
Influence of the channel size of isostructural 3d–4f MOFs on the catalytic aerobic oxidation of cycloalkenes†
Patricio
Cancino
 ab,
Luis
Santibañez
a,
Christian
Stevens
a,
Pablo
Fuentealba
ab,
Nathalie
Audebrand
c,
Daniel
Aravena
d,
Julia
Torres
e,
Sebastian
Martinez
e,
Carlos
Kremer
e and
Evgenia
Spodine
*ab
ab,
Luis
Santibañez
a,
Christian
Stevens
a,
Pablo
Fuentealba
ab,
Nathalie
Audebrand
c,
Daniel
Aravena
d,
Julia
Torres
e,
Sebastian
Martinez
e,
Carlos
Kremer
e and
Evgenia
Spodine
*ab
aFacultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Santiago, Chile. E-mail: espodine@ciq.uchile.cl
bCentro para el Desarrollo de la Nanociencia y la Nanotecnología, CEDENNA, Santiago, Chile
cUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France
dDepartamento de Química y Biología, Universidad de Santiago de Chile, Santiago, Chile
eDepartamento Estrella Campos, Facultad de Química, Universidad de la República, Montevideo, Uruguay
First published on 11th June 2019
Abstract
The present work reports a new group of heterogeneous catalysts with a 3D structure, CuLnIDA, {[Cu3Ln2(IDA)6]·8H2O} (Ln: LaIII, GdIII or YbIII), with an organic linker (H2IDA: iminodiacetic acid). Different sets of O2 pressure and time were used in order to obtain the optimal reaction conditions at 75 °C. The reaction was found to depend on the [aldehyde]/[substrate] ratio. The best results, with a conversion of 73% for CuLaIDA as the catalyst, were obtained for the smallest ratio of 0.2. Finally, the importance of the pore size was analysed by comparing the catalytic activity of the as formed catalyst with that of the thermally activated one. The conversion increased ca. 26–35% for the different catalysts when they were previously activated. In addition, the selectivity increased towards cyclohexenone. The use of molecular oxygen as the oxidizing agent in a system where an auxiliary solvent is not used, as the cyclohexene substrate and products play the role of a solvent, permitted us to generate a more friendly environmental system for the oxidation of cycloalkenes under mild conditions.
Introduction
The oxidation of olefins has become a topic of interest among many research groups.1 The oxidation of cycloalkenes is particularly investigated, due to the products that this reaction yields, which are used in the agrochemical and pharmaceutical industry, as well as in the manufacture of perfumes and glues, among others.2–5 Thus, efforts aim to find new efficient, economical and clean catalytic routes to obtain these oxidation products. In some of the catalytic routes, the use of a solvent implies an environmental impact. A solvent-free oxidation reaction decreases the effect on the environment. Besides, the high cost of the mostly used environmentally “green” oxidants, such as hydrogen peroxide and organic hydroperoxides, makes molecular oxygen a perfect and optimal oxidant to be considered in the oxidation reactions of cycloalkenes.6In catalytic systems, the use of homogeneous and supported catalysts7 presents well known problems: the inability of recovering the catalyst in homogeneous systems, and leaching or decomposition of supported catalysts.
In the heterogeneous catalysis area, the use of homometallic inorganic polymers has been reported in the oxidation of cycloalkenes,8–15 proving their low solubility and stability under the reaction conditions.16 Some examples related to catalytic systems based on homonuclear metal–organic frameworks (MOFs) based on copper(II),17–19 cobalt(II),20,21 vanadium(IV),22,23 manganese(II,III) or iron(III)24 ions have been reported.25–27
Heterometallic polymers have been also used as catalysts in reactions such as hydrogenation of small olefins using RhII polymers containing CuII, NiII and PdII metalloporphyrins.28 Degradation of methyl blue and rhodamine B can be produced using heterometallic uranium based polymers, with NiII as the secondary ion for methyl blue degradation,29 and AgI for the rhodamine B one.30 In the area of oxidation reactions, heterometallic MOFs show catalytic activity for different kinds of substrates. The studied catalytic systems were based on the use of the existing empty spaces in the MOFs to incorporate metallic nanoparticles, which can cooperate in the reaction mechanism, together with the original active metal sites existing in the structure.31 For example, porous CuII MOFs, based on a pyridyl substituted diketonate ligand, with adsorbed Au0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 or Pd033 nanoparticles, have been used in the oxidation of benzylic alcohols. Furthermore, bimetallic catalysts of Pd0–Au0 nanoparticles encapsulated in an AlIII MOF were studied in the aerobic oxidative carbonylation of amines.34 Besides, Sun et al. report the catalytic performance of a CoII/NiII MOF-74, which corresponds to a partially Co-substituted Ni-MOF-74. The authors reported that the catalytic activity increased as the amount of incorporated CoII increased, suggesting that this ion was acting as the active site for the oxidation of cyclohexene.35,36 On the other hand, only a few 3d–4f heterometallic polymers have also been reported as catalytically active systems in the oxidation of alkenes and benzylic substrates.37–39
32 or Pd033 nanoparticles, have been used in the oxidation of benzylic alcohols. Furthermore, bimetallic catalysts of Pd0–Au0 nanoparticles encapsulated in an AlIII MOF were studied in the aerobic oxidative carbonylation of amines.34 Besides, Sun et al. report the catalytic performance of a CoII/NiII MOF-74, which corresponds to a partially Co-substituted Ni-MOF-74. The authors reported that the catalytic activity increased as the amount of incorporated CoII increased, suggesting that this ion was acting as the active site for the oxidation of cyclohexene.35,36 On the other hand, only a few 3d–4f heterometallic polymers have also been reported as catalytically active systems in the oxidation of alkenes and benzylic substrates.37–39
However, despite the fact of the numerous catalysts available for olefin oxidation, for many of them the reaction still does not occur without the assistance of an activator or a secondary oxidation agent. Depending on the mechanism of the reaction, these activators perform in diverse ways, proving themselves helpful as they allow the performance of reactions under less aggressive conditions, which is always a plus when trying to find a new catalytic route.
Some examples can be found in the literature such as the use of isobutyraldehyde as an activator in the epoxidation of olefins,40 or the use of the 2,2,6,6-tetramethyl-1-piperidinyloxy radical (TEMPO) as an activator for the oxidation of benzylic alcohols.41 Another widely used initiator is tert-butylhydroperoxide (TBHP), also reported for the oxidation of alkenes.42,43
The present work reports the importance of the channel size existing in the structure of a series of isostructural heterogeneous catalysts, CuLnMOFs, {[Cu3Ln2(IDA)6]·8H2O}n (Ln: LaIII, GdIII or YbIII (H2IDA: iminodiacetic acid), on the observed activity in the oxidation of cycloalkenes under mild conditions.
Materials and methods
Synthesis of [Cu3Ln2(IDA)6]·8H2O (CuLnIDA) (Ln = LaIII, GdIII, YbIII)
The complexes were prepared by direct reaction of iminodiacetic acid with stoichiometric amounts of CuII and LnIII salts following previously reported procedures.2 CuCl2·6H2O (0.13 g, 0.75 mmole) and LnCl3·6H2O (0.5 mmole) were dissolved in 5 mL of water. Iminodiacetic acid (H2IDA) (0.20 g, 1.5 mmole) was dissolved in 5 mL of water and the pH value was adjusted to 9.0 with diluted ammonia. Both solutions were mixed and a blue solution was obtained. The pH value was readjusted to 4.8–5.1 with diluted ammonia. After 3–5 days, blue crystals were obtained, separated by filtration and washed with water. Yield 50–60%. Anal. for CuLaIDA: calc. N, 6.0; C, 20.7; H, 2.9. Found: N, 5.9; C, 20.1; H, 3.8%. Anal. for CuGdIDA: calc. N, 5.9; C, 20.2; H, 2.8. Found: N, 5.8; C, 19.8; H, 3.6%. Anal. for CuYbIDA: calc. N, 5.8; C, 19.7; H, 2.8. Found: N, 5.9; C, 19.8; H, 3.4%. IR peaks associated with the IDA ligand appear at ca.: 3230 (N–H), 2930 (C–H), 1620 and 1407 (COO), 1450 (C–N), 950 (C–C), 1115 (C–N–C) cm−1. TGA diagrams show a single weight loss between room temperature and 60 °C, corresponding to crystallization water molecules. Calc. for CuLaIDA: 10.3, found: 10.0%. Calc. for CuGdIDA: 10.0, found: 9.8%. Calc. for CuYbIDA: 9.9, found: 9.5%. Decomposition point, ca. 290 °C. It was possible to obtain single crystals of CuYbIDA by this procedure, which were characterized by X-ray diffraction.Chemicals and equipment
All laboratory chemicals were of reagent grade, purchased from commercial sources and used without further purification. The infrared spectra (as KBr pellets) were obtained with a Bomen MB 102 FTIR spectrophotometer. Elemental analysis (C, H, N) was performed on a Flash 2000 instrument. Thermal analyses were carried out on a Shimadzu TGA-50 instrument with a TA 50I interface, using a platinum cell and nitrogen atmosphere. The experimental conditions were a 0.5 °C min−1 temperature ramp rate from 20 °C until 700 °C and a 50 mL min−1 nitrogen flow rate.ICP spectroscopy
The solutions obtained after the catalytic reactions were analyzed by optical ICP spectroscopy, using a PerkinElmer Optima 2000 DV model spectrometer. The free copper concentrations were determined using standards of different concentrations.X-ray data collection and refinement
A summary of the crystal data, data collection, structure determination and refinement for CuYbIDA is given in Tables S1–S6 (ESI†).Diffraction data were collected at 100 K using a Bruker D8 Venture single crystal diffractometer equipped with a CuKα sealed tube X-ray source operating at 50 kV/30 mA and a PHOTON 100 area detector operating in shutterless mode. Unit cell determination, data collection and processing were performed using the Bruker APEX3 software suite.44 Structure determination and refinement were performed with the SHELXT and SHELXL (with the OLEX2 graphical user interface) programs respectively.45,46 Crystallization water molecules could not be refined adequately because of disorder. To avoid this problem a solvent masking procedure was applied to the disordered electron density in the cavities using the OLEX2 program.47 Hydrogen atoms on C1, C3 and N1 were located geometrically and refined allowing them to ride on their parent atoms Uiso(H) = 1.2Ueq(C) and Uiso(H) = 1.2Ueq(N) respectively. Supplementary crystallographic information, bond lengths, bond angles and torsion angles can be found in Tables S1–S6 (ESI†). CCDC 1878174.†
Powder X-ray diffraction data, used to check the catalyst stability after the catalytic reaction, were collected with a Bruker D8 Advance diffractometer, operating with Cu Kα1 monochromatic radiation, equipped with a LynxEye detector, in the range of 5 to 120° 2θ. The whole pattern matching was done using the FullProf software in the WinPLOTR interface (see the ESI†).
Catalytic reactions
Three different systems were used for the catalytic reactions (Scheme 1). | ||
| Scheme 1 Principal products obtained from the oxidation of cycloalkenes (a): epoxide (b); conjugated alcohol (c); conjugated ketone (d). 1,2-Cyclohexanediol was detected as a minor product under specific reactions conditions. | ||
In the first system, 20 mmole of substrate, 4 mmole of isobutyraldehyde as the activator and 0.0035 mmole of {[Cu3Ln2(IDA)6]·8H2O}n were all placed together in a high pressure resistant round bottom flask of 25 mL capacity and the mixture was stirred at 75 °C for 24 hours, with a charge of 1 or 5 atm of oxygen.
For the second system, 40 mmole of substrate (C6: cyclohexene, C7: cycloheptene or C8: cyclooctene), 8 mmole (20% in mole) of isobutyraldehyde as the activator and 0.007 mmole {[Cu3Ln2(IDA)6]·8H2O}n (Ln: LaIII, GdIII or YbIII) were all placed together in a double neck round bottom flask of 25 mL capacity. The mixture was stirred at 75 °C under a continuous flow of 1 atm of O2 for 24 hours.
For the third system the catalytic conditions were the same as used in the second system, the only difference was that the catalysts were previously thermally activated. The activation of the catalysts was done using a vacuum oven at 100 °C for two hours. Following this procedure, the samples were cooled in a desiccator for fifteen minutes, and used in the catalytic reactions.
Samples of all the catalytic reactions were characterized and quantified by gas chromatography using a 5890 model SERIES II Hewlett Packard gas chromatograph. Nitrobenzene was used as an external standard in the final stage, just before the injection of the sample. The amount was 20 mg for 1 mL of sample. The column was a capillary non-polar Equity-1.
Computational details
Discrete models of the CuII site for CuLaIDA, CuGdIDA and CuYbIDA were constructed from their crystallographic structures.48–50 To account for the chemical environment of the copper centre, the complete coordination sphere of the central CuII and its four neighbour CuII positions was kept intact from the crystal structure. The four lanthanides connecting the copper centres were also included. Dangling IDA groups at the end of the model were replaced by H2O ligands. Peripheral copper positions were replaced with ZnII atoms to force the reduction process to happen at the central CuII site. The structural model contains 201 atoms, charge –2, multiplicity 2. In the first step, the hydrogen positions were relaxed using the XTB program (GFN1 model, solvent water).51 Vertical electron affinities were calculated as the difference between the oxidized and reduced species using the ORCA 4.0.1.2 software package.52 Two density functionals were considered (BP8653,54 and B3LYP55,56); calculations were performed in the gas phase and under a continuum solvation model (CPCM, water). Two different basis sets were tested (def2-SVP and def2-TZVP57,58); both cases include a 28-electron ECP for the lanthanide atom.59 In all cases, the same trend in electron affinity was observed across the series. Results for Def2-TZVP in water are presented.Results and discussion
Description of the structure of {[Cu3Yb2(IDA)6]·8H2O}n
Complex {[Cu3Yb2(IDA)6]·8H2O}n (CuYbIDA) crystallizes in the trigonal crystal system, space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1. The structure can be described as [Cu(IDA)2] units which link YbIII ions in a neutral 3D structure (Fig. 1a). Each CuII ion is coordinated by two deprotonated tridentate IDA ligands. The geometry of the CuII ion can be described as distorted octahedral with two elongated axial bonds (Cu1–O3 distance, 2.429(3) Å), resulting from the Jahn–Teller effect (Fig. 1b). This is reflected in the bond distances around this metal ion and crystallographic data, as depicted in Tables S1–S6 (ESI†). Each [Cu(IDA)2] unit has four oxygen atoms not coordinated to copper (O1 and O4), which are used to bind to YbIII ions. The Ln ion is nine-coordinated in a distorted tricapped trigonal prism geometry, surrounded by nine carboxylic oxygen atoms from six neighboring IDA ligands (Fig. 1c).
c1. The structure can be described as [Cu(IDA)2] units which link YbIII ions in a neutral 3D structure (Fig. 1a). Each CuII ion is coordinated by two deprotonated tridentate IDA ligands. The geometry of the CuII ion can be described as distorted octahedral with two elongated axial bonds (Cu1–O3 distance, 2.429(3) Å), resulting from the Jahn–Teller effect (Fig. 1b). This is reflected in the bond distances around this metal ion and crystallographic data, as depicted in Tables S1–S6 (ESI†). Each [Cu(IDA)2] unit has four oxygen atoms not coordinated to copper (O1 and O4), which are used to bind to YbIII ions. The Ln ion is nine-coordinated in a distorted tricapped trigonal prism geometry, surrounded by nine carboxylic oxygen atoms from six neighboring IDA ligands (Fig. 1c).
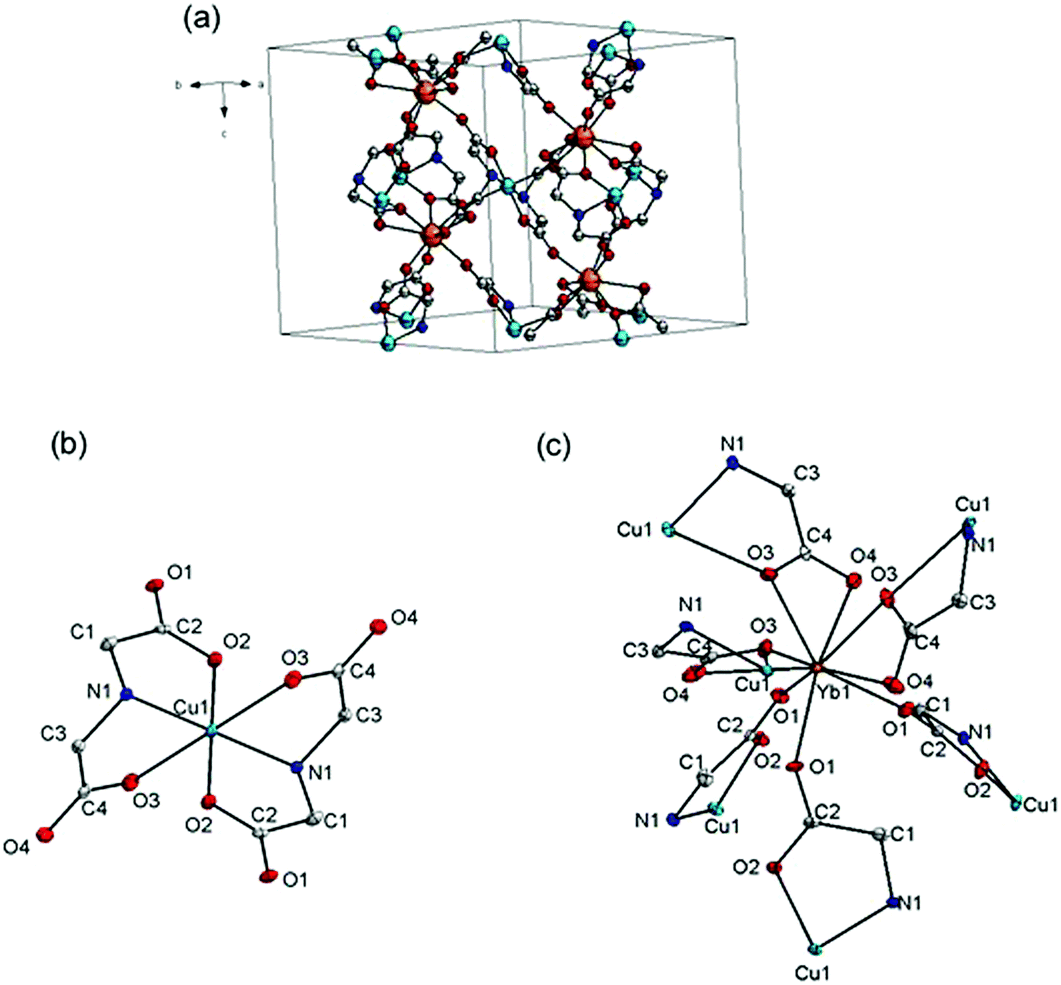 | ||
| Fig. 1 (a) Unit cell; (b) coordination environment around CuII and (c) coordination environment of YbIII with the labeling scheme. Thermal ellipsoids are shown at 50% probability. H atoms and crystallization water molecules are omitted for clarity. Color code: YbIII (dark orange); CuII (cyan); O (red); N (blue). | ||
The links are supported by two kinds of bridges: three μ2 anti–anti carboxylate bridges per LnIII ion (O1–C2–O2), and three μ2 oxygen (O3) bridges. Hexagonal channels are present in the structure, running along the crystallographic c axis (Fig. 2). The channels are delimited by six CuO4N2 polyhedra and six LnO9 polyhedra. The diameter of the channel is approximately 8.32(1) Å. A comparison with the average channel size along the series [Cu3Ln2(IDA)6] (Fig. S1, ESI†) shows that CuYbIDA presents the narrowest channel among the reported structures.
 | ||
| Fig. 2 Packing of CuYbIDA along the crystallographic c axis, showing the channels. Color code: YbIII, dark orange; CuII, cyan. | ||
The lattice water molecules are situated inside the channels. The TGA profile shows a single weight loss at 60 °C (Fig. S2, ESI†), which corresponds to the eight crystallization water molecules. This fact suggests that water molecules are only weakly bound to the network. The solvent accessible void was calculated using the PLATON program,60 being 26.5% of the total volume of the unit cell.
Catalytic results
The three isostructural catalysts {[Cu3Ln2(IDA)6]·8H2O}n (CuLaIDA, CuGdIDA and CuYbIDA) were used to study the oxidation reaction of cyclohexene in a system where an auxiliary solvent is not used at 75 °C, with molecular oxygen as the oxidant and isobutyraldehyde as the activator. The effect of the amount of molecular oxygen was tested. In the closed “batch” system a first set of reactions were carried out, loading 1 or 5 atm of molecular oxygen in the batch reactor, and permitting the catalytic system to react for 24 h.In preliminary studies, the reactions were carried out in the absence of isobutyraldehyde, and the results after 24 h showed a null conversion, thus demonstrating the importance of the co-oxidant for reactions done under mild conditions. Moreover, it is well known that oxygen activation occurs by direct electron transfer with the metal, or can also be achieved by using aldehydes in combination with metal ions, as was done in our work.61 The mechanism to activate molecular oxygen using isobutyraldehyde has been well described in the literature,61 and corresponds to the initial attack of the activator on the copper(II) centre in a redox reaction. After that, the next step involves the interaction of molecular oxygen with the species generated from the reduction of CuII and the activator to produce the copper–acylperoxy intermediate. This intermediate finally reacts with the substrate to generate the products by a series of radical chain reactions and regenerate the catalyst61 (Scheme S1, ESI†).
Table 1 summarizes the obtained results for 24 h of reaction with an initial pressure of 1 atm. It is possible to observe that the conversion, in the presence of the activator, is similar for the three catalytic systems, with a low value of ca. 25%. However, it is evident that the catalyst modulates the selectivity. Thus, the percentage of cyclohexenone, corresponding to the product of allylic oxidation, is greater for CuLaIDA than for CuGdIDA and CuYbIDA, while the percentage of cyclohexene oxide is the lowest when CuLaIDA is used as the catalyst. Due to the low conversion obtained after 24 h of reaction with an initial pressure of 1 atm, a second set of reactions were carried out, using a pressure of 5 atm of oxidant under the same reaction conditions as for the first set. These reactions were stopped after 3 and 24 hours, in order to study the effect of time on the conversion and selectivity.
| Catalyst | Conversion (%) | Products | Selectivity (%) | TOF (h−1) |
|---|---|---|---|---|
| Reaction conditions: temperature (75 °C), substrate (20 mmole), isobutyraldehyde (4 mmole), catalyst (0.0035 mmole), CuII per mole of catalyst (0.0005 mmole), reaction time (24 h), S/C ratio (2870/1 based on catalyst; 22077/1 based on CuII), [aldehyde]/[substrate] = 0.2. TOF values are calculated as moles of product per mole of active CuII centres. |
||||
| CuLaIDA | 25 | Cyclohexenone | 26 | 230 |
| Cyclohexenol | 35 | |||
| Cyclohexene oxide | 38 | |||
| CuGdIDA | 25 | Cyclohexenone | 20 | 230 |
| Cyclohexenol | 30 | |||
| Cyclohexene oxide | 50 | |||
| CuYbIDA | 25 | Cyclohexenone | 18 | 230 |
| Cyclohexenol | 39 | |||
| Cyclohexene oxide | 43 | |||
| Blank | 5 | Cyclohexenone | 18 | |
| Cyclohexenol | 35 | |||
| Cyclohexene oxide | 47 | |||
Both systems gave approximately the same conversion of ca. 70% (Table 2). The percentage of cyclohexenol remained similar for both reactions (3 and 24 h), regardless of the nature of the used catalyst. However, while the selectivity of cyclohexene oxide was similar for the three catalysts for the 3 h reaction, a slight decrease in the amount of this product was detected for that of 24 h, as the catalyst was changed from the LaIII to the YbIII based catalyst. Besides, a new product was detected after 24 h of reaction for all three catalysts; this was identified as cyclohexanediol.
| Catalyst | Conversion (%) | Product | Selectivity (%) | TOF (h−1) | |||
|---|---|---|---|---|---|---|---|
| 3 h | 24 h | 3 h | 24 h | 3 h | 24 h | ||
| Reaction conditions: temperature (75 °C), substrate (20 mmole), isobutyraldehyde (4 mmole), catalyst (0.0035 mmole), CuII per mole of catalyst (0.0005 mmole), reaction time (24 h), S/C ratio (2870/1 based on catalyst; 22077/1 based on CuII), [aldehyde]/[substrate] = 0.2. TOF values are calculated as moles of product per mole of CuII active centres. |
|||||||
| CuLaIDA | 70 | 73 | Cyclohexenone | 29 | 24 | 7727 | 667 |
| Cyclohexenol | 44 | 41 | |||||
| Cyclohexene oxide | 27 | 23 | |||||
| 1,2-Cyclohexanediol | — | 12 | |||||
| CuGdIDA | 70 | 73 | Cyclohexenone | 34 | 25 | 7727 | 667 |
| Cyclohexenol | 38 | 42 | |||||
| Cyclohexene oxide | 28 | 20 | |||||
| 1,2-Cyclohexanediol | — | 13 | |||||
| CuYbIDA | 70 | 73 | Cyclohexenone | 36 | 31 | 7727 | 667 |
| Cyclohexenol | 37 | 40 | |||||
| Cyclohexene oxide | 27 | 16 | |||||
| 1,2-Cyclohexanediol | — | 13 | |||||
| Blank | 20 | Cyclohexenone | 25 | 28 | |||
| Cyclohexenol | 30 | 35 | |||||
| Cyclohexene oxide | 45 | 37 | |||||
| 1,2-Cyclohexanediol | — | . | |||||
In order to have a more complete idea of the influence of the concentration of molecular oxygen on the conversion and selectivity of the oxidation products, a continuous flow of 1 atm of oxygen was used during 24 h of reaction. These systems presented a conversion between 39 and 70% depending on the used catalyst, as shown in Table 3. As compared with the “batch” experiments the continuous flow systems showed a dramatic dependence on the nature of the catalyst.
| Catalyst | Conversion (%) | Product | Selectivity (%) | TOF (h−1) |
|---|---|---|---|---|
| Reaction conditions: temperature (75 °C), substrate (40 mmole), isobutyraldehyde (8 mmol), catalyst (0.007 mmol), CuII per mol of catalyst (0.0009 mmole), reaction time (24 h), S/C ratio (5740/1 based on catalyst; 44000/1 based on CuII). [aldehyde]/[substrate] = 0.2. TOF values are calculated as moles of product per mole of active CuII centres. |
||||
| CuLaIDA | 73 | Cyclohexenone | 66 | 1343 |
| Cyclohexenol | 30 | |||
| Cyclohexene oxide | 4 | |||
| CuGdIDA | 45 | Cyclohexenone | 26 | 819 |
| Cyclohexenol | 52 | |||
| Cyclohexene oxide | 22 | |||
| CuYbIDA | 39 | Cyclohexenone | 30 | 718 |
| Cyclohexenol | 46 | |||
| Cyclohexene oxide | 24 | |||
| Blank | 15 | Cyclohexenone | 17 | |
| Cyclohexenol | 33 | |||
| Cyclohexene oxide | 50 | |||
A high catalytic activity is appreciated for the CuLaIDA catalyst (73%) with a selectivity favoring the formation of cyclohexenone (66%). In contrast, as the size of the lanthanide ion becomes smaller, the conversion decreases, showing the lowest conversion for the CuYbIDA catalyst (39%). The selectivity is also altered, as the main product for both the CuGdIDA and CuYbIDA catalysts was the conjugated alcohol (52 and 46% respectively). Thus, CuLaIDA is the best catalyst among the studied MOFs for the attainment of the conjugated ketone. These results can be explained by considering the size of the pores present in the studied catalysts, since along the c axis the catalysts present channels. As reported by Kremer et al.62 the size of the pore depends of the secondary metal ion present in the network (Fig. S1, ESI†). Thus when the lanthanide ion radius decreases the pore size decreases, and makes the interaction of the activator and oxygen with the metal centres less feasible. The catalyst with the biggest pore size is CuLaIDA and presents the better catalytic results (conversion: 73%; selectivity: 66% to ketone). Using CuLaIDA, the reusability of this heterometallic polymer in the oxidation reaction was studied in order to determine if it could be considered as a good heterogeneous catalyst. After three catalytic cycles, the activity decreased ca. 15%. ICP analyses were done after each cycle to determine the amount of CuII in solution. After the first cycle the amount of CuII in solution was less than 0.13%, for the second cycle 0.10% and for the third cycle 0.20%. With these results it is possible to infer that the catalytic reactions proceed by a heterogeneous mechanism.
Powder X-ray diffractograms were measured using pristine ground single crystals, and after the first and the third catalytic cycle (Fig. S3, ESI†). The diffractograms of the pristine catalyst and the recycled catalyst, after the first cycle, were indexed with unit cell parameters closely related to the ones obtained from single crystal data (Table S7 and Fig. S4, S5, ESI†), proving the stability of the catalyst.
However, after the third cycle the diffractogram of the reused catalyst showed additional and broader peaks, indicating a modification of the crystal structure. Furthermore, the FTIR spectra of the solid obtained after the third cycle also showed differences, as compared with the one of the single crystals of the pristine catalyst and the one after the first cycle (Fig. S6, ESI†). Vibration bands due to the ligand (IDA) indicated a modification of the catalyst.
When comparing the “batch” results at 1 atm with those obtained for the constant flow of molecular oxygen at 1 atm, the first conclusion is that the amount of oxygen is determinant for the conversion and the selectivity percentages obtained with the different catalysts. For example, for CuLaIDA the conversion increased from 24 to 73%, while the percentage of cyclohexenone from 23 to 66%. On the other hand, the percentage of cyclohexenol remains invariable, while that of cyclohexene oxide decreased dramatically from 38 to 4%. For CuYbIDA the effect is less evident, since the conversion increases only from 26 to 39%. In addition, the selectivity to cyclohexenone also increases from 18 to 31%, while that of cyclohexenol from 39 to 46%, and cyclohexene oxide decreases from 43 to 24%. Thus, the results of the “batch” and flow systems are affected by the way that the oxidant is provided to the reacting mixture, the effect being more evident for CuLaIDA than for CuYbIDA, the latter with the smallest channels.
Analogous reactions for 24 h were studied in order to determine the influence of the nature of the substrate on the conversion and selectivity of the studied system. For this purpose, cycloheptene (C7) and cyclooctene (C8) were also oxidized under a continuous oxygen flow, using CuLaIDA as the catalyst. The results are summarized in Table S8 (ESI†). A similar conversion value ca. 50% was observed for both substrates; however, a difference in the selectivities is well defined. For C7 the presence of “over-oxidation” products was detected, unlike for C8 where these products were not observed. Nevertheless, the high selectivity for cyclooctene epoxide (90%) proved the influence of the ring size over the reactivity of the double bond, favouring the epoxidation for larger rings such as C8, and the allylic oxidation for smaller rings like C7 or C6.27,63
Previous reports have shown an interesting dependence of the conversion and the selectivity on the [aldehyde]/[substrate] ratio used in the catalytic reactions.61,63–65 The authors reported that when the ratio is greater than two, the epoxidation is first-order dependent on the aldehyde, and the conversion and selectivity to epoxide are increased. In order to corroborate this trend, experiments using [aldehyde]/[cyclohexene] ratios from 0.2 to 3.0 were done (Fig. 3).
 | ||
| Fig. 3 Dependence of the conversion on the [aldehyde]/[cyclohexene] ratio. Reaction conditions: temperature (75 °C), isobutyraldehyde (variable), catalyst (0.007 mmole), CuII per mole of catalyst (0.13), reaction time (24 h), S/C ratio (5740/1 based on catalyst; 44000/1 based on CuII). | ||
The obtained results were opposite to those reported in the literature; the conversion decreased as the [aldehyde]/[substrate] ratio increased. The results have been rationalized by assuming that as the concentration of aldehyde in the reacting mixture increased the pores were occupied by the activator, making the diffusion of the substrate into the pores more difficult. In this way the activation process would be slowed down.
The selectivity was as expected, as when the [aldehyde]/[substrate] ratio increased, the selectivity to epoxide was enhanced. Thus, when the ratio used was three, the selectivity to cyclohexene oxide reached 60% (Table S9, ESI†). However, when the ratio was less than one, the main product was different from epoxide. The oxidation occurred preferably over the allylic position, the main products being cyclohexenol (41%, for a ratio of 0.5), and cyclohexanone (66%, for a ratio of 0.2).
Since the TGA analyses showed that the water molecules present in the channels were lost at ca. 60 °C (Fig. S2, ESI†), and the reactions were done at 75 °C, it was assumed that the water molecules present inside the channels were partially removed.
To determine the relevance of the pore size, the reactions were also done with catalysts having a previous thermal treatment at 100 °C to ensure that the water molecules present inside the pores were completely removed before the catalytic reaction started. The obtained results are summarized in Table 4.
| Catalyst | Conversion (%) | Selectivity (%) | TOF (h−1) | |
|---|---|---|---|---|
| Reaction conditions: temperature (75 °C), substrate (40 mmole), isobutyraldehyde (8 mmole), catalyst (0.007 mmole), CuII per mole of catalyst (0.0009 mmole), reaction time (24 h), S/C ratio (5740/1 based on catalyst; 44000/1 based on CuII). [aldehyde]/[substrate] = 0.2. TOF values are calculated as moles of product per mole of active CuII centres. |
||||
| CuLaIDA | 99 | Cyclohexenone | 76 | 1821 |
| Cyclohexenol | 18 | |||
| Cyclohexene oxide | 0 | |||
| 1,2-Cyclohexanediol | 6 | |||
| CuGdIDA | 77 | Cyclohexenone | 43 | 1417 |
| Cyclohexenol | 37 | |||
| Cyclohexene oxide | 15 | |||
| 1,2-Cyclohexanediol | 5 | |||
| CuYbIDA | 74 | Cyclohexenone | 37 | 1361 |
| Cyclohexenol | 40 | |||
| Cyclohexene oxide | 18 | |||
| 1,2-Cyclohexanediol | 5 | |||
The systems maintained the tendency observed for the reactions without the activation of the catalysts. However, the conversion increased ca. 26–35% for the different catalysts, when these were previously thermally activated.
Besides, all the studied systems became slightly more oxidizing as compared with the one with the catalyst without thermal treatment, evidenced by the appearance of a small amount of 1,2-cyclohexanediol among the oxidation products, and also by the increase of the selectivity of cyclohexenone. The catalyst CuLaIDA remains the most active catalyst for the family with a conversion of 99% after 24 h of reaction, and a selectivity of 76% towards the generation of cyclohexenone. These results confirm that the pore size is determinant in the activity of this family of catalysts, and that the partial or total occupancy of the pores by the water molecules affects the catalytic results.
Electronic structure calculations
Besides the inorganic polymer pore size, there could be another structural and/or electronic factor related to the higher catalytic activity of CuLaIDA in comparison to the GdIII and YbIII derivatives. To check this point, electronic structure calculations were performed for the three structures. Considering the CuII centre as the active site, models describing the internal surface of the channels for CuLaIDA, CuGdIDA and CuYbIDA were built. The model considers a central CuII ion surrounded by four LnIII centres and four distant CuII positions, which were replaced by redox inactive ZnII ions. In this way, calculations for the reduction process of CuII focus on the central ion of the model (technical details are presented in the computational details section and the model structure is depicted in Fig. S8, ESI†).After geometry optimization of the hydrogen positions, the vertical electron affinity was calculated using one GGA and one hybrid density functional (BP86 and B3LYP, respectively). In all cases, the reduced state associated with the formation of copper(I) stabilizes when advancing from LaIII to YbIII (Table 5). In this way, the higher catalytic activity of CuLaIDA cannot be related to changes in the redox properties of the copper centre. Thus, the pore size appears as the key factor determining the differences in reactivity observed for CuLaIDA, CuGdIDA and CuYbIDA.
| DFT functional | ||
|---|---|---|
| B3LYP | BP | |
| CuLaIDA | 2.00 | 2.44 |
| CuGdIDA | 3.49 | 4.64 |
| CuYbIDA | 3.66 | 5.13 |
Conclusions
For the studied catalysts, the amount of oxygen is determinant when the reactions were done in a batch reactor. On the other hand, the nature of the catalyst was important when the reactions were done with a constant oxygen flow of 1 atm.The [aldehyde]/[substrate] ratio was shown to control the catalytic parameters. When the reaction was done using a ratio of less than 1, the conversion increased and the main product was cyclohexenone or cyclohexenol. On the other hand, when the ratio was higher than 1, the conversion decreased and the main product was the epoxide.
The pore size was found to be the main factor to explain the different results. DFT calculations showed that the higher catalytic activity of CuLaIDA could not be related to changes in the redox properties of the copper centre. Besides, when the reactions were done using thermally activated catalysts, the conversion increased. Thus, when the water molecules present in the channels were eliminated by the thermal treatment, the catalytic reactions were enhanced.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Proyecto Anillo ACT 1404, and Proyecto Basal CEDENNA FB0807, LIA 3M-CNRS 1207 grant. C. K. thanks CSIC (Uruguay) for financial support through Programa de Apoyo a Grupos de Investigación. Powered@NLHPC: this research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02).References
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. – Eur. J., 2016, 22, 8012–8024 CrossRef CAS.
- A. R. Sheldon and J. K. Kochi, Metal Complex Catalyzed Oxidation of Organic Compounds, Academic P., 1981 Search PubMed.
- G. Cainelli and G. Cardillo, Chromium Oxidation in Organic Chemistry, Springer-V., 1984 Search PubMed.
- J. I. Kroschwitz and K. Othmer, Encyclopedia of Chemical Technology Wiley-Interscience, Wiley-Interscience, New York, 1992 Search PubMed.
- F. Ullmann, Ullmann's Encyclopedia of Industrial Chemistry, 2003 Search PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2011, 1, 856–867 RSC.
- M. Liniger, Y. Liu and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 13944–13949 CrossRef CAS PubMed.
- D. Dang, P. Wu, C. He, Z. Xie and C. Duan, J. Am. Chem. Soc., 2010, 132, 14321–14323 CrossRef CAS PubMed.
- S. Goswami, H. S. Jena and S. Konar, Inorg. Chem., 2014, 53, 7071–7073 CrossRef CAS PubMed.
- J. Long, H. Liu, S. Wu, S. Liao and Y. Li, ACS Catal., 2013, 3, 647–654 CrossRef CAS.
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS.
- F. Ke, L. Wang and J. Zhu, Nanoscale, 2015, 7, 1201–1208 RSC.
- H. L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 CrossRef CAS PubMed.
- D. Sun, L. Ye and Z. Li, Appl. Catal., B, 2015, 164, 428–432 CrossRef CAS.
- Y. Yang, H. F. Yao, F. G. Xi and E. Q. Gao, J. Mol. Catal. A: Chem., 2014, 390, 198–205 CrossRef CAS.
- A. Santiago-Portillo, S. Navalón, F. G. Cirujano, F. X. L. I. Xamena, M. Alvaro and H. Garcia, ACS Catal., 2015, 5, 3216–3224 CrossRef CAS.
- I. Luz, F. X. Llabrés I Xamena and A. Corma, J. Catal., 2010, 276, 134–140 CrossRef CAS.
- T. T. Hoang, T. A. To, V. T. T. Cao, A. T. Nguyen, T. T. Nguyen and N. T. S. Phan, Catal. Commun., 2017, 101, 20–25 CrossRef CAS.
- T. A. Fernandes, C. I. M. Santos, V. André, J. Kłak, M. V. Kirillova and A. M. Kirillov, Inorg. Chem., 2016, 55, 125–135 CrossRef CAS PubMed.
- Z. Wang, G. Laddha, S. Kanitkar and J. J. Spivey, Catal. Today, 2017, 298, 209–215 CrossRef CAS.
- G. Zhang, Y. Shi, Y. Wei, Q. Zhang and K. Cai, Inorg. Chem. Commun., 2017, 86, 112–117 CrossRef CAS.
- K. Leus, M. Vandichel, Y. Y. Liu, I. Muylaert, J. Musschoot, S. Pyl, H. Vrielinck, F. Callens, G. B. Marin, C. Detavernier, P. V. Wiper, Y. Z. Khimyak, M. Waroquier, V. Van Speybroeck and P. Van Der Voort, J. Catal., 2012, 285, 196–207 CrossRef CAS.
- J. W. Brown, Q. T. Nguyen, T. Otto, N. N. Jarenwattananon, S. Glöggler and L. S. Bouchard, Catal. Commun., 2015, 59, 50–54 CrossRef CAS.
- S. A. Sotnik, R. A. Polunin, M. A. Kiskin, A. M. Kirillov, V. N. Dorofeeva, K. S. Gavrilenko, I. L. Eremenko, V. M. Novotortsev and S. V. Kolotilov, Inorg. Chem., 2015, 54, 5169–5181 CrossRef CAS PubMed.
- Y. S. Raupp, C. Yildiz, W. Kleist and M. A. R. Meier, Appl. Catal., A, 2017, 546, 1–6 CrossRef CAS.
- D. Ruano, M. Díaz-García, A. Alfayate and M. Sánchez-Sánchez, ChemCatChem, 2015, 7, 674–681 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 836–840 CrossRef CAS.
- T. Sato, W. Mori, C. N. Kato, E. Yanaoka, T. Kuribayashi, R. Ohtera and Y. Shiraishi, J. Catal., 2005, 232, 186–198 CrossRef CAS.
- Z. Yu, Z. Liao, Y. Jiang, G. Li, G. Li and J. Chen, Chem. Commun., 2004, 1814–1815 RSC.
- Z. T. Yu, Z. L. Liao, Y. S. Jiang, G. H. Li and J. S. Chen, Chem. – Eur. J., 2005, 11, 2642–2650 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. A. Asiri and H. Garcia, Catal. Sci. Technol., 2016, 6, 5238–5261 RSC.
- J. S. Wang, F. Z. Jin, H. C. Ma, X. B. Li, M. Y. Liu, J. L. Kan and G. J. Chen, and Y. Bin Dong, Inorg. Chem., 2016, 55, 6685–6691 CrossRef CAS PubMed.
- G. J. Chen, J. S. Wang, F. Z. Jin, M. Y. Liu, C. W. Zhao, Y. A. Li and Y. B. Dong, Inorg. Chem., 2016, 55, 3058–3064 CrossRef CAS PubMed.
- H. Duan, Y. Zeng, X. Yao, P. Xing, J. Liu and Y. Zhao, Chem. Mater., 2017, 29, 3671–3677 CrossRef CAS.
- D. Sun, F. Sun, X. Deng and Z. Li, Inorg. Chem., 2015, 54, 8639–8643 CrossRef CAS PubMed.
- Y. Fu, D. Sun, M. Qin, R. Huang and Z. Li, RSC Adv., 2012, 2, 3309 RSC.
- P. Cancino, A. Vega, A. Santiago-Portillo, S. Navalon, M. Alvaro, P. Aguirre, E. Spodine and H. García, Catal. Sci. Technol., 2016, 6, 3727–3736 RSC.
- P. Cancino, V. Paredes-García, C. Aliaga, P. Aguirre, D. Aravena and E. Spodine, Catal. Sci. Technol., 2017, 7, 231–242 RSC.
- P. Cancino, L. Santibáñez, P. Fuentealba, C. Olea, A. Vega and E. Spodine, Dalton Trans., 2018, 47, 13360–13367 RSC.
- A. Zhang, L. Li, J. Li, Y. Zhang and S. Gao, Catal. Commun., 2011, 12, 1183–1187 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 48–53 CrossRef CAS.
- I. Y. Skobelev, A. B. Sorokin, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2013, 298, 61–69 CrossRef CAS.
- Y. Ha, M. Mu, Q. Liu, N. Ji, C. Song and D. Ma, Catal. Commun., 2018, 103, 51–55 CrossRef CAS.
- U. Bruker-AXS, APEX3. 2016, Bruker AXS: Madison, Wisconsin.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 2008–2010 CrossRef.
- C. S. Manna, S. Konar, E. Zangrando, J. Ribas and N. Ray, Polyhedron, 2007, 26, 2507–2516 CrossRef.
- M. Xu, F. Liao, J. Li, H. Sun, Z. Li and S. Gao, Inorg. Chem. Commun., 2003, 6, 841–844 CrossRef CAS.
- R. Yan-ping, L. La-Sheng, M. Bing-Wei, Y. You-Zhu, H. Rong-bin and Z. Lan-Sun, Angew. Chem., Int. Ed., 2003, 42, 1999–2002 Search PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, e1327 Search PubMed.
- A. D. Becke, Phys. Rev., 1988, 38, 3098–3100 CAS.
- E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher and J. Pleiss, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 5648, 5648 CrossRef.
- J. P. Perdew, Phys. Rev., 1986, 33, 8822–8824 Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 22, 1057–1065 RSC.
- F. Weigend, R. Ahlrichs and F. K. Gmbh, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Dolg, H. Stoll, H. Preuss, M. Oolg, H. Stoll, H. Preuss and W. Germany, J. Chem. Phys., 1989, 90, 1730 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., 2009, D65, 148–155 CrossRef PubMed.
- P. Chotmongkolsap, T. Bunchuay, W. Klysubun and J. Tantirungrotechai, Eur. J. Inorg. Chem., 2018, 703–712 CrossRef CAS.
- J. Torres, P. Morales, S. Domínguez, J. González-platas, R. Faccio, J. Castiglioni, A. W. Mombrú and C. Kremer, J. Mol. Struct., 2011, 1004, 215–221 CrossRef CAS.
- U. Junghans, C. Suttkus, J. Lincke, D. Lässig, H. Krautscheid and R. Gläser, Microporous Mesoporous Mater., 2014, 216, 151–160 CrossRef.
- Z. Li, S. Wu, H. Ding, D. Zheng, J. Hu, X. Wang, Q. Huo, J. Guan and Q. Kan, New J. Chem., 2013, 5, 1561–1568 RSC.
- B. B. Wentzel, P. A. Gosling, M. C. Feiters and R. J. M. Nolte, J. Chem. Soc., Dalton Trans., 1998, 2241–2246 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1878174. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj02091h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |