assembly process and the nanostructures formed (fibrils, twisted ribbons, and spheres), which was indicative of a functional relationship between AMP self-assembly and antimicrobial activity. A better understanding of the effect of AMP self-assembly on their antimicrobial activity will enable a greater understanding of their mechanism of action, a more efficient design of AMPs, and processing routes to obtain supramolecular nanomaterials made of AMPs to prevent infection in regenerative medicine and other biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Wei Zhang and Dr Bob Hafner, Characterization Facilities, University of Minnesota (UMN) for technical assistance with TEM imaging. The authors also acknowledge Dr James Marti, Minnesota Nano Center, UMN for technical assistance with titration curve assessment; Nicholas Fischer, MDRCBB, UMN for technical assistance with the fluorescence microplate reader; and Ms Ruoqiong Chen, School of Dentistry, UMN for technical assistance with MIC analysis. The authors also acknowledge Professor Sven Gorr, School of Dentistry, UMN for constructive discussions regarding the effect of proteases on the activity of L- and D-GL13K peptides and for donating the S. gordonii strain used here. The authors acknowledge Erik Skoe for language editing of the manuscript. This research study was supported by the National Institute for Dental and Craniofacial Research of the National Institutes of Health [grant number RO1DE026117 to C. A.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S. A. acknowledges financial support from the Ministry of Economy, Industry and Competitiveness of the Spanish Government through a travel fellowship for graduate students [Ref. ID: EEBB-I-18-13053]. The funding bodies had no role in study design, the collection, analysis and interpretation of data, in the writing of the report, and in the decision to submit the article for publication. Parts of this work were carried out in the University of Minnesota I.T. Characterization Facility, which receives partial support from NSF through the MRSEC program. Portions of this work were conducted in the Minnesota Nano Center, which is supported by the National Science Foundation through the National Nano Coordinated Infrastructure Network (NNCI) under Award Number ECCS-1542202.References
- R. E. W. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- A. L. Hilchie, K. Wuerth and R. E. W. Hancock, Nat. Chem. Biol., 2013, 9, 761–768 CrossRef CAS PubMed.
- M. R. Yeaman, Pharmacol. Rev., 2003, 55, 27–55 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS PubMed.
- X. Tian, F. Sun, X. R. Zhou, S. Z. Luo and L. Chen, J. Pept. Sci., 2015, 21, 530–539 CrossRef CAS PubMed.
- E. Eiríksdóttir, K. Konate, Ü. Langel, G. Divita and S. Deshayes, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1119–1128 CrossRef PubMed.
- K. Pagel, S. C. Wagner, K. Samedov, H. Von Berlepsch, C. Böttcher and B. Koksch, J. Am. Chem. Soc., 2006, 128, 2196–2197 CrossRef CAS.
- M. Zhang, J. Zhao and J. Zheng, Soft Matter, 2014, 10, 7425–7451 RSC.
- S. Gour, V. Kaushik, V. Kumar, P. Bhat, S. C. Yadav and J. K. Yadav, J. Pept. Sci., 2016, 22, 201–207 CrossRef CAS.
- A. N. Calabrese, Y. Liu, T. Wang, I. F. Musgrave, T. L. Pukala, R. F. Tabor, L. L. Martin, J. A. Carver and J. H. Bowie, ChemBioChem, 2016, 17, 239–246 CrossRef CAS.
- D. Xu, W. Chen, Y. J. Tobin-Miyaji, C. R. Sturge, S. Yang, B. Elmore, A. Singh, C. Pybus, D. Greenberg, T. J. Sellati, W. Qiang and H. Dong, ACS Infect. Dis., 2018, 4, 1327–1335 CrossRef CAS PubMed.
- L. L. Martin, C. Kubeil, S. Piantavigna, T. Tikkoo, N. P. Gray, T. John, A. N. Calabrese, Y. Liu, Y. Hong, M. A. Hossain, N. Patil, B. Abel, R. Hoffmann, J. H. Bowie and J. A. Carver, Pept. Sci., 2018, e24052 CrossRef.
- V. Castelletto, R. H. Barnes, K. A. Karatzas, C. J. C. Edwards-Gayle, F. Greco, I. W. Hamley, R. Rambo, J. Seitsonen and J. Ruokolainen, Biomacromolecules, 2018, 19, 2782–2794 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 14246–14253 CrossRef CAS PubMed.
- M. Beter, H. K. Kara, A. E. Topal, A. Dana, A. B. Tekinay and M. O. Guler, Mol. Pharm., 2017, 14, 3660–3668 CrossRef CAS PubMed.
- L. Schnaider, S. Brahmachari, N. W. Schmidt, B. Mensa, S. Shaham-Niv, D. Bychenko, L. Adler Abramovich, L. J. W. Shimon, S. Kolusheva, W. F. Degrado and E. Gazit, Nat. Commun., 2017, 8, 1365 CrossRef PubMed.
- S.-U. Gorr, M. Abdolhosseini, A. Shelar and J. Sotsky, Biochem. Soc. Trans., 2011, 39, 1028–1032 CrossRef CAS PubMed.
- M. Abdolhosseini, S. R. Nandula, J. Song, H. Hirt and S. U. Gorr, Peptides, 2012, 35, 231–238 CrossRef CAS PubMed.
- H. Hirt and S. U. Gorr, Antimicrob. Agents Chemother., 2013, 57, 4903–4910 CrossRef CAS PubMed.
- K. V. Holmberg, M. Abdolhosseini, Y. Li, X. Chen, S. U. Gorr and C. Aparicio, Acta Biomater., 2013, 9, 8224–8231 CrossRef CAS PubMed.
- X. Chen, H. Hirt, Y. Li, S. U. Gorr and C. Aparicio, PLoS One, 2014, 9, e111579 CrossRef PubMed.
- N. Harmouche, C. Aisenbrey, F. Porcelli, Y. Xia, S. E. D. Nelson, X. Chen, J. Raya, L. Vermeer, C. Aparicio, G. Veglia, S. U. Gorr and B. Bechinger, Biochemistry, 2017, 56, 4269–4278 CrossRef CAS PubMed.
- V. Balhara, R. Schmidt, S. U. Gorr and C. Dewolf, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2193–2203 CrossRef CAS PubMed.
- H. Hirt, J. W. Hall, E. Larson and S.-U. Gorr, PLoS One, 2018, 13, e0194900 CrossRef.
- C. De La Fuente-Núñez, F. Reffuveille, S. C. Mansour, S. L. Reckseidler-Zenteno, D. Hernández, G. Brackman, T. Coenye and R. E. W. Hancock, Chem. Biol., 2015, 22, 196–205 CrossRef PubMed.
- M. Melchionna, K. Styan and S. Marchesan, Curr. Top. Med. Chem., 2016, 16, 1–1 CrossRef.
- S. Marchesan, C. D. Easton, K. E. Styan, L. J. Waddington, F. Kushkaki, L. Goodall, K. M. McLean, J. S. Forsythe and P. G. Hartley, Nanoscale, 2014, 6, 5172–5180 RSC.
- Z. Luo, X. Zhao and S. Zhang, PLoS One, 2008, 3, e2364 CrossRef PubMed.
- N. Sreerama and R. W. Woody, Anal. Biochem., 2000, 287, 252–260 CrossRef CAS PubMed.
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- C. L. Towse, G. Hopping, I. Vulovic, V. Daggett and A. Fersht, Protein Eng., Des. Sel., 2014, 27, 447–455 CrossRef CAS PubMed.
- S. L. Porter, S. M. Coulter, S. Pentlavalli, T. P. Thompson and G. Laverty, Acta Biomater., 2018, 77, 96–105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: High-resolution TEM micrographs of L-GL13K self-assembled structures; CD spectra of L-GL13K peptides at high pH that were titrated back to low pH. See DOI: 10.1039/c8nr07334a |
| This journal is © The Royal Society of Chemistry 2019 |