DOI:
10.1039/C8NJ05212C
(Paper)
New J. Chem., 2019,
43, 77-84
Red-emitting dyes based on phenothiazine-modified 2-hydroxychalcone analogues: mechanofluorochromism and gelation-induced emission enhancement†
Received
13th October 2018
, Accepted 14th November 2018
First published on 14th November 2018
Abstract
New D–π–A type phenothiazine-modified 2-hydroxychalcone analogues PTNn (n = 2, 4, 8, 12, and 16) have been synthesized, and they exhibited reversible mechanofluorochromism. Notably, the emission of PTNn in different solid-states was in the range of 600–800 nm, and red-emitting mechanofluorochromic (MFC) materials were seldom reported. The as-synthesized crystals of PTN16 emitted red light centered at 667 nm, and were changed into deep red emitting ground powders after grinding. The fluorescence emission could be recovered to the original state when the ground powders were fumed with the DCM vapor or heated. The MFC processes were accompanied by the transformation between crystalline and amorphous states, which could be confirmed based on the results of XRD patterns in different solid-states. Additionally, it was found that the length of the alkyl chain affected the performance in mechanofluorochromism for PTNn. The dye bearing a short carbon chain showed high-contrast MFC behavior compared with the one bearing a long carbon chain. Moreover, PTN12 and PTN16 bearing long carbon chains could self-assemble into organogels in a certain amount of organic solvents, and gelation-induced emission enhancement was observed during organogelation.
Introduction
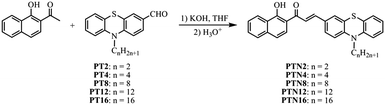
Recently, solid-state fluorescent materials have been receiving increasing attention in the fields of organic light emitting diodes (OLEDs), optoelectronic devices, solid-state organic lasers and fluorescent sensors.1 Particularly, as a kind of emitter with tuneable solid-state emission adjusted by external forces such as grinding, compression, shearing and so on, mechanofluorochromic (MFC) materials have attracted attention recently due to their potential applications in mechanosensors, optical recording, security papers, data storage, and optoelectronic devices.2,3 Generally, the changes in molecular packing modes in different solid-states are deemed to be responsible for the MFC behavior.4 To date, it is still a challenge to design MFC dyes with high performance and to reveal the relationship between molecular structures and properties. Recently, some π-conjugated organic molecules have been found to exhibit MFC properties. For example, Araki et al. and Kato et al. found that 10,10′-bis(phenylethynyl)-9,9′-bianthryl derivatives were responsive to force stimuli.5 Harima designed a series of benzofuro[2,3-c]oxazolo[5,4-a]carbazole-based fluorescent dyes with mechanofluorochromism, and suggested that D–π–A type emissive compounds contributed to the realization of fluorescence changes stimulated by external forces.6 In addition, nonplanar tetraphenylethene, 9,10-divinylanthracene, and triphenylamine derivatives are preferentially considered to act as MFC materials.7 Phenothiazine is also an excellent functional block for the construction of organic luminescent materials,8 and has been introduced into MFC materials.9 It should be noted that the solid red-emitting MFC materials were seldom reported since the molecules with low-lying excited states often contain extended conjugated systems or donor/acceptor moieties, which would lead to strong π–π interactions as well as an ACQ effect.10 The development of red-emitting MFC materials remains a challenge. Following these considerations, we designed new D–π–A type conjugated compounds PTNn (n = 2, 4, 8, 12, and 16, Scheme 1), in which phenothiazine acted as a donor and a 2-hydroxychalcone analogue was used as an acceptor, which were bridged by a vinyl group. Although the emission of PTNn in solutions was weak, they showed red fluorescence in the aggregated states. Interestingly, their emitting colors were changed between red and deep red upon the treatment of grinding/fuming with DCM. We also found that PTNn bearing a short carbon chain showed high-contrast performance in mechanofluorochromism compared with the one bearing a long carbon chain.11 Besides, the red-emitting organogels of PTN12 and PTN16 could be generated driven by van der Waals forces and π–π interactions, and gelation-induced emission enhancement was detected.
 |
| | Scheme 1 Synthetic routes to PTNn. | |
Results and discussion
Synthesis
The synthetic routes to 3-phenothiazinyl-1-(1-hydroxynaphthalenyl)prop-2-en-1-one PTNn (n = 2, 4, 8, 12, and 16) are shown in Scheme 1. Firstly, compounds PTn were synthesized according to a reported method.12PTNn (n = 2, 4, 8, 12, and 16) were obtained via Knoevenagel reactions of 1-(1-hydroxynaphthalen-2-yl)ethanone and PTn catalyzed by KOH in dry THF. The target compounds were characterized by 1H NMR, 13C NMR, MALDI-TOF mass spectrometry and FT-IR. They are soluble in common solvents cyclohexane, toluene, THF, DCM and DMF.
Photophysical properties
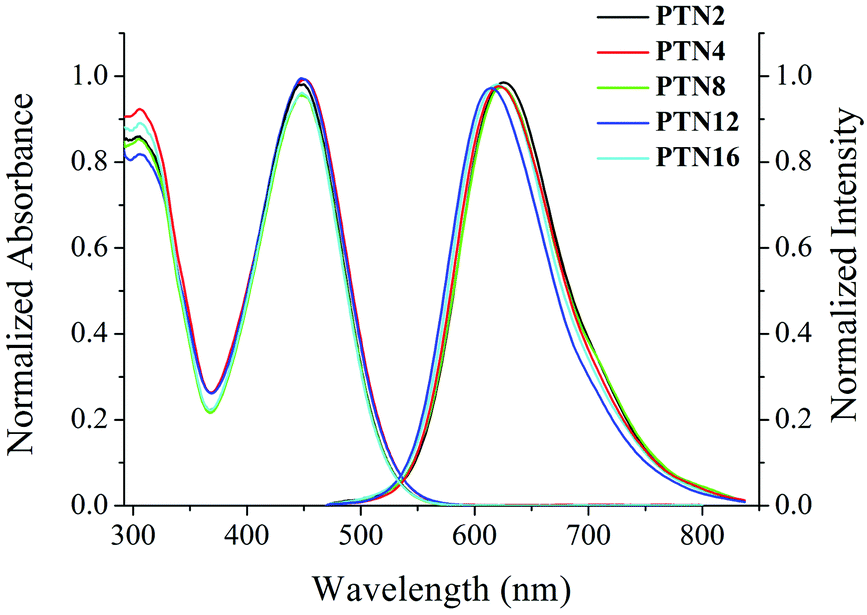
The UV-vis absorption and fluorescence emission spectra of PTNn (n = 2, 4, 8, 12, and 16) in toluene (1.0 × 10−5 M) are shown in Fig. 1. Since the conjugated skeletons are the same, PTNn exhibited quite similar absorption and emission spectra. The absorption bands of PTNn appeared at ca. 305 nm and ca. 449 nm, wherein, the first one might be derived from π–π* transition, and the second one could be ascribed to intramolecular charge-transfer (ICT) transition. In addition, the emission band of PTNn was located at ca. 620 nm in toluene. Besides, we found that the ICT absorption of the PTNn was red-shifted in more polar solvents. For example, the ICT absorption of PTN2 was red-shifted to 457 nm in DMF from 435 nm in cyclohexane (Fig. S1a, ESI†).13 Upon increasing the polarity of the solvents the significant solvatochromism in fluorescence was detected. As shown in Fig. S1b (ESI†), the maximal emission of PTN2 was located at 589 nm in cyclohexane and was red-shifted to 715 nm in DMF. Thus, the Stokes shifts of PTN2 increased obviously with increasing solvent polarities, and they were 6011 cm−1 and 7895 cm−1 in cyclohexane and DMF, respectively. This suggests that the emission of PTNn in polar solvents has resulted from ICT transitions.14 It should be noted that the emission of PTNn was weak in solutions, and the fluorescence quantum yields (ΦF) were in the range of 0.01–0.05 using Rhodamine 6G (1.0 × 10−5 M) in ethanol (ΦF = 0.94) as the standard.
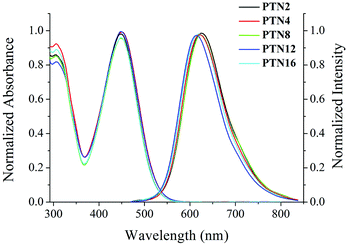 |
| | Fig. 1 Normalized UV-vis absorption and fluorescence emission (λex = 450 nm) spectra of PTNn in toluene (1.0 × 10−5 M). | |
MFC properties of PTNn
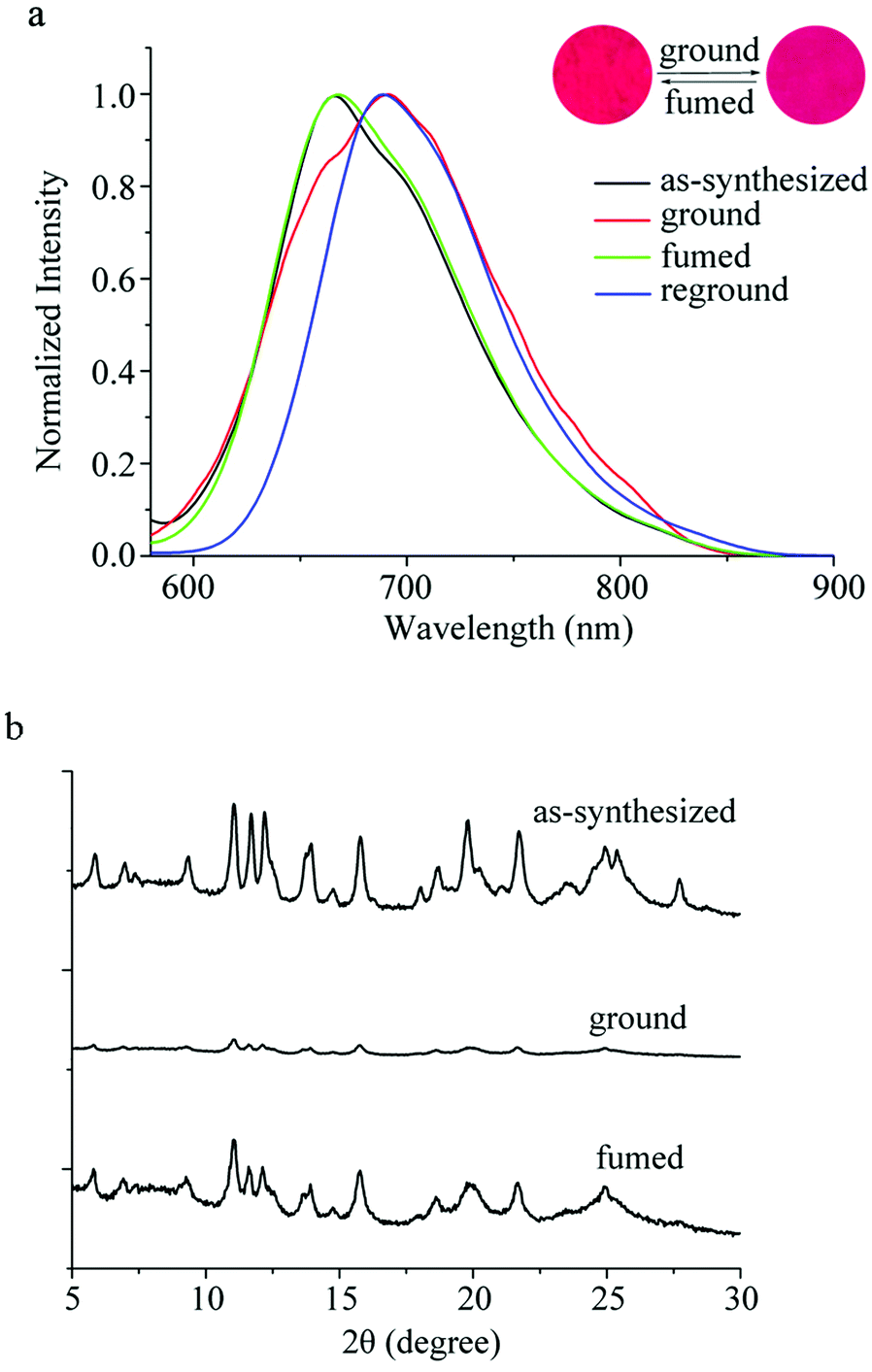
It has been found that some phenothiazine derivatives exhibit MFC behavior,12 so we investigated the solid-state fluorescence changes of the synthesized compounds stimulated by external mechanical forces. As shown in Fig. 2a and Table S2 (ESI†), the as-synthesized sample of PTN16 shows an emission band at 667 nm with a fluorescence quantum yield of 0.07, and emitted red light under UV irradiation. The grinding of the as-synthesized sample resulted in the emitting color to turn deep red, accompanied by the red-shift of the emission to 690 nm. When the ground powders were exposed to the DCM vapor for 4 s or heated at 150 °C for 3 s, the emission recovered to 668 nm. Such an MFC process could be reversibly repeated several times (Fig. S2, ESI†)
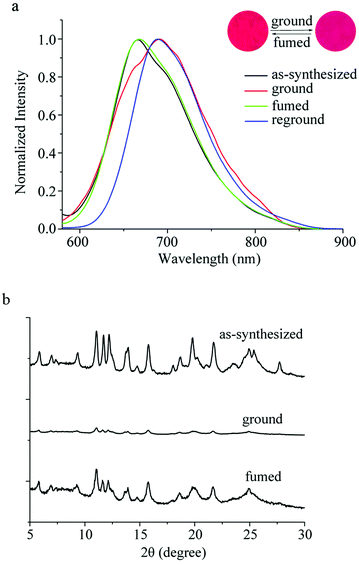 |
| | Fig. 2 (a) Fluorescence emission spectra (λex = 450 nm) and (b) XRD patterns of PTN16 in different solid-states. Inset: Photos of PTN16 in different solid-states under 365 nm light. | |
In order to reveal the MFC mechanism, we measured the UV-vis spectra and the XRD patterns of PTN16 in different solid-states as shown in Fig. S3 (ESI†) and Fig. 2b. As shown in Fig. S3 (ESI†), compared with the as-synthesized sample, a red-shift of 18 nm for the ICT absorption of PTN16 was observed in the ground powders. After fuming the ground powders, it was blue-shifted from 502 nm to 485 nm. Moreover, the absorption of PTN16 in solution was located at ca. 450 nm. We deemed that the aggregated degree was different in the as-synthesized sample and in the ground powders. As shown in Fig. 2b, the as-synthesized sample of PTN16 exhibited sharp and intense diffraction peaks, indicative of the well-ordered microcrystalline structures. In contrast, only quite weak diffraction peaks were observed for the ground powders, indicating the amorphous state.15 After fuming by the DCM vapor, sharp and strong diffraction peaks similar to those of the as-synthesized crystals appeared again, suggesting the regeneration of the crystalline structures. Therefore, the phase transition between crystalline and amorphous states could be responsible for the MFC behavior of PTN16.16 The MFC behaviors of PTNn (n = 2, 4, 8 and 12) were also studied to reveal the effect of the length of the alkyl chain on their MFC properties. It was found that they also exhibited reversible MFC processes, accompanied by the transition between crystalline and amorphous states (Fig. S2–S5, ESI†). Notably, the shifts of the emission bands for PTNn between the as-synthesized crystals and the ground powders were different, and they were 38 nm, 36 nm, 30 nm, 26 nm and 23 nm for PTN2, PTN4, PTN8, PTN12 and PTN16, respectively. This illustrates that the shorter the N-alkyl chain of PTNn, the more remarkable the MFC performance.17 To reveal the reason for the different MFC behaviors of PTNn, the UV-vis spectra in different solid-states are shown in Fig. S3 (ESI†) and the absorption wavelengths are listed in Table S2 (ESI†). The absorption of the as-synthesized samples for PTN2 and PTN4 bearing short carbon chains appeared at ca. 470 nm, but those for PTN8, PTN12 and PTN16 bearing long carbon chains emerged at ca. 480 nm. This meant that the π–π interactions in the as-synthesized samples of PTNn with long carbon chains were stronger than those with short carbon chains.17a–c Upon grinding, the red-shifts of the absorption bands were observed, and the shift values decreased upon increasing the length of the carbon chains (Table S2, ESI†), because changing the aggregated states of PTNn with long carbon chains might be more difficult than others. This work has demonstrated that the subtle manipulation of alkyl groups of phenothiazine could endow these compounds with unique and tunable solid-state optical properties.
Gelation properties
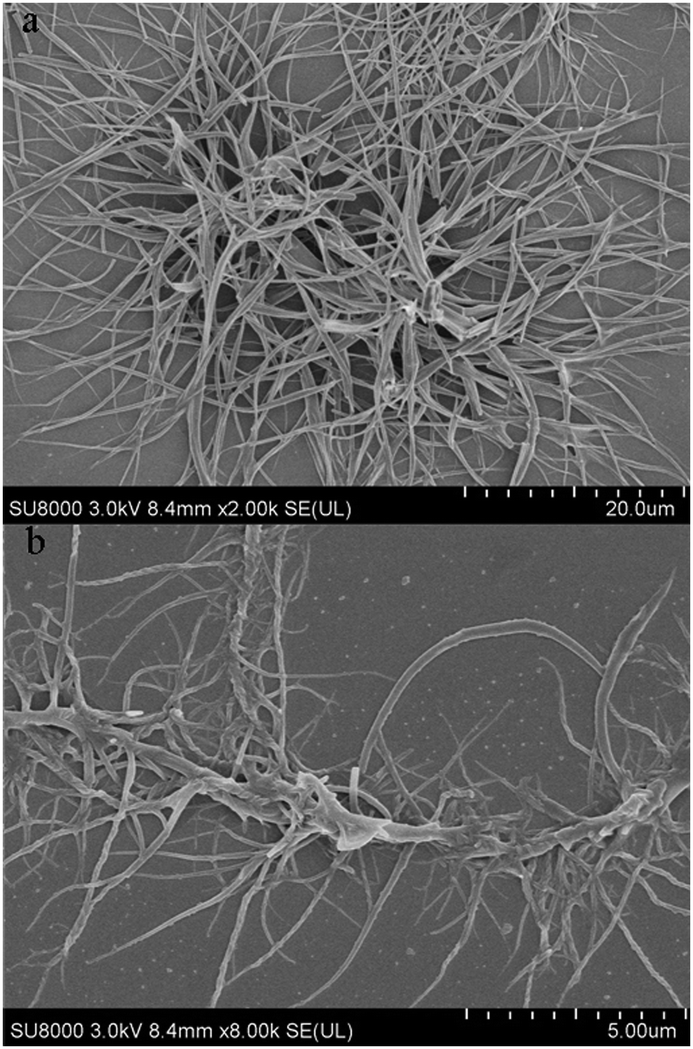
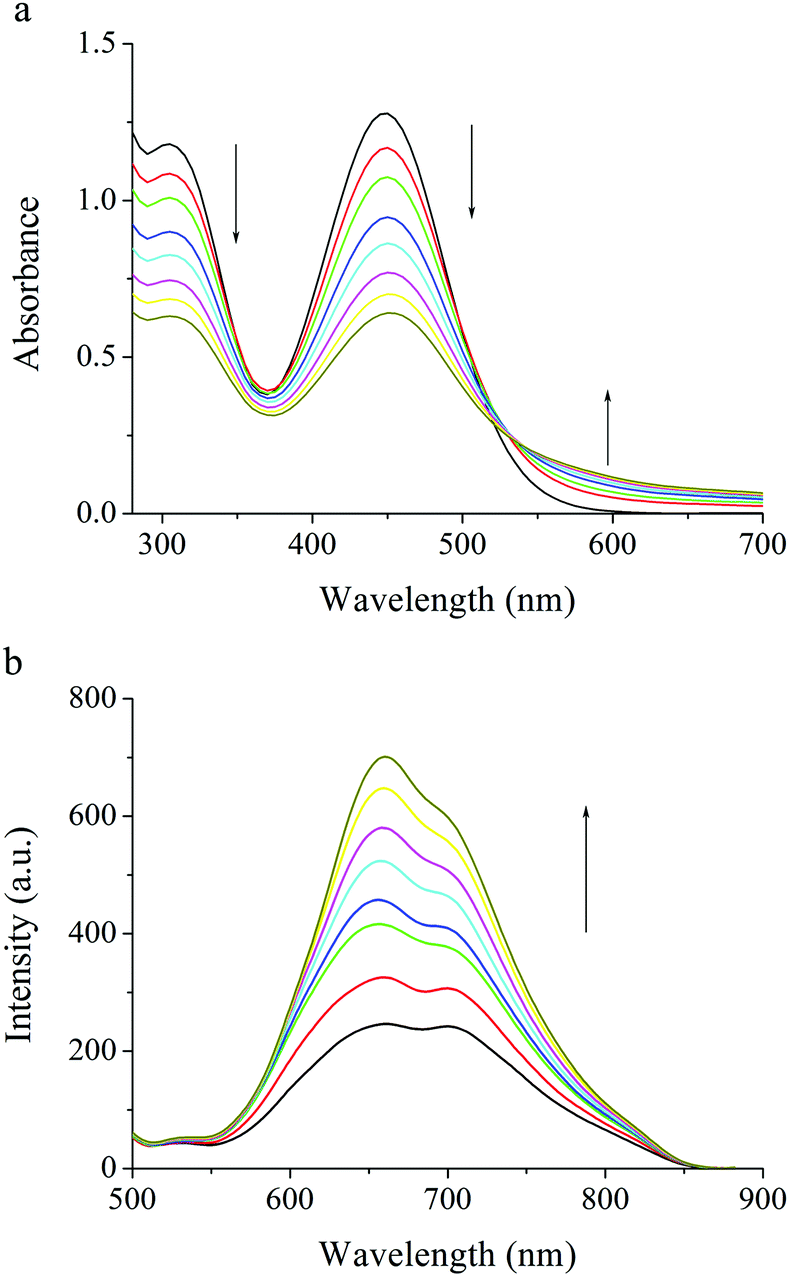
The gelation properties of PTNn were tested in the selected solvents by means of the “stable to inversion of a test tube” method,18 and the results are listed in Table 1. It was found that PTN12 and PTN16 could form organogels in solvents n-hexane, n-heptane, cyclohexane, methylcyclohexane, methanol, n-butanol, iso-butanol, tert-butanol, n-pentanol, tert-pentanol, DMSO and acetonitrile under ultrasound stimulation. The critical gelation concentrations (CGCs) of PTN12 and PTN16 depended on the solvents and varied in the ranges of 5.0–20.0 mg mL−1 and 1.1–10.0 mg mL−1, respectively. It was clear that in the same solvent the CGC for PTN16 was lower than that for PTN12. For instance, the CGCs were 1.1 mg mL−1 and 6.7 mg mL−1 for PTN16 and PTN12 in n-butanol, respectively, meaning that the gelation ability of PTN16 was stronger than that of PTN12. It was found that the obtained n-butanol gel of PTN16 was stable for several days at room temperature, and that of PTN12 was destroyed for several hours at room temperature, further indicating the stronger gelation ability of PTN16 than PTN12. The gained gels could be transformed into solutions upon heating, which could recover to organogels upon stimulation by ultrasound, followed by cooling to room temperature. However, PTN2, PTN4 and PTN8 could not form any gel in the selected solvents. Therefore, we deduced that the van der Waals forces between long carbon chains might play key roles in the gelation of PTN12 and PTN16. The morphologies of the xerogels PTN16 and PTN12 obtained from n-butanol are depicted in Fig. 3. It is clear that 3D networks constructed from 1D nanofibers with diameters of 100–350 nm are formed from PTN16. PTN12 also self-assembled to form 3D networks consisting of a large number of entangled nanofibers with diameters of 0.2–1.5 μm in n-butanol. In order to reveal the driving forces for the gelation, the time-dependent UV-vis absorption and fluorescence emission spectra during the gelation of PTN16 are shown in Fig. 4. We could find that the absorption peaks at ca. 300 nm and 444 nm were decreased gradually upon gelation, and the maximal absorption was red-shifted to 451 nm in the gel state. Meanwhile, the absorption over ca. 530 nm increased gradually, suggesting the molecular aggregation during the gelation of PTN16.19 As depicted in Fig. 4b, PTN16 gave weak emission at 660 nm in hot n-butanol. During the gelation process, the emission intensities were increased gradually. PTN12 exhibited similar electronic spectral changes during gelation in n-butanol (Fig. S6, ESI†). Such gelation-induced emission enhancement originated from molecular aggregation, which will be discussed below. Therefore, π–π interactions played an important role in the formation of the gel.
Table 1 Gelation ability of PTNn organic solventsa
| Solvent |
PTN2
|
PTN4
|
PTN8
|
PTN12 (CGC)a |
PTN16 (CGC) |
|
I: insoluble; S: soluble; G: stable gel; P: precipitate. CGC: critical gelation concentration (mg mL−1).
|
|
n-Hexane |
P |
P |
P |
G (6.7) |
G (4.0) |
|
n-Heptane |
P |
P |
P |
G (5.0) |
G (3.3) |
| Cyclohexane |
P |
P |
P |
G (10.0) |
G (6.7) |
| Methylcyclohexane |
S |
S |
S |
G (10.0) |
G (6.7) |
| Methanol |
P |
P |
P |
G (10.0) |
G (4.0) |
|
n-Butanol |
P |
P |
P |
G (6.7) |
G (1.1) |
| Iso-butanol |
P |
P |
P |
G (8.0) |
G (3.3) |
|
tert-Butanol |
P |
P |
P |
G (10.0) |
G (4.0) |
|
n-Pentanol |
P |
P |
P |
G (8.0) |
G (6.7) |
|
tert-Pentanol |
P |
P |
P |
G (10.0) |
G (3.3) |
| DMSO |
S |
S |
S |
G (20.0) |
G (10.0) |
| Acetonitrile |
S |
S |
S |
G (6.7) |
G (4.0) |
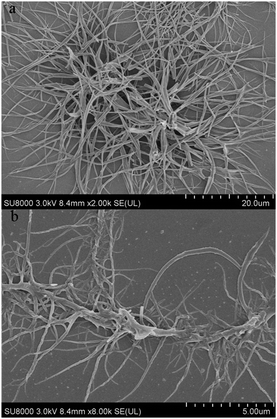 |
| | Fig. 3 SEM microscopy images of xerogels PTN16 and PTN12 obtained from n-butanol. | |
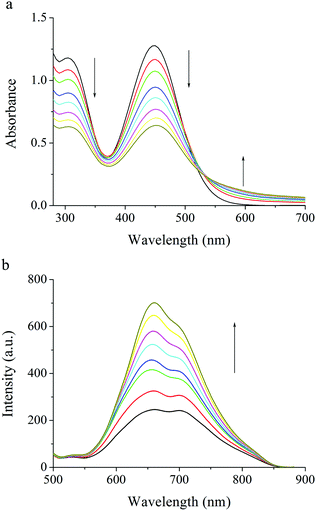 |
| | Fig. 4 Time-dependent (a) UV-vis spectra and (b) fluorescence emission spectra of PTN16 (λex = 450 nm) upon aging the hot solution in n-butanol, which is first stimulated by ultrasound. | |
The FT-IR spectra of PTN16 in chloroform (1 wt%), in xerogels obtained from n-butanol and in the powders are shown in Fig. S7b (ESI†). It was found that the antisymmetric (νas) and symmetric (νs) stretching frequencies of CH2 appeared at 2925 cm−1 and 2861 cm−1 in solution. They were shifted to 2919 cm−1 and 2849 cm−1 in xerogels, and shifted to 2920 cm−1 and 2851 cm−1 in the powders. Therefore, the fluidity of the hydrophobic chains was decreased due to the formation of aggregates driven by van der Waals forces.20 In addition, based on the FT-IR spectra of PTN12 in different states it is also illustrated that van der Waals force was one of the driving forces for the gelation (Fig. S7a, ESI†). The XRD patterns of xerogels PTN12 and PTN16 obtained from n-butanol are shown in Fig. S8 (ESI†), and we could find several intense diffraction peaks. Although the molecular stacking mode cannot be deduced, we deemed that the molecules arranged into ordered structures in gel states.
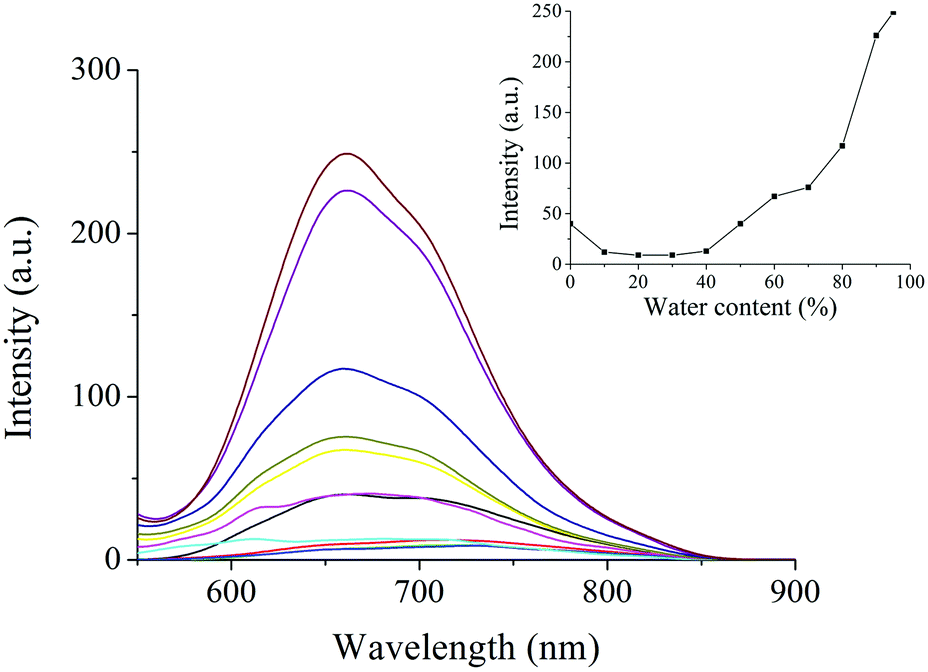
To further understand the gelation-induced emission enhancement, the fluorescence spectra of PTN16 in THF/water with different water contents are shown in Fig. 5. It was clear that PTN16 was almost non-emissive in THF/water when the water content was below 50%. If the water content was over 50%, the emission intensity was increased upon increasing the water content. When the water content was 95%, the emission intensity at 660 nm for PTN16 was ca. 6 times higher than that in THF. We deemed that the intramolecular rotations would be restricted in aggregated states, thus turning on the emission.21PTN12 in THF/water also showed similar AIE activity (Fig. S9, ESI†).
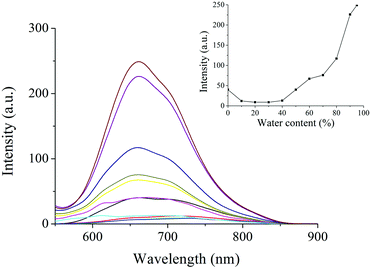 |
| | Fig. 5 Fluorescence emission spectra of PTN16 in THF/water with different water contents (λex = 450 nm). Inset: The plot of the emission intensities at 660 nm vs. water contents. | |
Conclusions
In summary, we have synthesized new phenothiazine-modified 2-hydroxychalcone analogues PTNn (n = 2, 4, 8, 12, and 16). Interestingly, these compounds showed red fluorescence in the solid-state and exhibited reversible mechanofluorochromism. XRD patterns in different solid-states suggested that the transformation between crystalline and amorphous states was responsible for the MFC behavior. In addition, the shifts of the maximal emission peak during MFC processes decreased with the increasing of the length of the carbon chain in PTNn on account of the strong π–π interactions in the as-synthesized crystals of PTNn with long carbon chains. Additionally, the organogels with red-emission could be generated from PTN12 and PTN16 driven by π–π and van der Waals interactions. This demonstrates that the red emission could be tuned by the phase transitions of D–π–A type 2-hydroxychalcone analogues.
Experimental section
Measurement and characterization
1H NMR and 13C NMR spectra were recorded using a Varian Mercury Plus instrument at 400 MHz and 100 MHz in DMSO-d6 and CDCl3 as solvents and tetramethylsilane (TMS) as the internal standard in all cases. FT-IR spectra were recorded on a Nicolet-360 FT-IR spectrometer by incorporation of samples into KBr disks. The UV-vis absorption spectra were obtained using a Shimadzu UV-3100 spectrophotometer. Fluorescence emission spectra were measured using a Cary Eclipse fluorescence spectrophotometer. Mass spectra were obtained using Agilent 1100 MS series and AXIMA CFR MALDI-TOF (Compact) mass spectrometers. Scanning electron microscopy (SEM) was performed using a JEOL JSM-6700F (operating at 5 kV). The samples were prepared by casting the organogels on silicon wafers and dried at room temperature, followed by coating with gold. The XRD patterns were obtained using an Empyrean X-ray diffraction instrument equipped with graphite-monochromatized Cu-Kα radiation (λ = 1.5418 Å) by employing a scanning rate of 0.026° s−1 in the 2θ range of 5°–30°. The data were collected when the samples were heated for the first time. The fluorescence decay curves were measured using an Edinburgh FLS920 steady state fluorimeter equipped with an nF900 nanosecond flash lamp. The optimized molecular configuration was used to obtain the frontier orbitals by density functional theory (DFT) calculations at the B3LYP/6-31G(d) level with the Gaussian 09W program package.
THF was dried over sodium and benzophenone. The other reagents were used as received without further purification.
Synthesis
Compounds PTn (n = 2, 4, 8, 12, and 16) were synthesized according to a reported method.12
(E)-3-(10-Ethyl-10H-phenothiazin-3-yl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (PTN2).
Anhydrous potassium hydroxide (0.6 g, 10.8 mmol) was added to a solution of 1-(1-hydroxynaphthalen-2-yl)ethanone (0.50 g, 2.7 mmol) and 10-ethyl-10H-phenothiazine-3-carbaldehyde (PT2) (0.69 g, 2.7 mmol) in dry THF (20 mL). The mixture was stirred at 70 °C for 12 h. After cooling to room temperature, the mixture was acidified with hydrochloric acid to pH = 1 and extracted with DCM. The combined organic phases were washed with brine and dried with anhydrous sodium sulfate. After the solvent was removed, the residue was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 3/1) to afford 0.93 g of a wine solid (83% in yield). M.p.:160.7–162.5 °C. 1H NMR (DMSO-d6, 400 MHz, ppm) δ 15.25 (s, 1H), 8.38–8.33 (t, J = 9.8 Hz, 2H), 8.07 (d, J = 15.2 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.88 (dd, J = 8.4, 6.8 Hz, 2H), 7.73 (t, J = 7.4 Hz, 2H), 7.61 (t, J = 7.6 Hz, 1H), 7.46 (d, J = 9.2 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.07 (d, J = 8.4 Hz, 2H), 6.98 (t, J = 7.4 Hz, 1H), 3.99 (q, J = 6.8 Hz, 2H), 1.33 (t, J = 7.0 Hz, 3H) (Fig. S10, ESI†). 13C NMR (101 MHz, CDCl3, ppm) δ 192.94 (s), 164.30 (s), 147.22 (s), 144.01 (s), 143.59 (s), 137.30 (s), 130.06 (s), 129.32 (s), 128.98 (s), 127.70–127.29 (m), 126.57 (s), 125.85 (s), 125.54 (s), 124.52 (d, J = 9.2 Hz), 123.95 (s), 123.13 (d, J = 14.6 Hz), 118.03 (d, J = 17.4 Hz), 115.30 (s), 114.80 (s), 113.56 (s), 42.21 (s), 12.89 (s). (Fig. S11, ESI†) MALDITOF MS (m/z): calcd for C27H21NO2S: 423.1; found: 423.5, (Fig. S12, ESI†) FT-IR (KBr, cm−1): 3423, 2960, 2894, 1740, 1623, 1600, 1567, 1467, 1442, 1391, 1371, 1242, 1063, 801, 743.
(E)-3-(10-Butyl-10H-phenothiazin-3-yl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (PTN4).
PTN4 was synthesized according to the similar synthetic method for PTN2 from 1-(1-hydroxynaphthalen-2-yl)ethanone (0.50 g, 2.7 mmol), 10-butyl-10H-phenothiazine-3-carbaldehyde (PT4) (0.76 g, 2.7 mmol) and anhydrous potassium hydroxide (0.6 g, 10.8 mmol) in dry THF (20 mL). The crude product was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 3/1) to afford 0.95 g of a wine solid (80% in yield). M.p.: 146.3–147.9 °C; 1H NMR (DMSO-d6, 400 MHz, ppm) δ 15.26 (s, 1H), 8.36 (t, J = 7.6 Hz, 2H), 8.08 (d, J = 15.2 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 14.8 Hz, 2H), 7.78–7.71 (m, 2H), 7.61 (t, J = 7.2 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.22 (d, J = 6.8 Hz, 1H), 7.18 (dd, J = 7.6, 1.2 Hz, 1H), 7.10 (dd, J = 8.4, 5.2 Hz, 2H), 6.99 (t, J = 7.0 Hz, 1H), 3.95 (t, J = 7.0 Hz, 2H), 1.69 (dt, J = 14.4, 7.2 Hz, 2H), 1.42 (dd, J = 14.8, 7.4 Hz, 2H), 0.89 (t, J = 7.4 Hz, 3H). (Fig. S13, ESI†) 13C NMR (101 MHz, CDCl3) δ 192.95 (s), 164.28 (s), 147.66 (s), 144.02 (d, J = 6.3 Hz), 137.31 (s), 130.06 (s), 129.26 (s), 129.02 (s), 127.75–127.25 (m), 126.69 (s), 125.85 (s), 125.54 (s), 125.14 (s), 124.48 (s), 123.85 (d, J = 17.7 Hz), 123.09 (s), 118.04 (d, J = 14.8 Hz), 115.67 (s), 115.19 (s), 113.56 (s), 47.49 (s), 28.89 (s), 20.14 (s), 13.81 (s) (Fig. S14, ESI†). MALDITOF MS (m/z): calcd for C29H25NO2S: 451.2; found: 451.3 (Fig. S15, ESI†). FT-IR (KBr, cm−1): 3424, 2958, 1742, 1625, 1560, 1497, 1469, 1442, 1408, 1373, 1243, 1059, 807, 749.
(E)-1-(1-Hydroxynaphthalen-2-yl)-3-(10-octyl-10H-phenothiazin-3-yl)prop-2-en-1-one (PTN8).
PTN8 was synthesized according to the similar synthetic method for PTN2 from 1-(1-hydroxynaphthalen-2-yl)ethanone (0.50 g, 2.7 mmol), 10-octyl-10H-phenothiazine-3-carbaldehyde (PT8) (0.92 g, 2.7 mmol) and anhydrous potassium hydroxide (0.6 g, 10.8 mmol) in dry THF (20 mL). The crude product was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 3/1) to afford 1.07 g of a wine solid (78% in yield). M.p.: 134.2–135.9 °C; 1H NMR (DMSO-d6, 400 MHz, ppm) δ 15.25 (s, 1H), 8.38–8.33 (t, J = 9.8 Hz, 2H), 8.07 (d, J = 15.6 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 14.4 Hz, 2H), 7.74 (t, J = 7.8 Hz, 2H), 7.61 (t, J = 7.4 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.22(t, J = 7.8 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 7.07 (dd, J = 8.8, 4.8 Hz, 2H), 6.99 (t, J = 7.4 Hz, 1H), 3.94 (t, J = 6.8 Hz, 2H), 1.70 (dt, J = 14.4, 7.0 Hz, 2H), 1.36 (m, 2H), 1.20 (m, 8H), 0.82 (t, J = 6.6 Hz, 3H) (Fig. S16, ESI†) 13C NMR (101 MHz, CDCl3, ppm) δ 192.95 (s), 164.30 (s), 147.64 (s), 144.01 (d, J = 9.2 Hz), 137.31 (s), 130.06 (s), 129.26 (s), 129.01 (s), 127.87–127.29 (m), 126.68 (s), 125.85 (s), 125.55 (s), 125.11 (s), 124.48 (s), 123.94 (s), 123.73 (s), 123.08 (s), 118.04 (d, J = 15.6 Hz), 115.66 (s), 115.18 (s), 113.56 (s), 47.82 (s), 31.74 (s), 29.20 (d, J = 2.2 Hz), 26.84 (d, J = 10.4 Hz), 22.63 (s), 14.11 (s). (Fig. S17, ESI†). MALDI-TOF MS (m/z): calcd for C33H33NO2S: 507.2; found: 507.7 (Fig. S18, ESI†). FT-IR (KBr, cm−1): 3432, 2923, 2852, 1743, 1624, 1564, 1468, 1439, 1370, 1245, 1208, 1062, 799, 750.
(E)-3-(10-Dodecyl-10H-phenothiazin-3-yl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (PTN12).
PTN12 was synthesized according to the similar synthetic method for PTN2 from 1-(1-hydroxynaphthalen-2-yl)ethanone (0.50 g, 2.7 mmol), 10-dodecyl-10H-phenothiazine-3-carbaldehyde (PT12) (1.07 g, 2.7 mmol) and anhydrous potassium hydroxide (0.6 g, 10.8 mmol) in dry THF (20 mL). The crude product was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 3/1) to afford 1.21 g of a wine solid (80% in yield). M.p.: 114.2–116.0 °C; 1HNMR (DMSO-d6, 400 MHz, ppm) δ 15.25 (s, 1H), 8.36 (t, J = 9.6 Hz, 2H), 8.07 (d, J = 15.2 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 14.4 Hz, 2H), 7.73 (t, J = 8.0 Hz, 2H), 7.61 (d, J = 7.6 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 7.07 (dd, J = 8.4, 4.8 Hz, 2H), 6.98 (t, J = 7.4 Hz, 1H), 3.93 (t, J = 6.8 Hz, 2H), 1.69 (dt, J = 14.0, 6.8 Hz, 2H), 1.42–1.35 (m, 2H), 1.27–1.18 (m, 16H), 0.82 (t, J = 6.6 Hz, 3H) (Fig. S19, ESI†). 13C NMR (101 MHz, CDCl3, ppm) δ 192.95 (s), 164.30 (s), 147.66 (s), 144.02 (d, J = 8.2 Hz), 137.31 (s), 130.06 (s), 129.26 (s), 129.00 (s), 127.69–127.29 (m), 126.67 (s), 125.85 (s), 125.55 (s), 125.12 (s), 124.48 (s), 123.94 (s), 123.74 (s), 123.07 (s), 118.03 (d, J = 16.4 Hz), 115.65 (s), 115.17 (s), 113.56 (s), 47.80 (s), 31.93 (s), 29.74 – 29.08 (m), 26.83 (d, J = 9.8 Hz), 22.71 (s), 14.15 (s). (Fig. S20, ESI†). MALDI-TOF MS (m/z): calcd for C37H41NO2S: 563.3; found: 563.9 (Fig. S21, ESI†). FT-IR (KBr, cm−1): 3440, 2920, 2852, 1743, 1625, 1602, 1567, 1470, 1372, 1247, 1064, 800, 750.
(E)-3-(10-Hexadecyl-10H-phenothiazin-3-yl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (PTN16).
PTN16 was synthesized according to the similar synthetic method for PTN2 from 1-(1-hydroxynaphthalen-2-yl)ethanone (0.50 g, 2.7 mmol), 10-hexadecyl-10H-phenothiazine-3-carbaldehyde (PT16) (1.22 g, 2.7 mmol) and anhydrous potassium hydroxide (0.6 g, 10.8 mmol) in dry THF (20 mL). The crude product was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 3/1) to afford 1.32 g of a wine solid (79% in yield). M.p.: 97.5–99.0 °C; 1H NMR (DMSO-d6, 400 MHz, ppm) δ 15.26 (s, 1H), 8.35 (t, J = 8.4 Hz, 2H), 8.07 (d, J = 15.6 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 15.2 Hz, 2H), 7.73 (t, J = 8.0 Hz, 2H), 7.61 (t, J = 7.4 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.22 (t, J = 8.0 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 7.07 (dd, J = 8.0, 5.6 Hz, 2H), 6.99 (t, J = 7.4 Hz, 1H), 3.94 (t, J = 6.8 Hz, 2H), 1.69 (dt, J = 14.4, 7.2 Hz, 2H), 1.43–1.34 (m, 2H), 1.28–1.18 (m, 24H), 0.81 (t, J = 6.6 Hz, 3H) (Fig. S22, ESI†). 13C NMR (101 MHz, CDCl3, ppm) δ 192.94 (s), 164.30 (s), 147.65 (s), 144.02 (d, J = 8.1 Hz), 137.30 (s), 130.05 (s), 129.26 (s), 129.00 (s), 127.69–127.24 (m), 126.67 (s), 125.84 (s), 125.55 (s), 125.11 (s), 124.48 (s), 123.94 (s), 123.73 (s), 123.07 (s), 118.03 (d, J = 16.6 Hz), 115.65 (s), 115.17 (s), 113.56 (s), 47.80 (s), 31.94 (s), 29.56 (ddd, J = 32.9, 17.8, 9.6 Hz), 26.83 (d, J = 9.6 Hz), 22.71 (s), 14.15 (s). (Fig. S23, ESI†) MALDITOF MS (m/z): calcd for C41H49NO2S: 619.4; found: 619.9 (Fig. S24, ESI†). FT-IR (KBr, cm−1): 3441, 2921, 2849, 1743, 1623, 1568, 1470, 1441, 1371, 1210, 1064, 800, 750.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51773076 and 21503090) and the Open Project of State Key Laboratory of Supramolecular Structure and Materials (SKLSSM201813).
Notes and references
-
(a) C. Wang, X. L. Li, Y. Y. Pan, S. T. Zhang, L. Yao, Q. Bai, W. J. Li, P. Lu, B. Yang, S. J. Su and Y. G. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 3041–3049 CrossRef CAS;
(b) J. S. Wu, W. M. Liu, J. C. Ge, H. Y. Zhang and P. F. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC;
(c) Y. Q. Hong, L. Meng, S. Chen, C. W. T. Leung, L. T. Da, M. Faisal, D. A. Silva, J. Liu, J. W. Y. Lam, X. Huang and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 1680–1689 CrossRef CAS;
(d) S. X. Li, Q. Q. Zhu, L. Wang, D. M. Tang, Y. J. Cho, X. J. Liu, N. Hirosaki, T. Nishimura, T. Sekiguchi, Z. R. Huang and R. J. Xie, J. Mater. Chem. C, 2016, 4, 8197–8205 RSC;
(e) D. Li, H. Y. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC;
(f) H. Xin, W. Yan and S. A. Jenekhe, ACS Omega, 2018, 3, 12549–12553 CrossRef CAS;
(g) P. Pander, S. Gogoc, M. Colella, P. Data and F. B. Dias, ACS Appl. Mater. Interfaces, 2018, 10, 28796–28802 CrossRef CAS.
-
(a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS;
(b) Z. G. Chi, X. Q. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. W. Liu and J. R. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC;
(c) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175–5178 CrossRef CAS;
(d) S. Umar, A. K. Jha, D. Purohit and A. Goel, J. Org. Chem., 2017, 82, 4766–4773 CrossRef CAS;
(e) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS;
(f) P. C. Xue, J. P. Ding, P. P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688–6706 RSC;
(g) C. D. Dou, L. Han, S. S. Zhao, H. Y. Zhang and Y. Wang, J. Phys. Chem. Lett., 2011, 2, 666–670 CrossRef CAS;
(h) P. Wen, Z. X. Gao, R. Zhang, A. R. Li, F. Zhang, J. Li, J. J. Xie, Y. Z. Wu, M. Wu and K. P. Guo, J. Mater. Chem. C, 2017, 5, 6136–6143 RSC.
-
(a) M. P. Aldred, G. F. Zhang, C. Li, G. Chen, T. Chen and M. Q. Zhu, J. Mater. Chem. C, 2013, 1, 6709–6716 RSC;
(b) A. Pucci and G. Ruggeri, J. Mater. Chem., 2011, 21, 8282–8291 RSC;
(c) M. Louis, A. Brosseau, R. Guillot, F. Ito, C. Allain and R. Metivier, J. Phys. Chem. C, 2017, 121, 15897–15907 CrossRef CAS;
(d) F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC;
(e) X. Cheng, D. Li, Z. Y. Zhang, H. Y. Zhang and Y. Wang, Org. Lett., 2014, 16, 880–883 CrossRef CAS PubMed;
(f) Y. Q. Dong, J. W. Y. Lan and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429–3435 CrossRef CAS;
(g) P. C. Xue, Z. C. Yang and P. Chen, J. Mater. Chem. C, 2018, 6, 4994–5000 RSC.
-
(a) Y. J. Zhang, J. W. Sun, G. L. Zhuang, M. Ouyang, Z. W. Yu, F. Cao, G. X. Pan, P. S. Tang, C. Zhang and Y. G. Ma, J. Mater. Chem. C, 2014, 2, 195–200 RSC;
(b) Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 52, 12268–12272 CrossRef CAS;
(c) F. Liu, J. Tu, X. R. Wang, J. Q. Wang, Y. B. Gong, M. M. Han, X. X. Dang, Q. Y. Liao, Q. Peng, Q. Q. Li and Z. Li, Chem. Commun., 2018, 54, 5598–5601 RSC;
(d) C. D. Dou, D. Chen, J. Iqbal, Y. Yuan, H. Y. Zhang and Y. Wang, Langmuir, 2011, 27, 6323–6329 CrossRef CAS PubMed;
(e) M. S. Kwon, J. Gierschner, J. Seo and S. Y. Park, J. Mater. Chem. C, 2014, 2, 2552–2557 RSC;
(f) K. P. Guo, F. Zhang, S. Guo, K. Li, X. Q. Lu and J. Li, Chem. Commun., 2017, 53, 1309–1312 RSC.
-
(a) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS;
(b) S. Yamane, Y. Sagara, T. Mutai, K. Araki and T. Kato, J. Mater. Chem. C, 2013, 1, 2648–2656 RSC.
-
(a) Y. Ooyama, G. Ito, H. Fukuoka, T. Nagano, Y. Kagawa, I. Imae, K. Komaguchi and Y. Harima, Tetrahedron, 2010, 66, 7268–7271 CrossRef CAS;
(b) Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372–8380 RSC.
-
(a) P. C. Xue, P. Chen, J. H. Jia, Q. X. Xu, J. B. Sun, B. Q. Yao, Z. Q. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569–2571 RSC;
(b) R. Misra, T. Jadhav, B. Dhokale and S. Mobin, Chem. Commun., 2014, 50, 9076–9078 RSC;
(c) X. Q. Zhang, Z. G. Chi, Y. Zhang, S. W. Liu and J. R. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC;
(d) C. P. Ma, B. J. Xu, G. Y. Xie, J. J. He, X. Zhou, B. Y. Peng, L. Jiang, B. Xu, W. J. Tian, Z. G. Chi, S. W. Liu, Y. Zhang and J. R. Xu, Chem. Commun., 2014, 50, 7374–7377 RSC;
(e) S. S. Zhao, L. Chen, L. Wang and Z. G. Xie, Chem. Commun., 2017, 53, 7048–7051 RSC;
(f) Q. K. Sun, K. Zhang, Z. Z. Zhang, L. L. Tang, Z. L. Xie, Z. G. Chi, S. F. Xue, H. C. Zhang and W. J. Yang, Chem. Commun., 2018, 54, 8206–8209 RSC;
(g) R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2014, 20, 8320–8324 CrossRef CAS;
(h) J. B. Wei, B. Y. Liang, X. Cheng, Z. L. Zhang, H. Y. Zhang and Y. Wang, RSC Adv., 2015, 5, 71903–71910 RSC.
-
(a) J. H. Jia, K. Y. Cao, P. C. Xue, Y. Zhang, H. P. Zhou and R. Lu, Tetrahedron, 2012, 68, 3626–3632 CrossRef CAS;
(b) S. F. Xue, Y. J. Wu, Y. S. Lu, X. Xu, Q. K. Sun and W. J. Yang, J. Mater. Chem. C, 2017, 5, 11700–11707 RSC;
(c) Y. Y. Gong, Y. Zhang, W. Z. Yuan, J. Z. Sun and Y. M. Zhang, J. Phys. Chem. C, 2014, 118, 10998–11005 CrossRef CAS;
(d) C. Arivazhagan, P. Malakar, R. Jagan, E. Prasad and S. Ghosh, CrystEngComm, 2018, 20, 3162–3166 RSC.
-
(a) P. C. Xue, B. Q. Yao, J. B. Sun, Q. X. Xu, P. Chen, Z. Q. Zhang and R. Lu, J. Mater. Chem. C, 2014, 2, 3942–3950 RSC;
(b) X. A. Zhang, Z. G. Chi, J. Zhang, H. Li, B. J. Xu, X. Li, S. W. Liu, Y. Zhang and J. R. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef CAS PubMed;
(c) C. Arivazhagan, P. Malakar, R. Jagan, E. Prasad and S. Ghosh, CrystEngComm, 2018, 20, 3162–3166 RSC;
(d) M. Okazaki, Y. H. Takeda, P. Data and P. Pander, Chem. Sci., 2017, 8, 2677–2686 RSC.
-
(a) M. Shimizu, R. Kaki, Y. Takeda, T. Hiyama, N. Nagai, H. Yamagishi and H. Furutani, Angew. Chem., Int. Ed., 2012, 51, 4095–4099 CrossRef CAS;
(b) G. Han, D. Kim, Y. Park, J. Bouffard and Y. Kim, Angew. Chem., Int. Ed., 2015, 54, 3912–3916 CrossRef CAS;
(c) M. Strobl, A. Walcher, T. Mayr, I. Klimant and S. M. Borisov, Anal. Chem., 2017, 89, 2859–2865 CrossRef CAS;
(d) J. Wang, Q. Wu, C. Yu, Y. Wei, X. Mu, E. Hao and L. Jiao, J. Org. Chem., 2016, 81, 11316–11323 CrossRef CAS.
-
(a) Y. Han, H. T. Cao, H. Z. Sun, Y. Wu, G. G. Shan, Z. M. Su, X. G. Hou and Y. Liao, J. Mater. Chem. C, 2014, 2, 7648–7655 RSC;
(b) L. Y. Bu, M. X. Sun, D. T. Zhang, W. Liu, Y. L. Wang, M. Zheng, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2013, 1, 2028–2035 RSC;
(c) B. R. Crenshaw and C. Weder, Chem. Mater., 2003, 15, 4717–4724 CrossRef CAS;
(d) S. Y. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS;
(e) Y. J. Dong, B. Xu, J. B. Zhang, X. Tan, L. J. Wang, J. L. Chen, H. G. Hu, S. P. Wen, B. Li and W. J. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS.
- P. C. Xue, B. Q. Yao, X. L. Liu, J. B. Sun, P. Gong, Z. Q. Zhang, C. Qian, Y. Zhang and R. Lu, J. Mater. Chem. C, 2015, 3, 1018–1025 RSC.
-
(a) Z. H. Guo, Z. X. Jin, J. Y. Wang and J. Pei, Chem. Commun., 2014, 50, 6088–6090 RSC;
(b) Y. Y. Gong, Y. Q. Tan, J. Liu, P. Lu, C. F. Feng, W. Z. Yuan, Y. W. Lu, J. Z. Sun, G. F. He and Y. M. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC;
(c) Y. Sagra and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef.
-
(a) C. Qian, M. Y. Liu, G. H. Hong, P. C. Xue, P. Gong and R. Lu, Org. Biomol. Chem., 2015, 13, 2986–2998 RSC;
(b) J. Hancock, A. Gifford, Y. Zhu, Y. Lou and S. Jenekhe, Chem. Mater., 2006, 18, 4924–4932 CrossRef CAS.
-
(a) B. Zhang, C. H. Hsu, Z. Q. Yu, S. Yang and E. Q. Chen, Chem. Commun., 2013, 49, 8872–8874 RSC;
(b) T. Wen, D. Zhang, J. Liu, R. Lin and J. Zhang, Chem. Commun., 2013, 49, 5660–5662 RSC.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS.
-
(a) S. F. Xue, X. Qiu, Q. K. Sun and W. J. Yang, J. Mater. Chem. C, 2016, 4, 1568–1578 RSC;
(b) Y. L. Wang, W. Liu, L. Y. Bu, J. F. Li, M. Zheng, D. T. Zhang, M. X. Sun, Y. Tao, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2013, 1, 856–862 RSC;
(c) Q. K. Sun, W. Liu, S. A. Ying, L. L. Wang, S. F. Xue and W. J. Yang, RSC Adv., 2015, 5, 73046–73050 RSC;
(d) M. Zheng, M. P. Sun, Y. P. Li, J. F. Wang, L. Y. Bu, S. F. Xue and W. J. Yang, Dyes Pigm., 2014, 102, 29–34 CrossRef CAS;
(e) L. Y. Bu, Y. P. Li, J. F. Wang, M. X. Sun, M. Zheng, W. Liu, S. F. Xue and W. J. Yang, Dyes Pigm., 2013, 99, 833–838 CrossRef CAS;
(f) X. Q. Zhang, Z. G. Chi, B. J. Xu, L. Jiang, X. Zhou, Y. Zhang, S. W. Liu and J. R. Xu, Chem. Commun., 2012, 48, 10895–10897 RSC;
(g) W. Liu, Y. Wang, L. Y. Bu, J. Li, M. X. Sun, D. Zhang, M. Zheng, C. Yang, S. F. Xue and W. J. Yang, J. Lumin., 2013, 143, 50–55 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
-
(a) J. Zhou, Z. F. Chang, B. R. He, M. Du, P. Lu, Z. J. Zhao and B. Z. Tang, Chem. Commun., 2013, 49, 2491–2493 RSC;
(b) H. G. Lu, B. Xu, Y. Dong, F. Chen, Y. Li, Z. Li, J. He, H. Li and W. J. Tian, Langmuir, 2010, 26, 6838–6844 CrossRef CAS PubMed.
- T. Kar, S. Debnath, D. Das, A. Shome and P. K. Das, Langmuir, 2009, 25, 8639–8648 CrossRef CAS.
- S. Kaur, A. Gupta, V. Bhalla and M. Kumar, J. Mater. Chem. C, 2014, 2, 7356–7363 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, and MALDI-TOF MS spectra; photophysical data; UV-vis absorption spectra; fluorescence emission spectra. See DOI: 10.1039/c8nj05212c |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Jiang
Peng
a,
Lu
Zhai
a,
Meng
Sun
a,
Jingbo
Sun
a and
Ran
Lu
*b,
Jiang
Peng
a,
Lu
Zhai
a,
Meng
Sun
a,
Jingbo
Sun
a and
Ran
Lu