
Biologically generated carbon dioxide: nature's versatile chemical strategies for carboxy lyases
Christopher T.
Walsh

ChEM-H Institute, Stanford University, Stanford, CA 94305, USA. E-mail: cwalsh2@stanford.edu
First published on 10th May 2019
Abstract
Covering: up to 2019
Metabolic production of CO2 is natural product chemistry on a mammoth scale. Just counting humans, among all other respiring organisms, the seven billion people on the planet exhale about 3 billion tons of CO2 per year. Essentially all of the biogenic CO2 arises by action of discrete families of decarboxylases. The mechanistic routes to CO2 release from carboxylic acid metabolites vary with the electronic demands and structures of specific substrates and illustrate the breadth of chemistry employed for C–COO (C–C bond) disconnections. Most commonly decarboxylated are α-keto acid and β-keto acid substrates, the former requiring thiamin-PP as cofactor, the latter typically cofactor-free. The extensive decarboxylation of amino acids, e.g. to neurotransmitter amines, is synonymous with the coenzyme form of vitamin B6, pyridoxal-phosphate, although covalent N-terminal pyruvamide residues serve in some amino acid decarboxylases. All told, five B vitamins (B1, B2, B3, B6, B7), ATP, S-adenosylmethionine, manganese and zinc ions are pressed into service for specific decarboxylase catalyses. There are additional cofactor-independent decarboxylases that operate by distinct chemical routes. Finally, while most decarboxylases use heterolytic ionic mechanisms, a small number of decarboxylases carry out radical pathways.
 Christopher T. Walsh | Christopher T. Walsh, completed a biology degree at Harvard (1965), a PhD at The Rockefeller University (1970), and after postdoctoral work at Brandeis with Robert Abeles, spent 15 years on the faculty at MIT, in both the biology and chemistry departments (1972–1987). From 1987–2013 he was in the department of biological chemistry and molecular pharmacology at Harvard Medical School. Since 2014 he has been a member of the ChEM-H Interdisciplinary Institute at Stanford University. His research interests center around mechanistically novel enzymatic transformations, with particular emphasis on deciphering the chemical logic and molecular machinery of metabolite interconversions. |
1. Introduction
Respiring organisms release CO2 as metabolic end product in complement to plants and photosynthetic microorganisms that fix CO2 by photosynthesis. On a global scale these processes occur in billions of tons of CO2 produced and taken up yearly. The source of all that metabolically generated CO2 is carboxylic acids, present as the anionic carboxylates at physiologic pH. A wide variety of carboxylate metabolite structures pose distinct chemical challenges to C–COO carbon–carbon bond cleavages.Also, the catabolic (degradative) branch of metabolism and the anabolic (biosynthetic) branches have distinct strategies for throwing off CO2 molecules. In the catabolic, energy-yielding reactions, decarboxylations of both α- and β-keto acids are the central energy-producing steps, and require coenzyme forms of vitamin B1 (thiamin) and vitamin B3 (nicotinamides), respectively. In biosynthetic pathways, the generation of CO2 may drive biosynthetic equilibria (fatty acid biosynthesis) and/or represent trimming of the maturing metabolite frameworks: cholesterol and heme assembly release 8 and 14 CO2 molecules along the way.
Organisms have evolved a variety of mechanistic strategies to lower energy barriers for decarboxylation of different classes of carboxylate-containing substrates, including several varieties of coenzymes and/or metal ions as required cofactors for distinct sets of decarboxylases. Most decarboxylase catalysts operate in two electron steps and therefore need to stabilize incipient carbanion density at the carbon center where the C–COO bond is breaking. Other decarboxylases, often in microbial metabolism, use one electron reaction manifolds, generating radical intermediates during net decarboxylation.
This review examines the different mechanistic challenges offered by distinct molecular frameworks of carboxylate-containing substrates that undergo CO2 release. The separate strategies of several classes of decarboxylase catalysts allow prediction of reaction mechanisms for the wide variety of metabolites that lead to metabolic CO2 production.
2. The logic and reach of one carbon metabolism
2.1 Five one carbon metabolites
One carbon units, the simplest of organic structures, are nonetheless central molecules in all forms of life. Five molecules, methane, methanol, formaldehyde, formic acid and carbon dioxide (Fig. 1A) span four different oxidation states, with removal (oxidation) or addition (reduction) of two electrons at a time at each stage. The most reduced of the one carbon units, methane, is restricted to microbial metabolism,1,2 where certain anaerobic archaeal single cell organisms evolve methane from CO2. In microbial communities methane oxidizing bacteria3 make a complementary living, removing the electron pairs on the way back up to CO2.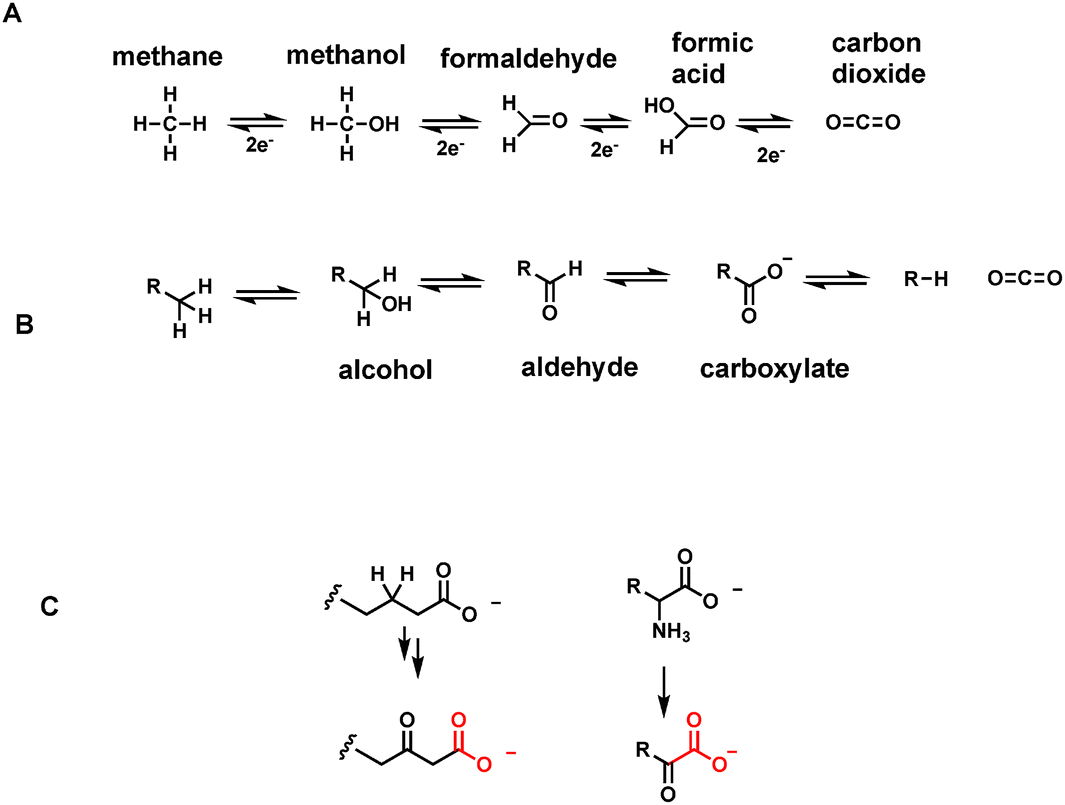 | ||
| Fig. 1 (A) Oxidation states of one carbon molecules between methane and carbon dioxide; (B) oxidation states for hydrocarbons, primary alcohols, aldehydes, carboxylic acid anions and carbon dioxide; (C) two of the most common structural types that are enzymatically decarboxylated are β-keto acid anions and α-keto acid anions. | ||
The one carbon units at the intermediate three oxidation states of methanol, formaldehyde and formate are rarely allowed to roam freely in cells of higher organisms such as humans. Instead they are carried around on coenzyme molecules as covalent adducts4 that dampen down the intrinsic reactivity of methanol as solvent, formaldehyde as crosslinking agent, and formate as an organic acid (formic acid spraying ants and carabid beetles are exceptions).5,6
The job of ferrying these one carbon units inside cells falls largely to two vitamins – folic acid (vitamin B9) and vitamin B12.4 Folic acid, in its four electron reduced tetrahydro form, acts as a formaldehyde sponge, sopping up nascent aldehyde molecules as they are formed from the cleavage of L-serine in the active site of the enzyme serine transhydroxymethylase.7,8 What emerges from the active site is the N-5,N-10-methylene-tetrahydrofolate, with the one carbon bridging methylene (CH2) a latent form of formaldehyde, to be delivered back up only in the active site of other target enzymes. Most notable is the enzyme deoxythymidylate synthase, using that one carbon formaldehyde equivalent to install it as a methyl substituent at carbon 5 of dUMP and thereby create the unique DNA building block dTMP.9
To access the formal oxidation states of methanol (two electrons reduced) and formate (two electrons oxidized), humans and other eukaryotes have enzymes that either reduce N-5,N-10-CH2-tetrahydrofolate to the N-5-CH3-folate adduct, or oxidize it by two electrons to the N-10-CHO-tetrahydrofolate (Fig. 2). Two distinct regioisomers hold one carbon four electrons apart in their redox states.
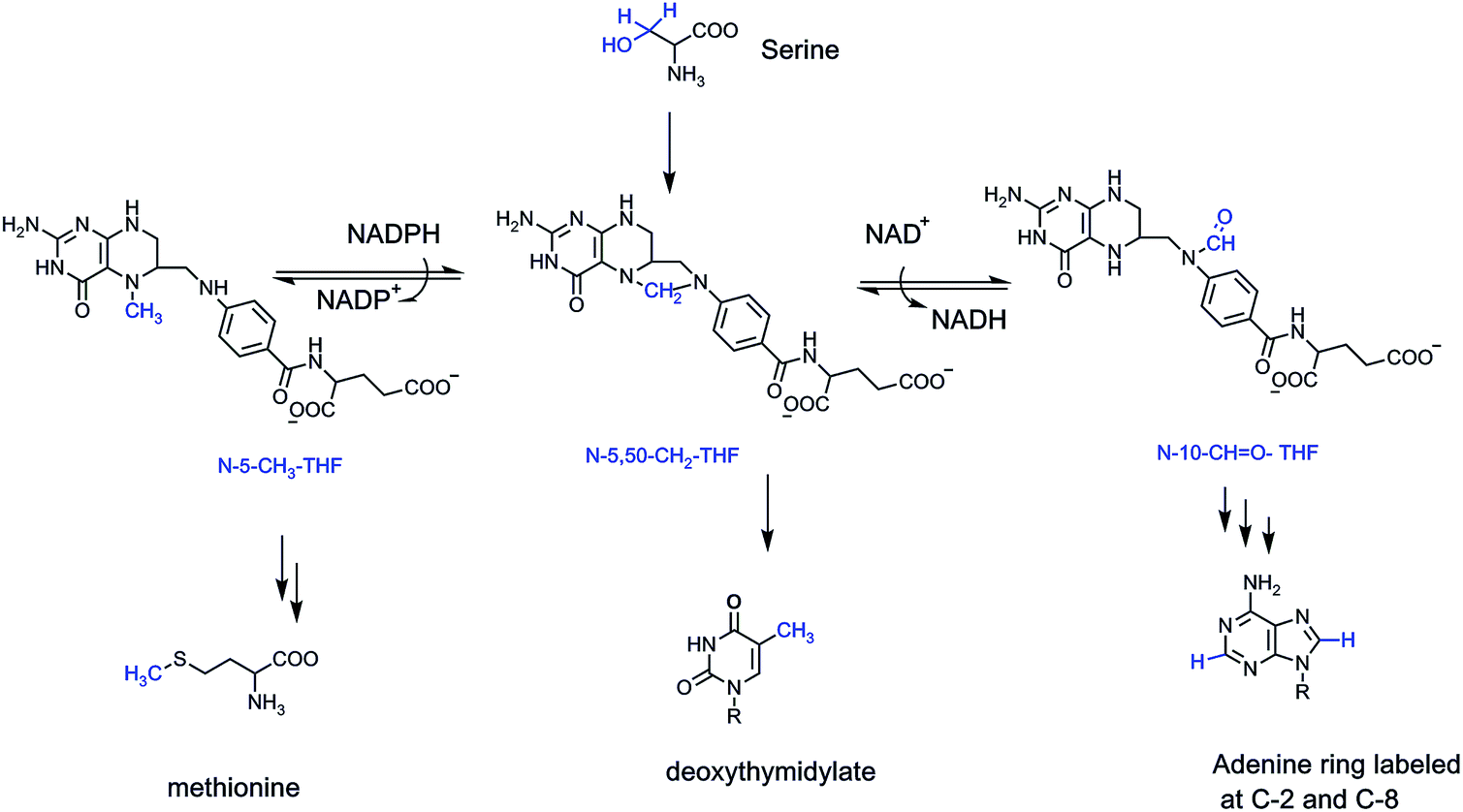 | ||
| Fig. 2 Carbon 3 of L-serine is the donor of the formaldehyde equivalent that is captured by N-5,10-tetrahydrofolate and carried around in cells in latent form. The one carbon unit can be enzymatically reduced to N-5-CH-THF or enzymatically oxidized to N-10-CHO-THF. As shown, the formaldehyde equivalent is the source of the C-5-methyl group in the DNA precursor dTMP, the CH3 group for methionine (and ultimately S-adenosylmethionine), and the formyl group as the source of both C-2 and C-8 in purine ring assembly. | ||
The formyl group is delivered twice to enzymes assembling purine ring building blocks for RNA and DNA, so several billions of such transfers occur every cell cycle to build the 3 billion base pairs of human DNA and the myriad RNA transcriptome constituents. The N-5-CH3-tetrahydrofolate transfers its methyl to vitamin B12, which in turn methylates homocysteine to yield the proteinogenic amino acid methionine. Methionine is also the precursor to the cell's premier methylating reagent, S-adenosylmethionine (SAM). SAM methylates dozens of low molecular weight metabolites in cells, trimethylates phosphatidylethanolamine to phosphatidylcholine in cell membranes, methylates lysine side chains in proteins, most famously histone tails, and methylates RNA species and CpG sequences at C5 of cytosine in DNA.10,11
2.2 Carbon dioxide: what goes in comes out
Clearly, the one carbon units at the oxidation states of methanol (N-5-CH3-THF), formaldehyde (N-5,10-CH2-THF), and formate (N-10-CHO-THF), are essential in almost every facet of anabolic metabolism. Which brings us to the fifth and most oxidized of the one carbon organic molecules, carbon dioxide. CO2 has a double existence in global metabolism. Plants famously fix CO2 into organic matter, literally creating plant biomass that humans and other organisms consume. The key enzyme is ribulose-bisphosphate carboxylase, generating a carbanionic intermediate to capture the electrophilic carbon of CO2, generating a new C–C bond12 (Fig. 3). The transient six carbon nascent product is then cleaved hydrolytically to two identical three carbon molecules of 3-phosphoglycerate, a primary building block in glycolysis and other central metabolic pathways. Estimates of the amount of CO2 fixed into biomass carbon on the planet are ∼250 billion tons.13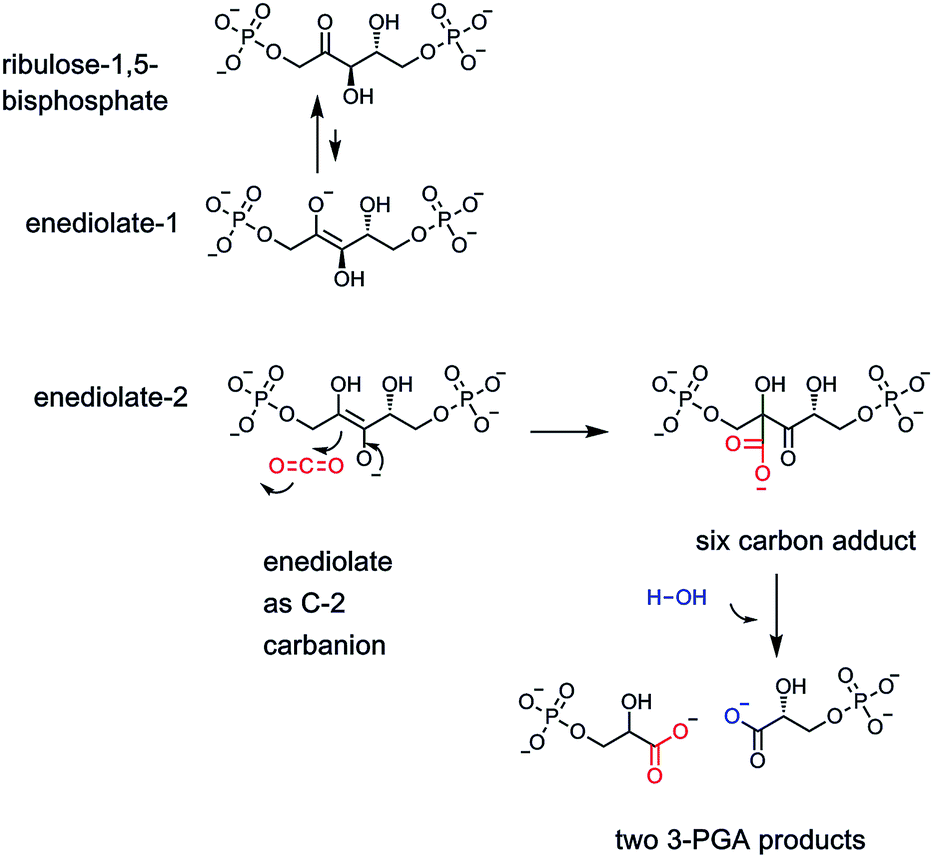 | ||
| Fig. 3 The enzyme ribulose bisphosphate carboxylase is the catalyst responsible for global fixation of CO2 by photosynthetic organisms, including plants. An enediolate tautomer of ribulose substrate provides the C3 carbanion necessary for attack on the electrophilic CO2 cosubstrate. The initial six carbon carboxy adduct is fragmented hydrolytically to release two molecules of 3-phosphoglycerate, one of which has incorporated the CO2 as the carboxylate group. | ||
Humans, animals, fungi, non-photosynthetic prokaryotes including bacteria, equally famously cannot fix CO2 into organic molecules on any comparably large scale (they do encode carboxylases that we shall note in the next to last section below for specific cellular reactions). Instead, carbon dioxide is the final product of energy metabolism. Foods such as proteins, carbohydrates, fatty acids, all have carbon chains at partially reduced states (e.g. one or more hydrogen atoms). The way organisms make a living is to sequentially oxidize the reduced carbon sites eventually all the way to CO2. The treasure is the electrons removed in those oxidations, stored in molecules such as NADH, NADPH, FADH2,14 (and converted to ATP) that can do chemical work, most notably the biosynthesis of the major biopolymers-proteins, polysaccharides, membranes, RNA and DNA. Essentially all CO2 in exhaled air derives from carboxylic acid-containing organic metabolites undergoing decarboxylation (Fig. 4A).
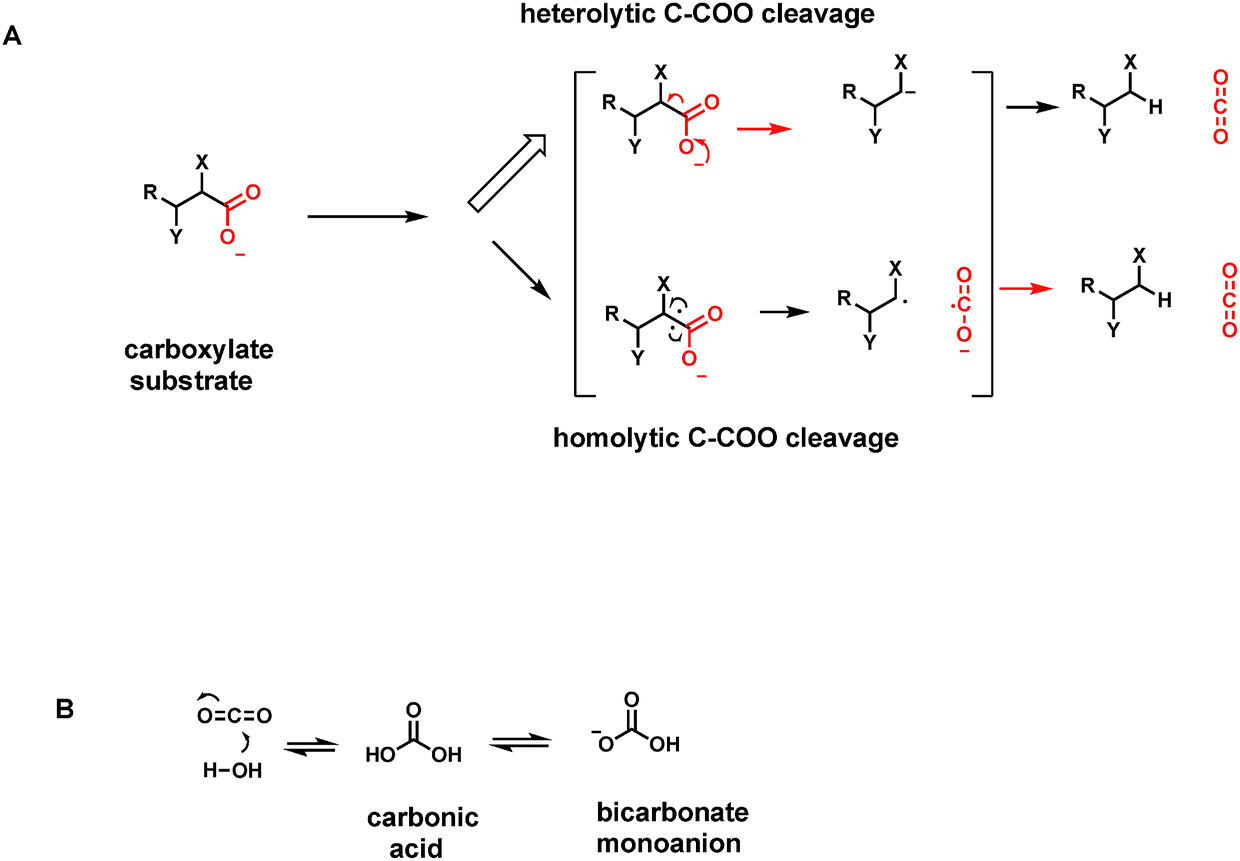 | ||
| Fig. 4 (A) Two models of C–C bond cleavage by decarboxylase action. The most common heterolytic route yields transient carbanionic electron density at the carbon that had been connected to the carboxylate. The rarer homolytic cleavage route generates one unpaired electron on the two carbon centers, a pair of intermediate radicals including the CO2˙ radical; (B) CO2 dissolved in water is hydrated to carbonic acid, which is largely present as the monoanionic bicarbonate contributor (B). The CO2-HCO3− system is the major buffer in human blood pH homeostasis. | ||
CO2 is soluble in aqueous solutions and hydrates to a small extent (about 0.1–0.2%) to carbonic acid H2CO3. That equilibrium is rapidly established by the enzyme carbonic anhydrase.15 The first pKa of this diprotic acid occurs at pH 6.1 so carbonic acid is mostly dissociated as the bicarbonate monoanion (Fig. 4B). The bicarbonate/CO2 system is the major buffer maintaining blood pH at pH 7.4. This may be the most important physiologic function of CO2 as an end point, one carbon metabolite.
2.3 CO2 generation is large scale chemical biology
Thus, CO2 is largely a waste product of energy metabolism. Humans, for example, breathe in air that has about 0.04% CO2 content. On average 15 times a minute, they exhale air with 10 fold higher CO2 content (∼0.4%).16 That order of magnitude increase comes from the CO2 molecules that have accumulated as every cell practices oxidation of reduced substrate molecules. Cells that run anaerobically, completing the glycolytic pathway but not the citric acid cycle, incompletely oxidize their substrates, get less energy and produce less CO2 (it is the citrate cycle that for example releases four of the six carbons of glucose as CO2).The estimate of total CO2 exhaled by an adult human of average physical activity is about 900 grams per day.17 Given a molecular weight of 44 g mol−1 for CO2, this is ∼22 moles of carbon dioxide (about 1025 molecules) from each of 7 billion humans per day.18 Over a year, this reaches between 2-3 billion tons of global CO2 exhaled by the human population, less than the estimated 250 million tons of CO2 fixed into biomass by action of that one plant enzyme ribulose-bisphosphate carboxylase (Fig. 3).
2.4 Carboxy lyases are decarboxylases
How do humans release these billions of tons of CO2 as end products of energy metabolism? Specifically, we analyze what kinds of metabolites, low molecular weight molecules that are enzyme substrates, yield CO2 when operated on by biocatalysts. The general strategy is to act on a molecule with a C–COO− bond, all as carboxylic acid anions at physiologic pH, and cleave it to CO2, leaving the electron pair from the single C–C bond that has broken with the carbon atom left behind (a very small number of decarboxylases break the C–COO bond homolytically generating carbon radical species as transient intermediates/transition states as we note in Section 4) (Fig. 4A).The formal term for the dozens/hundreds of enzymes that carry out this chemistry are carboxy-lyases – carrying out a lytic transformation on the carboxylate-bearing substrate (Enzyme Commission terminology, category E.C.4.1.1; 136 Protein Data Base entries as of 01/01/2019). More familiarly, they are also known as decarboxylases and that is the terminology we shall employ. Two questions arise. (1) What kinds of low molecular weight metabolites are typical substrates for decarboxylases: what are the structural features and so the challenges they pose to the respective enzyme catalysts. (2) What are the catalytic strategies that decarboxylases employ to selectively lower the energy barriers such that these otherwise stable C–COO linkages are susceptible to cleavage: with stabilization of the electron rich atoms left behind in the remainder portion of the decarboxylated product scaffold (Fig. 4B). Three billion tons of CO2 released per year from decarboxylases is about 3 × 1015 grams and thus ∼2 × 1013 moles of CO2 or about 7 × 1037 molecules. This is the yearly throughput of human decarboxylases: large scale chemistry.
2.5 Sources and roles of carboxylate groups in metabolites
The most abundant functional groups in organic molecules that are substrates for oxidation in food stuffs are the oxygen functionalities in carbohydrates, in the glycerol backbone of triglycerides and phospholipids, and in the carboxylic acid groups of fatty acids. Amino acid constituents released by breakdown of proteins are oxidatively deaminated to α-keto acids. Thus, in parallel to the oxidation states of the one carbon molecules methane, methanol, formaldehyde, and formate, organic metabolites go from hydrocarbons, to alcohols, to aldehydes (or ketones for secondary alcohols), and to carboxylic acids in comparable oxidative progressions (Fig. 1B).2.6 Pathways that generate many CO2 molecules
Carboxylate functional groups in metabolites have two major roles. On the one hand, they can be converted to acyl thioesters as kinetically stable, thermodynamically active acyl transfer and carbanion stabilizing agents.14 On the other hand, they can be released as carbon dioxide by decarboxylases. One can look beyond single decarboxylases and assess the loss of carbons as CO2 throughout particular metabolic pathways. In addition to the tandem action of glycolysis and the citrate cycle that oxidizes all six carbons of glucose to 6 CO2, enzymatic oxidation of palmitate, the common saturated C16 fatty acid, yields 16 CO2 molecules in mitochondrial metabolism (Fig. 5).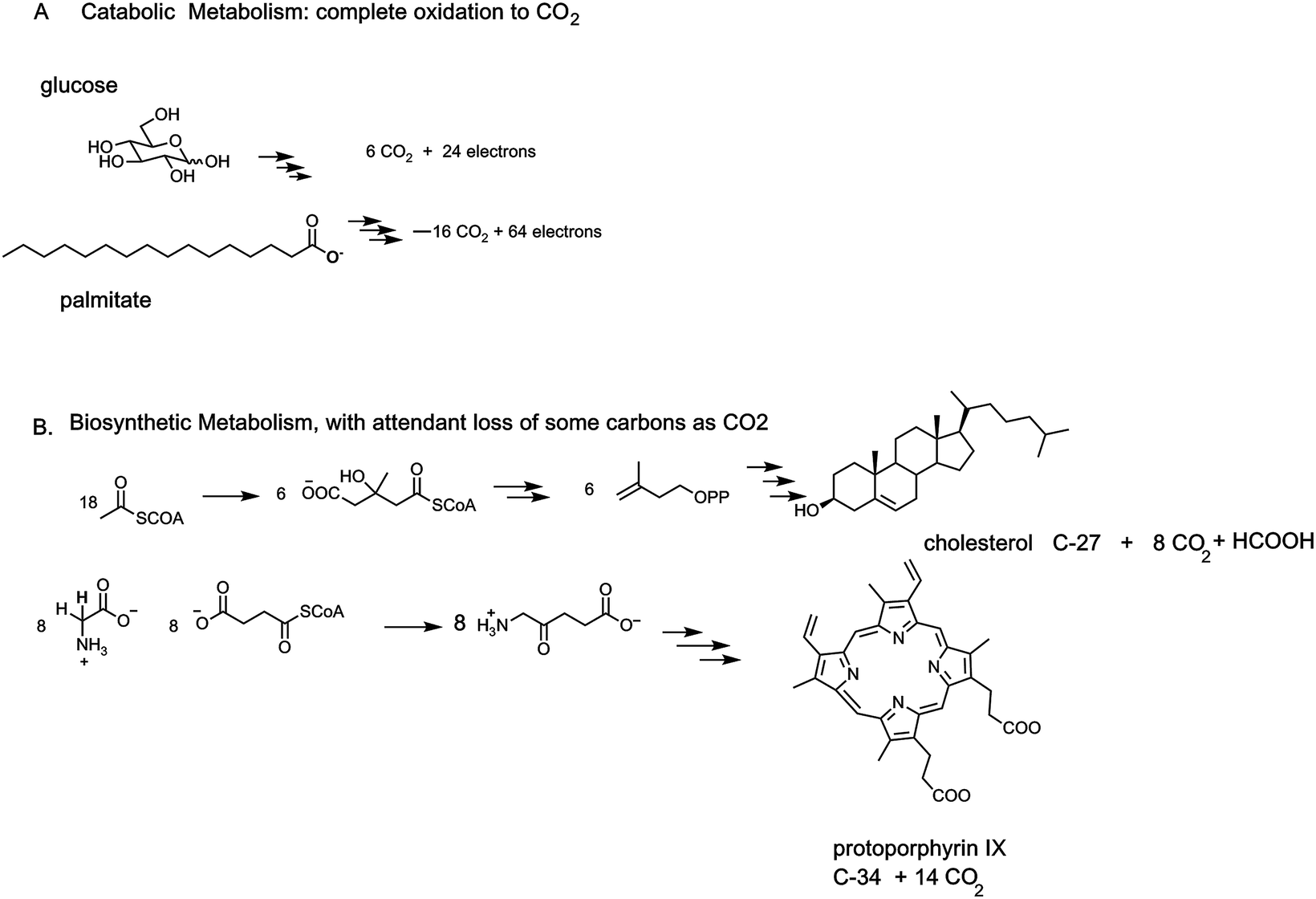 | ||
| Fig. 5 Examples of two catabolic and two anabolic pathways. (A) All six carbons of glucose are converted to 6 CO2. Analogously the sixteen carbon fatty acid palmitate is catabolized as an energy source to 16 CO2 molecules. (B) By contrast anabolic pathways may see some carbon atoms carved out as CO2 but the main purpose is to build the final steroid or tetrapyrrole macrocyclic structure. Trimming of intermediates is largely achieved by decarboxylases, hence the quantitative jettisoning of carbons as CO2. | ||
The cholesterol and heme biosynthetic pathways involve controlled jettisons of carbons trimmed away as CO2 molecules. Thus 18 molecules of acetyl CoA (36 carbons in the acetyl groups) give the 27 carbons of cholesterol, with loss of nine carbons as 8 CO2 (and one as formate). Analogously eight molecules of succinyl CoA and eight molecules of glycine are starting point for biogenesis of the 34 carbon scaffold tetrapyrrolic macrocycle protoporphyrin IX into which FeII insertion gives heme. Fourteen of the starting 48 carbon atoms are converted to CO2 by action of three of the eight enzymes in the pathway, acting as decarboxylases (Fig. 5).
Some enzymes such as pyruvate carboxylase, initiating gluconeogenesis, and acetyl CoA carboxylase, producing malonyl CoA for fatty acid biosynthesis, run in the opposite direction, fixing CO2 as carboxylate groups at the start of those two metabolic pathways. In each of these two cases the next enzymes in each pathway act as decarboxylases to remove those just introduced carboxylate groups.
2.7 Some common C2–C6 carboxylate metabolites in primary pathways
Scaffolds of 2–6 carbons bearing an array of these oxygen functional groups form the prevalent network of intermediates in primary metabolic pathways. Thus, two carbon carboxylates include frameworks where the carbon adjoining the carboxylate can be a CH3, CH2OH, CHO or even COO as shown, referring respectively to acetate, glycolate, glyoxalate, and oxalate, respectively (Fig. 6). In the three carbon scaffolds, abundant carboxy metabolites are 3-P-glycerate (the primary product in plant action of ribulose bisphosphate carboxylase), pyruvate, and the dicarboxylate malonate. At the four carbon framework, the diacids succinate, fumarate, malate, and the 2-keto acid oxaloacetate are key components of the tricarboxylate cycle. Acetoacetate, the 3-ketoacid, is a hydrolysis metabolite of acetoacetyl CoA in early stages of steroid biosynthesis. The proteinogenic amino acids at the four carbon level are threonine and the dicarboxy amino acid aspartate (from transamination of oxaloacetate).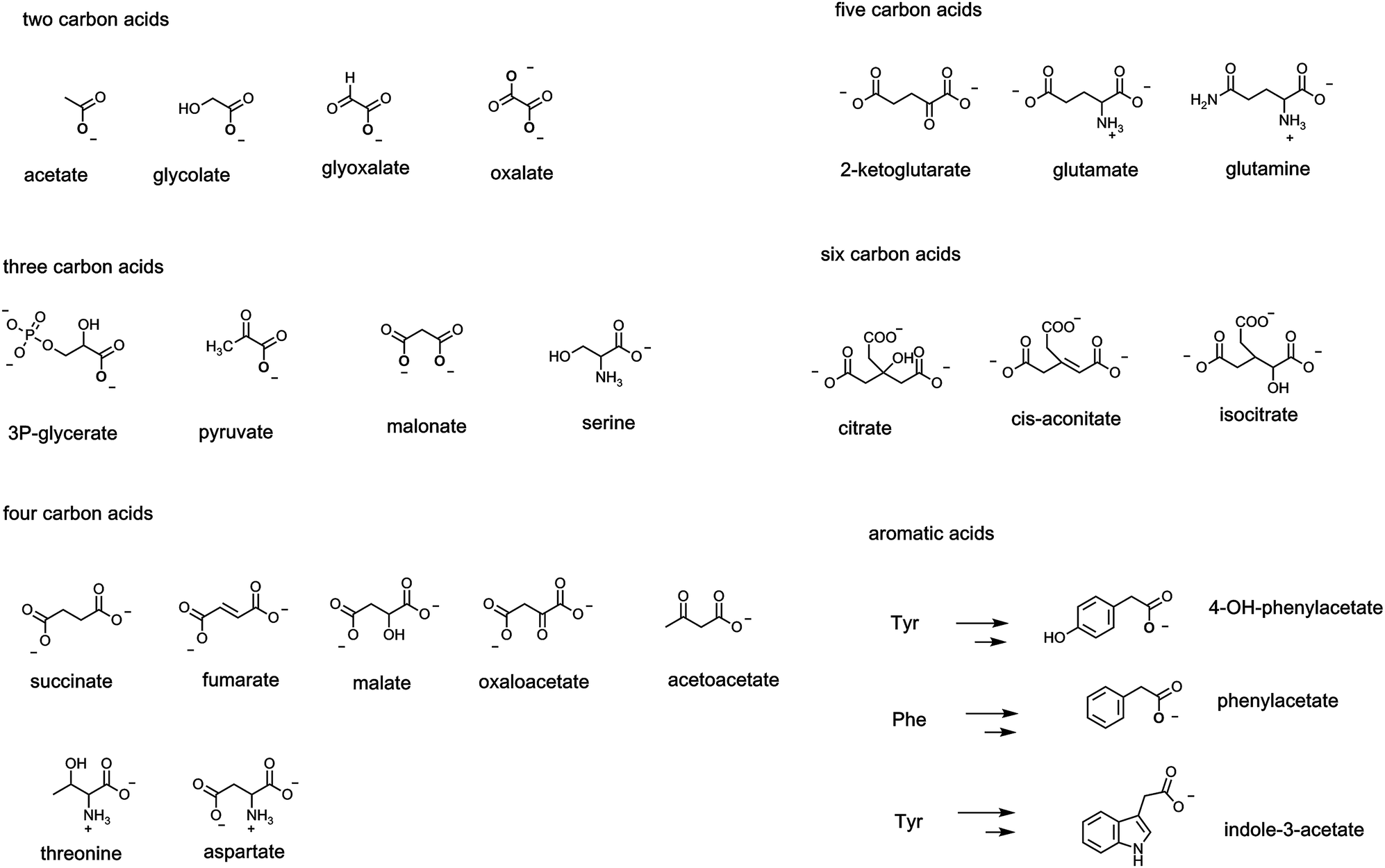 | ||
| Fig. 6 A list of common metabolites between two to six carbons that bear one or more carboxylate groups, including carboxylates in the enzymatic degradations of Phe, Tyr, Trp. | ||
Fewer metabolites at the C5 level are central but 2-ketoglutarate in the citrate cycle and the proteinogenic amino acids glutamate and glutamine are notable. The six carbon level encompasses the three tricarboxylates in the citrate cycle: citrate, cis-aconitate, and isocitrate are most prevalent.
A large range of bacteria and fungi degrade aromatic and heteroaromatic ring systems to create acyclic carbon fragments that feed in to central metabolic pathways. Among the many degradative routes are those for the three proteinogenic aromatic amino acids phenylalanine (phe), tyrosine (tyr) and tryptophan (trp). The three carbon amino acid side chains of each can be enzymically transaminated to the pyruvyl side chains and then subjected to TPP-dependent decarboxylations to the acetate side chains, e.g. 4-OH-phenylacetate, phenylacetate, and indole-3-acetate, as shown in Fig. 6. Each of those acetyl side chains can be further decarboxylated to the corresponding alkanes, e.g. para-cresol, toluene, and skatole by microbial decarboxylases noted in a subsequent section.
2.8 Mechanistic routes for decarboxyases
Given the ∼3 billion tons of CO2 exhaled by humans globally each year, the question arises what mechanisms have enzyme evolved to break all those C–C bonds in the various C–COO substrates that are otherwise chemically stable. As always, one can parse C–C bond cleavage into heterolytic bond-breaking vs. homolytic bond-breaking routes (Fig. 4).A heterolytic (two electron) route for decarboxylation would leave both electrons in the C–COO bond being cleaved on the incipient hydrocarbon as CO2 is formed. This amounts to the need for stabilizing carbanion density at least transiently on the remaining carbon center. We shall note a variety of mechanisms that enzymes have evolved to deal with the electronic requirements of distinct types of carboxylate-containing organic substrates.
Homolytic routes to decarboxyation are much less common but do exist for certain substrates in certain organisms that have ways to generate radical initiators. We shall note both radical SAM decarboxylases and also glycyl radical enzymes that carry out substrate decarboxylations.
3. Coenzymes and cofactors utilized by different decarboxylases
3.1 A plethora of chemical strategies for decarboxylases
Although at first glance one might anticipate the cleavage of the C–C bond in carboxylate metabolites would be among the more straightforward chemical reactions in cells and organisms, the extant diversity of the requisite catalysts is surprising and ultimately instructive. A variety of cofactors are utilized by different decarboxylases, based on the electronic demands of specific substrates. Table 1 summarizes the set of distinct carboxylase cofactors.| Cofactor | Substrate type | Enzyme example |
|---|---|---|
| Heterolytic cleavage of C–COO | ||
| Thiamin-PP | α-Keto acid | Pyruvate decarboxylase |
| Pyridoxal-P | α-Amino acid | Histidine decarboxylase |
| Pyruvamide | α-Amino acid | SAM decarboxylase aspartate α-decarboxylase |
| Flavins (FMN, FAD) | α-Amino acid | Pantetheinylcysteine decarboxylase |
| Prenylated flavin | Aryl acids | Cinnamate decarboxylase |
| NAD+ | Prephenate | Prephenate dehydrogenase |
| ATP | Mevalonate-1-PP | Mevalonate kinase/decarboxylase |
| Biotin | Oxaloacetate | Oxaloacetate sodium pump |
| Zinc2+ | 2,6-Dihydroxybenzoate | 2,6-DHB decarboxylase |
| –Lys–NH2 side chain | β-Keto acids | Acetoacetate decarboxylase |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Homolytic cleavage of C–COO | ||
| SAM radical | Propionic acid side chain | Coproporphyrinogen oxidase |
| Glycyl radical | Indole acetate | Indole acetate decarboxylase |
| MnII/MnIII | Oxalate | Oxalate decarboxylase |
|
||
| Cofactor independent C–COO cleavage | ||
| β-Keto acid | Lanosterol 4α–COO decarboxylase | |
| sp2 C–COO | OMP decarboxylase, decarboxylase uroporphyrinogen III | |
| cis-Aconitate | cis-Aconitate decarboxylase | |
| Aryl acids | Pyrrole-2-COO-decarboxylase | |
The distinct electronic demands of the many types of carboxylated metabolites that get cleaved to CO2 and residual scaffold are reflected in the several distinct cofactors required. The coenzyme forms of five B vitamins (B1, B2, B3, B6, B7)4 are put into play in the decarboxylase classes noted above. This widespread use of B vitamin coenzymes indicates invention/evolution/recruitment of decarboxylases multiple times.
There are also examples where ATP, SAM, Mn2+ and Zn2+ are obligate cofactors. Radical SAM and glycine radical enzymes carry out rare homolytic cleavages of C–COO bonds during decarboxylations. The mangano-enzyme oxalate decarboxylase uses an O2-dependent radical-based decarboxylation manifold19 in contrast to the thiamin-PP-dependent oxalyl CoA decarboxylase.20 Finally, there are a number of cofactor-independent decarboxylases, among which are several β-keto acid decarboxylases, generation of itaconate from the citrate cycle intermediate cis-aconitate,21 and reversible decarboxylation/carboxylations of electron rich aromatic/heteroaromatic scaffolds.22
3.2 β-Keto acid decarboxylation: imine/enamine catalysis
The easiest chemical route for metabolite decarboxylation may be presented by β-keto acids (Fig. 1C). The incipient negative charge development on Cα can be delocalized into the adjacent carbonyl by participation of a resonance contributor with the negative charge on the more basic oxygen atom – the enolate part structure. Charge delocalization leads to lower energy, more accessible carbanion formation. The prototypic β-ketoacid decarboxylase subjected to mechanistic investigation was microbial acetoacetate decarboxylase which yields CO2 and the useful solvent acetone23,24 (Fig. 7). Acetoacetate is a metabolite formed from hydrolysis of acetoacetyl-CoA, the first committed metabolite in isoprenoid/steroid biosynthesis via the mevalonate pathway (but not the methylerythritol pathway to isoprenyl-PPs) and also the oxidation product from 3-hydroxybutyrate, an intermediate in fatty acid metabolism.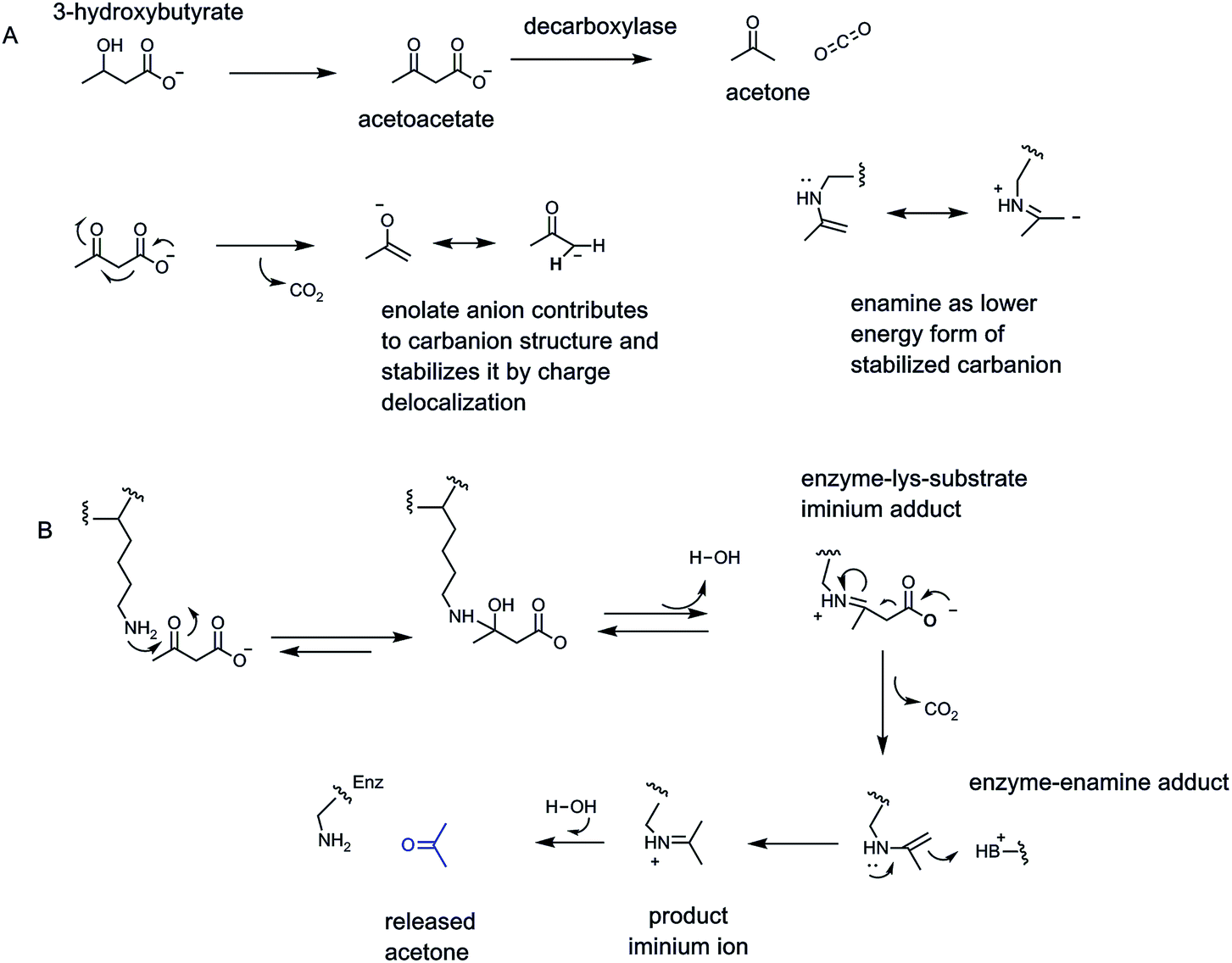 | ||
| Fig. 7 (A) Acetoacetate decarboxylase as paradigm for β-keto acid enzymatic decarboxylation via (B) iminium/enamine catalysis via participation of an active site lysine-NH2 group. The enolate and enamine contributors provide stabilization of the Cα carbanionic intermediates in β-keto acid decarboxylations. | ||
Examination of the reaction manifold revealed an active site lysine residue with low pKa,23 such that the free amine form accumulated and served as catalytic nucleophile towards the C3 ketone of substrate to form a covalent acetoacetate-imine adduct at the start of each catalytic cycle. The resultant C2 carbanion form of the developing acetone product was stabilized as the enamine contributor. Nitrogen-based enamines are more stable forms of oxygen-based enolates (nitrogen is a more basic atom than oxygen) and lower the aggregate energy for formation of the associated stabilized carbanions (Fig. 7). Protonation at carbon (now C1 of the nascent three carbon acetone product) yields the product imine. Hydrolysis regenerates the active site lysine for the next catalytic cycle and releases the acetone product.
3.3 α-Keto acid decarboxylation: thiamin-PP gives a β-iminium equivalent
Organisms have a set of enzymes that process α-keto acid substrates, such as pyruvate and 2-ketoglutarate for decarboxylation to the corresponding aldehydes (Fig. 1C). This is substantially more difficult than decarboxylation of β-keto acids because there is no ready way to stabilize the resulting incipient α-carbanion and so lower its energy14 (Fig. 8A). Whereas β-keto acids lead to enolate anion intermediates during decarboxylation, the α-keto acids would generate acyl anions, too high in energy to form in biologic systems. Thus, organisms have evolved a lower energy route by using the coenzyme form of vitamin B1, thiamin-pyrophosphate as obligate cofactor4,25 (Fig. 8B). The thiazolium heterocycle is the key reactive portion of thiamin-PP via the acidic C2-H on that ring.26 The TPP-recognizing decarboxylases accelerate thiazolium C2 carbanion formation some 1012 fold,27 which then acts as the nucleophile towards the keto group of bound α-keto acid. The resultant tetrahedral adduct now has a positively charged iminium moiety in the thiazolium ring of the coenzyme in a position β to the substrate carboxylate, providing a newly created low energy path to decarboxylation. On cleavage of the C–COO bond, the developing negative charge can delocalize into the thiazolium nitrogen. In effect this is an iminium/enamine system, notionally akin, but structurally coming from very different chemical space, to the molecular logic for β-keto acid decarboxylation in Fig. 7 above.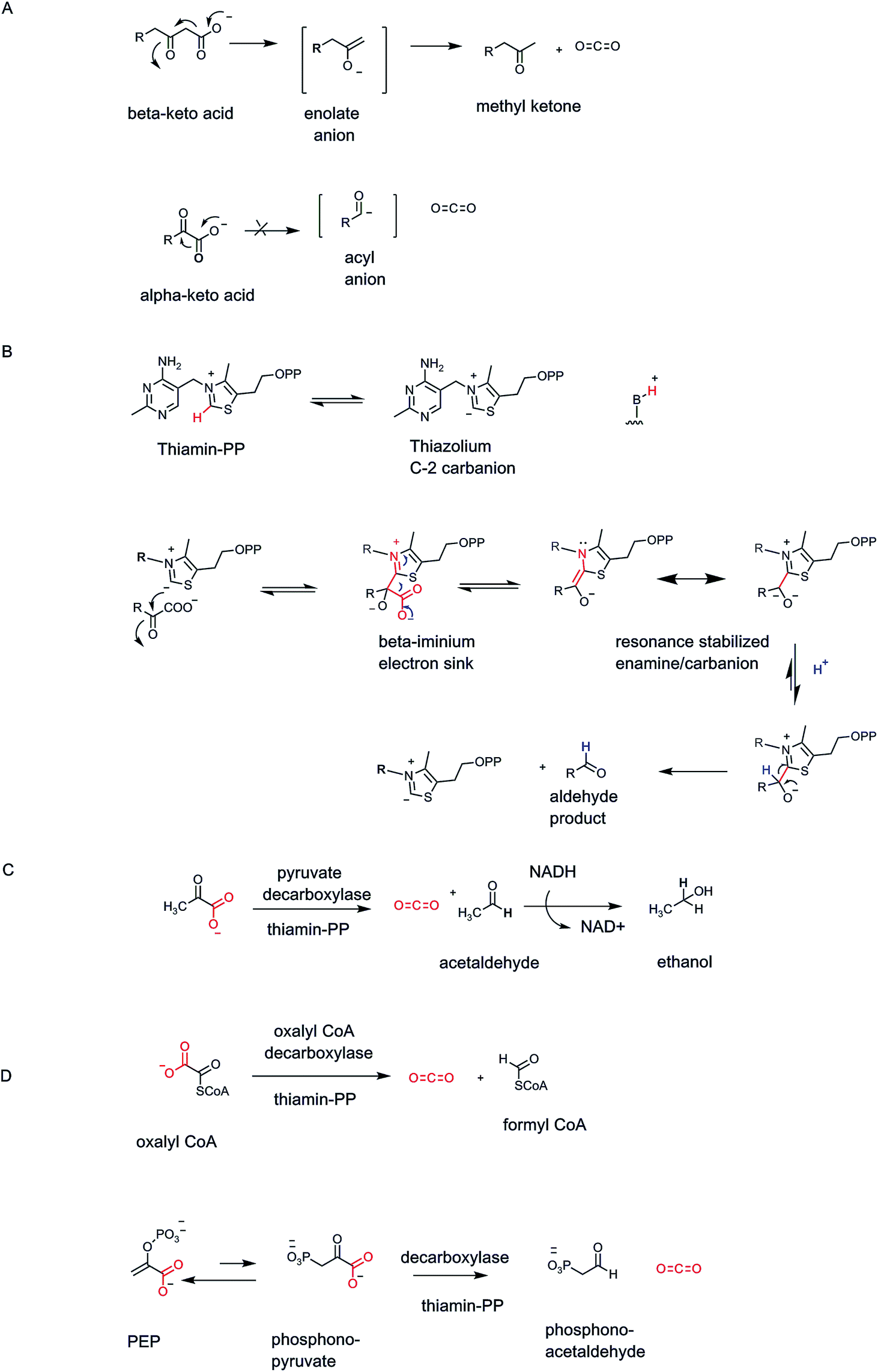 | ||
| Fig. 8 (A) Comparison of enolate vs. acyl anion structures in β- vs. α-keto acid decarboxylations; (B) the C-2 thiazolium ion of the coenzyme form of vitamin B1, thiamin-PP, acts as initial nucleophile towards α-keto acid cosubstrate and generates an adduct that has a β-iminium linkage, lowering the energy for decarboxylation; (C) yeast pyruvate decarboxylase gives CO2 and acetaldehyde; (D) fungal oxalyl CoA decarboxyase also requires TPP and yields CO2 and formyl CoA. | ||
Brewers yeast use TPP-containing pyruvate decarboxylase to convert pyruvate to acetaldehyde and CO2 (ref. 28) (Fig. 8C). The acetaldehyde is produced in large amount and is quantitatively reduced to the two carbon alcohol, ethanol. The ethanol passes out of the yeast cells passively and can accumulate to ∼5% in the medium before the yeasts succumb.29
The biologic role of thiamin-PP (vitamin B1) is always stabilization of otherwise high energy substrate carbanion species.4 A variant of α-keto acid decarboxylases is oxalyl CoA decarboxylase found in fungi that cleaves the one C–C bond in the substrate to CO2 and formyl CoA20 (Fig. 8D). This detoxifies the oxalate framework and gives an activated formylating metabolite for biosynthetic one-carbon transfers.
Many other TPP-dependent decarboxylases occur, in pathways from prokaryotes to complex eukaryotes. For example, bacteria that make direct C–P bonds from PEP use a mutase to generate phosphonopyruvate.30 The C–P linkage is disfavored at equilibrium but the next step is a TPP-dependent phosphonopyruvate decarboxylase that irreversible generates the phosphonoacetaldehyde31 (Fig. 8D).
3.4 PLP-amino acid decarboxylases: neurotransmitters and beyond
The coenzyme form of vitamin B6 is the pyridine aldehyde pyridoxal-phosphate.4 It is synonymous with amino acid metabolism and enables enzymatic cleavage of any of the four bonds to Cα (C-2) of the many α-amino acids in primary metabolic pathways (Fig. 9A). Notable in human metabolism are PLP-containing decarboxylases that generate neurotransmitter amines from the free amino acids.32 These include decarboxylation of 5-OH-tryptophan to 5-OH-tryptamine (serotonin), tyrosine to tyramine, 3,4-dihydroxyphenylalanine (DOPA) to dopamine, histidine to histamine, and glutamate to γ-aminobutyrate (GABA) (Fig. 9A and B).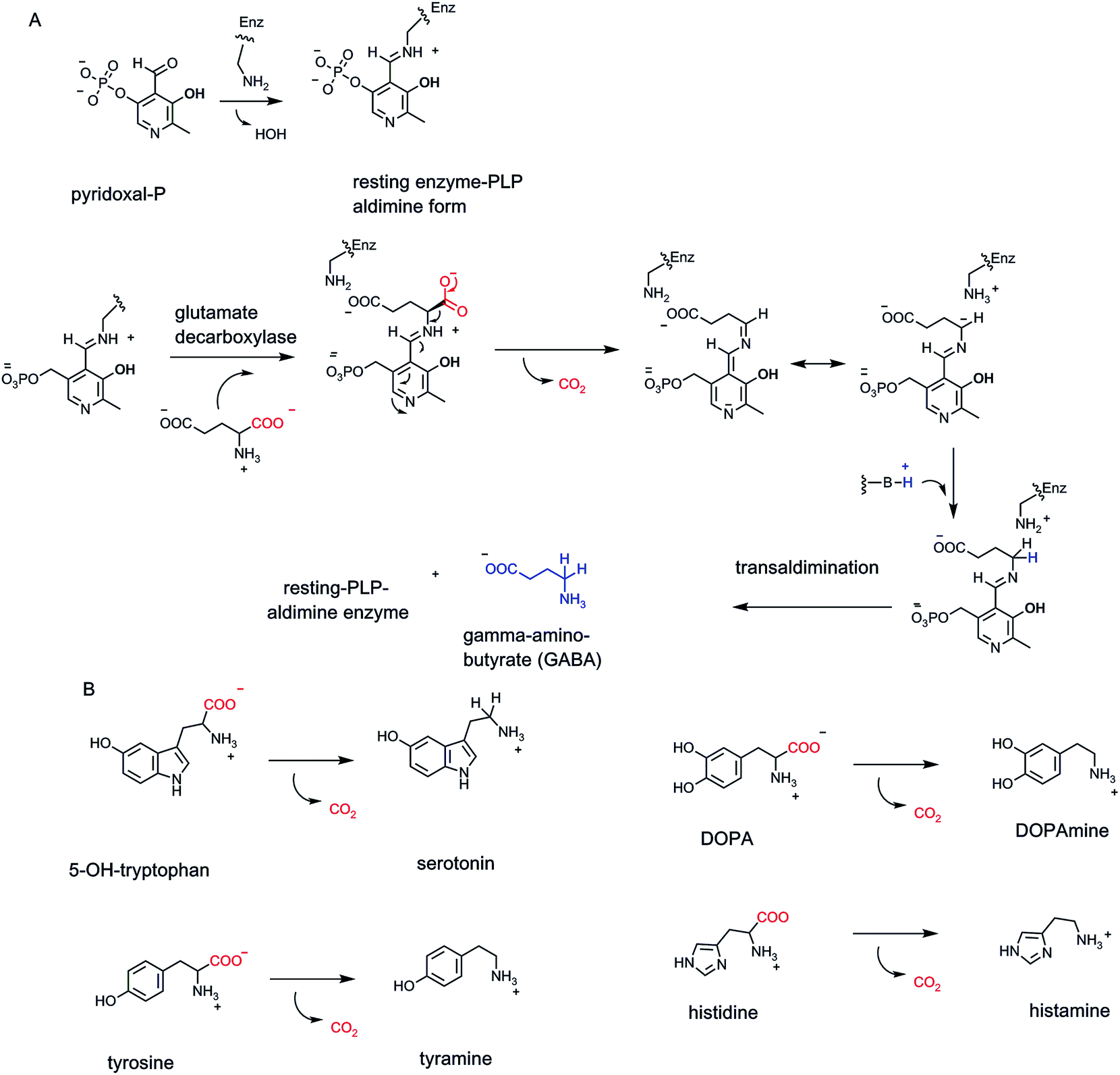 | ||
| Fig. 9 (A) Pyridoxal-P is bound to apoenzymes in aldimine linkage to an active site lysine residue. The reaction mechanism for decarboxylation of L-glutamate to the inhibitory neurotransmitter γ-aminobutyrate (GABA) involves a stabilized covalent adduct to PLP; (B) four other PLP-containing decarboxylases that yield neurotransmitter and/or inflammatory amines produce serotonin, dopamine, tyramine, and histamine. | ||
The aldehyde group in PLP is the key functional group for coenzyme function. Resting forms of PLP enzymes keep the coenzyme tightly associated by virtue of an imine linkage between an active site lysine residue ε-amine side chain and the pyridine aldehyde carbonyl.33 Then, the first catalytic step is attack of the amino group of an incoming amino acid substrate on the aldimine linkage to PLP. The resultant tetrahedral adduct can expel the Enz–Lys residue-NH2 as it forms a substrate-PLP aldimine linkage (Fig. 9A). The PLP coenzyme is now an electron sink to stabilize incipient carbanion electron density that develops at Cα. Members of the decarboxylase family orient the substrate carboxyl group so it is orthogonal to the plane of the aldimine-PLP linkage.34 This gives maximal orbital overlap during C–COO bond cleavage as CO2 forms. The carbanion developing at Cα is delocalized and stabilized by resonance forms that accommodate that negative charge at the benzylic carbon of the PLP coenzyme and also as the para-quinoid resonance form, thereby lowering the energy barrier to its formation. Regiospecific and stereospecific delivery of a proton to Cα in the active site yields the product-PLP aldimine adduct. Transaldimination by attack back in of the active site Lys-NH2 regenerates the resting enzyme-PLP aldimine and releases the product amine.
All three of these types of decarboxylases, the cofactor-independent β-keto acid decarboxylases, the thiamin-PP (vitamin B1) requiring α-keto acid decarboxylases, and the PLP (vitamin B6)-dependent α-amino acid decarboxylases, act by a common underlying chemical strategy: stabilization of an incipient carbanion in the residual framework as CO2 is formed by C–C bond cleavage. The three enzymes use distinct but convergent strategies, adapted to the functional groups presented in the substrates undergoing the metabolic decarboxylation. We can compare these three strategies, shaped by substrate structural demands (Fig. 10).
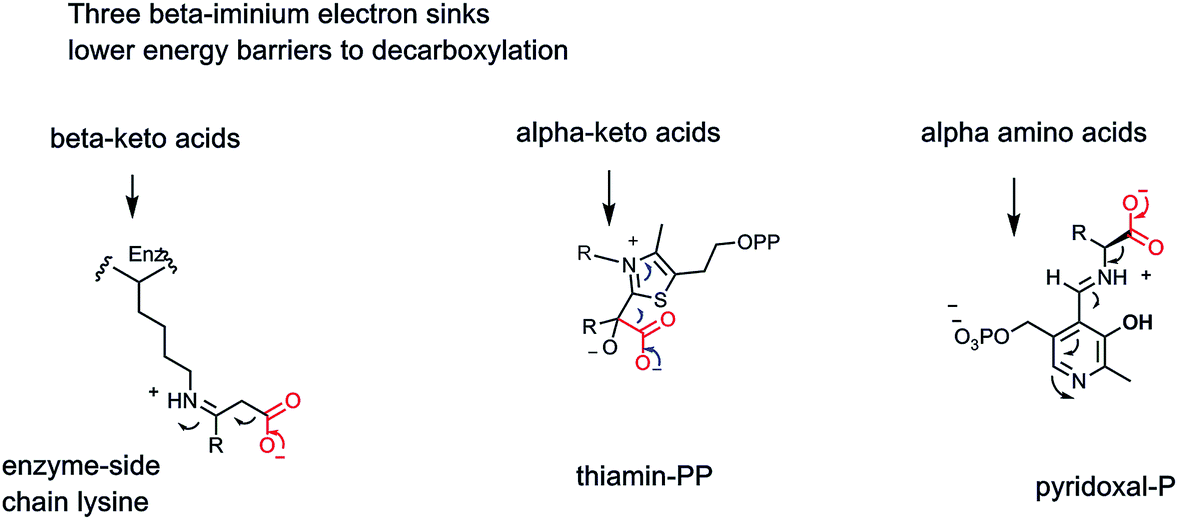 | ||
| Fig. 10 Three enzymatic strategies lower energy barriers for decarboxylation by providing beta iminium intermediates to stabilize incipient α-carbanionic species. | ||
3.5 Pyruvamide decarboxylases for SAM, aspartate, phosphatidylethanolamine
The genius of the coenzyme form of vitamin B6 is that it brings a new functional group, an aldehyde, to the catalytic armamentarium of protein catalysts. None of the 20 proteinogenic amino acids that are the building blocks for protein biosynthesis in all forms of life bring an aldehyde or ketone carbonyl to the inventory. One might ask: what did organisms do before PLP was invented? A hint may be found in a small class of enzymes35,36 that undergo autocatalytic cleavage at an internal X-ser–peptide bond, fragmenting the single polypeptide chain into a two chain form in which the previously internal serine residue has initiated cleavage by attack of its side chain-OH on the immediately upstream peptide bond (Fig. 11A). The resulting ester can fragment as shown, producing two polypeptide chains from one, while generating an enamino N-terminus of the newly formed, second polypeptide chain.35 The enamine is the unstable tautomer of the imino pyruvamide. Thus, tautomerization and imine hydrolysis generates an N-terminal pyruvamide in the new subunit. The two polypeptide chains remain together by noncovalent interactions and the newly uncovered ketone in the pyruvamide can be considered a primordial analog of the PLP carbonyl group.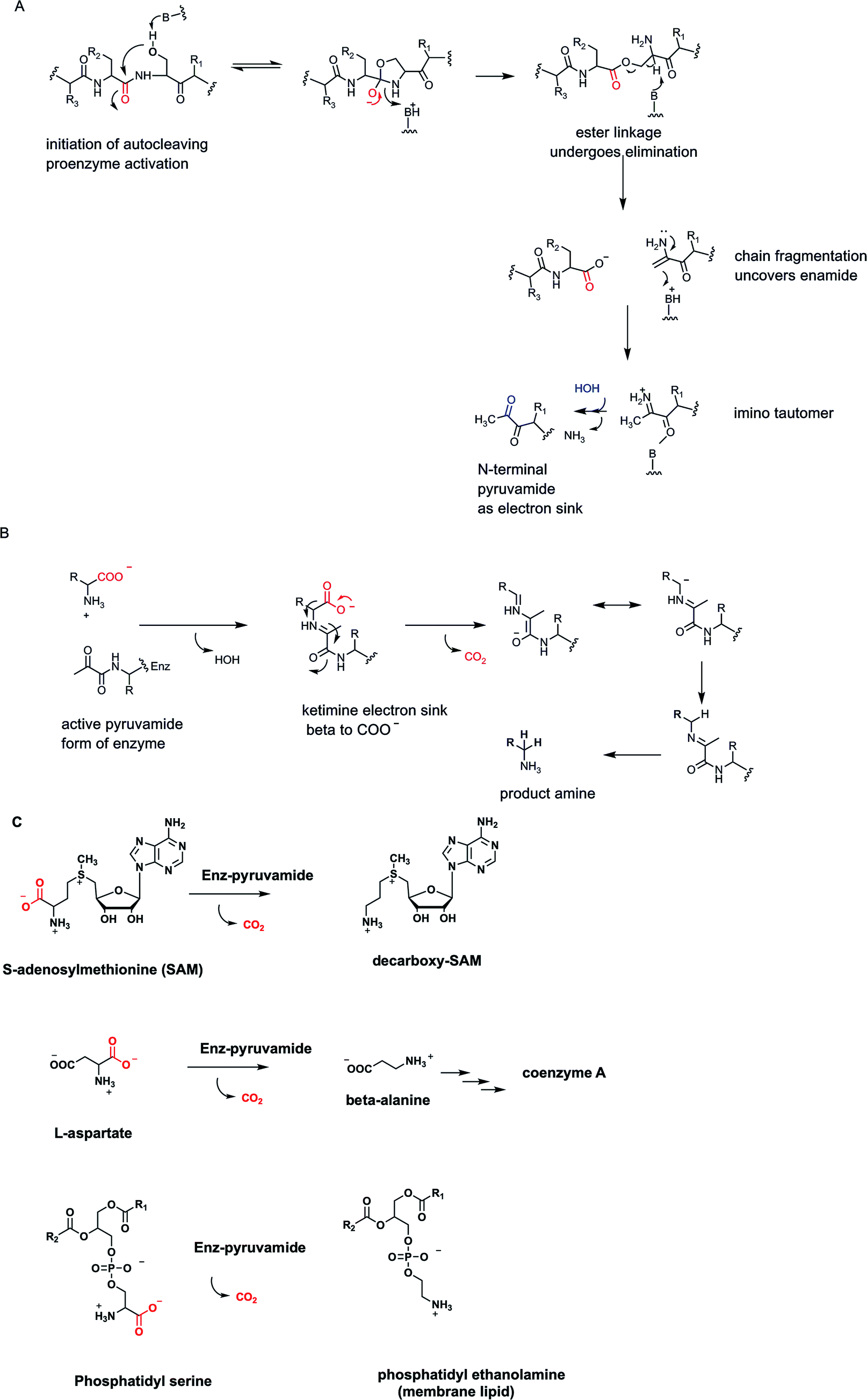 | ||
| Fig. 11 (A) N-Terminal pyruvamide electron sink forms by autocleavage at a serine side chain of a single chain proenzyme to a two chain active enzyme; (B) the pyruvamide keto group then forms a ketimine with an amino acid substrate to lower energy for decarboxylation; (C) three pyruvamide-dependent enzymes decarboxylate SAM, aspartate, and phosphatidyl serine, respectively. | ||
In these active enzyme forms the pyruvamide residue ketone forms a ketimine with a partner amino acid substrate and can serve as electron sink and stabilizing enamide contributor to lower the energy barriers for Cα carbanion formation during decarboxylation (Fig. 11B). Subsequent protonation of that stabilized carbanion at Cα and imine hydrolysis yields the amine product and the two chain form of active enzyme for the next catalytic cycle.
Three enzymes that fall in to this auto-activating, pyruvamide utilizing electron sink category are noted here35 (Fig. 11C). One is S-adenosylmethionine decarboxylase, converting the aminobutyryl chain of SAM to the propionyl amine side chain in the decarboxy SAM product. In the next metabolic step this molecule is the donor of the propionyl group to a molecule of 1,4-diaminobutane (putrescine) to yield the C7 triamine spermidine, one of the organic counterions to shield the phosphate negative charges on DNA. Incidentally, the 1,4-diaminobutane arises from standard PLP-dependent ornithine decarboxylase action.
A second pyruvamide-containing multiple chain catalyst is aspartate decarboxylase. Directed loss of CO2 gives β-alanine as the amine coproduct.36 This is famously one of the building blocks of coenzyme A, the ubiquitous thiol-containing coenzyme that combines with acyl groups to form activated acyl-CoA metabolites (acetyl CoA, succinyl CoA, hydroxymethylglutaryl CoA, palmitoyl-CoA).37 The third pyruvamide-containing decarboxylase of note is phosphatidyl serine decarboxylase35,38 which along with CO2 yields the coproduct phosphatidyl ethanolamine, one of the main constituents of phospholipid bilayers in cell membranes, made in tens of millions of copies in each cell cycle. All three of these presumably “primordial” enzyme catalysts sit at important biosynthetic nodes of metabolism.
3.6 Oxidative vs. nonoxidative TPP-decarboxylase nanomachinery
We noted two sections above that yeast strains use thiamin-PP as obligate cofactor for pyruvate decarboxylase, where acetaldehyde is the product in what one would term a nonoxidative decarboxylation process. The C-1-acetaldehyde carbonyl is at the same oxidation state as the C-2 ketone in the starting substrate pyruvate process. However, some of the TPP-enzymes in higher eukaryotes are part of protein nanomachines that oxidize the starting C-2 ketone group up to the level of a carboxylic acid.39 The two electrons removed are captured and stored in coproduct NADH, the mobile electron carrier that is thermodynamically activated but kinetically stable enough to be a diffusible cosubstrate for dozens of cellular electron transfer reactions. Simultaneously, the decarboxylated acyl group product is not the free carboxylate but instead an acyl-CoA, activated for acyl transfer reactions in the cell.4 Thus, these enzyme nanomachines remove two electrons in oxidative decarboxylation of α-keto acids, trapping some of the oxidation energy in two of the three thermodynamically activated, kinetically stable workhorse molecules of cell metabolism-NADH, and acyl CoAs.14The two companion enzymes that make up the oxidative decarboxylase nanomachinery are a lipoamide-containing enzyme and an NAD+-reducing flavoprotein dehydrogenase.39 Thus, the coenzyme forms of vitamins B1, B2, and B3 are involved as well as the disulfide form of lipoamide covalently tethered to enzyme 2 of the three enzyme machine.
Using pyruvate dehydrogenase as an example, the TPP-containing decarboxylase (enzyme 1) carries out the generation of CO2 from the TPP-pyruvate covalent adduct to yield the carbanion of hydroxyethyl-TPP as the nascent product in the enzyme 1 active site (Fig. 12). That carbanion can attack the lipoamide disulfide presented on a 20 Å long arm by enzyme 2 and generate a tetrahedral adduct connecting the TPP thiazolium to one of the lipoamide sulfur atoms. That can collapse to liberate back the TPP C-2-thiazolium carbanion, readying enzyme 1 for the next catalytic cycle. Inspection of the two carbon substrate-derived adduct transferred to lipoamide reveals it as an acetyl group in thioester linkage. Thus, the oxidation step has now occurred. It has happened as a thiohemiacetal is converted to an acyl thioester. That acetyl thioester is tethered covalently to the prosthetic group of enzyme 2. Now the cosubstrate coenzyme A (CoASH) can engage in thioester exchange, via tetrahedral adduct, with net transfer of the acetyl group out to observed product acetyl CoA, leaving enzyme 2 in the dithiolipoamide form-two electron reduced.
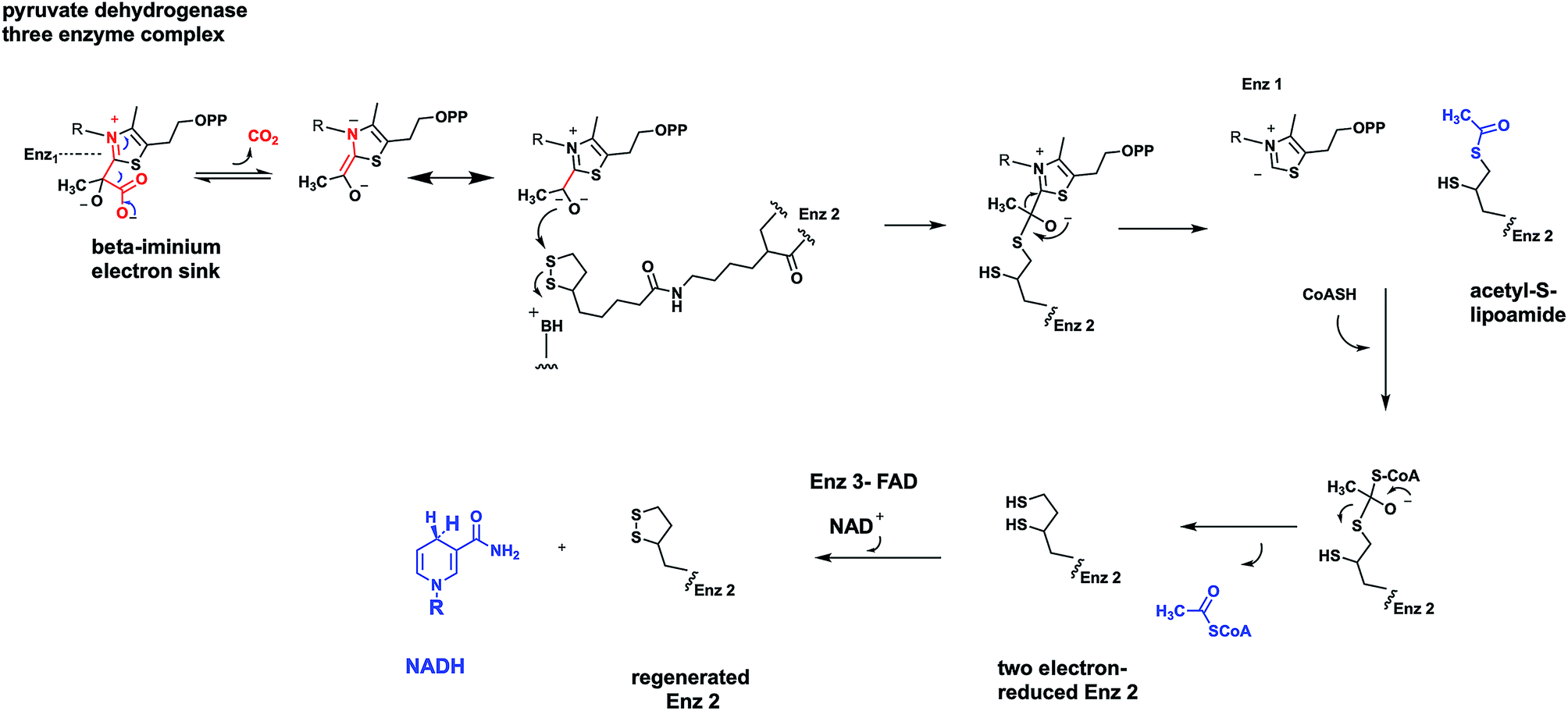 | ||
| Fig. 12 Three component oxidative α-keto acid decarboxylases contain TPP in enzyme 1, lipoamide in enzyme 2, and FAD in enzyme 3. NAD is the substrate that reoxidizes the FADH2 in enzyme 3. CoASH engages in thiol exchange with acetyl-lipoamide in enzyme 2. | ||
From the point of view of pyruvate, the reaction is complete. CO2 was released by enzyme 1, acetyl CoA released by enzyme 2. However enzyme 2 is not ready for another catalytic cycle. That requires the oxidized, disulfide form of the tethered lipoamide. That reoxidation is the job of enzyme 3 and its tightly associated FAD coenzyme.40 The mobile dithiolipoamide can reach into the enzyme 3 active site and be reoxidized by two electrons back to the disulfide, while reducing the FAD by two electrons to FADH2. Enzyme 2 is now regenerated in active form but enzyme 3 is down by two electrons in the form of FADH2. Now NAD+ can be reduced by enzyme 3 to NADH, as soluble product, while the bound FADH2 is reoxidized back to starting FAD.
All three enzymes in the nanomachine that is pyruvate dehydrogenase have been returned to their starting oxidation states and can act again, over and over, catalytically. The two electrons that started in substrate pyruvate end up in mobile NADH. The C-2 ketone of pyruvate becomes the C-1 acyl thioester of acetyl CoA. The original C-2 and C-3 of pyruvate are released as the activated acetyl thioester, not the low energy resonance stabilized acetate anion. The three enzyme complex is termed pyruvate dehydrogenase,39 emphasizing the redox chemistry that has occurred during the decarboxylation.
Two other TPP oxidative decarboxylase nanomachines show up in nodal points of primary metabolism, all in mitochondrial compartments. One is in the middle of the citrate metabolic cycle, α-ketoglutarate dehydrogenase, generating CO2, along with succinyl CoA, and NADH as the energy capture products.41 The second three enzyme complex is branched chain α-keto acid decarboxylase/dehydrogenase, involved in harvesting energy from the three α-keto acids generated by transaminase action on the three proteinogenic amino acids, leucine, isoleucine, and valine (Fig. 13).
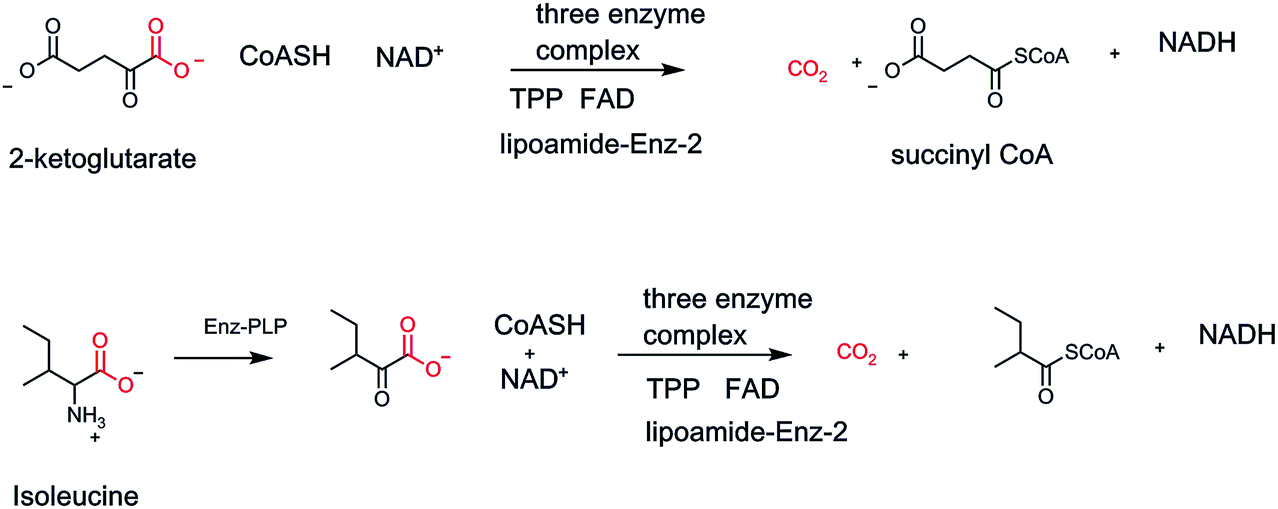 | ||
| Fig. 13 Two forms of oxidizing α-keto acid decarboxylation three component complexes; the α-ketoglutarate dehydrogenase functions in the citric acid cycle. The branched chain α-keto acid decarboxylase is essential for routing carbons from leucine, isoleucine and valine during their degradative metabolism. | ||
From the point of view of chemical strategy, these decarboxylase-dehydrogenase enzyme nanomachines are marvels of efficiency and strategy at energy harvesting. The CO2 is the “throw away” byproduct of trapping the remainder of the organic scaffolds as activated acyl thioesters.
The serial involvement of coenzyme forms of vitamins B1, B2, and B3 provide conduits for the pair of electrons removed in oxidation to end up in the mobile cellular electron currency NADH. Three attributes of heterocyclic chemical biology4 are employed in those three coenzymes to lower energy barriers, transfer electrons, and then store them in the kinetically stable dihydropyridine scaffold of NADH/NADPH.
3.7 Coupling β-hydroxy acid oxidation to β-keto acid formation and decarboxylation: NAD+ requirement as preamble
A variant of the above enzyme strategy involves oxidation of β-hydroxy acids to β-keto acids transiently and then decarboxylating them before release. The oxidation step can use NAD+ or NADP+ as electron acceptor, as in isocitrate dehydrogenase isoenzyme forms, or NADP+ as in 6-P-gluconate dehydrogenase and malic enzyme (malate to pyruvate, CO2 and NADPH)42,43 (Fig. 14). In each case the nascent β-keto acid is decarboxylated before release from the enzyme active site. In the six carbon isocitrate case, the released product is the five carbon α-ketoglutarate, revealing that the citrate cycle uses tandem decarboxylation of β-keto acid, then α-keto acid, to yield the two CO2 molecules for each turn of the citrate cycle. In so doing, those two enzymes harvest the two pairs of electrons removed in two NADHs and produce the four carbon, activated succinyl CoA as remainder scaffold.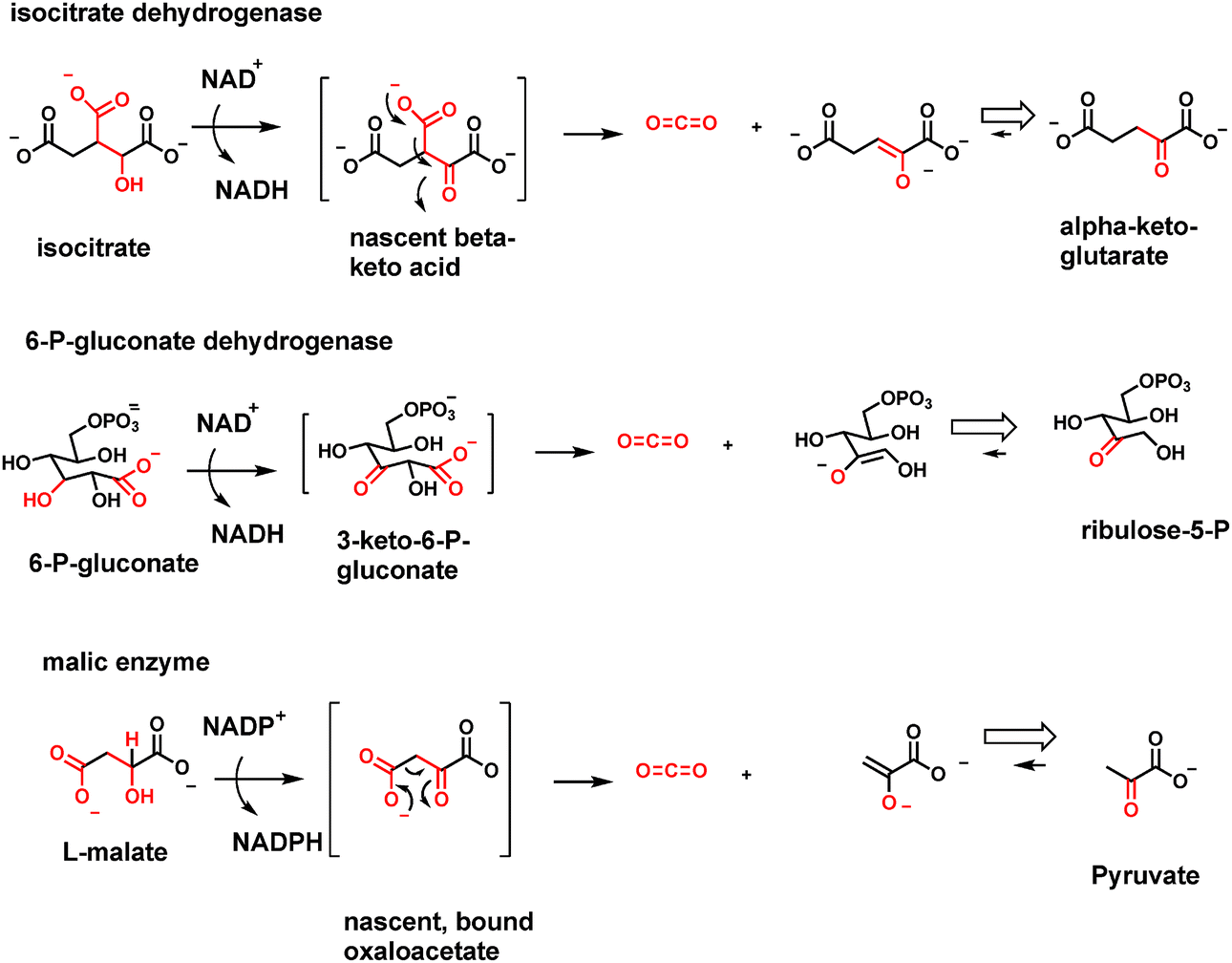 | ||
| Fig. 14 Three enzymes that oxidize β-hydroxy acids to β-keto acids via nicotinamide cosubstrates: isocitrate dehydrogenase functions in the citric acid/Krebs cycle. 6-P-Gluconate dehydrogenase functions in the pentose phosphate shunt pathway. Malic enzyme generates NADPH in cell cytoplasm. Each of these named dehydrogenases are also decarboxylases. The nascent β-keto acids are NOT released but instead decarboxylated in situ in each enzyme's active site. | ||
In the 6-P-gluconate case, the product that emerges is the five carbon ketose ribulose-5-P, a subsequent isomerization away from ribose-5-P, the building block sugar for RNA and DNA. This is a major nodal point for diverting glucose to ribose billions of times per cell cycle.
A variant of this chemistry occurs in plant cell wall construction, specifically in assembly of xylan polysaccharide chains. UDP glucose is progenitor to UDP-glucuronate (the C6′-carboxylate).44 Oxidation of the C4-OH to the ketone by bound NAD+, facilitates loss of the C-6 carboxylate as CO2. Re-reduction of the 4-keto sugar by the bound NADH, produces UDP-xylose as the activated form of the xylose monomeric building block for plant xylan chain extensions.
Late in the cholesterol biosynthetic pathway, specifically in the conversion of the tetracyclic thirty carbon lanosterol to the twenty seven carbon cholesterol, double bonds are saturated and rearranged and most notably the gem dimethyl groups at C-4 and the methyl group at C-14 are removed45 (Fig. 15A). The C-14 one-carbon unit is ejected as formate, while the two methyl groups at C-4 are lost as two molecules of carbon dioxide. The strategy at each of these three methyl groups is to use hemeprotein monooxygenases of the cytochrome P450 superfamily, to oxygenate each methyl group ad seriatim, to the C–CH2OH and then the C–CHO levels. Each oxygenation event spends an NADPH molecule, as initial reductant of a P450 reductase.
 | ||
| Fig. 15 (A) Lanosterol to cholesterol involves double bond reductions and isomerizations and loss of three pendant methyl groups, one at C-14 and two at C-4. (B) Mechanistic scheme for methyl group oxidative removal during conversion of lanosterol to cholesterol. The methyl at carbon 14 is oxygenated twice to the aldehyde and then released in a third oxygenation step to give formate. At carbon 4, three consecutive oxygenations of the α-methyl give the carboxylate substituent. Oxidation of the 3-β-alcohol to ketone creates the β-keto acid grouping to facilitate decarboxylation. Enzymatic isomerization of the remaining C-4-β-CH3 to the α-position then sets up the triple oxygenation to the pendant β-keto-α-carboxylate for decarboxylation. Then reduction of the 3-keto back to the 3-β-OH completes the trimming away of the three methyl groups of lanosterol. | ||
At that point the oxygenase strategies diverge (Fig. 15B). The C-14-CHO is lost as formate, probably by net oxygen insertion into the C–CHO carbon–carbon bond and then bond cleavage46 (Fisher et al., 1989). The oxygenase chemistry at C4 proceeds a third time converting each of the C-4–CHO to C-4–COO groups. Now, the 3-β-OH group, a signature functional group of lanosterol and cholesterol, is oxidized via action of an NAD+-dependent dehydrogenase to the 3-keto group. This new carbonyl is beta to the C-4–COO− group and serves as electron sink for cofactor-free decarboxylation.
In terms of timing, the 4-α-methyl group is oxygenated three times and decarboxylated via the 3-keto steroid first. Then the β-methyl is enzymatically isomerized into the α-position and again oxygenated three times to get to the α-carboxylate,45 and then lost as CO2. When both of the methyl groups at C-4 have been eliminated, the 3-keto, having done its job as electron sink, is reduced back to the 3-β-OH by a molecule of reduced nicotinamide coenzyme (Fig. 15B). The tell tale β-keto acid intermediate is cryptic in this double decarboxylase-tailoring of the C-4 locus of lanosterol, but is hinted at by the requirement for NAD+ as catalytic partner.
3.8 Flavin coenzymes in decarboxylases
Organisms press into decarboxylase service the FAD coenzyme form of vitamin B2 (riboflavin) only rarely but when they do, the chemical strategy is quite illuminating and fits the substrate electronic demands.The bacteria enzyme lactate monooxygenase contains FMN, and requires O2 as cosubstrate for oxidative decarboxylation of L-lactate to acetate and CO2.47 With O2, one of the oxygen atoms ends up in the carboxylate of acetate (Fig. 16A). Mechanistic investigation has indicated the expected bound nascent products pyruvate and the peroxide anion. Pyruvate is retained in the active site long enough for OOH− to add into the pyruvate carbonyl. Decarboxylation from that tetrahedral adduct can proceed with loss of CO2 concomitant with fission of the weak O–O single bond. Then the acetate product is released.
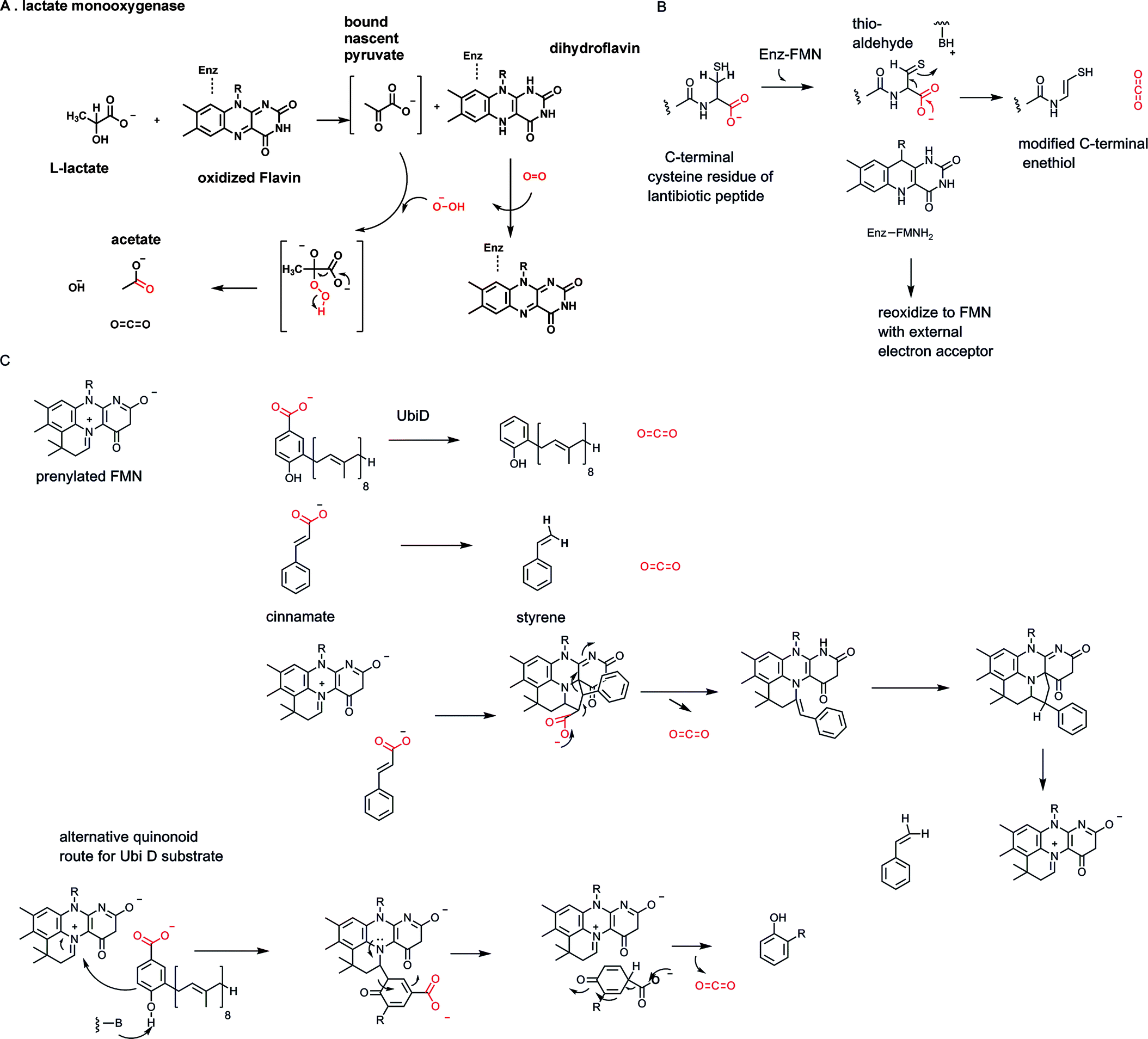 | ||
| Fig. 16 Three flavin-dependent decarboxylases: (A) bacterial FMN-dependent lactate monooxygenase; (B) bacterial pantetheinylcysteine decarboxylase generates pantetheine, vitamin B5, in the coenzyme A biosynthetic pathway; generation of a terminal enethiol moiety at the C-terminus of some lantibiotic peptides; (C) prenylated flavin coenzyme in aryl acid decarboxylases: possible mechanistic alternatives. | ||
In a sense that decarboxylation is an epiphenomenon to the buildup of pyruvate and peroxide anion in a confined volume of space. On the other hand, it illustrates that enzymes can use the nucleophilicity of the peroxy anion and the low energy for O–O bond cleavage to drive decarboxylation.
The second example of a flavin-dependent decarboxylase occurs in microbial biosynthesis of vitamin B5, pantetheine (Fig. 16B). In turn, ingested pantetheine is converted by humans in four enzymatic steps to coenzyme A. The immediate progenitor to phosphopantetheine is phosphopantothenoyl-cysteine. Enzymatic conversion to P-pantetheine involves decarboxylation of the carboxylate group in the cysteine moiety.48 While one might have anticipated a PLP-dependent enzyme, given so many PLP-amino acid decarboxylase metabolic precedents, that is not the case. The enzyme contains FAD. To lower the energy barrier for decarboxylation the enzyme reversibly oxidizes the cysteinyl thiol in the substrate to a thioaldehyde, storing the two electrons in the flavin coenzyme as FADH2. That C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S thio carbonyl is now β to the carboxylate to be eliminated. C–COO bond breakage allows delocalization of the electron pair available from that bond cleavage into that thio-aldehyde, producing the enethiolate tautomer. The enethiolate can isomerize back to the thio-aldehde with protonation at what had been C2 of the cysteinyl moiety. Now re-reduction of the thioaldehyde group by FADH2 regenerates the starting form of the enzyme and covers up the fact that there was an intermediate with a thio-aldehyde electron sink. This is an ingenious reversible redox strategy that is independent and parallel to vitamin B6 decarboxyation logic.
S thio carbonyl is now β to the carboxylate to be eliminated. C–COO bond breakage allows delocalization of the electron pair available from that bond cleavage into that thio-aldehyde, producing the enethiolate tautomer. The enethiolate can isomerize back to the thio-aldehde with protonation at what had been C2 of the cysteinyl moiety. Now re-reduction of the thioaldehyde group by FADH2 regenerates the starting form of the enzyme and covers up the fact that there was an intermediate with a thio-aldehyde electron sink. This is an ingenious reversible redox strategy that is independent and parallel to vitamin B6 decarboxyation logic.
Close variants of this flavin-dependent decarboxylase strategy are found in the posttranslational modification of some peptide antibiotics that derive from lanthionine cross-linked protein precursors.49 A subset has been processed at the terminal cysteine residue by the same intermediate oxidation of the thiol side chain to the thioaldehyde as electron sink for decarboxylation.50 In these cases the resultant enethiol is not re-reduced by FMNH2 but persists in the mature lantibiotic framework as C-terminal enethiol (Fig. 16B).
The third example of evolution of vitamin B2-dependent redox catalysts for substrate decarboxylation has been discovered recently in microbial decarboxylation of a variety of both acrylic acids such as cinnamates and aryl carboxylate substrates including p-OH benzoate, by enzymes in the UbiD family51 (Fig. 16C). Unexpectedly, the flavin FMN coenzyme has been modified to bear a fourth ring in iminium linkage to N-5 and C-6 of the tricyclic flavin nucleus.52 This prenylated FMN (PrFMN) is inactive for normal flavin redox chemistry but instead supports decarboxylation by a proposed cycloaddition/elimination mechanism.51
The newly introduced fourth ring grafted on the tricyclic flavin core can be written as an azamethine ylide, a functional group known to engage in 1,3-dipolar cycloadditions.51 The acrylic acid substrates can behave as dipolarophiles. Formation of a cyclic adduct would then place the carboxylate to be eliminated beta to the FAD. Transient storage of the electrons in the FAD framework is then akin to the role that pyridoxal-P and thiamin-PP each play in lowering energy barriers for their respective decarboxylases.51 It is less clear that aryl acids such as phenolic carboxylates will be equivalent dipolarophiles and/or undergo comparable cycloadditions. Thus, it is also unclear whether a universal cycloaddition route obtains for the UbiD family of decarboxylases. UbiD itself converts 3-octaprenyl-4-hydroxybenzoate to 2-octaprenylphenol during the assembly of the benzoquinone electron transfer coenzyme ubiquinone52,53 (Fig. 16B and C).
Flavins thus enable three kinds of redox modes for decarboxylation: generation of peroxide anion, a rare, sometimes reversible oxidation of thiol to thioaldehyde as transient electron sink, and the use of a tetracyclic flavin iminium scaffold as electrophile.
3.9 Nicotinamide-dependent decarboxylases
Leaving aside carbonic acid (H2CO3) as the hydrated species in equilibrium with dissolved carbon dioxide, formic acid is the simplest organic carboxylic acid, 99.9% as the formate carboxylate anion at neutral, physiologic pH. Emphasizing that the conversion of formate to CO2 involves loss of two electrons, there are formate dehydrogenases that use NAD+ as cosubstrate, reducing it to NADH54 (Fig. 17A). Consistent with the role of NAD+/NADH as mobile cellular electron currency, this redox reaction is formulated as a hydride transfer to C-4 of the pyridinium ring, with electron flow to quench the positive charge on the pyridinium nitrogen. The net result is generation of the thermodynamically activated dihydropyridine ring in NADH. Although these enzymes are named formate dehydrogenases, they are also hydride-ejecting decarboxylases.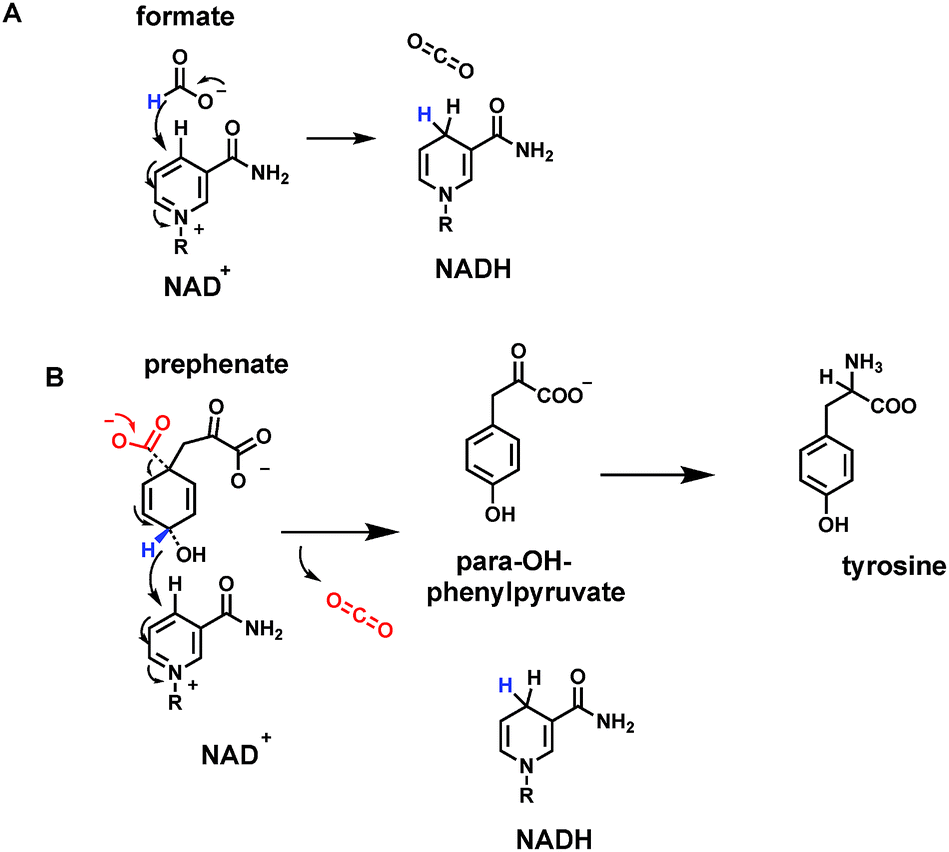 | ||
| Fig. 17 Two nicotinamide-dependent CO2 producing enzymes (A) formate dehydrogenase uses NAD+; (B) prephenate dehydrogenase on the way to tyrosine biosynthesis, uses NADP+ as electron acceptor. | ||
Another example of such a hydride-ejecting decarboxylase occurs as one of the metabolic fates of a key aromatic metabolite precursor, the dicarboxycyclohexadiene prephenate. Prephenate dehydrogenases couple decarboxylation with vinylogous loss of a hydride equivalent at C-4 of the aromatizing ring55 (Fig. 17B). The cosubstrate recipient is NADP+ and the driving force may be the aromatization of the cyclohexadiene to the resonance-stabilized phenyl ring system. The coproduct, along with NADPH and CO2, is para-hydroxyphenylpyruvate. One of the two carboxylates from prephenate is retained as the pyruvate side chain. The next enzymatic step, transamination, leads to L-tyrosine, a key aromatic proteinogenic amino acid. Tyrosine is also a substrate for the PLP-dependent tyrosine decarboxylase, by mechanisms outlined above, to yield tyramine (as in Fig. 9). Thus, in three consecutive metabolic steps, prephenate to tyramine, both carboxylates are removed as CO2, by two very different types of cofactor-dependent decarboxylases. We will take up two additional, alternate forms of prephenate decarboxylases in the cofactor-independent sections below.
3.10 Biotin-dependent decarboxylases
One cofactor-dependent decarboxylase type atypically uses biotin, vitamin B7 (ref. 56) (Fig. 18A). This was a surprising finding since biotin is instead the classic cofactor for enzymes that work in the opposite direction, splitting ATP to activate CO2. Actually, it is the HCO3− equilibrium partner, for C–C bond formation in such enzymes as acetyl CoA carboxylase and pyruvate carboxylase4,57 (Fig. 30 and 31 below). In those canonical carboxylases, the biotin is covalently tethered in amide linkage through a carrier protein domain that visits the carboxylation domain/subunit where it reacts with the anhydride of carbonic acid and phosphoric acid to produce N-1-carboxybiotin.57 This modified biotinyl group then moves to the carboxy transfer domain and offers up the carboxy group to a carbanion equivalent in the cosubstrate, e.g. C-3 of pyruvate or C-2 of acetyl-CoA, to fix the CO2 unit as a new carboxylate group.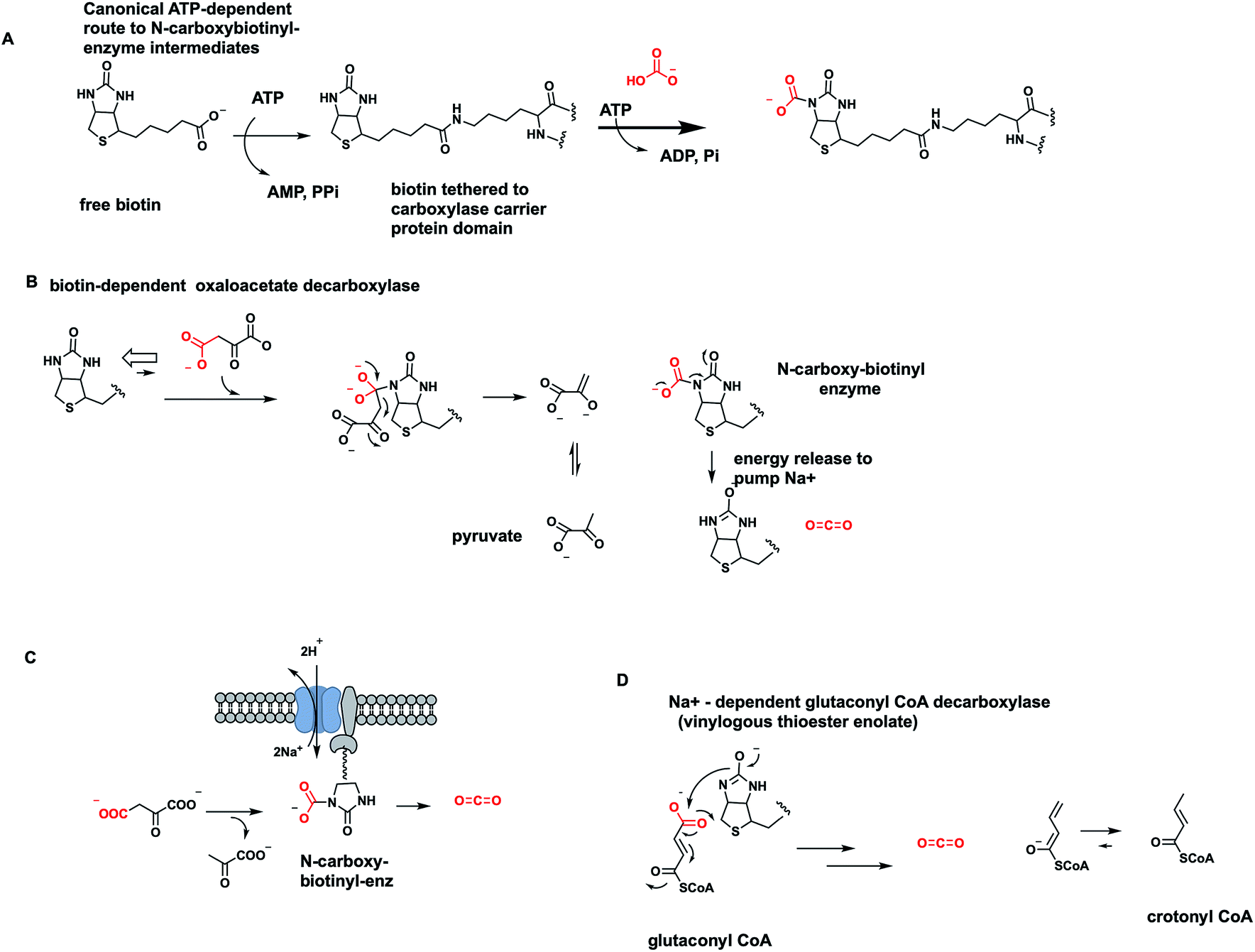 | ||
| Fig. 18 Biotin-dependent oxaloacetate- and glutaconyl decarboxylases are membrane-bound sodium pumps. (A) ATP-dependent canonical formation of N1-carboxybiotinyl-enzymes; (B) reverse reaction involves decarboxylation of oxaloacetate to pyruvate and N-carboxybiotin that is hydrolyzed in each catalytic cycle to free CO2. (C) Schematic for vectorial pumping of Na+ ions by these membranous decarboxylases; (D) vinylogous decarboxylation and Na+ ion pumping by a homologous glutaconyl CoA decarboxylase. | ||
The question therefore arose in certain bacterial biotin-containing enzymes58 that act to decarboxylate oxaloacetate to pyruvate and CO2, whether this thermodynamically favored dissociation of N-carboxybiotin to biotin and free CO2 would be coupled to some cellular function, such as mechanical work. Indeed, it turns out these oxaloacetate decarboxylases are sodium ion-dependent and membrane-bound.58 The catalytic decarboxylation cycle drives Na+ ions across the bacterial membrane (Fig. 18C). Thus, the physiologic function of these proteins is primarily as vectorial pumps for sodium ion transport. Rather than using a familiar energy source such as ATP to drive ion flux, they use decomposition of the N-carboxybiotin adduct.
Acidaminococcus fermentans is another microbe using a membranous multi-subunit complex to decarboxylate, in this case, glutaconyl CoA while pumping sodium ions across the membrane to generate an electrochemical potential.59 The C–C bond cleaving step involves transfer of the migrating COO− unit to the biotin prosthetic group on one of the enzyme subunits. The remaining C-4 carbanion scaffold is stabilized as a vinylogous thioester enolate, with reprotonation to give 2-butenoyl-CoA as released product (Fig. 18D). Dissociation of nascent N-carboxybiotinyl enzyme to CO2 and biotinyl enzyme drives ion flux across the membrane.
One further example of a microbial biotin-containing decarboxylase acting as vectorial Na+ pump is malonyl CoA decarboxylase.58 This occupies a different physiologic niche from the comparable use of cofactor-independent malonyl thioester decarboxylation to drive fatty acid biosynthesis noted in Section 7.1, and also from a free-standing malonyl CoA decarboxylase.60
In these Na+ ion-pumping enzyme nanomachines, the biotin-mediated C–C-bond cleaving decarboxylations provide the driving force for the transport cycle, with a stoichiometry of 1–2 sodium ions transported out across the bacterial cell membrane in each catalytic cycle.58 We will examine other modes for physiologic coupling of carboxylases and decarboxylases in Sections 7.1 and 7.2.
3.11 An ATP-dependent decarboxylase: phosphomevalonyl-1-PP decarboxylase (olefin-forming)
A rare example of ATP-dependent hydrolysis to ADP and Pi, coupled to decarboxyation occurs early in the biosynthesis of steroids and other isoprenoids (e.g. farnesyl-, geranyl-, geranylgeranyl lipids). The enzyme mevalonate-PP decarboxylase acts also as a kinase at the 3-OH of mevalonate-1-pyrophosphate61,62 (Fig. 19A). The 3-phospho-1-pyrophosphomevalonate does not get released from the active site. Instead this enzyme-bound nascent product undergoes decarboxylation of the C-6-carboxylate. Loss of CO2 is coupled to ejection of the newly derivatized-OH group, in the form of inorganic phosphate. That sets up a double bond in the product Δ3-isopentenyl diphosphate. The kinase reaction is cryptic in this formal elimination reaction of CO2 and –OH to create the new double bond.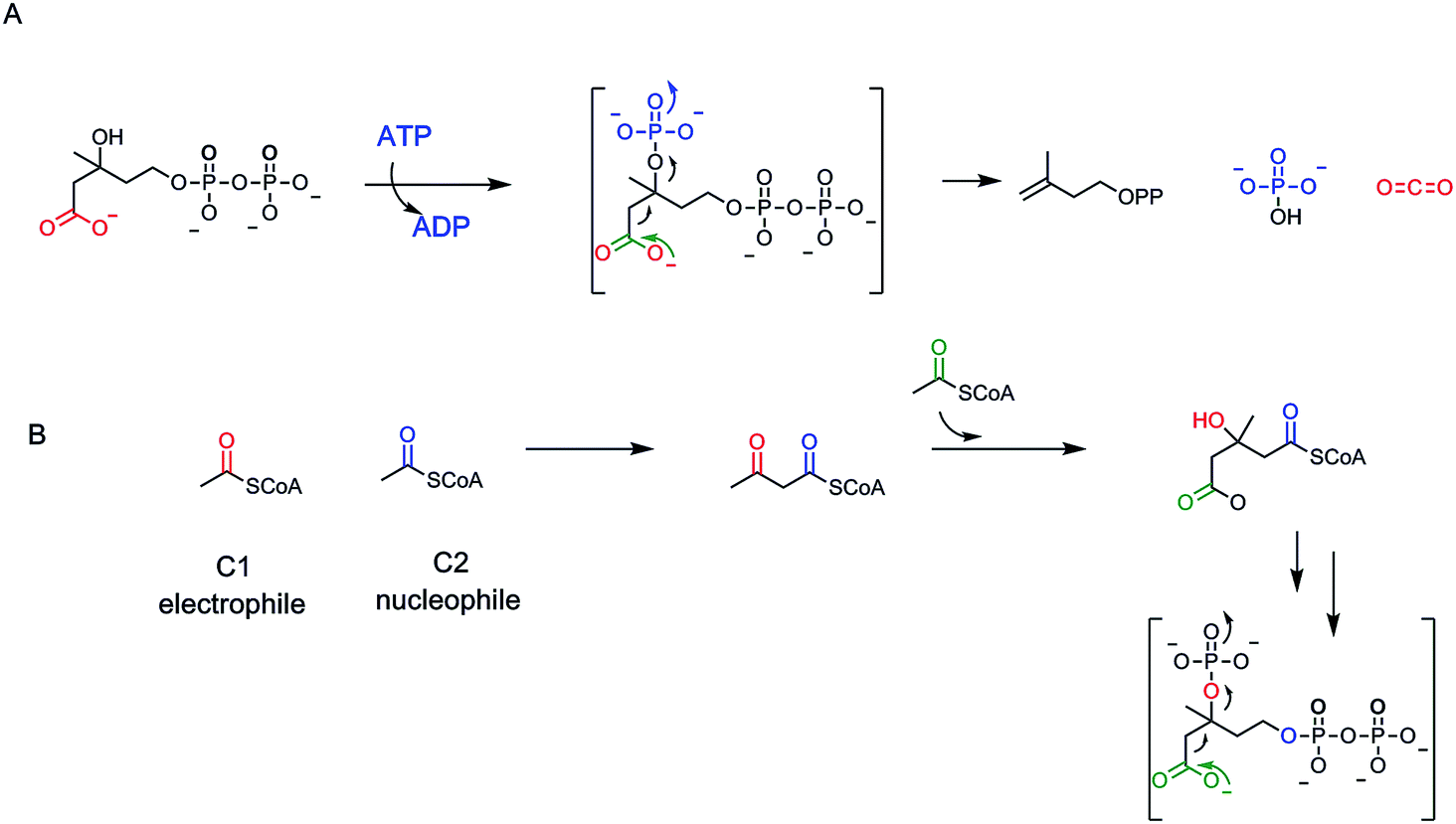 | ||
| Fig. 19 (A) Mevalonate-1-PP decarboxylase is an ATP-dependent kinase. The kinase action is cryptic as the 3-OPO32− nascent product undergoes decarboxylation and phosphate elimination before release, to yield the Δ3-isopentenyl diphosphate product in isoprenoid biosynthetic pathways; (B) Three molecules of acetyl CoA are condensed, two as nucelophile, one as electrophile, to set up the hydroxymethylglutaryl CoA branched scaffold that is carried forward to the isopentenyl diphosphate. | ||
A subsequent isomerase interconverts the Δ3 and the Δ2-olefinic isomers63 (Fig. 19). Those are the two five carbon isoprene building blocks for all ∼55000 known isoprenoid/steroid natural product scaffolds.11 The cryptic kinase action on mevalonyl-1-PP converts the 3-OH to the 3-OPO32− ester and thereby creates a low energy path for both phosphate elimination and decarboxylation. Elimination of –OPO32− is a much lower energy pathway than elimination of OH−. By pKa values, the second pKa for phosphoric acid is at pH 6 while pKa for water is at pH 14, for a difference of ∼8 powers of 10, facilitating C3–OX bond cleavage. As CO2 forms from the C-6–COO− and the pair of electrons flow into the adjacent C-5, they serve as neighboring group to facilitate cleavage of the C–O–PO3 bond (Fig. 19). One might expect this to be a more common route of dehydrative decarboxylation, albeit it expends an ATP in each catalytic cycle and so is energetically costly. Presumably, the gain of the isoprene building blocks are worth the energy expenditure to cells and organisms.
Let's look at how the 5-P-mevalonate framework got constructed. In the essence of simplicity the isoprenoid/steroid pathway begins with back to back condensations of three acetyl CoA molecules63 (Fig. 19B). The first enzyme uses the orthogonal reactivity of the acetyl group of this ubiquitous acyl thioester metabolite: C-1 as electrophilic carbonyl, C-2 as stabilized carbanion (via the thioester enolate contributor) to produce CoASH and acetoacetyl CoA, with the new bond between C-2 and C-3 of the four carbon product. The second enzyme in the pathway, hydroxymethylglutaryl CoA synthase, takes a third molecule of acetyl CoA and uses it as C-2 nucleophile to add in to the C-3-ketone of acetoacetyl CoA. Loss of CoASH from that tetrahedral adduct drives the C–C bond formation and has created a branched chain six carbon 3-hydroxy-dicarboxylate, one carboxylate still activated as the CoA thioester.14
This is hydroxymethylglutaryl CoA and has the branched framework that is harbinger to the isoprenyl diphosphate scaffolds. The third enzyme does a double reduction of the thioester carbonyl to intermediate aldehyde and then alcohol. Now three ATPs are spent.63 The first two ATPs consecutively phosphorylate the C-1-alcohol to the phosphate and then the pyrophosphate (this pyrophosphate grouping is the key to facile allylic carbocation formation11 later on, from Δ2-IPP in the isoprenoid chain elongation pathways). The third ATP is the one that cryptically phosphorylates the C-3-OH to set it up as a good leaving group on concerted decarboxylation of the C-6- carboxylate. The pendant C-5,6–CH2–COO− is converted to the terminal CH3 as the double bond forms between C-3 and C-4. The six enzyme sequence is an elegantly efficient route to convert three acetyl groups from acetyl CoA to the branched chain olefinic alcohol-PP.63
3.12 Zinc in 2,6-dihydroxybenzoate decarboxylation
Many bacteria can chew up aryl acids, with several examples of substituted benzoic acids undergoing enzymatic decarboxylation.64–66 While some enzymes are cofactor-free as noted below in Section 5, there are enzymes that contain a tightly coordinated zinc ion in the active site, as in 2,6-dihydroxybenzoate decarboxylase. The zinc2+ participates in decarboxylation by coordinating the phenolic C-2–OH. One can write participation by the minor cyclohexadienone tautomer in providing a transient electron sink beta to the carboxylate, facilitating the C-C- bond cleavage step (Fig. 20).66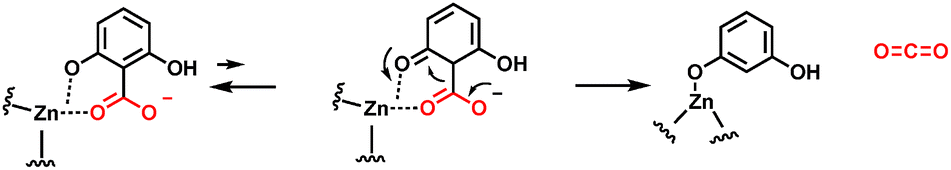 | ||
| Fig. 20 Zinc-dependent 2,6-dihydroxybenzoate decarboxylase. The zinc divalent cation serves as a super acid and chelator, enabling cyclohexadienyl tautomer formation as an intermediate. | ||
4. Homolytic C–CO bond cleavage routes
4.1 Radical SAM decarboxylases
All the classes of cofactor-dependent decraboxylases noted in the preceding sections, differing as they are in cofactor requirements and electron sink generations to lower C–COO− cleavage energies, use some form of heterolytic C–C bond cleavage routes. As the developing CO double bond in incipient CO2 forms, the pair of electrons in the remaining carbon center, place excess electron density on the Cα carbon in the transition state. The thiamin-PP- and PLP-requiring decarboxylases are stabilizing carbanionic transition states, while the NAD+-dependent enzymes may be stabilizing hydride ion transition states.
In principle, one can also imagine homolytic cleavage instead at the C–COO carbon–carbon bond, generating a transient pair of radicals (see Fig. 4). Recent evidence suggests that two distinct types of enzymes can indeed generate radical-based intermediates during decarboxylation of specific substrates.67,68 The best studied may be the HemN enzyme69 in the biosynthetic pathway to tetrapyrrole macrocycles, most notably heme, where FeII has been inserted in the equatorial plane of the conjugated protoporphyrin IX macrocycle (Fig. 21).
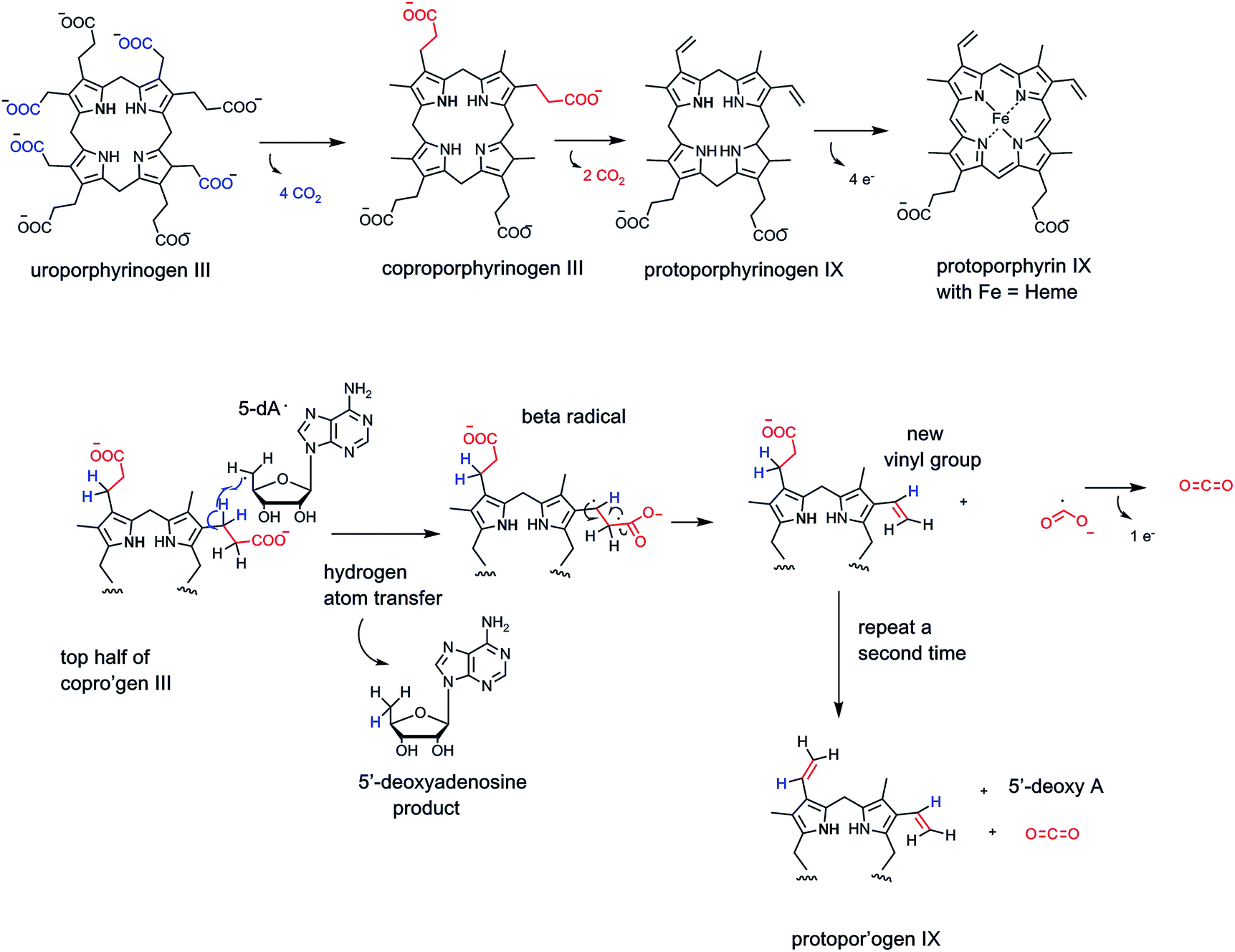 | ||
| Fig. 21 Uroporphyrinogen III conversion to protoporphyrin IX and heme involves loss of six carboxylates as CO2. The oxidative decarboxylation of the two propionate side chains to vinyl groups is catalyzed by coproporphyrinogen oxidase, an iron/sulfur cluster, radical S-adenosylmethionine (radical SAM) enzyme. The pathway involves homolytic cleavage of the C–COO with the 5′-deoxyadenosyl radical as initiator of substrate-derived one electron reaction manifold. | ||
Late in that pathway the tetracarboxy tetrapyrrolic intermediate coproporphyrinogen undergoes enzymatic decarboxylation of two of the four propionyl side chains to yield CO2 and two carbon vinyl side chains-clearly an oxidative decarboxylation process.70
The anaerobic bacterial route is mediated by Hem N which is a member of the superfamily of radical SAM enzymes.67 There are over 100000 bioinformatically predicted members of this superfamily.70,71 All radical SAM enzymes are predicted to contain a 4Fe/4S cubane type cluster that has the special property that while three of the irons have prototypic side chain cysteinyl residue thiol groups, the fourth iron atom of the cubane is bis-coordinated by the amine and carboxylate of substrate S-adenosylmethionine (SAM).72
On one electron input to the Fe/S cluster, homolysis of the C-5′–S bond of the coordinated SAM occurs, yielding metal cubane-liganded methionine and the 5′-deoxyadenosyl radical (dA˙) (Fig. 21). This carbon-centered radical is robust enough to abstract a hydrogen atom (H˙) from neighboring unactivated carbon atoms, e.g. on the propionyl side chain of cosubstrate coproporphyrinogen. Regiospecific H˙ abstraction from the C-3 methylene carbon of one of the propionyl chains would yield the carbon radical. Next, if the C-2–C-1–OO bond fragments homolytically, the CO2˙ radical would form transiently along with the C–CH2˙ radical. The two odd electrons at C-2 and C-3 go on to form the vinyl double bond. The CO2˙ would have to give up one more electron, either back to the one electron-oxidized Fe/S cubane cluster or to an external electron acceptor. Repetition at the second propionyl side chain would give the dicarboxy-, divinyl-tetrapyrrole product protoporphyrinogen IX.
Note in this proposed reaction manifold, the cosubstrate SAM is cleaved to 5′-deoxyadenosine (dA-H) and methionine in every turnover.11,72
4.2 Glycyl radical-containing indole acetate decarboxylase
SAM radical enzymes are not the only category of enzymes that carry out homolytic catalysis on their specific substrates. A small family of enzymes, working in anaerobic conditions, contain glycyl radicals in their activated forms68 (Fig. 22A). Typically there are partner activases that convert inactive precursor forms to the glycyl residue radical in the protected active site microenvironment.73 As in the dA˙-generating enzyme category above, the glycyl radical is a sufficiently robust oxidant to abstract an H˙ atom from a carbon site on a bound substrate, or preferably to generate an enzyme thiol radical (–S˙), not shown, that then acts on bound substrates.68 In the case of indole acetate, abstraction of an electron˙ from the C-2 acetate side chain carboxyl oxygen, transiently quenches the glycyl side chain radical and generates the carboxy radical anion on the acetyl side chain74 (Fig. 22A). Next, homolysis of the C-2–COO bond would yield CO2 and the CH2˙ radical form of skatole, transfer of a hydrogen atom equivalent back from the active site glycine residue, via cys radical side chain participation, regenerates the active radical form of the enzyme and produces the methyl group of the skatole product. We will compare this enzyme-glycyl radical route for conversion of an acetate side chain to CO2 and a terminal methyl group with a cofactor-independent route in uroporphyrinogen decarboxylase below. Indoleacetate acts as a hormone (an auxin) in plant metabolism so control of its decarboxylation is part of programmed physiologic responses in plants.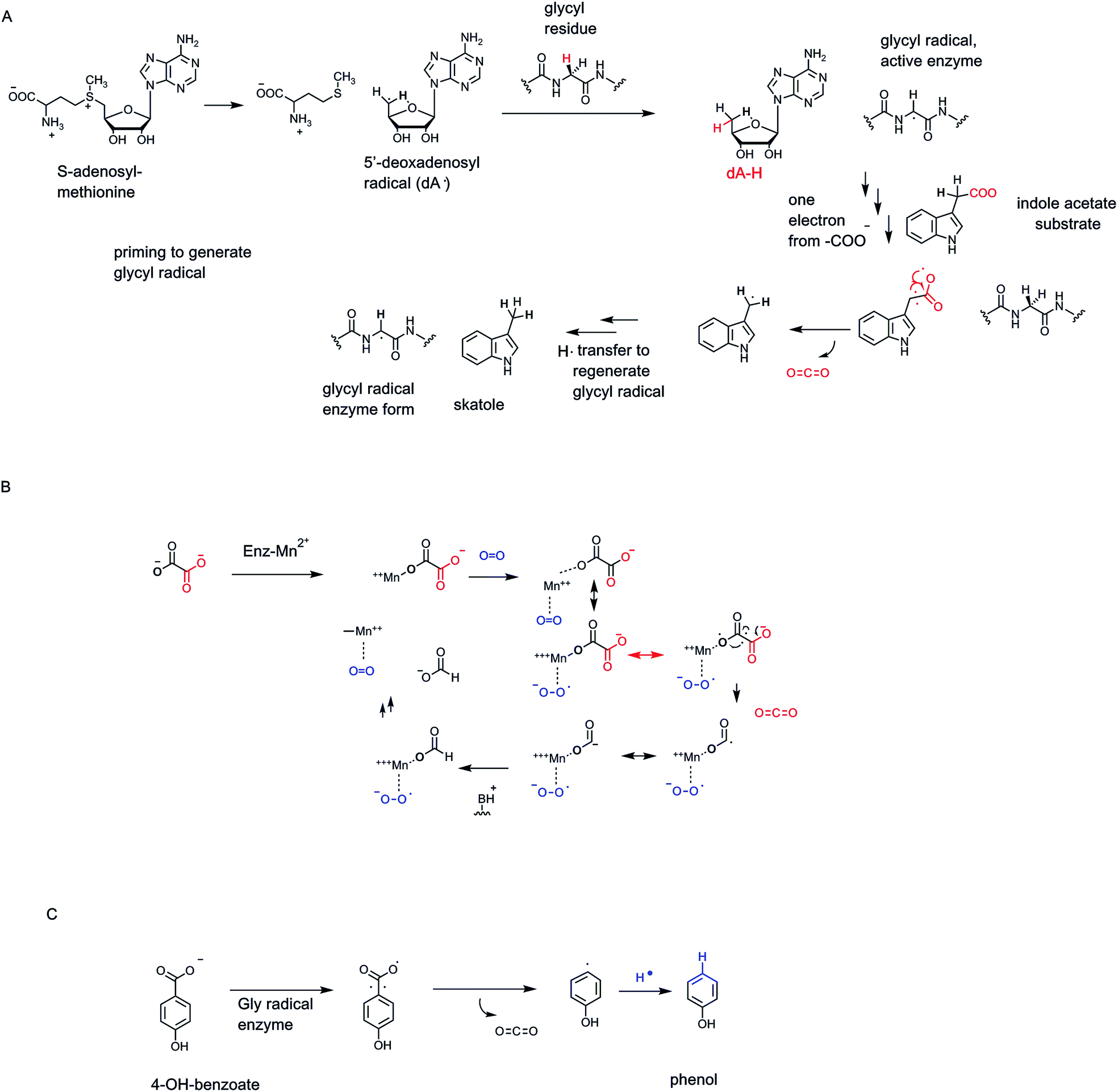 | ||
| Fig. 22 (A) Tandem action of 5′-deoxyadenosyl radical and enzyme-glycyl residue radical in catalysis of decarboxylation of indole-3-acetate to skatole. (B) A proposed radical mechanism for oxalate decarboxylation by a bacterial MnII and O2-requiring oxalate decarboxylase. (C) 4-Hydroxybenzoate decarboxylase by radical path. | ||
4.3 Mangano-oxalate decarboxylase
Bacteria such as Bacillus subtilis encode an oxalate decarboxylase that is distinct in cofactor-dependence, and reaction manifold19 from the oxalyl CoA decarboxylase,20 noted above as a thiamin-PP-dependent decarboxylation catalyst. One distinction between the thioester linkage in oxalyl CoA and the free carboxylate in dianionic oxalate is the electrophilicity of the two carbonyl carbons. Thioester carbonyls are more electrophilic, more “ketone-like” in electrophilicity compared to oxoester carbonyls, in large part due to the lessened overlap of sulfur molecular orbitals vs. oxygen orbitals in those carbonyl groups. Thus, the TPP thiazolium carbanion adds in to the oxalyl CoA thioester carbonyl much as it would add into the ketone of α-keto acids. Not so for the carboxylate anions of oxalate.Instead oxalate decarboxylase requires an O2 molecule catalytically, proposed to be a ligand to the MnII that is already coordinating an oxalate carboxylate in the resting enzyme state19 (Fig. 22B). Transfer of one electron to O2 from the manganese ion oxidizes it to MnIII and reduces O2 by one electron to superoxide anion. Back transfer of an electron from the coordinated oxalate carboxyl group regenerates MnII while generating a one electron oxidized oxalate. Homolytic fragmentation of the oxalate radical C–C bond releases CO2, while leaving one electron on the coordinated CO2. Electron transfer within the complex from MnII would cycle it yet back to MnIII and produce the coordinated formate anion. Protonation gives formate as the second product (along with the CO2) and leaves an MnIII-superoxide, which can transfer an electron one last time and return the enzyme to resting MnII and O2. Not surprisingly, this redox cycle can sometimes be derailed by irreversible transfer to O2.19 This enzyme has presumably evolved to use the one electron redox properties of manganese and the behavior of O2 as one electron acceptor to evolve a radical-based route to cleave oxalate to two one-carbon products at different oxidation states.
4.4 4-Hydroxyphenylacetate decarboxylase: radical convergences
Some bacteria can decarboxylate 4-hydroxyphenyl acetate to CO2 and 4-methylphenol (para-cresol).75,76 This is a case of a glycyl radical enzyme catalyzing homolytic cleavage of the CH2–COO side chain to a CH2˙ on the way to the final –CH3 side chain (Fig. 22C). The glycyl radical in the decarboxylase is, in turn, created by an activating enzyme that is a radical SAM protein.77 Thus, it is the 5 dA˙ in the activating enzyme that creates the radical form of the glycyl residue to activate the decarboxylase. This exemplifies nature's convergent strategy to try out reaction manifold combinations to effect substrate chemical transformations.5. Cofactor-independent decarboxylation logic
Nature's chemical inventiveness in fashioning decarboxylation catalysts that have evolved to fit the demands of specific substrates shows the broad sweep of molecular strategies to lower barriers for controlled decarboxylation in active site microenvironments.5.1 Dehydrative decarboxylations. Prephenate dehydratase/decarboxylase: aromatizing
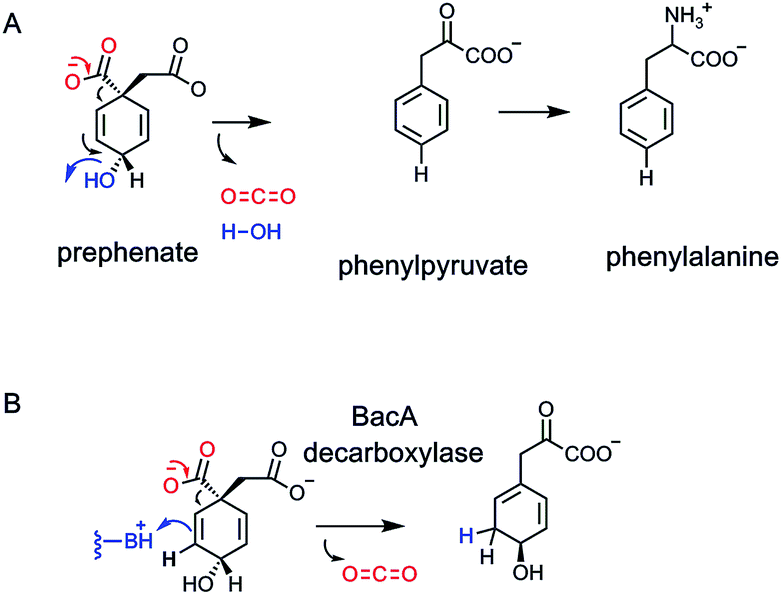
One example of catalytic logic is for enzymes to couple decarboxylation with net dehydration, loss of OH−, OH2. We have just discussed the variant of dehydrative decarboxylation in which an ATP is spent to modify an alcoholic group to a phosphate ester in isoprenoid biosynthesis.61,62 We look now at a coupled dehydrative decarboxyation that does not require ATP, but is presumed to be driven by gain in aromatization energy.This harks back to the metabolic fates of the cyclohexadiene dicarboxylate prephenate.55 We noted in the subsection on NAD+-dependent decarboxylases that action of prephenate dehydrogenase couples loss of CO2 to ejection of a hydride at C-4 of the aromatizing phenyl ring (Fig. 18). Thus, para-OH phenylpyruvate, the immediate precursor to tryptophan, is formed by dumping the two electrons released into the cyclohexadienyl ring, as the C-1–COO bond breaks. At C-4 the hydrogen substituent departs as a hydride to NAD+.
The second and parallel enzyme catalyzing aromatizing decarboxylation of prephenate, on the way to phenylalanine not tyrosine, is prephenate dehydratase78,79 (Fig. 23A). The same problem is surmounted. As CO2 forms and the C–COO bond breaks, electrons flow into the cyclohexadiene ring and out with departure of OH− (if protonated on the way out, the net release of HOH is a lower energy event than loss of OH−). Again, the name dehydratase focuses on the loss of water, but it is the aromatizing decarboxylation that drives the formation of phenylpyruvate, the phenylalanine precursor. Both the dehydrogenase and dehydratase outcomes are crucial for generating the pair of aromatic proteinogenic amino acids, phenylalanine and tyrosine, and all the myriad downstream aromatic metabolites from each.80
 | ||
| Fig. 23 Cofactor-independent decarboxylations of prephenate. (A) Prephenate dehydratase aromatizing action to phenylpyruvate in the phenylalanine biosynthetic pathway; (B) BacA in bacilysin biosynthetic pathway decarboxylates prephenate without aromatization. | ||
5.2 Nonaromatizing prephenate decarboxylation
For many years it had been assumed that loss of CO2 and ejection either of the C-4-hydride ion (dehydrogenase) or the C-4-OH (dehydratase outcome) might occur in a single concerted transition state. Recently, a third class of prephenate decarboxylase has been characterized, in the biosynthetic pathway to the antibiotic anticapsin and its dipeptide bacilysin.81–83 The first enzyme in that pathway is a non-aromatizing decarboxylase. CO2 is lost, one of the double bonds migrates and is protonated at the new olefinic terminus. The result is a decarboxylated, rearranged cyclohexadiene83 (Fig. 23B). Inter alia, it shows that enzymes decarboxylating prephenate do not need the gain in resonance energy as the driving force for CO2 ejection, and that a properly placed conjugate acid group in the enzyme active site can intercept the rearranging vinylogous cyclohexadienyl carbanion.5.3. Anthranilate decarboxylation in indole glycerol-3-P synthase
Tryptophan, one of the 20 proteinogenic amino acids, and also precursor to many indole-containing natural product frameworks, is an essential amino acid in the diet of humans. Daily intake amounts may be in the range of 250 mg per day for a 70 kg male adult.84 The microbial and plant biosynthetic pathway goes through anthranilate, aka ortho-aminobenzoate, in turn an amination, aromatization product from the nodal metabolite chorismate, precursor to prephenate, noted in above sections85 (Fig. 24A). Anthranilate is first converted to N-phosphoribosyl-anthranilate and then isomerized to the open chain isomer (Fig. 24B). This is the substrate for indoleglycerol-3-phosphate synthase80 which creates the pyrrole ring fused to the benzene ring of substrate as the bicyclic indole nucleus is created. A new C–C bond is formed between C-1 of the aminobenzene moiety and the C-2′ ketone of the ring-opened ribulose to give an iminium intermediate. Then decarboxylation from C-1 and loss of water from C2′ completes the aromatization of the indole ring.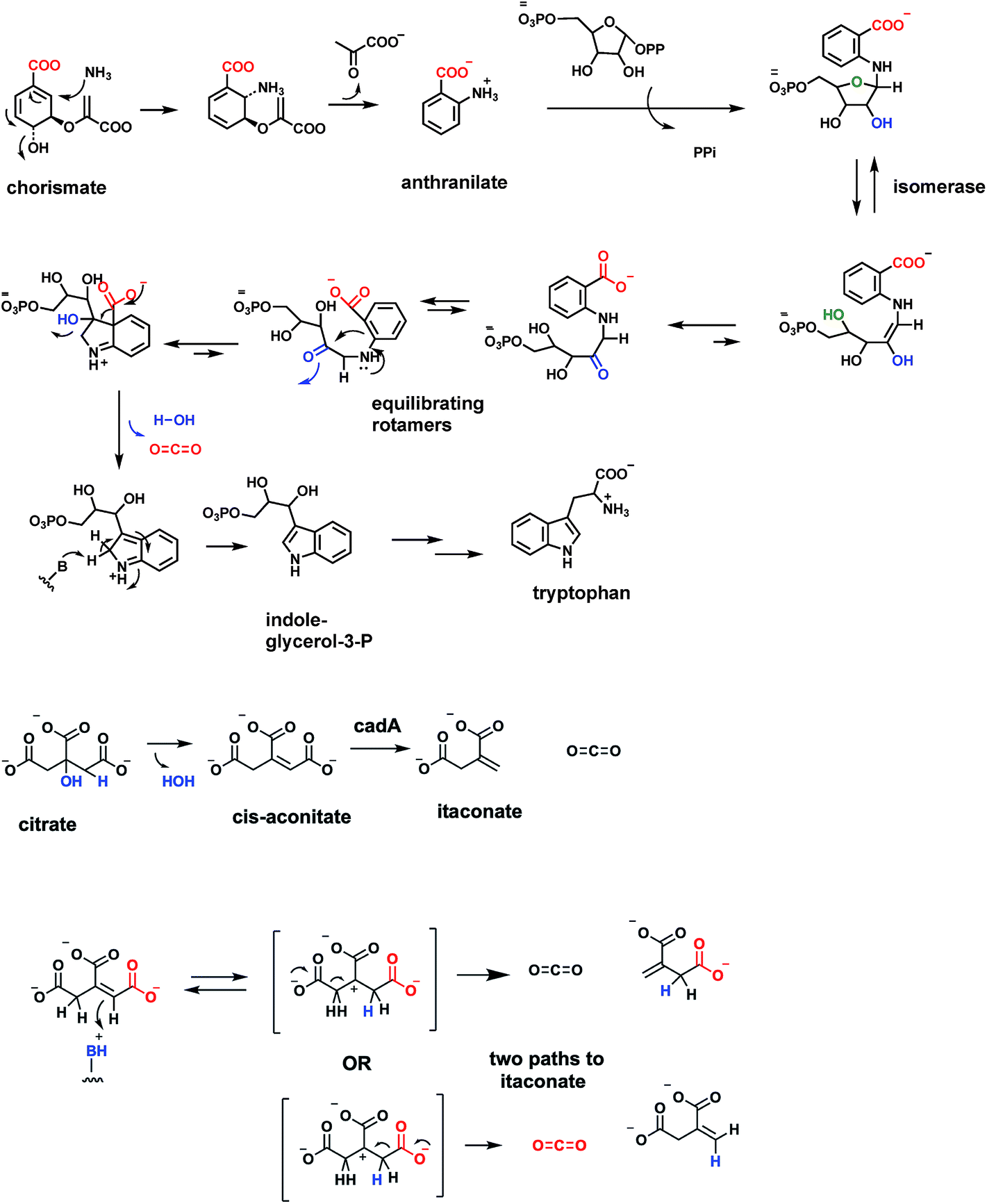 | ||
| Fig. 24 Anthranilate formation and decarboxylation in the pathway from chorismate to tryptophan. Decarboxylation of cis-aconitate by a proposed carbocationic transition state to the four carbon methylene diacid itaconate. Note the proposed cation has C2 symmetry. | ||
It is of interest to compare the underlying chemical logic for construction of the two pyrrole rings found in nodal metabolites: the porphobilinogen aminopyrrole building block and the fused pyrrole in the ubiquitous indole scaffold. Decarboxylations occur in each pathway. For porphobilinogen, the decarboxylation occurs in construction of the C–C bond in aminolevulinate that joins C-1 of succinyl CoA to C-2 of the glycine fragment (see Fig. 28 below). Then, dimerizing double dehydration leads to the aminopyrrole scaffold. In formation of the pyrrole ring of the bicyclic indole scaffold, it is dehydrative decarboxylation at the last step that aromatizes the five membered ring in the 6,5-bicyclic indole (Fig. 24).
5.4 Decarboxylation of cis-aconitate to itaconate
An unanticipated decarboxylation of cis-aconitate, the unsaturated tricarboxylate intermediate between citrate and isocitrate in the citrate/Krebs cycle, is catalyzed by the cofactor-free CadA cis-aconitate decarboxylase21,86 (Fig. 24B). The enzyme is found from humans to microbes. The reaction product is itaconic acid a five carbon diacid with a side chain methylene group. The bacterial enzymes have been well-characterized and are cofactor-free. The presumed mechanism is to initiate catalysis by protonation of the aconitate double bond (π electrons acting as nucleophile towards a proton), with regiospecificity of protonation to yield the most stable trisubstituted, tertiary carbocation as shown. If the proton transfer to the double bond is fully complete before decarboxylation, this intermediate/transition state has C2 symmetry and either carboxylate could be lost. If the enzyme activates a specific carboxylate for CO2 formation the enzyme may act regiospecifically.5.5 Cofactor-free decarboxylases operating at sp2 carbon centers
Many of the carboxy-containing metabolites listed in the above sections have the COO group connected to an sp3 (tetrahedral) carbon. The α-keto acids are an exception in that C-2 is an sp2 carbonyl. However, decarboxylation only proceeds after addition of the thiazolium C-2 carbanion of cofactor thiamin-PP, to form a tetrahedral sp3 carbon center.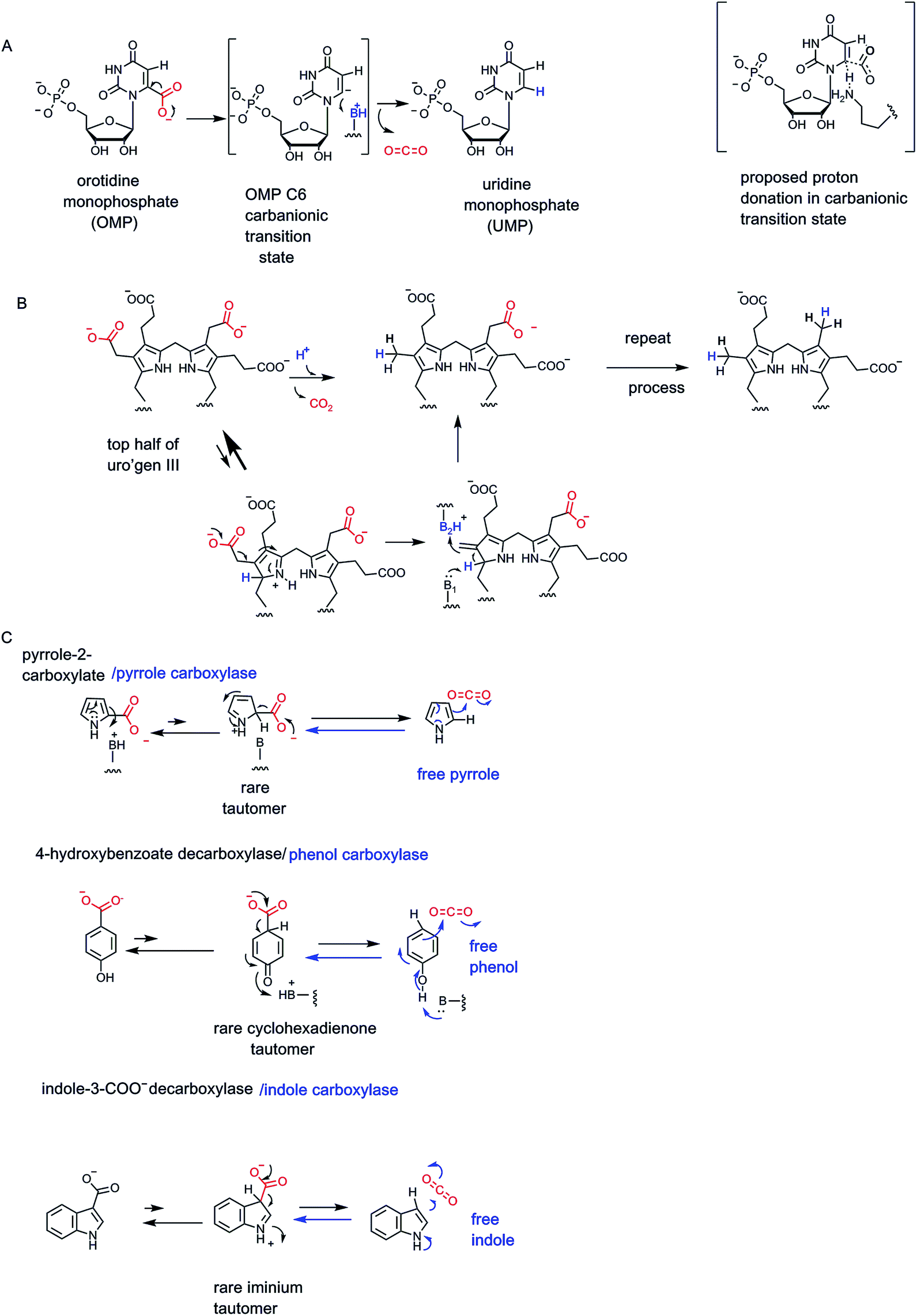 | ||
| Fig. 25 Cofactor-independent decarboxylation at sp2 centers. (A) Orotidine monophosphate decarboxylase generates UMP in pyrimidine biosynthesis. (B) Uroporphyrinogen III decarboxylase converts the four acetate side chains into methyl groups (first two are shown). (C) Reversible decarboxylation/carboxylation by bacterial enzymes acting on pyrrole-2-carboxylate, 4-hydroxybenzoate, indole-3-carboxylate. | ||
The chemical problem to solve is that these –CH2–COO side chains look to be unactivated. Indeed the enzyme is definitely on the slow side, with only about 10 turnover events per minute.88 On the other hand, as indication of the chemical stability of those acetate side chains, the nonenzymatic rate for their decarboxylation is estimated to give a half-life of 3.3 billion years. Thus, by catalytic proficiency measures, the uroporphyrinogen decarboxylase is one of the best enzymes that has evolved in contemporary organisms,88 although the evolutionary path to such proficiency is obscure.
It is presumed that in the active site, there may be an enzyme-assisted increase in the fraction of pyrrole ring tautomer(s) that have a proton at C-2 and a cationic nitrogen89 (Fig. 25B). In that structural contributor, the isopyrrolic nitrogen is a vinylogous electron sink for stabilizing the C-2-carbanion that would arise as decarboxylation proceeds. The corresponding azafulvene nascent product isomer would isomerize to the more stable methyl pyrrole.
5.6 Aromatic and heteroaromatic decarboxylases
Microbial degradative metabolism of aromatic and heteroaromatic molecules is replete with decarboxylases to work on such substrates as hydroxy-benzoate isomers, indole 3-carboxylate, and pyrrole-2-carboxylate.65,90 Following up the discussion above on uroporphyrinogen III decarboxylase, that removes CO2 from each of the acetate side chains at the 3-position of each of the four pyrrole rings, is the report of pyrrole-2-carboxylate decarboxylase from Bacillus megaterium.91 The purified enzyme shows activity in a reducing, anaerobic atmosphere and likely proceeds by a similar rare isopyrrole tautomer (Fig. 25C). Intriguingly, the reaction reaches an equilibrium, suggesting the free pyrrole can be carboxylated in the back direction. This surprising carboxylase activity was validated in initial rate studies in the back direction91 (blue arrow in Fig. 25C). This is a measure of the chemical competency of C-2 of the pyrrole ring as a nucleophile.Analogously, a variety of microbial hydroxybenzoates on purification are likewise cofactor-independent. More intriguingly, the phenol products will act as carboxylation substrates as shown for phenol to 4-hydroxybenzoate64,90 in Fig. 25D. The rare cyclohexadienone tautomer is invoked as proximate intermediate for front direction decarboxylation and also nascent product in the carboxylation back direction. A third example, arguing for generality of mechanism, is shown for indole-3-carboxylate decarboxylase.22 The pyrrole ring of the bicyclic indole is known to have nucleophilic character at C-3. This can be manifested as protonation prior to decarboxylation in the front direction and as nucleophile in the back, carboxylation direction.
Microbiologists studying degradation of electron rich aromatic hydrocarbons such as pyrrole, phenol, and indole, among others, have argued that the bacteria may actually use the carboxylase activities physiologically to add a water-soluble carboxylate to the insoluble hydrocarbon framework.64 Aromatic/heteroaromatic ring oxygenases then follow to set up the resultant catecholic acids for ring-cleaving dioxygenases to return the aliphatic fragments to primary metabolic pools.
6. Pathway perspectives: glucose, fatty acids, pyrimidine, steroids, heme
6.1 Decarboxylation of glucose: glycolysis and the citrate cycle
The classic strategy for energy production in the catabolic (degradative) arms of metabolism is the complete oxidation of the ur-metabolite glucose into 6 CO2 molecules (Fig. 26). Twenty four electrons are removed from glucose, in twelve pairs, as each carbon is oxidized to CO2. Inspection of the CO2-generating steps indicates that all occur in the mitochondria, are oxidative in nature, and the two electrons removed are captured in NADH. The eight enzymes of glycolysis, taking glucose to two identical molecules of the three-carbon pyruvate involve zero decarboxylations and removal of only four of the 24 electrons from the glucose scaffold. The heavy lifting occurs in eukaryotic mitochondria.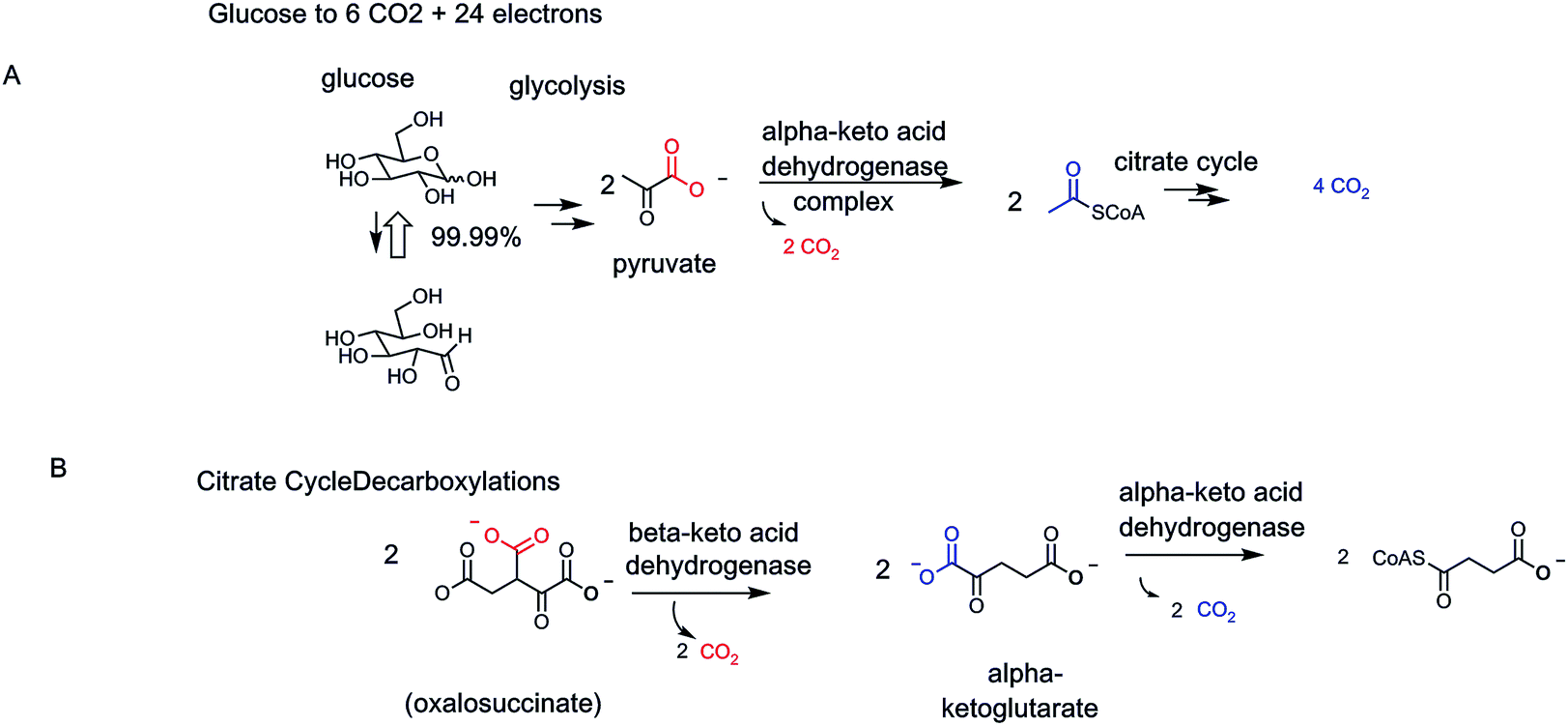 | ||
| Fig. 26 Decarboxylation in catabolic, energy-generating metabolic pathways. (A) Glucose is completely converted to 6 CO2 by three oxidative decarboxylases working on each of two molecules of pyruvate within mitochondria. (B) Two of the enzymes are three component α-keto acid decarboxylases while the third is the β-keto acid dehydrogenase/decarboxylase. The tandem action of isocitrate dehydrogenase and α-ketoglutarate dehydrogenase in the citrate cycle carry out a β-keto acid then an α-keto acid decarboxylation to take the six carbon isocitrate to the four carbon succinyl CoA. The two carbons released are two CO2 product molecules and the four electrons are captured as two NADH molecules. | ||
Three enzymes each perform oxidative decarboxylations: two on α-keto acid, one on a β-keto acid. The pyruvate dehydrogenase complex mediates the transition from glycolysis to citrate cycle. Consistent with the α-keto acid structure of pyruvate, the thiamin-PP coenzyme is required. Likewise, consistent with the oxidative outcome, the lipoamide- and FAD-containing pair of cocatalysts are part of the PDH complex.
This logic is mirrored in the second α-keto acid dehydrogenase–decarboxylase complex, the α-ketoglutarate-processing dehydrogenase of the citrate cycle. The third CO2-producing enzyme is the β-OH acid-oxidizing isocitrate dehydrogenase. The nascent oxalosuccinate product is decarboxylated in situ to yield the above-mentioned α-ketoglutarate (previous Fig. 13 and 14).
Since glucose yields two molecules of pyruvate, as two molecules of pyruvate undergo full decarboxylation by these three oxidative decarboxylases, 6 CO2 molecules have been produced. Glucose metabolism leaves nothing to waste. Also note that only two types of decarboxylases, for two α-keto acids and one β-keto acid, are employed to take all carbons of glucose to CO2
6.2 Palmitoyl-CoA to 16 CO2
Saturated long chain fatty acids are the preferred storage form of electrons in cells. Because the carbons are reduced to CH2 groups, six electrons are available for ultimate transfer to NAD+ or FAD on their oxidation to CO2. The prevalent sixteen carbon fatty acid palmitate is converted to palmitoyl CoA and then subjected to the enzymes of fatty acid degradation, cleaving off acetyl CoA units during the sequential beta oxidation cycles, eventually producing 8 acetyl-CoAs in the mitochondria (Fig. 27). These enter the citrate cycle, undergo the sequential β-, then α-decarboxylations by the tandem actions of isocitrate dehydrogenase and α-ketoglutarate dehydrogenase just noted above.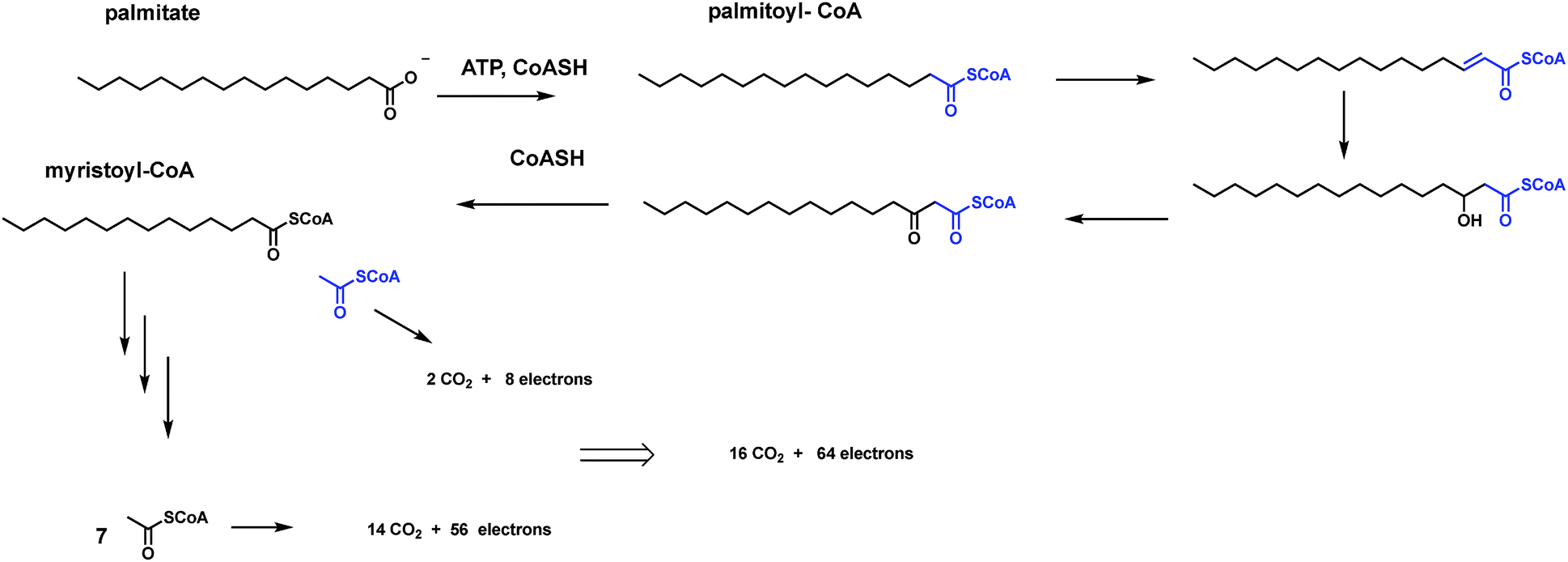 | ||
| Fig. 27 Beta oxidation of fatty acids. The strategy for oxidizing all sixteen carbons in palmitate is to release them two at a time as acetyl CoA fragments. Then each of the 8 acetyl CoAs are fed into the citrate cycle for tandem action of isocitrate dehydrogenase and α-ketoglutarate dehydrogenase to produce 2 CO2 in each of eight turns of the cycle. Thus, both carbohydrates and fatty acids are ultimately and economically decarboxylated by the same two core enzymes of the citrate cycle. | ||
This is the simple route to 16 CO2 molecules and 64 electrons removed from each molecule of palmitoyl CoA utilized as energy source. Again, only two types of decarboxylation machinery are needed: isocitrate dehydrogenase and 2-ketoglutarate dehydrogenase in their oxidative decarboxylation modes. The isothermal burning of glucose and of fatty acids to CO2 represent the major catabolic routes for energy generation in organisms. We next note three biosynthetic pathways where CO2 molecules are generated along the way, but not for the main purpose of energy generation.
6.3 Decarboxylation in steroid biogenesis: loss of 8 carbons as CO2
In biosynthetic pathways carbons are not oxidized to CO2 for energy generation. Instead, carboxylates may be used as functional groups whose ejection as CO2 may drive irreversible processes and/or bias equilibria in favor of the decarboxy product accumulation. The early steps to isoprenoids and steroids use three acetyl CoA molecules to first create a branched six carbon scaffold. Then decarboxylation irreversibly provides the branched five carbon framework AND installs the olefin essential to all subsequent prenyl chain elongation reactions.If one scans all the way to the steroid biosynthetic end product cholesterol, that molecules has a C27 scaffold (Fig. 15). Six molecules of isopentenyl-PPs (6 × 5 = 30 carbons) are used to build that framework. In turn these came from six molecules of hydroxymethyl glutaryl CoA (6 × 6 = 36 carbons), and one step back from a total of 18 acetyl CoAs (18 × 2 = 36 carbons). Going from 36 starting carbons to 27 carbons in cholesterol shows that nine carbons were jettisoned along the way. Six of them are released as CO2 in the 5-P-mevalonate-1-PP decarboxylase just discussed.
We have noted in a section above that two of the other three are also lost as CO2, very late in the pathway, as the C-30 tetracyclic lanosterol is converted to C-27 cholesterol. The late pathway strategy is a variant of the NAD+-dependent β-hydroxy acid oxidation, then decarboxylation of the resultant keto acid (Fig. 15). From this carbon accounting perspective, cholesterol formation is accompanied by trimming eight carbon atoms from the scaffold as CO2. And the methyl at C-14 is ejected as formate.
6.4 Heme biosynthesis: jettison of 14 CO2
At three points in the eight enzyme conversion of eight succinyl CoA and eight glycine molecules to protoporphyrin IX, and on FeII insertion, to heme, decarboxylases are the catalysts. Three distinct decarboxylation strategies are employed, reflecting the substrate demands for the reaction manifolds employed (Fig. 28).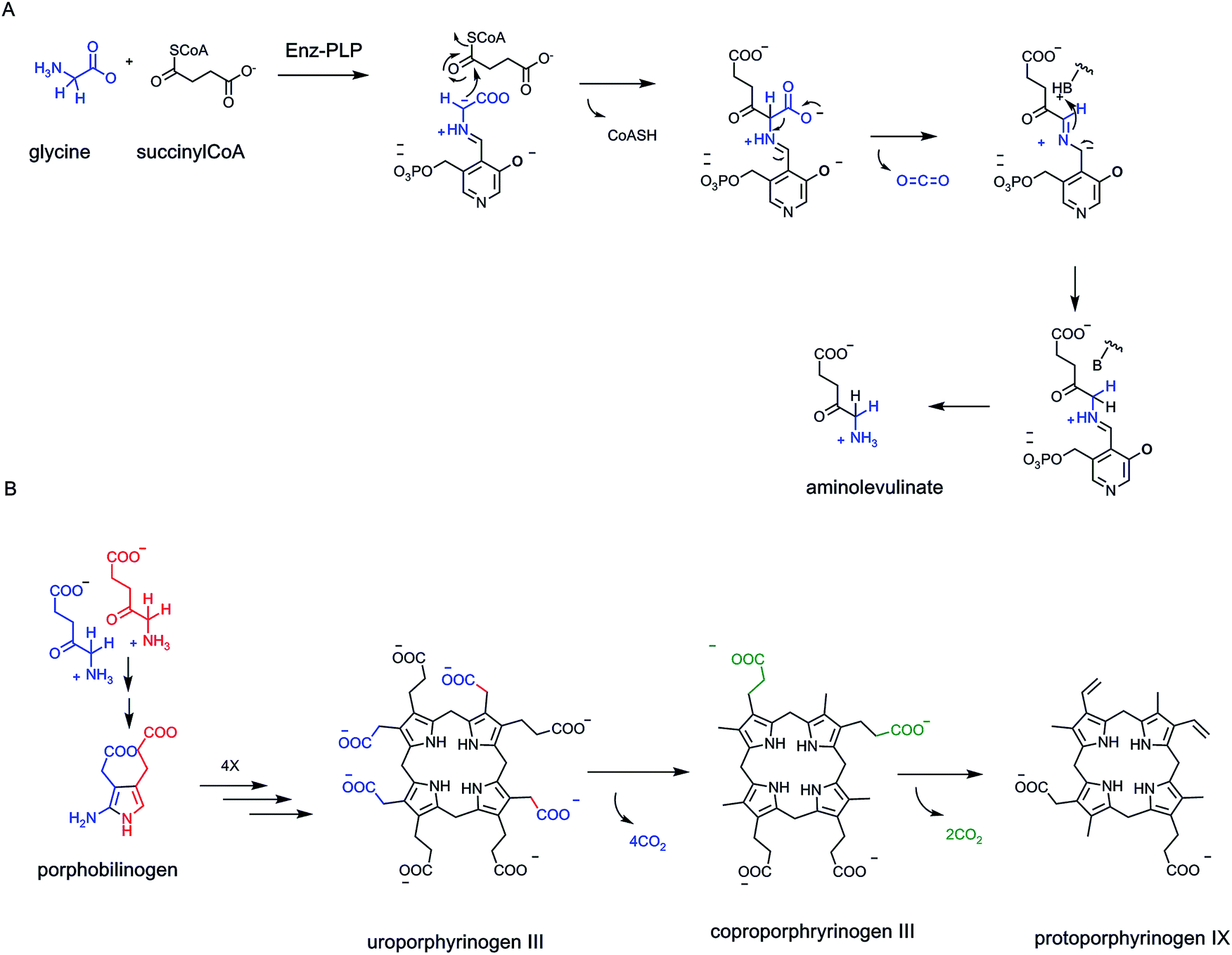 | ||
| Fig. 28 Three decarboxylations in the eight enzyme pathway to protoporphyrin IX release 14 CO2 product molecules. (A) Aminolevulinate synthase is a PLP enzyme that utilizes glycine for both C–C bond formation (Gly C-2 to succinyl CoA) and for C-1 decarboxylation. Eight glycines yield eight aminolevulinates. (B) Porphobilinogen synthase condenses two aminolevulinates so the eight aminolevulinates give the four porphobilinogen monomers needed to build the tetrapyrrolic framework. Uroporphyrinogen III is substrate for the cofactor-independent decarboxylase that removes four acetate side chain carboxylates as CO2. The third decarboxylase is the radical SAM-dependent coproporphyrinogen oxidase, converting two of the four propionyl side chains to vinyl groups of protoporphyrin IX, and accounting for the total of 14 CO2 trimmed away. | ||
In the first step, formation of the C–C bond between C-2 of glycine and C-1 of succinyl CoA, the PLP coenzyme in the active site of aminolevulinate synthase generates the requisite low energy carbanion on C-2 of glycine.92,93 Decarboxylation and hydrolysis of the CoA thioester may be steps that draw off the equilibrium in the forward direction. Eight glycine molecules are consumed and eight CO2 produced to get to the tetrapyrrole macrocycle of uroporphyrinogen III. This is an unusual variant of a PLP-dependent decarboxylase, coupling C–C bond formation to loss of CO2.
Another four molecules of CO2 are then released by the uroporphyrinogen III decarboxylase that converts the acetate side chain on each of the four pyrrole rings to the methyl substituent. This is a cofactor-free catalyst even though it operates at unactivated –CH2COO− side chains. We noted above that it has been hypothesized that the pyrroline C2 tautomeric form may enable formation of the C-2 pyrrole carbanion.
The third and last decarboxylase is the O2-requiring coproporphyrinogen oxidase, removing the carboxylate from two of the four propionate side chains and desaturating each of those two carbon substituents to vinyl groups. This is a radical SAM-Fe/S cluster enzyme72 and utilizes homolytic rather than heterolytic cleavage of the –CH2CH2–COO− on the way to the –CHCH2 products. The final tetracyclic scaffold of protoporphyrin IX retains four of the eight original glycine nitrogens and 34 of the 48 carbons from the starting metabolite pair. Only two of the fourteen decarboxylations are oxidative, reflecting that heme biosynthesis is not an energy-generating pathway. Rather, this is biosynthetic strategy for assembly of a molecule crucial for cell and organism physiology. The extrusions of the first eight carboxylates as CO2 build the key aminolevulinate frameworks. Loss of the next six carboxylates may be more about conversion of a highly charged polyanion with eight negative charges to a final tetrapyrrole scaffold closer to neutrality and with a hydrophobic/hydrophilic balance on the way to a membrane-soluble cofactor, e.g. in chlorophyll.
6.5 Pyrimidine and purine biosynthesis: one decarboxylation vs. one CO2 fixation
The heterocyclic pyrimidine and purine building blocks for RNA and DNA assembly are rich in nitrogen compared to most other primary metabolites and the biosynthetic pathways reflect reliance on nitrogen-containing building blocks. There is only one decarboxylation in pyrimidine assembly and none in the formation of the 6,5-fused bicyclic purines (Fig. 25).The single decarboxylase in pyrimidine maturation is orotidine monophosphate decarboxylase, converting OMP to UMP.88 The carboxylate at C-6 of OMP originated as the C-1–COO of aspartate (the C4 carboxylate of aspartate has become the sp2 C-4 of the orotate ring). Decarboxylation, as noted in the cofactor-independent decarboxylase section above, occurs at an sp2 trigonal carbon with no obvious mechanism to lower the energy of a carbanionic transition state. Despite the apparent mechanistic difficulty, this enzyme presumably evolved early on and has a high catalytic efficiency of 7 × 1016 over the extremely sluggish nonenzymatic OMP decarboxylation.88
In the C5N4 bicyclic purine scaffold, the sources of the five carbons are: both carbons of glycine, two molecules of formate (at C-2 and C-8) from N-10-CHO-tetrahydrofolate, and one molecule of free CO2. Consistent, perhaps, with an early evolution, three of the five carbons in purines are from one-carbon units. The CO2 fixation itself is a rare biologic carboxylation without any cofactors, reflecting cyclizing capture of a possible nonenzymatically formed N-carboxy adduct.
7. Tandem carboxylations and decarboxylations: to what purpose
This review has focused on the span and scope of decarboxylase enzymes as they go about their multiple tasks and recruit their particular coenzyme and cosubstrate forms to perform a broad range of decarboxyation chemistry. We have barely raised the complementary subject of the reverse reaction, substrate carboxylation, other than in the introductory remarks about how CO2 is actually the most precious building block of plant metabolism and life cycles. There are two cases in mammalian metabolism of tandem action of a carboxylase and then immediately of a decarboxylase, removing the just added CO2. One might wonder at apparent futile cycles logic. Instead, these tandem reactions show the use of CO2 both as a temporary protecting group and its elimination as a driving force on accumulation of the co-product for a key physiologic process.7.1 Acetyl CoA carboxylase and β-keto acyl synthase
The first tandem pair of carboxylase/decarboxylase (Fig. 29A and B) occur in the first two steps of fatty acid biosynthesis and, in a mechanical analogy, provide the power stroke for building up the elongating fatty acid carbon chains. The carboxylase is the biotin-dependent acetyl CoA carboxylase (Fig. 29A). It cleaves an ATP substrate molecule to activate CO2 (actually its hydrated HCO3− form) to accumulate enough of the mixed carbonic-phosphoric anhydride for it to accumulate in an enzyme subsite and be captured by N1 of the tethered biotin group. The N1-carboxybiotin diffuses to a second subsite on the enzyme and the COO− transferred to the C-2 carbanion/thioester enolate of acetyl CoA. Malonyl CoA, the three carbon half thioester of the dicarboxy acid malonate, is the released product, and is then the major building block for fatty acids.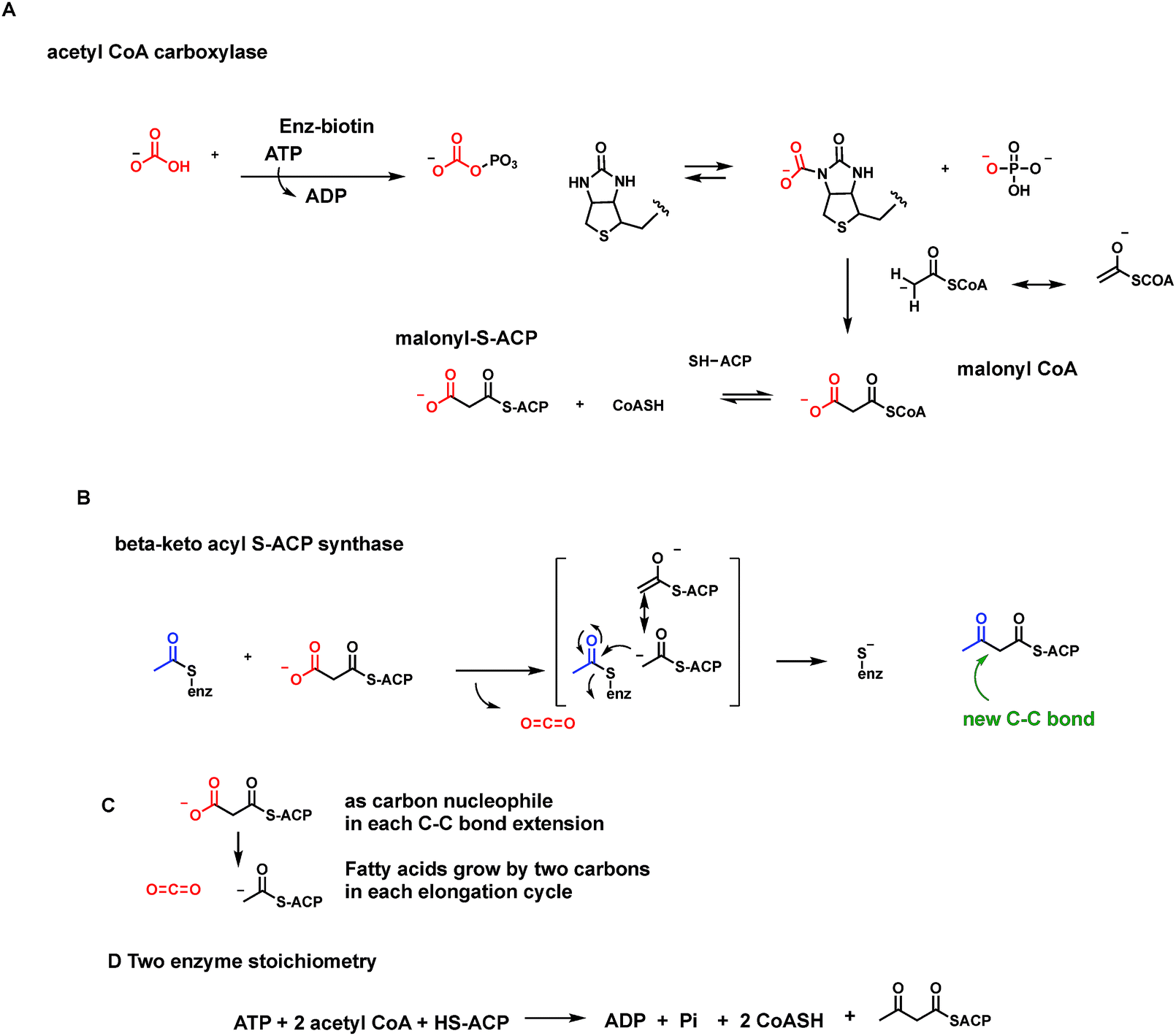 | ||
| Fig. 29 Tandem action of a carboxylase/decarboxylase pair to drive fatty acid biosynthesis. (A) The first committed step in fatty acid biosynthesis is executed by the ATP- and biotin-dependent acetyl CoA carboxylase, with C–C bond formation between N-carboxybiotinyl enzyme and the enolate-stabilized carbanion of pyruvate. (B) The next enzyme is the C–C bond-forming catalyst in fatty acid chain elongations, the β-ketoacyl-ACP synthase. (C) It decarboxylates malonyl-S-ACP only when the partner electrophilic acetyl-S-enzyme covalent intermediate is presented, building the elongating chain by two carbons as a nascent β-ketoacyl-ACP adduct. (D) In the two enzyme stoichiometry, the CO2 drops out of the equation, as it is used catalytically, a transient activating group strategy. | ||
The tandem enzyme, the first component in the fatty acid synthase array, is a β-keto acyl thioester synthase94 (Fig. 29B). This is the crucial C–C bond-forming enzyme for all fatty acid elongations, two carbons at a time. The soluble CoA group is substituted for, in an energy neutral thioester exchange, by the thiol of a 10 kDa acyl carrier protein (ACP). The malonyl-S–ACP reacts with an acetyl-S-enzyme form of the synthase. Decarboxylation of the malonyl CoA (via the stabilized thioester enolate) yields the C-2 carbanion to attack the acetyl-S-enzyme C-1 carbonyl. That acetyl group is transferred to C-2 of the decarboxylated malonyl-S-ACP. The C–C bond-forming step constitutes a two carbon chain elongation step and yields a β-ketoacyl four carbon chain in thioester linkage to the ACP protein. The cycle of malonyl-S decarboxylation to the acetyl C-2 carbanion as carbon nucleophile is iterated up to seven and eight cycles to generate sixteen and eighteen carbon fatty acyl chains. A three-carbon malonyl thioester is the addend in each cycle but is decarboxylated to release CO2 to form the carbon nucleophile in the condensation so that the three-carbon input results in a two-carbon elongation (Fig. 29C and D).
Considering the two enzymes together, what has happened. Cells have spent an ATP to ADP and Pi, for a 105 boost in equilibrium for making malonyl CoA out of CO2 and acetyl CoA. Then, the three-carbon malonyl CoA is decarboxylated in every fatty acid chain elongation reaction, releasing back the CO2. From a thermodynamic point of view, ATP is cleaved to influence the equilibria of each C–C bond-forming step in fatty acid chain elongation. Fatty acids are the most efficient form of energy storage in cells so this is a key strategy for lean times to come.
From a mechanistic point of view, one uses the C-2 carbanion form of acetyl CoA/acetyl-S-ACP in both the first and the second enzymes in tandem. It may be that the C-2 carbanion of free acetyl CoA is too unstable a species to diffuse from one enzyme active site to the other. It may be more efficient to decarboxylate the malonyl thioester on demand: only when the growing acyl chain is already bound and in place to serve as electrophile in the C–C bond-forming step in the keto acyl-ACP synthase active site.
Finally, from the point of view of CO2: it disappears from the two enzyme stoichiometry (Fig. 29D). It has been catalytic not stoichiometric, a transient activating group, installed at the expense of ATP cleavage, to create a carbanion when needed for fatty acid chain elongations. Every chain elongation in fatty acid biosynthesis is a decarboxylation-thioclaisen condensation.94 Every one of them consumes an ATP in the first of the two tandem enzyme steps.
7.2 Pyruvate carboxylase and PEP carboxykinase
The second tandem carboxylase/decarboxylase pair acts to convert pyruvate to its trapped, phosphorylated enol phosphoenolpyruvate, PEP (Fig. 30). PEP can then be used to run glycolysis in the back direction to produce glucose-6-P in the endoplasmic reticulum. Cleavage of the G-6-P phosphoester yields free glucose at high concentrations to be exported to the blood. This is the diurnal pathway of gluconeogenesis undertaken by liver cells during nocturnal fasting to maintain glucose homeostasis in the blood and other tissues. This is clearly a crucial physiologic mechanism. At a chemical level it depends on surmounting the energy barrier for converting pyruvate to the enolate in high percentage and trapping it by phosphorylation to generate the thermodynamically activated enol phosphate PEP.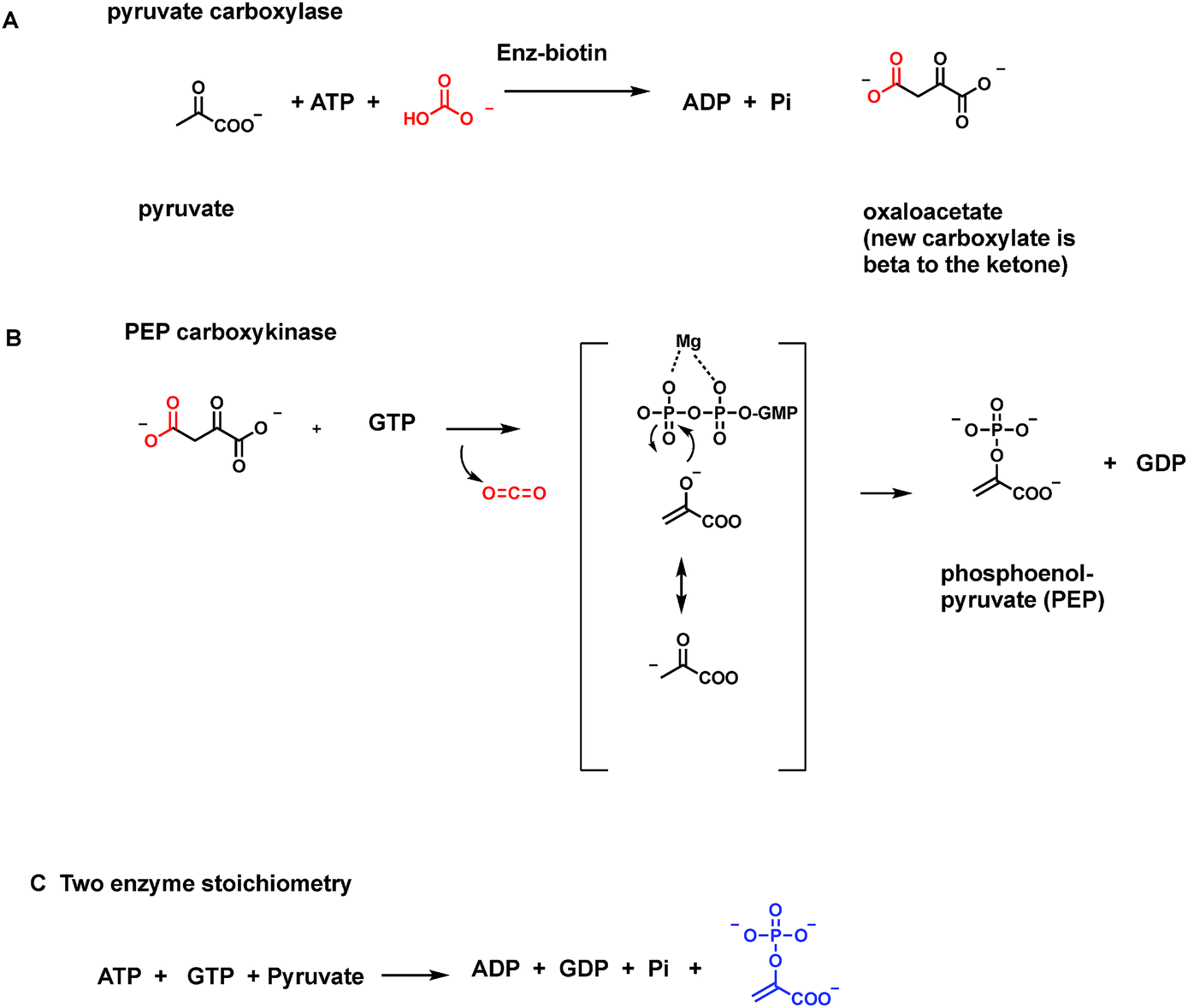 | ||
| Fig. 30 Tandem action of a carboxylase/decarboxylase pair to drive gluconeogenesis. Liver cells use a comparable two enzyme strategy to accumulate the high energy enol phosphate metabolite phosphoenolpyruvate (PEP) to drive glycolytic enzymes in the reverse direction. This is the process of gluconeogenesis and provides enough glucose for secretion into the blood during fasting to maintain adequate blood glucose to feed other tissues. (A) Again, an ATP- and biotin-dependent enzyme, pyruvate carboxylase, acts, to produce oxaloacetate by CO2 fixation. (B) The next enzyme PEP carboxykinase, uses GTP as the phosphoryl donor to the enol of pyruvate, generated from oxaloacetate decarboxylation. (C) CO2 does not show up in the two enzyme stoichiometry, which reflects the need to consume two high energy phosphoric anhydride bonds to drive accumulation of PEP. | ||
Hepatocytes and muscle cells use a variant of the tandem carboxylation–decarboxylation strategy to achieve PEP in quantity to run gluconeogenesis to feed all the extrahepatic tissues. The first enzyme is again an ATP-cleaving biotin carboxylase, in this case utilizing pyruvate as cosubstrate.95 Pyruvate carboxylase thus makes oxaloacetate, from presumably trace amounts of the pyruvate enolate anion in its active site (Fig. 30A). The enzyme in humans is mitochondrial.
The second enzyme, PEP carboxykinase, spends another molecule of nucleoside triphosphate, this time GTP96 (Fig. 30B). There are isozymes both in hepatic mitochondria and cytoplasm. The enzyme decarboxylates the just formed β-keto acid oxaloacetate (without need for external cofactors) to CO2 and the pyruvate C-3 carbanion. This nascent product has its major structural contributor with the negative charge on the more basic oxygen, e.g. as the enolate anion, and the carbanion as minor resonance form. That enolate oxygen anion now attacks the electrophilic γ-phosphorus atom of bound GTP, cleaving it to GDP and trapping the enolate as the activated enol phosphate PEP. In the two enzyme stoichiometry, both CO2/HCO3 and oxaloacetate, disappear, revealing they act catalytically (Fig. 30C).
Again the CO2 has acted as a transient grouping, put on by one enzyme and taken off by the next. This time the key carbanion is used in the second enzyme active site as the oxyanion contributor. An O–P bond, not a C–C bond, has been made (Fig. 31). One molecule of ATP and one molecule of GTP have been split, each providing 105 boost in the front direction of each of enzyme. The group transfer potential of PEP is thus much higher than that of the more common ATP and GTP phosphoric anhydride side chains, and can drive gluconeogenesis.
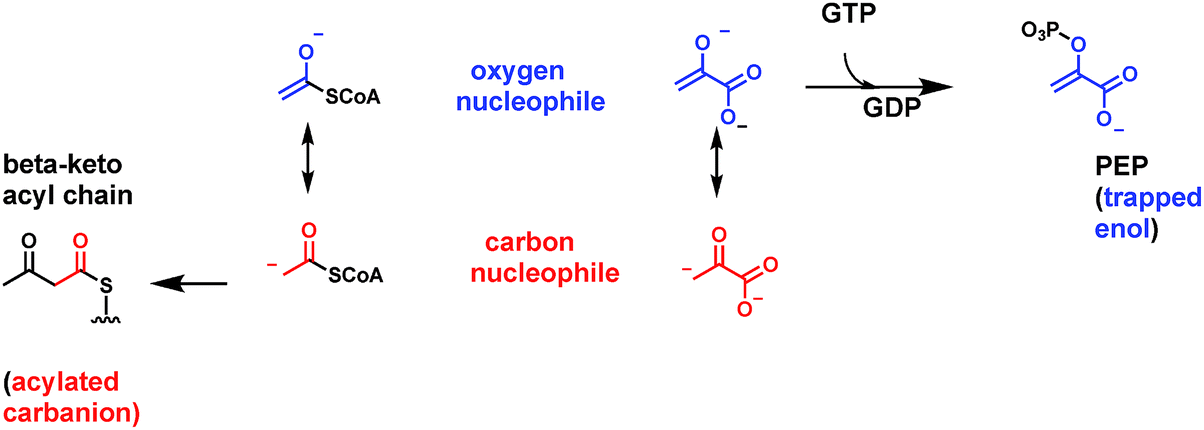 | ||
| Fig. 31 Stabilized carbanion/thioester enolate (from acetyl CoA) vs. carbanion/enolate (from pyruvate) and reaction of distinct resonance contributors. C–C bond formation in fatty acid synthase/chain elongation is via net acylation of the C-2 carbanion equivalent (red). In the PEP case it is the oxygen anion (blue) form of the C-3 carbanion equivalent that reacts towards the electrophilic γ-phosphate of GTP to trap the enolate as the phosphoryl ester in PEP. | ||
Nature thus uses CO2 as a vanishing protecting group at two biosynthetic nodal points by the tandem carboxylation/decarboxylation strategy. These reaction sequences hint at the utility of this one carbon, fully oxidized CO2 molecule for biosynthetic strategies in nucleophilic chemical biology.
8. Summary
The ∼3 billion tons of CO2 generated by the 7 billion humans planetary wide come from enzymatic decarboxylations of organic carboxylic metabolites. Much of those ∼1039 molecules of CO2 are released by humans, and the myriad other organisms on the planet, get energy out of ingested food by oxidizing the reduced carbon atoms (one or more C–H bond) in organic molecules to CO2. It is the electrons that are valuable, become stored in molecules that can do useful work, and the CO2 is generally the throw away product (but see Section 7 above), diffusing to, and excreted by the lungs. It is just the reverse in plants and microbes that fix CO2 into organic molecular frameworks to provide the foods that are those organic metabolites.In a very real sense the CO2 molecule is the bridge between the photosynthetic carbon fixers and the CO2-producing respirers. The gaseous nature of CO2 aids in its loss by exhalation. Its high aqueous solubility on the one hand allows efficient transport to the lungs through the blood. On the other hand, that provides the solution-based CO2, available as needed for ribulose-bisphosphate carboxylase action in plants.
The precursors to CO2 release are always carboxylic acids in the ionized carboxylate state. Primary metabolites range mostly between two and seven carbons in which carbonyl groups – aldehydes and ketones – are the key functional groups. The enzymatic oxidation/dehydrogenation of aldehydes to acids are the ultimate enzymatic steps that produce the carboxylate groups in those low molecular weight frameworks.
Some generic structures of organic acids that undergo enzymatic decarboxylation to release CO2 are shown in Fig. 32, with X-substituents at Cα and Y-substituents at Cβ. Most common in the high flux catabolic pathways for energy generation are X = O or YO. Analogously X = OH and YOH are typically an NAD (P)+-dependent oxidative step away from the keto acids that are so often the immediate substrates for decarboxylation. When X and Y substituents = H, that is saturated Cα and Cβ carbons, the energy barriers to be surmounted by decarboxylases are typically higher and chemically more difficult. In two key biosynthetic pathways, pyrimidine biogenesis and protoporphyrin IX tetrapyrrole macrocycle assembly, decarboxylations at carboxylates attached to heterocycles occur and may be chemically the most difficult of all the types of decarboxylations surveyed in this review.
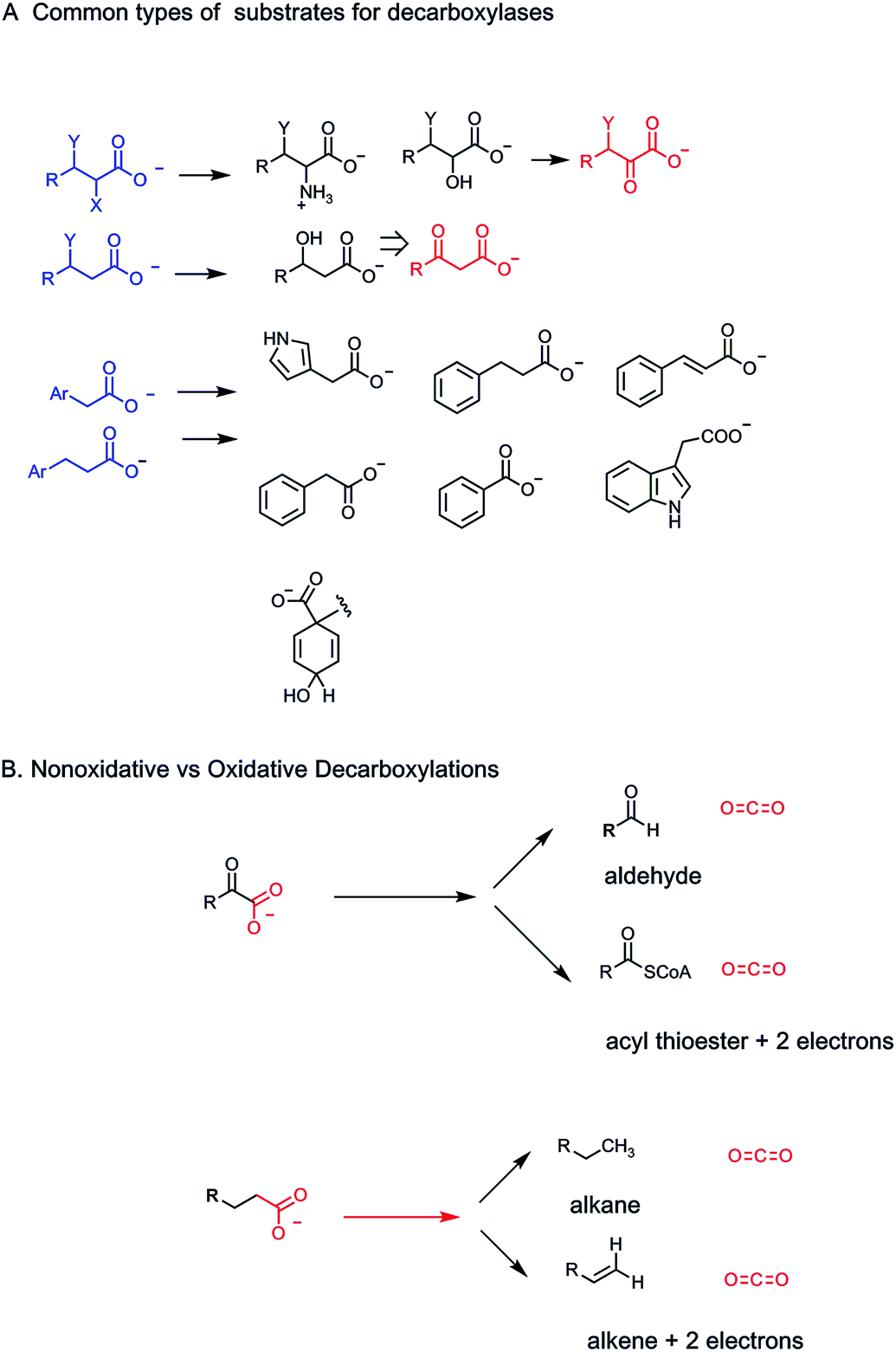 | ||
| Fig. 32 (A) Schematic of the structural variants of carboxylated metabolites that undergo enzymatic decarboxylations. α- and β-carboxylate scaffolds are the most common and reflect the core strategy of energy-generating decarboxylation in the citrate cycle. A variety of other aliphatic, aromatic, and heteroaromatic scaffolds are decarboxylated. (B) Nonoxidative vs. oxidative decarboxylation of two kinds of carboxylate substrates. | ||
Most decarboxylases break the C–COO carbon–carbon bond in carboxylate substrates heterolytically and have the task of stabilizing the two electron/carbanion density at the Cα carbon left behind as CO2 forms. A few decarboxylases carry out homolytic C–COO bond cleavage and they use radicals from glycine residues (and transient cys thiol radicals therefrom) and/or from 5′-deoxyadenosyl radical fragments generated from S-adenosylmethionine. There are multiple cases where the substrates contain adjacent functional groups that stabilize carbanion intermediates. We have noted β-keto groups as most common structural elements that lower energy of Cα–COO cleavage by enolate formations. Analogously, thioesters via participation of thioester enolates are stabilizing, as in malonyl CoA decarboxylation. They can be vinylogous as in glutaconyl CoA decarboxylation.
Most decarboxylases recruit cofactors or cosubstrates to lower energy barriers for the great bulk of the other organic acid metabolites to be coaxed into giving up CO2. These range through the coenzyme forms of five vitamins: B1 (α-keto acids), vitamin B2 (P-pantothenoylcysteine), B3 (the first half of isocitrate dehydrogenase and the actual hydride transfer step of prephenate dehydrogenase), B6 (amino acid decarboxylases) and B7 (Na+-pumping oxalacetate decarboxylase). There are decarboxylases that require the prior kinase action of ATP, the prior dehydrogenase action of NAD+, the radical cleavageability of SAM, and the chelating ability of Zn2++.
We have also noted nonoxidative vs. oxidative decarboxylation outcomes (Fig. 32B). The metabolic paradigm is the handling of the α-keto acid pyruvate. Nonoxidative decarboxylation generates acetaldehyde: oxidative decarboxylation converts the substrate ketone to the acid oxidation product as the acyl thioester. Two electrons are removed, typically captured in NADH. The other noted variant is in the saturated acetyl and propionyl side chains of tetrapyrrole macrocycle biosynthetic intermediates. The acetyl chain is decarboxylated nonoxidatively to the terminal methyl group. The propionyl side chain gives a terminal vinyl group and two electrons.
The cofactor-independent OMP decarboxylase and uroporphyrinogen III decarboxylase each act on a heterocycle with a direct Cα–COO bond, where Cα is trigonal and part of an aromatic/heteroaromatic π electron system. The catalytic mechanistic advantage is still obscure but these two enzymes are among the most efficient of catalysts compared to the remarkably slow nonenzymatic decarboxylation rates. These are reflections on how stable OMP and uroporphyrinogen III molecules are in physiologic conditions, and how far these catalysts have come down their evolutionary paths to catalytic efficiency and proficiency.
CO2 is not just the tail end detritus of animal one carbon metabolism nor are decarboxylases just creating the gaseous end product of energy metabolism. One needs only to invert perspective and think like a plant to see how CO2 is the sole building block for every organic metabolite. Rather, one should regard the decarboxylases as the gatekeepers to freeing back up the one carbon CO2 for its endless biologic recycling to make the molecules of life.
9. Conflicts of interest
There are no conflicts of interest to declare.10. References
- R. Thauer, Biochemistry of Methanogenesis: a Tribute to Marjory Stephenson, Microbiology, 1988, 144, 2377–2406 Search PubMed.
- J. C. Murrell and M. Jetten, The microbial methane cycle, Environ. Microbiol. Rep., 2009, 1, 279–284 CrossRef CAS PubMed.
- R. S. Hanson and T. E. Hanson, Metabotrophic Bacteria, Microbiol. Rev., 1996, 60, 439–471 CAS.
- C. Walsh and Y. Tang, The Chemical Biology of Human Vitamins, Royal Chemical Society Press, London UK, 2018 Search PubMed.
- B. Hobdoller and E. O. Wilson, The Ants, Harvard University Press, Cambridge MA, 1990 Search PubMed.
- C. Rossini, A. Attygalle, A. Gonzalez, S. M. Smedley, M. Eisner, J. Meinwald and T. Eisner, Defensive production of formic acid (80%) by a carabid beetle (Galerita lecontei), Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6792–6797 CrossRef CAS PubMed.
- P. M. Jordan and M. Akhtar, The mechanism of action of serine transhydroxymethylase, Biochem. J., 1970, 116, 277–286 CrossRef CAS PubMed.
- R. Florio, M. L. de Salvo, M. Vivoli and R. Contestabile, Serine hydroxymethyltransferase: a model enzyme for mechanistic, structural and evolutionary studies, Biochim. Biophys. Acta, 2011, 1814, 1489–1496 CrossRef CAS PubMed.
- C. W. Carreras and D. Santi, The catalytic mechanism and structure of thymidylate synthase, Annu. Rev. Biochem., 1995, 64, 721–762 CrossRef CAS PubMed.
- M. Fontecave, M. Atta and E. Mulliez, S-Adenosylmethionine: nothing goes to waste, Trends Biochem. Sci., 2004, 29, 243–249 CrossRef CAS PubMed.
- C. Walsh and Y. Tang, Natural Product Biosynthesis: Chemical Logic and Enzymatic Machinery, Royal Society of Chemistry Press, London UK, 2017 Search PubMed.
- A. R. Portis and M. A. J. Parry, Discoveries in Rubisco: an historical perspective, Photosynth. Res., 2007, 94, 121–143 CrossRef CAS PubMed.
- Wikipedia, downloaded 1/8-19 Carbon Fixation.
- C. Walsh, B. Tu. and Y. Tang., Eight kinetically stable but thermodynamically activated molecules that power cell metabolism, Chem. Rev., 2018, 118, 1460–1494 CrossRef CAS PubMed.
- S. Lindskog, Structure and mechanism of carbonic anhydrase, Pharmacol. Ther., 1997, 74, 1–20 CrossRef CAS.
- M. Slobal, Monitoring Exhaled Carbon Dioxide, Respir. Care, 2016, 61, 1397–1416 CrossRef.
- G. S. Blog, posted 08/11/2008 by peggy, Release of carbon dioxide by individual humans, Scientist's blog downloaded 01/01/2019.
- B. Palmer, 7 Billion Carbon Sinks, Explainer in Slate.Com, 2009, August 13 Search PubMed.
- A. Tanner, L. Bowater, S. A. Fairhurst and S. Bornemann, Oxalate Decarboxylase Requires Manganese and Dioxygen for Activity, J. Biol. Chem., 2001, 276, 43627–43634 CrossRef CAS PubMed.
- C. L. Berthold, P. Moussatche, N. G. J. Richards and Y. Lindqvist, Structural Basis for Activation of the Thiamin Diphosphate-dependent Enzyme Oxalyl-CoA Decarboxylase by Adenosine Diphosphate, J. Biol. Chem., 2005, 280, 41645–41654 CrossRef CAS PubMed.
- N. C. Williams and L. A. J. O'Neill, A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation, Front. Immunol., 2018, 9, 141 CrossRef PubMed.
- T. Yoshida, K. Fujita and T. Nagasawa, Novel Reversible Indole-3-carboxylate Decarboxylase Catalyzing Nonoxidative Decarboxylation, Biosci., Biotechnol., Biochem., 2002, 66, 2388–2394 CrossRef CAS PubMed.
- G. A. Hamilton and F. Westheimer, On the mechanism of enzymatic decarboxylation of acetoacetate, J. Am. Chem. Soc., 1959, 81, 6332 CrossRef CAS.
- M.-C. Ho, J. F. Menetret, H. Tsurata and K. N. Allen, The origin of the electrostatic perturbation in acetoacetate decarboxylase, Nature, 2009, 459, 393–397 CrossRef CAS PubMed.
- R. Breslow, On the mechanism of thiamin action IV evidence from model studies, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
- R. Breslow, The mechanism of thiamin action 2. Rapid deuterium exchange in thiazolium salts, J. Am. Chem. Soc., 1957, 79, 1762–1763 CrossRef CAS.
- F. Jordan and N. S. Nemeria, Progress in the experimental observation of thiamin diphosphate-bound intermediates on enzymes and mechanistic information derived from these observations, Bioorg. Chem., 2014, 57, 251–262 CrossRef CAS PubMed.
- F. Dyda, W. Furey, S. Swaminathan, M. Sax, B. Farenkopf and F. Jordan, Catalytic centers in the thiamin diphosphate dependent enzyme pyruvate decarboxylase at 2.4-A resolution, Biochemistry, 1993, 32, 6165–6170 CrossRef CAS PubMed.
- L. Blieck, G. Toye, F. Dumortier, K. J. Verstrepen, F. R. Delvaux, J. M. Thevelein and V. Dijck, Isolation and characterization of brewer's yeast variants with improved fermentation performance under high-gravity conditions, Appl. Environ. Microbiol., 2007, 73, 815–824 CrossRef CAS PubMed.
- G. Zhang, J. Dai, Z. Lu and D. Dunaway-Mariano, The phosphonpyruvate decarboxylase from Bacteroides fragilis, J. Biol. Chem., 2003, 278, 41302–41308 CrossRef CAS PubMed.
- H. Nakashita, K. Watanabe, O. Hara, T. Hidaka and H. Seto, Studies on the Biosynthesis of Bialaphos. Mechanism of C–P Bond Formation: Discovery of Phosphonpyruvate Decarboxylase which Catalyzes the Formation of Phosphonoacetaldehyde from Phosphonopyruvate, J. Antibiot., 1997, 50, 212–219 CrossRef CAS PubMed.
- E. Sandmeier, T. I. Hale and P. Christen, Multiple evolutionary origin of pyridoxal-5-phosphate-dependent amino acid decarboxylases, Eur. J. Biochem., 1994, 221, 997–1002 CrossRef CAS PubMed.
- Y. Morino and F. Nagashima, Pyridoxal phosphate-binding site in enzymes: reduction and comparison of sequences, Methods Enzymol., 1984, 106, 116–137 CAS.
- H. Dunathan, Conformation and reaction specificity in pyridoxal-P enzymes, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 712–716 CrossRef CAS PubMed.
- P. D. vanPoelje and E. E. Snell, Pyruvoyl-Dependent Enzymes, Annu. Rev. Biochem., 1990, 59, 29–59 CrossRef CAS PubMed.
- J. M. Williamson and G. Brown, Purification and properties of aspartate decarboxylase an enzyme that catalyzes the formation of beta alanine, J. Biol. Chem., 1979, 254, 8074–8082 CAS.
- T. J. Grevengoed, E. L. Klett and R. A. Coleman, Acyl-CoA metabolism and partitioning, Annu. Rev. Nutr., 2014, 34, 1–30 CrossRef CAS PubMed.
- I. Schuiki and G. Dam, Phosphatidylserine Decarboxylases: Key Enzymes of Lipid Metabolism, IUBMB Life, 2009, 61, 151–162 CrossRef CAS PubMed.
- H. Zhou, D. B. McCarthy, M. O'Connor, J. Reed and K. Stoops, The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14802–14807 CrossRef PubMed.
- L. Sahlman and C. H. Williams Jr, Lipoamide dehydrogenase from Escherichia coli: steady state kinetics of the physiologic reaction, J. Biol. Chem., 1989, 264, 8039–8045 CAS.
- S. Yeaman, The 2-oxo acid dehydrogenase complexes: recent advances, Biochem. J., 1989, 257, 625–632 CrossRef CAS PubMed.
- W. J. Rutter and H. Lardy, Purification and properties of pigeon liver malic enzyme, J. Biol. Chem., 1958, 233, 374–382 CAS.
- M. Rippa, P. P. Giovanni, M. P. Barrett, F. Dallocchio and S. Hanau, 6-Phosphogluconate dehydrogenase: the mechanism of action investigated by a comparison of the enzyme from different species, Biochim. Biophys. Acta, 1998, 1429, 83–92 CrossRef CAS.
- H. Ankel and D. S. Feingold, Biosynthesis of UDP-D-xylose: UDP glucuronate carboxy-lyase of wheat germ, Biochemistry, 1965, 2468–2475 CrossRef CAS.
- J. Gaylor, Membrane-Bound Enzymes of Cholesterol Synthesis from Lanosterol, Biochem. Biophys. Res. Commun., 2002, 292, 1139–1146 CrossRef CAS.
- R. T. Fischer, S. H. Stam, P. R. Johnson, S. S. Ko, R. L. Magdola, J. L. Gaylor and J. M. Trzaskos, Mechanistic studies of lanosterol 14-alpha methyl demethylase: substrate requirements for the component reactions catalyzed by a single cytochrome P450 isozyme, J. Lipid Res., 1989, 30, 1621–1633 CAS.
- O. Lockridge, V. Massey and P. A. Sullivan, Mechanism of action of the flavoenzyme lactate oxidase, J. Biol. Chem., 1972, 247, 8097–8106 CAS.
- E. Strauss, H. Zhai, L. A. Brand, F. W. McLafferty and T. Begley, Mechanistic Studies on Phosphopantothenoylcysteine decarboxylase: Trapping of an Enethiolate Intermediate with a Mechanism-Based Inactivating Agent, Biochemistry, 2004, 43, 15520–15533 CrossRef CAS PubMed.
- O. McAuliffe, R. P. Ross and C. Hill, Lantibiotics: structure, biosynthesis, and mode of action, FEMS Microbiol. Rev., 2001, 25, 285–308 CrossRef CAS.
- F. Majer, D. G. Schmid, K. Altenam, G. Bierbaum and T. Kupke, The Flavoprotein MrsD Catalyzes the Oxidative Decarboxylation Reaction Involved in Formation of the Peptidoglycan Biosynthesis Inhibitor Mersacidin, J. Bacteriol., 2002, 184, 1234–1243 CrossRef CAS PubMed.
- D. Leys, Flavin metamorphosis: cofactor transformation through prenylation, Curr. Opin. Chem. Biol., 2018, 47, 117–125 CrossRef CAS PubMed.
- S. A. Marshall, K. A. P. Payne and D. Leys, The UbiX-UbiD system: the biosynthesis and use of prenylated FMN, Arch. Biochem. Biophys., 2017, 632, 209–221 CrossRef CAS PubMed.
- M. W. Bhuiya, S. G. Lee, J. M. Jez and O. Yu, Structure and Mechanism of Ferulic Acid Decarboxylase (FDC1) from Saccharomyces cerevisiae, Appl. Environ. Microbiol., 2015, 81, 4216–4223 CrossRef CAS PubMed.
- A. Alekseeva, S. S. Savin and V. I. Tishkov, NAD+-dependent formate dehydrogenase from plants, Acta Naturae, 2011, 3, 38–54 CrossRef CAS PubMed.
- F. Dosselaere and J. Vanderlyden, A metabolic node in action: chorismate-utilizing enzymes in microorganisms, Crit. Rev. Microbiol., 2001, 27, 75–131 CrossRef CAS PubMed.
- J. Zempleni, S. S. K. Wijeratne and Y. I. Hassan, Biotin, BioFactors, 2009, 35, 36–46 CrossRef PubMed.
- J. Knowles, The mechanism of biotin-dependent enzymes, Annu. Rev. Biochem., 1989, 58, 195–221 CrossRef CAS PubMed.
- W. Buckel, Sodium ion-translocating decarboxylases, Biochim. Biophys. Acta, 2001, 1505, 15–27 CrossRef CAS.
- K. Bendrat, S. Berger, W. Buckel, W. A. Etzel and K. H. Rohm, Carbon-13 labeled biotin-a new probe for the study of enzyme-catalyzed carboxylation and decarboxylation reactions, FEBS Lett., 1990, 277, 156–158 CrossRef CAS.
- D. Aparicio, R. Perez-Luque, X. Carpena, M. Diaz, J. C. Ferrer, P. C. Lowen and I. Fita, Structural asymmetry and disulfide bridges among subunits modulate the activity of human malonyl CoA decarboxylase, J. Biol. Chem., 2013, 288, 11907–11919 CrossRef CAS PubMed.
- M. L. Barta, W. J. McWhorter, H. M. Miziorko and B. V. Geisbrecht, Structural basis for nucleotide binding and reaction catalysis in mevalonate diphosphate decarboxylase, Biochemistry, 2012, 51, 5611–5621 CrossRef CAS.
- N. E. Voynova, F. Zu, K. P. Battaile, T. J. Herdendorf, J. J. KIm and H. M. Miziorko, Human mevalonate-PP decarboxylase: characterization, investigation of the mevalonate diphosphate binding site, Arch. Biochem. Biophys., 2008, 480, 58–67 CrossRef CAS PubMed.
- H. Miziorko, Enzymes of the mevalonate pathway of isoprenoid biosynthesis, Arch. Biochem. Biophys., 2015, 505, 131–143 CrossRef PubMed.
- A. Tschech and G. Fuchs, Anaerobic degradation of phenol by carboxylation to 4-hydroxybenzoate, Arch. Microbiol., 1989, 152, 594–599 CrossRef CAS.
- D. J. W. Grant and J. C. Patel, Non-oxidative decarboxylation of p-hydroxybenzoic acid, gentisic acid, protocatechuic acid, and gallic acid by Klebsiella aerogenes (Aerobacter aerogenes), J. Microbiol. Serol., 1969, 35, 325–343 CAS.
- T. Yoshida, Y. Hawakawa, T. Matsui and T. Nagasawa, Purification and characterization of 2,6-dihydroxybenzoate decarboxylase reversibly catalyzing nonoxidative decarboxylation, Arch. Microbiol., 2004, 6, 391–397 CrossRef PubMed.
- H. J. Sofia, G. Chen, B. G. Hetzler, J. F. Reyes-Spindola and N. E. Miler, Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods, Nucleic Acids Res., 2001, 29, 1097–1106 CrossRef CAS PubMed.
- L. R. F. Backman, M. Funk, C. D. Dawson and C. L. Drennan, New tricks for the glycyl radical enzyme family, Crit. Rev. Biochem. Mol. Biol., 2017, 52, 674–695 CrossRef CAS PubMed.
- G. Layer, A. J. Pierik, M. Trost, S. E. Rigby, H. K. Leech, K. Grage, D. Breckau, I. Astner, L. Jansch, P. Heathcote, M. J. Warren, D. K. Heinz and D. Jahn, The substrate radical of Escherichia coli oxygen-independent coproporphyrinogen III oxidase Hem N, J. Biol. Chem., 2006, 281, 15727–15734 CrossRef CAS PubMed.
- J. M. Camadro, H. Chambon, J. Jolles and P. Labbe, Purification and properties of coproporphyrinogen oxidase from Saccharomyces cerevisiae, Eur. J. Biochem., 1986, 156, 579–587 CrossRef CAS PubMed.
- J. Bridwell-Rabb, T. A. J. Grell and C. L. Drennan, A rich man poor man story of S-adenosylmethionine and cobalamin revisited, Annu. Rev. Biochem., 2018, 87, 555–584 CrossRef CAS PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duchene and E. M. Shepard, Radical S-Adenosylmethionine Enzymes, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS PubMed.
- K. A. Shisler and J. Broderick, Glycyl radical-activating enzymes, Arch. Biochem. Biophys., 2014, 546, 65–71 CrossRef PubMed.
- J. Fan, Q. Liu, Q. Hao, M. Teng and L. Niu, Crystal Structure of Uroporphyrinogen Decarboxylase from Bacillus subtilis, J. Bacteriol., 2007, 189, 3573–3580 CrossRef CAS PubMed.
- B. Selvaraj, W. Buckel, B. T. Golding, G. M. Ullmann and B. M. Martins, Structure and Function of 4-Hydroxyphenylacetate Decarboxylase and its Cognate Activating Enzyme, J. Mol. Microbiol. Biotechnol., 2016, 26, 76–91 CrossRef CAS PubMed.
- T. Selmer and P. Andrei, p-Hydroxybenzoate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalyzing the formation of para cresol, Eur. J. Biochem., 2001, 268, 1363–1371 CrossRef CAS PubMed.
- K. A. Shisler and J. Broderick, Glyclyl radical activating enzymes: structure, mechanism, and substrate interactions, Arch. Biochem. Biophys., 2014, 546, 64–71 CrossRef CAS PubMed.
- J. Hermes, P. A. Tipton, M. Fisher, M. O'Leary, J. Morrison and W. W. Cleland, Mechanisms of Enzyme-catalyzed and Acid-catalyzed Decarboxylations of Prephenate, Biochemistry, 1984, 23, 6263–6275 CrossRef CAS PubMed.
- J. VanVleet, A. Kleeb, P. Kast, D. Hilvert and W. W. Cleland, Carbon-13 Isotope Effect on the Reaction Catalyzed by Prephenate Dehydratase, Biochim. Biophys. Acta, 2010, 1804, 752–754 CrossRef CAS PubMed.
- A. Parthasarathy, P. J. Cross, R. C. Dobson, L. E. Adams, M. A. Savka and A. O. Hudson, A Three Rig Circus: Metabolism of the Three Proteinogenic Aromatic Amino Acids and Their Role in the Health of Plants and Animals, Front. Mol. Biosci., 2018, 5, 29 CrossRef.
- S. A. Mahlstedt and C. Walsh, Investigation of anticapsin biosynthesis reveals a four enzyme pathway to tetrahydrotyrosine in Bacillus subtilis, Biochemistry, 2010, 49, 912–923 CrossRef CAS PubMed.
- S. Mahlstedt, E. N. Fielding, B. S. Moore and C. T. Walsh, Prephenate decarboxylases: a new prephenate-utilizing enzyme family that performs non-aromatizing decarboxylation en route to diverse secondary metabolites, Biochemistry, 2010, 49, 9021–9023 CrossRef CAS PubMed.
- J. B. Parker and C. Walsh, Stereochemical outcome at four stereogenic centers during conversion of prephenate to tetrahydrotyrosine by Bac ABGF in the bacilysin pathway, Biochemistry, 2012, 51, 5622–5632 CrossRef CAS PubMed.
- K. Bradley, in How Much Tryptophan a Day, Livestrong, 2018 Search PubMed.
- J. McMurry and T. Begley, The Organic Chemistry of Biological Pathways, WH Freeman, 2nd edn, 2015 Search PubMed.
- L. Dwiarti, K. Yamane, H. Yamatani, P. Kahar and M. Okabe, Purification and Characterization of Cis-Aconitic Acid Decarboxylase from Aspergillus terreus TN484-M1, J. Biosci. Bioeng., 2002, 94, 29–33 CrossRef CAS.
- T. C. Appleby, C. Kinsland, T. P. Begley and S. D. Ealick, The crystal structure and mechanism of orotidine-5′-monophosphate decarboxylase, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2005–2010 CrossRef CAS PubMed.
- B. G. Mller and R. Wolfenden, Catalytic Proficiency: The Unusual Case of OMP Decarboxylase, Annu. Rev. Biochem., 2002, 71, 847–885 CrossRef.
- C. A. Lewis and R. Wolfenden, Uroporphyrinogen decarboxylation as a benchmark for the catalytic proficiency of an enzyme, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17328–17333 CrossRef CAS.
- B. Lupa, D. Lyon, L. N. Shaw, M. Sieprawska-Lupa and J. Wiegel, Properties of the reversible nonoxidative vanillate/4-hydroxybenzoate decarboxylase from Bacillus subtilis, Can. J. Microbiol., 2008, 54, 75–81 CrossRef CAS.
- H. Omura, M. Wieser and T. Nagasawa, Pyrrole-2-carboxylate decarboxylase from Bacillus megaterium, an organic acid-requiring enzyme, Eur. J. Biochem., 1998, 253, 480–484 CrossRef CAS.
- G. A. Hunter and G. C. Ferreira, 5-Aminolevulinate synthase: catalysis of the first step in heme biosynthesis, Cell. Mol. Biol., 2009, 55, 102–110 CAS.
- G. A. Hunter and G. Ferreira, Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis, Biochim. Biophys. Acta, 2011, 1814, 1467–1473 CrossRef CAS.
- A. Witkoski, A. Joshi and S. Smith, Mechanism of the beta-ketoacyl synthase reaction catalyzed by the animal fatty acid synthase, Biochemistry, 2002, 41, 10877–10887 CrossRef.
- S. Jitrapakdee, M. S. Meurice, I. Rayment, W. W. Cleland, J. C. Wallace and P. V. Atwood, Structure, mechanism, and regulation of pyruvate carboxylase, Biochem. J., 2008, 413, 369–387 CrossRef CAS.
- T. Holyoak, S. M. Sullivan and T. Noak, Structural insights into the mechanism of PEPCK catalysis, Biochemistry, 2006, 45, 8254–8263 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |