
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModification of the glycosylation of extracellular vesicles alters their biodistribution in mice†
Felix
Royo‡
 a,
Unai
Cossío‡
b,
Ane
Ruiz de Angulo
c,
Jordi
Llop
*b and
Juan M.
Falcon-Perez
*ad
a,
Unai
Cossío‡
b,
Ane
Ruiz de Angulo
c,
Jordi
Llop
*b and
Juan M.
Falcon-Perez
*ad
aExosomes Laboratory, CIC bioGUNE, CIBERehd, Bizkaia Technology Park, Derio, 48160, Bizkaia, Spain. E-mail: jfalcon@cicbiogune.es
bRadiochemistry and Nuclear Imaging Group, CIC biomaGUNE, Paseo Miramón 182, 20014 Donosti, Spain. E-mail: jllop@cicbiomagune.es
cThenanostic Nanomedicine Laboratory, CIC biomaGUNE, Paseo Miramón 182, 20014 Donosti, Spain
dIKERBASQUE, Basque Foundation for Science, 48011, Bilbao, Spain
First published on 9th January 2019
Abstract
Extracellular vesicles (EVs) are considered sophisticated vehicles for cell-to-cell communication, thanks to the possibility of handling a variable cargo in a shell with multiple types of decoders. Surface glycosylation of EVs is a method that could be used to control their interaction with different cells and, consequently, the biodistribution of the vesicles in the body. Herein, we produced EVs derived from mouse liver proliferative cells, and we treated them with neuraminidase, an enzyme that digests the terminal sialic acid residues from glycoproteins. Afterwards, we labeled the EVs directly with [124I]Na and injected them in mice intravenously or into the hock. The amount of radioactivity in major organs was measured at different time points after administration both in vivo using positron emission tomography and ex vivo (after animal sacrifice) using dissection and gamma counting. The results showed that intravenous injection leads to the rapid accumulation of EVs in the liver. Moreover, after some hours the distribution led to the presence of EVs in different organs including the brain. Glycosidase treatment induced an accumulation in the lungs, compared with the intact EVs. Furthermore, when the EVs were injected through the hock, the neuraminidase-treated vesicles distributed better at the axillary lymph nodes than the untreated EVs. This result shows that modification of the glycosylated complexes on the EV surface can affect the distribution of these vesicles, and specifically removing the sialic acid residues allows more EVs to reach and accumulate at the lungs.
Extracellular vesicles (EVs) are cell-secreted nanovesicles that mediate cell-to-cell communication and modulate diverse biological processes including the immune response and cancer progression.1,2 Moreover, EVs play a pivotal role in stem cell plasticity and tissue regeneration,3 which has opened new avenues for regenerative therapy.4 Different experimental models and strategies have demonstrated that EVs are vehicles that give the cell the capacity to transfer signals and active molecules (lipids, proteins, mRNAs and microRNAs) to neighbouring and distant cells.5–10 However, to understand their physiological role and facilitate their application in biomedicine, a better knowledge of the mechanisms controlling the biodistribution of EVs is needed.4 Surface proteins determine the fate of EVs injected in vivo11 and, in particular, the integrin composition of the membrane can alter the affinity of EVs for certain tissues. Therefore, tailoring the surface of EVs could be used to specifically target selected tissues, and different strategies have been proposed for this aim.12 One of them is the manipulation of glycosylation, which is an important regulator of membrane-to-membrane interactions.13 The surface of EVs has a complex glycosylation pattern, which has been studied in depth to characterize and purify subpopulations of EVs,14–16 and it has also been shown that its manipulation increases their delivery into neuroblastoma cells.17
The study of the biodistribution of EVs after administration into living organisms has received great attention and various approaches have been designed for the in vivo tracking of EVs upon systemic delivery in different animal models.18–20 They have been labelled with either selective fluorescent dyes for nucleic acids21 or with lipophilic dyes to label membranes.22 Near-infrared (NIR) dyes such as carbocyanine DiOC18(7) (DiR) have been chosen for in vivo applications due to their high signal/noise ratio, the minimal autofluorescence of biological tissue in the 700–900 nm spectral range, and the enhanced tissue penetration of near-IR light.18 However, the poor penetration capacity of visible or infrared light limits the applicability of this technology to small rodents. Moreover, quantification of images is challenging, mainly due to attenuation and scattering effects. Besides this, one major limitation of lipophilic dyes is that they can promote EV aggregation and may give rise to artefacts, especially in vivo.23 Moreover, extensive washing steps, needed to remove the unbound dye, can cause a significant loss of EVs. Another commonly used imaging technique is magnetic resonance imaging (MRI);24,25 this technique, although it provides excellent anatomical images and superb spatial resolution, is limited by its poor intrinsic sensitivity. Additionally, MRI images are difficult to quantify and the acquisition of whole body images is challenging and time consuming. In this scenario, radiolabelling of EVs with a positron or gamma emitter followed by whole body imaging using positron emission tomography (PET) or single photon emission computed tomography (SPECT) might be anticipated as an ideal alternative. These minimally invasive and ultra-sensitive molecular imaging techniques are fully translational to the clinical field, as they rely on the detection of high-energy gamma rays, which have virtually no penetration limits, and have been proven to be ideally suited for the in vivo tracking of a wide variety of nanomaterials.26–29 The high energy of the emitted photons minimizes the attenuation and scattering effects while enabling the quantification of images at the whole body level. Despite these advantages, their use in the context of EVs has barely been reported in the literature.18,30,31 In these previous reports, EVs were labelled with 99mTc, the most commonly used radionuclide in nuclear medicine, although there is also a report with [125I]NaI.32 In both cases, images were acquired using SPECT due to the physical properties of the radionuclides. However, SPECT suffers from limited sensitivity; additionally, the acquisition of dynamic images is challenging and the quantification of images poses difficulties. For our study, we have selected PET as it provides improved spatiotemporal resolution and higher sensitivity with respect to SPECT, and enables absolute quantification of the images. Additionally, the selection of 124I as the radionuclide, with a half-life of >4 days, enables the in vivo tracking of labelled species from hours to days after administration. Unlike common labelling methods that use lipophilic compounds which migrate through the membrane and are entrapped in the EVs,31 our approach consists of the direct radiolabelling of proteins embedded in the membrane of the EVs by forming a covalent bond to tyrosine by a specific reaction facilitated by iodination tubes, thus preventing (or hampering) the release of the labelling agent during in vivo assays. To remove excess radiolabels, fractions of radio-labelled EVs were pooled, and were totally separated from the free iodine (Fig. S1, ESI Materials and methods†).
Extracellular vesicles were produced and isolated from a liver-derived mouse cell line named MLP2933 that presents progenitor features and releases EVs loaded with transcripts related to signalling and cellular differentiation.7 Surface glycosylation of the EVs was manipulated by treatment with neuraminidase to remove the terminal residues of sialic acid that have been reported to be involved in the cellular recognition of EVs.34 Thus, the EVs were produced and isolated (see ESI Materials and methods† for details) and, as previously reported, we could observe the vesicles by cryo-electron microscopy (Fig. S2A and B†). Then, the prepared EVs were treated with (termed MLP-Neu) or without (termed MLP-No Treat) the enzyme neuraminidase, and purified by a sucrose cushion (see ESI Materials and methods† for details). Exosomal markers including Lamp1, Flot1, Tsg101 and Cd81 were detected by western-blotting, which also confirmed the absence of intracellular organelle markers such as the mitochondrial protein Cox-IV (Fig. S2C†). The efficiency of the neuraminidase treatment was confirmed by the change of the electrophoretic movement of the glycosylated Lamp1 protein (Fig. S2C†). The treatment did not significantly change the size of the EVs that had an average size of 100 nm, as determined by NTA analysis (Fig. S2A and B†). No significant differences in protein concentration (23.7 ± 6.4 ng μL−1, n = 2) were found between the untreated and treated samples. Interestingly, while there was no alteration in the abundance of Tsg101, a reduction in the abundance of the Lamp1, Flotillin1 and Cd81 proteins was observed in the sample treated with neuraminidase, which could suggest a change in the composition of the vesicles. However, further analysis is needed to unravel the reason for this effect of the neuraminidase treatment. Afterwards, we proceeded with the [124I]NaI labelling of both the untreated and neuraminidase-treated EV preparations (see ESI Materials and methods† for details). The overall decay-corrected radiochemical yields after the purification process were 17 ± 2% and 19 ± 1% for MLP-Neu and MLP-No Treat, respectively. Bradford test analysis was performed on the EV solutions before and after radiolabelling and showed a loss in protein content <5% for both EVs. Specific activity values were 15 ± 2 MBq μg−1 protein at the end of the synthesis. iTLC analysis of the purified fraction confirmed a radiochemical purity above 95% at the injection time in all cases (Fig. S3†). Stability studies showed that the fraction of radioiodine that detached from the EVs after 72 hours of incubation in physiological saline solution was <10% in both cases (MLP-Neu and MLP-No Treat), which supports the suitability of the labelling method to approach subsequent in vivo studies.
Once the suitability of [124I]NaI for the direct labelling of EVs was validated, we studied in triplicate their biodistribution in wild-type mice after intravenous administration. The accumulation of radioactivity over time in different organs, expressed as % of injected dose per cm3 of tissue (%ID per cc), with different EVs (MLP-Neu and MLP-No Treat) and [124I]NaI (as the control) is shown in Fig. 1. The accumulation of radioactivity over time for the untreated or neuraminidase-treated EVs follows a similar trend in each organ. At short times (t = 15 min) the maximum accumulation of radioactivity was found in the liver and in the lungs. At this time point, the accumulation of free [124I]NaI in the same organs is clearly lower. Importantly, the biodistribution pattern of [124I]NaI is different, with major accumulation in the thyroid gland at short times after administration, which can be clearly visualised directly in the PET images (Fig. 2). Such fast accumulation is not observed after the administration of the EVs, supporting the in vivo stability of the labelled EVs. At long times after administration (t > 15 min), the amount of radioactivity decreases progressively in all organs for both MLP-Neu and MLP-No Treat, with the exception of the thyroid and bladder, in which the amount of radioactivity progressively increases with time. Indeed, the accumulation of radioactivity in urine increased over time and peaked at t = 480 minutes (8 hours). The presence of a radioactive signal in the brain was low but detectable. Interestingly, some differences in the accumulation of radioactivity could be observed at 72 hours, specifically in the lungs, where the accumulation of neuraminidase-treated EVs was higher than the accumulation of untreated EVs (Fig. 1). This effect was also observed in the ex vivo exploration of the organs, at the end of the experiment (Fig. 3). These data suggest that the removal of terminal sialic acid could improve the dynamic by reducing the charge of the EVs. This could result in improved mobility – better diffusion – and a better interaction with cells, which could increase their uptake and facilitate their travel in circulating cells. However, further experiments are required to certainly unravel the mechanism.
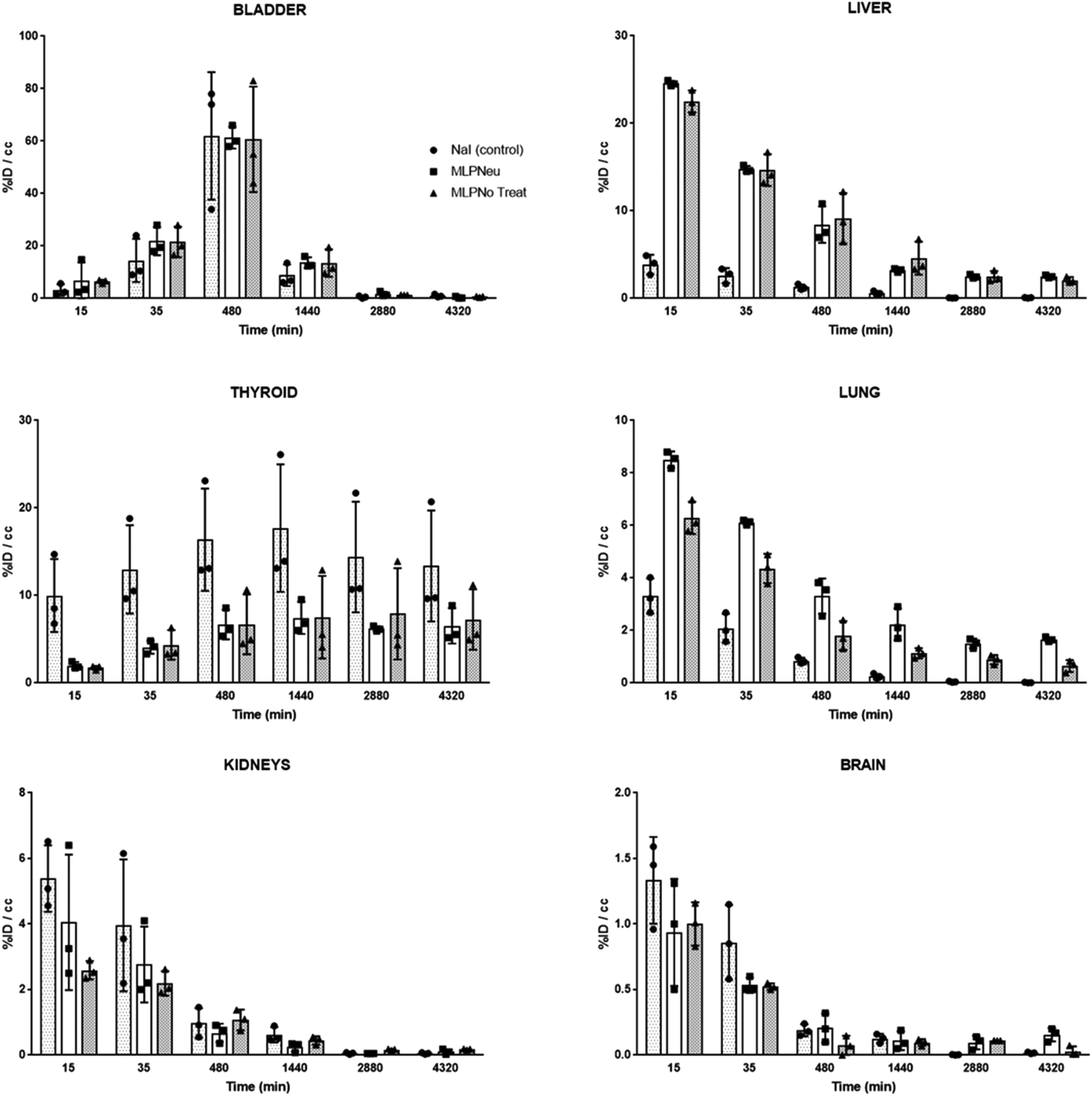 | ||
| Fig. 1 Accumulation of [124I]NaI (white bars; named Control), [124I]MLP29 neuraminidase-treated EVs (dark grey bars; named MLP-Neu) and [124I]MLP29 EVs (light grey bars; named MLP-No Treat) in different organs at different time points after intravenous administration, as determined by PET imaging. Results are expressed as % of injected dose per cm3 of tissue. Error bars correspond to the SD (n = 3), and t-tests were performed exclusively for the last time point between MLP-Neu and MLP-No Treat. | ||
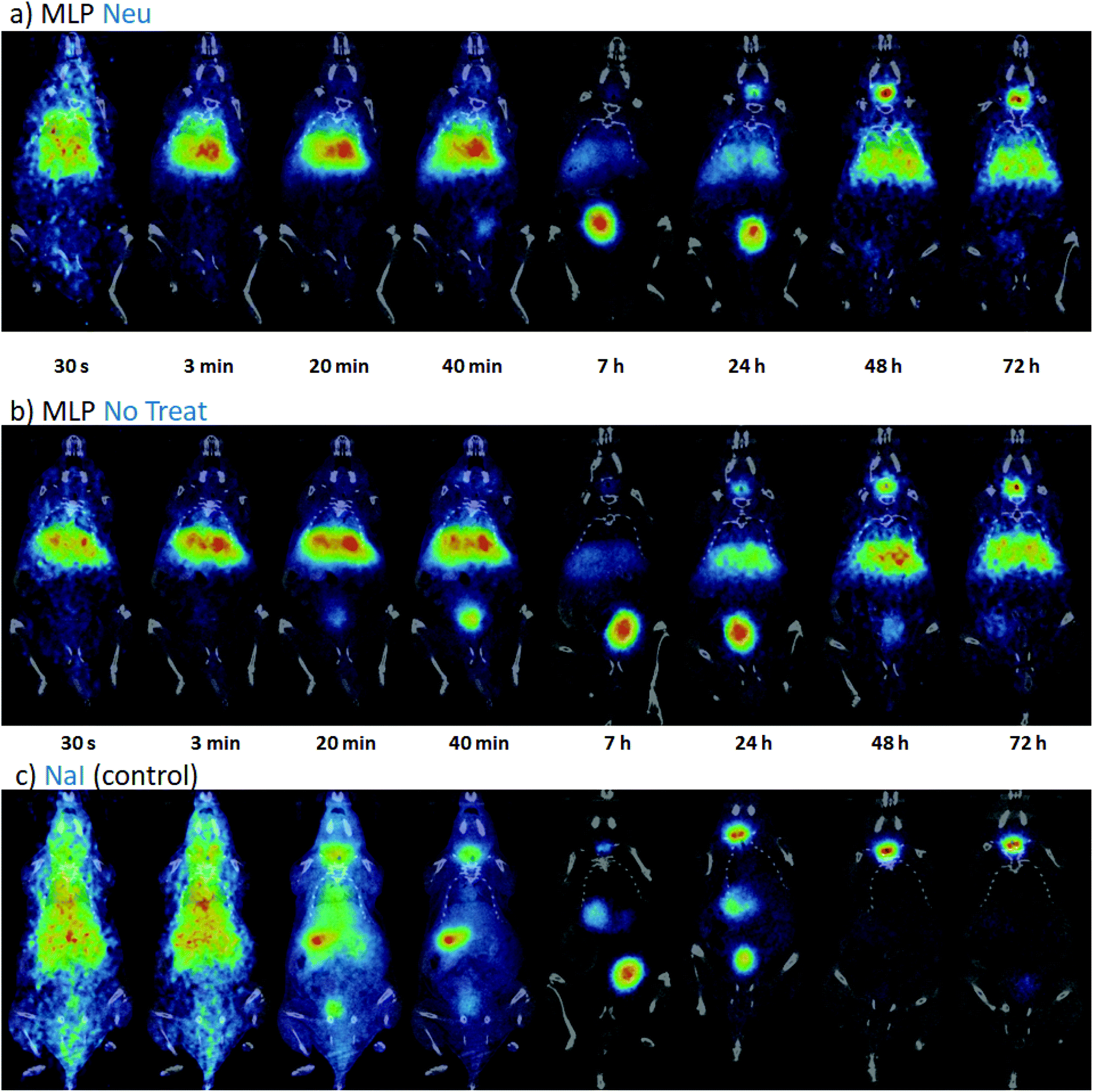 | ||
| Fig. 2 PET-CT coronal images obtained at different time points after intravenous administration of [124I]MLP-Neu EVs (a), [124I]MLP-No Treat (b) and [124I]NaI (c). PET images (coronal projections) have been co-registered with the CT images of the same animals for the location of the radioactive signal. | ||
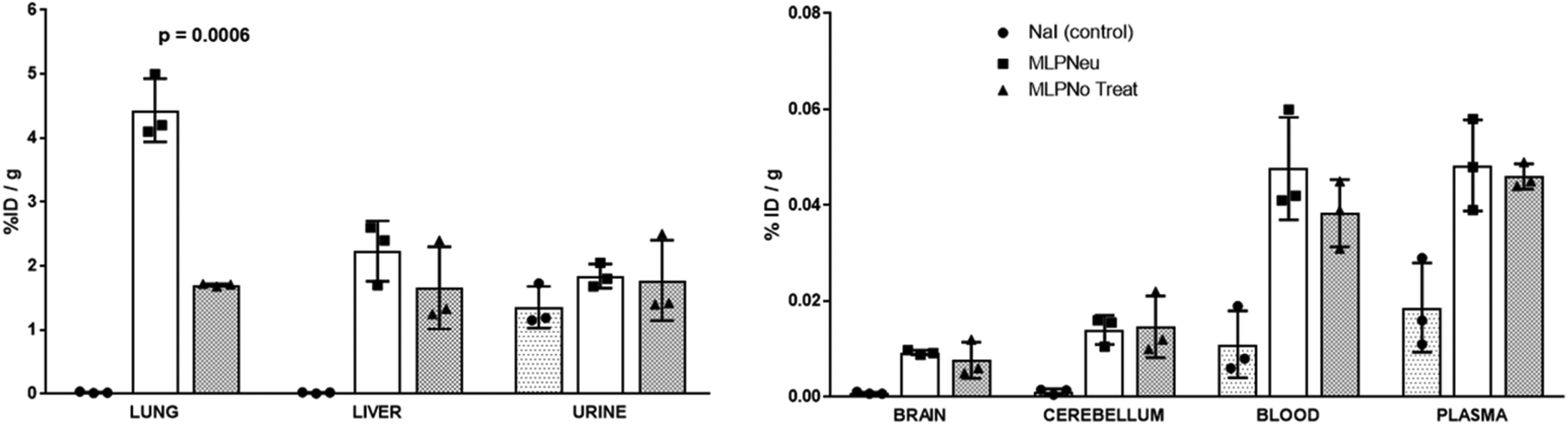 | ||
| Fig. 3 Accumulation of [124I]NaI (white bars; named Control), [124I]MLP29 neuraminidase-treated EVs (dark grey bars; named MLP-Neu) and [124I]MLP29 EVs (light grey bars; named MLP-No Treat) in different organs 72 hours after intravenous administration, measured ex vivo by dissection and gamma counting. Results are expressed as % of injected dose per gram of tissue. Error bars correspond to the SD (n = 3), and t-tests were performed exclusively between MLP-Neu and MLP-No Treat. | ||
We have also studied the accumulation of the EVs in different organs after hock administration (Fig. S4†). Similar to intravenous administration, the significant concentration of radioactivity in the urine suggested elimination via renal excretion. At 72 hours post administration, most of the remaining radioactivity was localized in the thyroid gland. Again, the distribution of the labelled EVs significantly differed from the distribution pattern observed after [124I]NaI. These differences can also be clearly visualised in the PET images (Fig. S5†). For the control ([124I]NaI), the presence of radioactivity could be detected only in the urine and the thyroid gland, while the presence of radioactivity was negligible in the other organs and lymph nodes. For the EVs, a relatively high uptake was observed in different lymph nodes after administration, as we quantified ex vivo at the end of the study (Fig. 4), suggesting that these labelled species migrated through the lymphatic system after subcutaneous administration. The labelled EVs were found not only in the nodes close to the site of injection but also in the more distal ones (for instance, the nodes located on the opposite side to the injection site). Again, neuraminidase treatment altered the EV biodistribution, and a remarkable accumulation in the axillary nodes with respect to the untreated EVs was observed (Fig. 4).
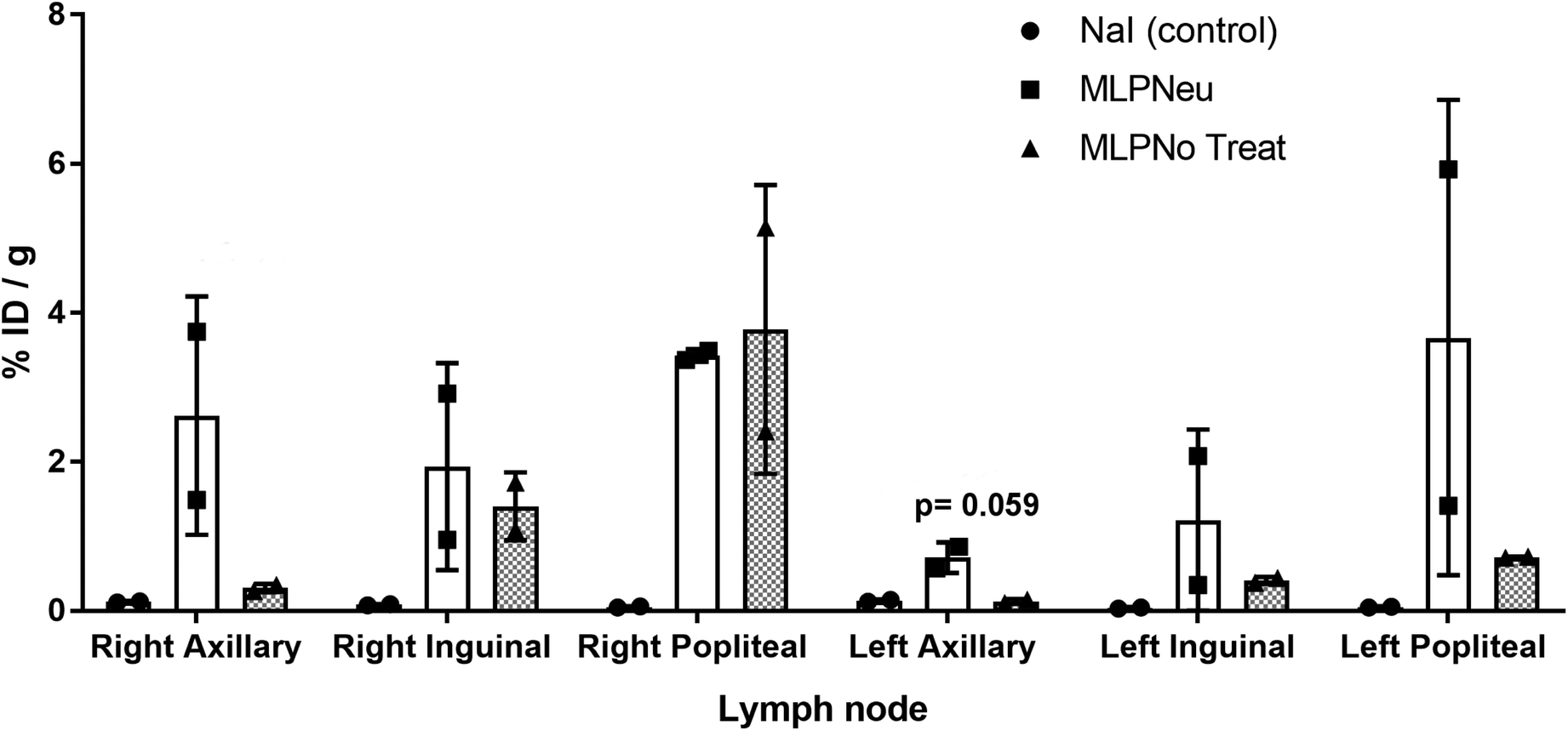 | ||
| Fig. 4 Accumulation of [124I]NaI (white bars; named Control), [124I]MLP29 neuraminidase-treated EVs (dark grey bars; named MLP-Neu) and [124I]MLP29 EVs (light grey bars; named MLP-No Treat) in different lymph nodes 72 hours after hock administration, measured ex vivo by dissection and gamma counting. Results are expressed as % of injected dose per gram of tissue. Error bars correspond to the SD (n = 2), and t-tests were performed exclusively between MLP-Neu and MLP-No Treat. | ||
Together, our results show that the administration route definitely determines the biodistribution pattern of EVs, as described in previous studies.25 In our case, and similar to the previously reported results for EVs injected in a foot pad,24 a subcutaneous injection through the hock leads to most of the EVs being delivered to the lymph nodes. Indeed, a wide distribution of the radioactive material through the lymphatic system could be observed. Although in the case of EVs derived from MLP29, a progenitor liver cell,35 the physiological meaning of this distribution is less interesting in the case of melanoma cells24 since MLP29-derived EVs are not expected to be found in the subcutaneous area. These results may trigger applications of our EVs as drug delivery agents when the lymphatic system is targeted, e.g. for immunotherapy.36,37
Unlike other reports, we employed EV preparations obtained by differential ultracentrifugation followed by a sucrose cushion-based enrichment, which resulted in the isolation of exosome-like vesicles as demonstrated in previous studies performed by our group.33 In our work, we have tested specifically the biodistribution of exosome-like vesicles secreted by a hepatic mouse cell line. These hepatic EVs were quickly taken up by different organs and cleared from the circulatory system within minutes, which was in agreement with a previous report that studied EVs secreted by melanoma cells.20Intravenous injection led to accumulation in the liver, kidney, spleen and lungs, which has also been observed for other studies.18,25,30,38 In our system, we observed a small although significant presence of these hepatic EVs in the brain. This shows that a small fraction of these vesicles could reach the brain which is a phenomenon previously reported as well, although in another model.39 Certainly, it has been acknowledged that EVs can cross the blood–brain barrier;40 moreover, EVs have been used for drug delivery to the brain by the modification of a surface receptor.41 Although the mechanism of the blood–brain barrier capacity under physiological conditions has not been fully elucidated, active transcellular crossing has been postulated.42 Our results are in good agreement with previous data and support the potential of EVs as vehicles which are able to cross the blood–brain barrier.
One of the main findings of our work lies in the fact that the treatment with neuraminidase actually alters the biodistribution of the EVs by increasing their capacity to accumulate in the lungs; additionally, the treatments seem to impose on the EVs a higher capacity to migrate through the lymphatic system. The alteration of the affinity of EVs for certain tissues by manipulating their integrin molecules is a well acknowledged phenomenon.11 However, other molecules could also be involved in the biodistribution of EVs. The role of neuraminidases has been studied by expressing them in HeLa cells, and the observed effect of this activity on the glycan in EVs was an enhancement of their dynamic biological behaviour. The authors proposed, as a mechanism, the modification of the negative charge and the steric hindrance of the glycocalyx.43 These results could explain the changes in distribution that we reported after EV treatment with neuraminidase, and confirm the importance of the glycoproteins that decorate the surface of the EVs. At the same time, they point out that the blood–brain barrier is within the scope of EV engineering in clinical therapy.
Conclusions
The reported results allow us to conclude that modification of the glycome of EVs affects their biodistribution in vivo. Moreover, the technique employed for the in vivo studies, PET, allowed for the accurate and quantitative tracking of radiolabelled EVs for 72 hours. It is also worth mentioning that intravenously injected EVs can reach the brain.Live subject statement
The animals were maintained and handled in accordance with the Guidelines for Accommodation and Care of Animals (European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes) and internal guidelines. All experimental procedures were approved by the ethical committee and the local authorities before conducting experimental work (Code: PRO-AE-SS-059).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the personnel at the Animal Facility of CIC biomaGUNE (Ainhoa Cano and Ander Arrieta) for the technical support during the in vivo experiments. In addition, we thank Dr David Gil from the Electron Microscope facility of CIC bioGUNE for his support in the electron microscope analysis. This work has been supported by the Health Basque Government (2015111149 to JMF-P), the Movember Foundation (GAP1 to JMF-P), the Ramón Areces Foundation to JMF-P, Instituto de Salud Carlos III (PI12/01604 to JMF-P), the Spanish Ministry of Economy and Competitiveness MINECO (SAF2015-66312 to JMF-P and PCIN-2015-116) and REDIEX (Excellence Network on Exosomes funded by MINECO). All of them were co-financed by ERDF (FEDER) Funds from the European Commission, “A way of making Europe”.References
- M. Tkach and C. Thery, Cell, 2016, 164, 1226–1232 CrossRef CAS PubMed.
- M. Yanez-Mo, P. R. Siljander, Z. Andreu, A. B. Zavec, F. E. Borras, E. I. Buzas, K. Buzas, E. Casal, F. Cappello, J. Carvalho, E. Colas, A. Cordeiro-da Silva, S. Fais, J. M. Falcon-Perez, I. M. Ghobrial, B. Giebel, M. Gimona, M. Graner, I. Gursel, M. Gursel, N. H. Heegaard, A. Hendrix, P. Kierulf, K. Kokubun, M. Kosanovic, V. Kralj-Iglic, E. M. Kramer-Albers, S. Laitinen, C. Lasser, T. Lener, E. Ligeti, A. Line, G. Lipps, A. Llorente, J. Lotvall, M. Mancek-Keber, A. Marcilla, M. Mittelbrunn, I. Nazarenko, E. N. Nolte-'t Hoen, T. A. Nyman, L. O'Driscoll, M. Olivan, C. Oliveira, E. Pallinger, H. A. Del Portillo, J. Reventos, M. Rigau, E. Rohde, M. Sammar, F. Sanchez-Madrid, N. Santarem, K. Schallmoser, M. S. Ostenfeld, W. Stoorvogel, R. Stukelj, S. G. Van der Grein, M. H. Vasconcelos, M. H. Wauben and O. De Wever, J. Extracell. Vesicles, 2015, 4, 27066 CrossRef PubMed.
- C. Tetta, S. Bruno, V. Fonsato, M. C. Deregibus and G. Camussi, Organogenesis, 2011, 7, 105–115 CrossRef PubMed.
- T. Lener, M. Gimona, L. Aigner, V. Borger, E. Buzas, G. Camussi, N. Chaput, D. Chatterjee, F. A. Court, H. A. Del Portillo, L. O'Driscoll, S. Fais, J. M. Falcon-Perez, U. Felderhoff-Mueser, L. Fraile, Y. S. Gho, A. Gorgens, R. C. Gupta, A. Hendrix, D. M. Hermann, A. F. Hill, F. Hochberg, P. A. Horn, D. de Kleijn, L. Kordelas, B. W. Kramer, E. M. Kramer-Albers, S. Laner-Plamberger, S. Laitinen, T. Leonardi, M. J. Lorenowicz, S. K. Lim, J. Lotvall, C. A. Maguire, A. Marcilla, I. Nazarenko, T. Ochiya, T. Patel, S. Pedersen, G. Pocsfalvi, S. Pluchino, P. Quesenberry, I. G. Reischl, F. J. Rivera, R. Sanzenbacher, K. Schallmoser, I. Slaper-Cortenbach, D. Strunk, T. Tonn, P. Vader, B. W. van Balkom, M. Wauben, S. E. Andaloussi, C. Thery, E. Rohde and B. Giebel, J. Extracell. Vesicles, 2015, 4, 30087 CrossRef PubMed.
- S. H. Kwon, K. D. Liu and K. E. Mostov, Curr. Biol., 2014, 24, 199–204 CrossRef CAS PubMed.
- K. Ridder, S. Keller, M. Dams, A. K. Rupp, J. Schlaudraff, D. Del Turco, J. Starmann, J. Macas, D. Karpova, K. Devraj, C. Depboylu, B. Landfried, B. Arnold, K. H. Plate, G. Hoglinger, H. Sultmann, P. Altevogt and S. Momma, PLos Biol., 2014, 12, e1001874 CrossRef PubMed.
- F. Royo, K. Schlangen, L. Palomo, E. Gonzalez, J. Conde-Vancells, A. Berisa, A. M. Aransay and J. M. Falcon-Perez, PLoS One, 2013, 8, e68693 CrossRef CAS PubMed.
- H. Valadi, K. Ekstrom, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lotvall, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed.
- A. Zomer, C. Maynard, F. J. Verweij, A. Kamermans, R. Schafer, E. Beerling, R. M. Schiffelers, E. de Wit, J. Berenguer, S. I. J. Ellenbroek, T. Wurdinger, D. M. Pegtel and J. van Rheenen, Cell, 2015, 161, 1046–1057 CrossRef CAS PubMed.
- S. Bala, T. Csak, F. Momen-Heravi, D. Lippai, K. Kodys, D. Catalano, A. Satishchandran, V. Ambros and G. Szabo, Sci. Rep., 2015, 5, 10721 CrossRef PubMed.
- A. Hoshino, B. Costa-Silva, T. L. Shen, G. Rodrigues, A. Hashimoto, M. Tesic Mark, H. Molina, S. Kohsaka, A. Di Giannatale, S. Ceder, S. Singh, C. Williams, N. Soplop, K. Uryu, L. Pharmer, T. King, L. Bojmar, A. E. Davies, Y. Ararso, T. Zhang, H. Zhang, J. Hernandez, J. M. Weiss, V. D. Dumont-Cole, K. Kramer, L. H. Wexler, A. Narendran, G. K. Schwartz, J. H. Healey, P. Sandstrom, K. J. Labori, E. H. Kure, P. M. Grandgenett, M. A. Hollingsworth, M. de Sousa, S. Kaur, M. Jain, K. Mallya, S. K. Batra, W. R. Jarnagin, M. S. Brady, O. Fodstad, V. Muller, K. Pantel, A. J. Minn, M. J. Bissell, B. A. Garcia, Y. Kang, V. K. Rajasekhar, C. M. Ghajar, I. Matei, H. Peinado, J. Bromberg and D. Lyden, Nature, 2015, 527, 329–335 CrossRef CAS PubMed.
- F. Shahabipour, N. Barati, T. P. Johnston, G. Derosa, P. Maffioli and A. Sahebkar, J. Cell Physiol., 2017, 232, 1660–1668 CrossRef CAS PubMed.
- C. Boscher, J. W. Dennis and I. R. Nabi, Curr. Opin. Cell Biol., 2011, 23, 383–392 CrossRef CAS PubMed.
- J. Echevarria, F. Royo, R. Pazos, L. Salazar, J. M. Falcon-Perez and N. C. Reichardt, ChemBioChem, 2014, 15, 1621–1626 CrossRef CAS PubMed.
- J. Q. Gerlach, A. Kruger, S. Gallogly, S. A. Hanley, M. C. Hogan, C. J. Ward, L. Joshi and M. D. Griffin, PLoS One, 2013, 8, e74801 CrossRef CAS PubMed.
- C. Williams, F. Royo, O. Aizpurua-Olaizola, R. Pazos, G. J. Boons, N. C. Reichardt and J. M. Falcon-Perez, J. Extracell. Vesicles, 2018, 7, 1442985 CrossRef PubMed.
- M. E. Hung and J. N. Leonard, J. Biol. Chem., 2015, 290, 8166–8172 CrossRef CAS PubMed.
- G. Di Rocco, S. Baldari and G. Toietta, Stem Cells Int., 2016, 2016, 5029619 Search PubMed.
- T. Smyth, M. Kullberg, N. Malik, P. Smith-Jones, M. W. Graner and T. J. Anchordoquy, J. Controlled Release, 2015, 199, 145–155 CrossRef CAS PubMed.
- Y. Takahashi, M. Nishikawa, H. Shinotsuka, Y. Matsui, S. Ohara, T. Imai and Y. Takakura, J. Biotechnol., 2013, 165, 77–84 CrossRef CAS PubMed.
- A. J. Ullal, D. S. Pisetsky and C. F. Reich 3rd, Cytometry, Part A, 2010, 77, 294–301 Search PubMed.
- E. J. van der Vlist, E. N. Nolte-'t Hoen, W. Stoorvogel, G. J. Arkesteijn and M. H. Wauben, Nat. Protoc., 2012, 7, 1311–1326 CrossRef PubMed.
- C. Grange, M. Tapparo, S. Bruno, D. Chatterjee, P. J. Quesenberry, C. Tetta and G. Camussi, Int. J. Mol. Med., 2014, 33, 1055–1063 CrossRef CAS PubMed.
- L. Hu, S. A. Wickline and J. L. Hood, Magn. Reson. Med., 2015, 74, 266–271 CrossRef CAS PubMed.
- O. P. Wiklander, J. Z. Nordin, A. O'Loughlin, Y. Gustafsson, G. Corso, I. Mager, P. Vader, Y. Lee, H. Sork, Y. Seow, N. Heldring, L. Alvarez-Erviti, C. I. Smith, K. Le Blanc, P. Macchiarini, P. Jungebluth, M. J. Wood and S. E. Andaloussi, J. Extracell. Vesicles, 2015, 4, 26316 CrossRef PubMed.
- J. Llop, V. Gómez-Vallejo and N. Gibson, Nanomedicine, 2013, 8, 1035–1038 CrossRef CAS PubMed.
- C. Pérez-Campaña, V. Gómez-Vallejo, M. Puigivila, A. Martín, T. Calvo-Fernández, S. E. Moya, R. F. Ziolo, T. Reese and J. Llop, ACS Nano, 2013, 7, 3498–3505 CrossRef PubMed.
- P. P. Di Mauro, V. Gomez-Vallejo, Z. Baz Maldonado, J. Llop Roig and S. Borros, Bioconjugate Chem., 2015, 26, 582–592 CrossRef CAS PubMed.
- J. Frigell, I. García, V. Gómez-Vallejo, J. Llop and S. Penadés, J. Am. Chem. Soc., 2014, 136, 449–457 CrossRef CAS PubMed.
- Z. Varga, I. Gyurko, K. Paloczi, E. I. Buzas, I. Horvath, N. Hegedus, D. Mathe and K. Szigeti, Cancer Biother.Radiopharm., 2016, 31, 168–173 CrossRef CAS PubMed.
- D. W. Hwang, H. Choi, S. C. Jang, M. Y. Yoo, J. Y. Park, N. E. Choi, H. J. Oh, S. Ha, Y. S. Lee, J. M. Jeong, Y. S. Gho and D. S. Lee, Sci. Rep., 2015, 5, 15636 CrossRef CAS PubMed.
- M. Morishita, Y. Takahashi, M. Nishikawa, K. Sano, K. Kato, T. Yamashita, T. Imai, H. Saji and Y. Takakura, J. Pharm. Sci., 2015, 104, 705–713 CrossRef CAS PubMed.
- J. Conde-Vancells, E. Rodriguez-Suarez, N. Embade, D. Gil, R. Matthiesen, M. Valle, F. Elortza, S. C. Lu, J. M. Mato and J. M. Falcon-Perez, J. Proteome Res., 2008, 7, 5157–5166 CrossRef CAS PubMed.
- A. Shimoda, Y. Tahara, S. I. Sawada, Y. Sasaki and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2017, 491, 701–707 CrossRef CAS PubMed.
- E. Medico, A. M. Mongiovi, J. Huff, M. A. Jelinek, A. Follenzi, G. Gaudino, J. T. Parsons and P. M. Comoglio, Mol. Biol. Cell, 1996, 7, 495–504 CrossRef CAS PubMed.
- A. Ruiz-de-Angulo, A. Zabaleta, V. Gomez-Vallejo, J. Llop and J. C. Mareque-Rivas, ACS Nano, 2016, 10, 1602–1618 CrossRef CAS PubMed.
- M. Cobaleda-Siles, M. Henriksen-Lacey, A. Ruiz de Angulo, A. Bernecker, V. Gomez Vallejo, B. Szczupak, J. Llop, G. Pastor, S. Plaza-Garcia, M. Jauregui-Osoro, L. K. Meszaros and J. C. Mareque-Rivas, Small, 2014, 10, 5054–5067 CrossRef CAS PubMed.
- D. Sun, X. Zhuang, X. Xiang, Y. Liu, S. Zhang, C. Liu, S. Barnes, W. Grizzle, D. Miller and H. G. Zhang, Mol. Ther., 2010, 18, 1606–1614 CrossRef CAS PubMed.
- C. P. Lai, O. Mardini, M. Ericsson, S. Prabhakar, C. Maguire, J. W. Chen, B. A. Tannous and X. O. Breakefield, ACS Nano, 2014, 8, 483–494 CrossRef CAS PubMed.
- G. Schiera, C. M. Di Liegro and I. Di Liegro, BioMed Res. Int., 2015, 2015, 152926 Search PubMed.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. Wood, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS PubMed.
- C. C. Chen, L. Liu, F. Ma, C. W. Wong, X. E. Guo, J. V. Chacko, H. P. Farhoodi, S. X. Zhang, J. Zimak, A. Segaliny, M. Riazifar, V. Pham, M. A. Digman, E. J. Pone and W. Zhao, Cell. Mol. Bioeng., 2016, 9, 509–529 CrossRef CAS PubMed.
- L. Paolini, F. Orizio, S. Busatto, A. Radeghieri, R. Bresciani, P. Bergese and E. Monti, Biochemistry, 2017, 56, 6401–6408 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods and supplementary Fig. 1 to 5 to complement the manuscript. See DOI: 10.1039/c8nr03900c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |