
A transition metal-free approach towards synthesis of β-carboline tethered 1,3,4-oxadiazoles via oxidative C–O bond formation†
Dharmender
Singh
a,
Sandip Kumar
Tiwari
b and
Virender
Singh
 *a
*a
aDepartment of Chemistry, Dr B R Ambedkar National Institute of Technology (NIT), Jalandhar, 144011, Punjab, India
bDrug Discovery and Molecular Synthesis Lab, Centre of Biomedical Research, SGPGIMS, Lucknow- 226014, Uttar Pradesh, India. E-mail: singhv@nitj.ac.in; Fax: +91 172-2214692
First published on 16th November 2018
Abstract
An efficient protocol has been developed for one-pot synthesis of biologically interesting β-carboline substituted 1,3,4-oxadiazoles via an I2-assisted oxidative C–O bond formation strategy. This metal-free sequential approach is found to be compatible with diversely substituted 1-formyl β-carbolines and aromatic as well as aliphatic hydrazides, providing access to a variety of multifunctional β-carboline linked 1,3,4-oxadiazole derivatives in good to excellent yields. The methodology was found to be applicable to gram scale synthesis of β-carboline substituted 1,3,4-oxadiazole derivatives. Additionally, β-carboline C1 linked 2-amino-1,3,4-oxadiazoles and bis-1,3,4-oxadiazoles were also synthesized using the same strategy.
Introduction
Naturally occurring β-carboline alkaloids and their synthetic derivatives are widely studied due to their broad spectrum of pharmacological properties, such as anticancer,1 antibacterial, antimalarial, anti-HIV, anti-Alzheimer etc.2 However, the majority of β-carboline alkaloids display anticancer properties via intercalation into DNA,3 and inhibition of cyclin-dependent kinases,4 topoisomerases5 and IkK kinase complexes.6 Importantly, the β-carboline alkaloids account for nearly one quarter of natural products (Fig. 1) and are also accommodated by several commercial drugs (Tadalfil, Abecarnil, Cipargamin etc.).7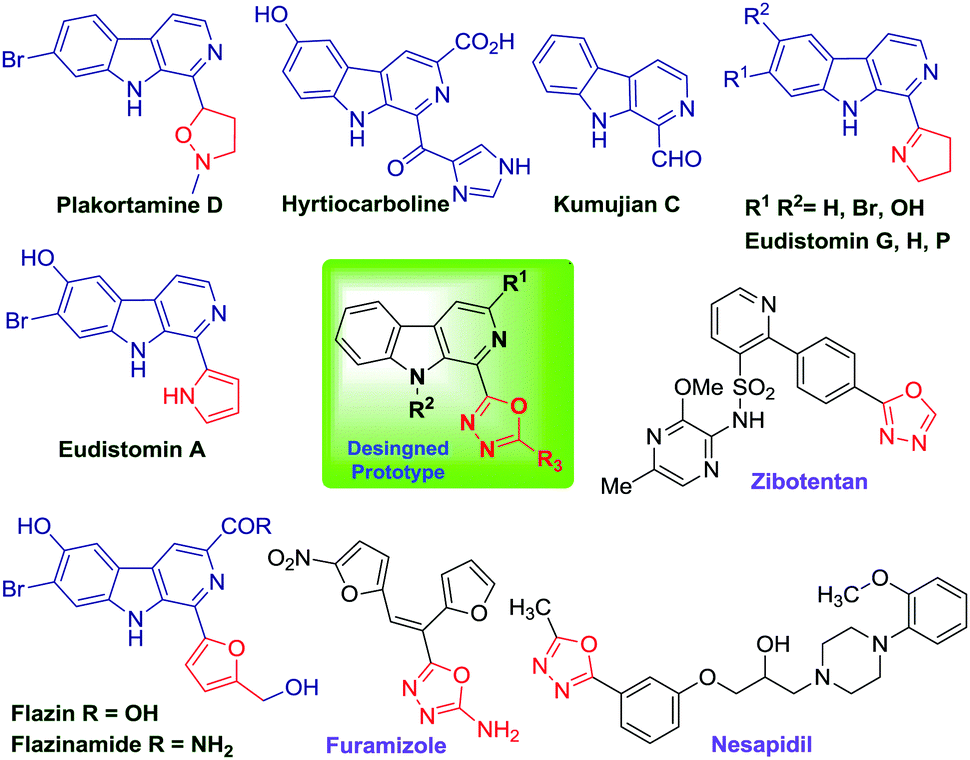 | ||
| Fig. 1 Representative examples of biologically active β-carboline and 1,3,4-oxadiazole derivatives. | ||
Similarly, 1,3,4-oxadiazole is another high value added scaffold which has attracted considerable attention for decades owing to its pharmaceutical and biological activities, such as anti inflammatory, antibacterial, anticancer, anti-diabetic, analgesic, antiviral, antifungal etc.8 Apart from this, 1,3,4-oxadiazole has demonstrated numerous applications in the field of materials science because of its excellent electron transporting and hole-blocking abilities.9 The optoelectronic properties10 of oxadiazole derivatives make them unique among the heterocyclic family. Moreover, the medicinal significance of this motif is evident from the fact that it is represented by numerous drugs such as Raltegravir11 (antiretroviral), Zibotentan12 (anticancer), Fenadiazole13 (hypnotic), Nesapidil14 (antihypertensive), ABT-751-oxadiazole and Furamizole15 (Fig. 1) used for the treatment of various ailments.
The medicinal potential of these two privileged scaffolds inspired us to construct a new molecular hybrid containing both the frameworks. Interestingly, an analysis of the literature revealed the presence of one narrowly related previous report (Fig. 2) in the form of a patent where Guo et al. have disclosed the 8-step synthesis and anti-diabetic properties of tetrahydro-β-carbolines containing 1,3,4-oxadiazole for the treatment of type 2 diabetes mellitus (Fig. 2).16 It was found that these molecular hybrids act as selective antagonists of the somatostatin subtype receptor 3 and could also be used for the treatment of depression, anxiety, insulin resistance, hyperglycemia, lipid disorders, obesity and hypertension. The literature revealed that dehydration of diacyl-hydrazines (POCl3′ SOCl2, PPA, H2SO4)17 and oxidative cyclization of acylhydrazones utilizing oxidants18 like HgO/I2, PbO2, IBX, DMP, chloramine T, KMnO4, CAN, I2, an Br2 or transition metal catalysts (Cu(OTf)2, PdCl2(dppp), CuI, and FeCl3) via C–O coupling reactions are two common approaches employed for the synthesis of 1,3,4-oxadiazoles.19 Recently, the Xu group reported a novel Pd-assisted synthesis of 2-amino-1,3,4-oxadiazoles through isocyanide insertions into hydrazides.20 Nevertheless, the major drawback associated with existing reports involves the use of expensive and hazardous reagents, harsh reaction conditions, multistep synthesis and non-scalability. Among these methods, annulation of acylhydrazides with carbonyl compounds is found to be a more effective approach21 as disclosed by Yu and co-workers.22 However, the substrate scope with acylhydrazides containing electron withdrawing substituents remains unexplored. Patel and co-workers have also developed a Cu(OTf)2 assisted excellent approach toward synthesis of symmetrical and unsymmetrical oxadiazoles.23 Our group has been involved in the development of chemical libraries of β-carboline based molecular hybrids using sustainable approaches. In a recent finding, we have demonstrated the I2-mediated synthesis of β-carboline N-fused imidazoles via decarboxylative oxidation of natural α-amino acids.24a In an extension to our research on the synthesis of β-carboline based privileged scaffolds, we have developed a surrogate approach for the synthesis of β-carboline substituted 1,3,4-oxadiazole derivatives via an iodine-mediated oxidative cyclisation process (Fig. 2). The details of these studies are presented here.
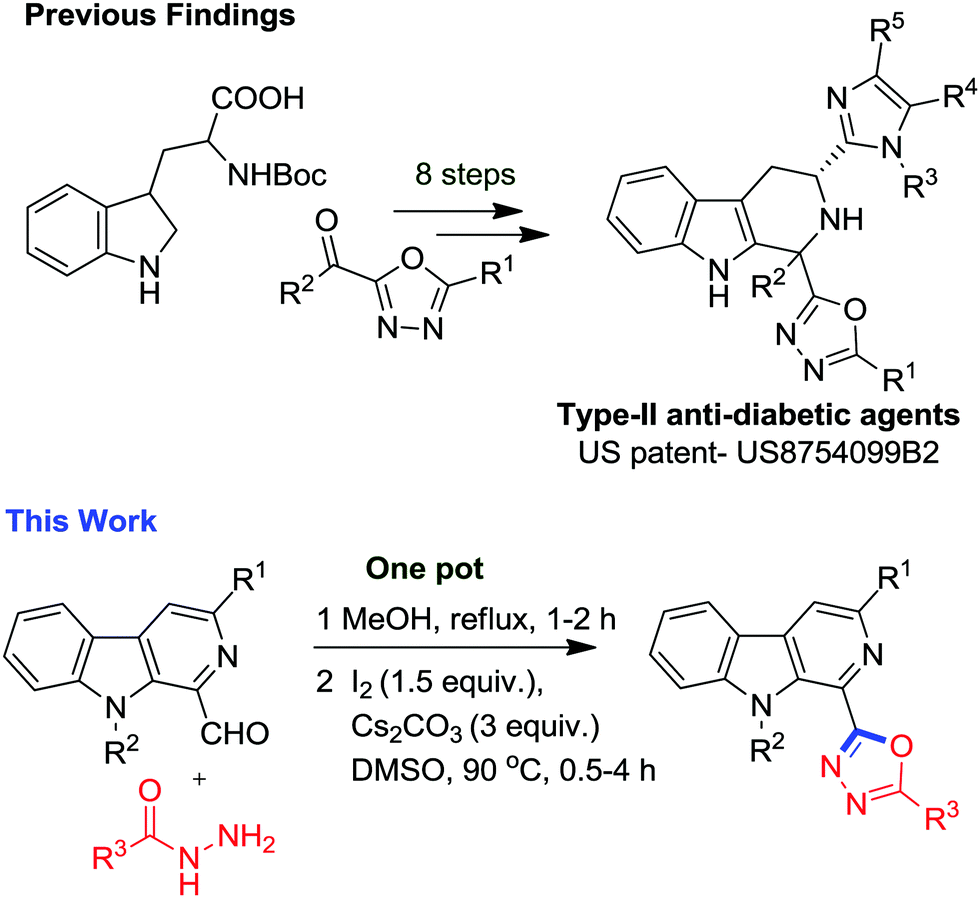 | ||
| Fig. 2 Previous finding related to the synthesis of a designed prototype. | ||
Results and discussion
To achieve the synthesis of the designed molecular hybrid, optimization of the reaction conditions was achieved using 1-formyl β-carboline 1a and benzohydrazide A as the model substrates. The condensation of 1a with A in methanol at 70 °C smoothly afforded the benzohydrazone 1aA in 89% yield without any assistance of Bronsted acid. Next, we explored the oxidative cyclization of the isolated hydrazone 1aA to generate the 1,3,4-oxadiazole framework. Recent studies have revealed that molecular iodine is one of the most versatile promoters and it offers numerous advantages over transition metal catalysts.22,24 Therefore, it has been extensively used in organic synthesis for a variety of transformations. Accordingly, the isolated benzohydrazone 1aA was treated with 1.5 equiv. of iodine in the presence of K2CO3 at 70 °C in MeOH. However, the reaction was very sluggish and it was not completed in 24 h (Table 1, entry 1). The purification of the reaction mixture via column chromatography afforded the desired product, β-carboline C1 substituted 1,3,4-oxadiazole, 2aA in 25% yield as confirmed on the basis of spectroscopic data. Nevertheless, 60% of the starting substrate 1aA was also recovered.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reagentsb | Basec | Solventd | Temp (°C) | Time (h) | Yieldse (%) 2aA |
| a Reaction conditions: All reactions were optimised with 0.134 mmol (1.0 equiv.) benzoylhydrazone 1aA in 1 mL of solvent. b Molecular iodine (1.5 equiv.), KI (2.0 equiv.), TBAI (2.5 equiv.), NCS (2.0 equiv.), DMP (2.5 equiv.), NH4Cl (2.0 equiv.) was used for oxidative cyclisation. c 3.0 equiv. of base was used. d Reactions were performed in anhydrous solvents (entries 1–21). e Isolated yields of the purified product. f NR = No reaction; the starting substrate 1aA was recovered. g 20 mol% catalyst was used and decomposition of benzoylhydrazone 1aA was observed. | ||||||
| 1 | I2 | K2CO3 | MeOH | 70 | 24 | 25% + 1a (60%) |
| 2 | I2 | K2CO3 | DMSO | 90 | 1 | 77 |
| 3 | I2 | NaHCO3 | DMSO | 90 | 1.5 | 70 |
| 4 | I2 | Cs2CO3 | DMSO | rt | 10 | 78 |
| 5 | I 2 | Cs 2 CO 3 | DMSO | 90 | 45 min | 85 |
| 6 | I2 | Cs2CO3 | DMSO | 120 | 45 min | 85 |
| 7 | I2 | Cs2CO3 | DMF | 90 | 1.5 | 65 |
| 8 | I2 | KOH | DMSO | 90 | 1 | 75 |
| 9 | I2 | — | DMSO | 90 | 16 | 66 + polar impurity |
| 10f | I2 | Et3N | DMSO | 90 | 24 | NR |
| 11f | I2 | DIPEA | DMSO | 90 | 24 | NR |
| 12f | I2 | — | AcOH | 90 | 16 | NR |
| 13f | TBAI | Cs2CO3 | DMSO | 90 | 16 | NR |
| 14 | NCS | K2CO3 | DMF | 90 | 12 | 32 + polar impurity |
| 15f | DMP | — | CH2Cl2 | 35 | 24 | NR |
| 16f | NH4Cl | — | DMSO | 90 | 12 | NR |
| 17f,g | FeCl3 | — | DMSO | 90 | 12 | NR |
| 18f,g | CuI | Cs2CO3 | DMSO | 90 | 12 | — |
| 19f,g | Cu(OTf)2 | Cs2CO3 | DMSO | 90 | 12 | — |
| 20f,g | La(OTf)2 | Cs2CO3 | DMSO | 90 | 12 | — |

In a quest to improve the yield of product, various reaction parameters like reagent, base, solvent and the temperature were modified to obtain the optimum yield (Table 1). Accordingly, when the reaction was examined in DMSO using K2CO3 as a base in the presence of iodine, 1aA was completely consumed within 1 h and the yield of the anticipated product 2aA was significantly improved (77%, Table 1, entry 2). Encouraged by these results, we screened various bases with DMSO as the solvent and molecular iodine as an oxidant. The reaction in the presence of NaHCO3 could not enhance the yield and generated the desired product in 70% yield with some unidentified polar impurities (Table 1, entry 3). The reaction with 1.5 equiv. of iodine and 3.0 equiv. of Cs2CO3 in DMSO at ambient temperature afforded a clean reaction (82% yield) but required 10 h for completion (Table 1, entry 4). Interestingly, when the reaction was performed under similar conditions at 90 °C, it was completed within 45 min to give the product in 85% yield (Table 1, entry 5). However, when the temperature was further increased to 120 °C, no visible improvement was observed in the reaction time or yield of the product (Table 1, entry 6). It was realized that the use of Cs2CO3 took less time as compared to other bases and a clean reaction was obtained to yield the product in excellent yield. More importantly, treatment of the reaction mixture with cold water furnished the desired product as a solid and the analytically pure product could be obtained by simple filtration under vacuum followed by washing with diethyl ether.
It was further observed that the reaction in anhydrous DMF at 90 °C in the presence of molecular iodine completed in 1.5 h but the product was generated in 65% yield (Table 1, entry 7). The combination of KOH with DMSO in the presence of iodine also efficiently produced 1,3,4-oxadiazole 2aA in 75% yield (Table 1, entry 8). To our surprise, I2-mediated oxidative cyclisation in the absence of any base also generated the desired product 2aA in 66% yield in 16 h but polar impurities were also formed (Table 1, entry 9). It was found that the use of organic bases (Et3N or DIPEA) failed to deliver the desired product (Table 1, entries 10 and 11). Similarly, the corresponding product was not generated when TBAI was used as an iodide source (I−) for this transformation instead of molecular iodine (Table 1, entry 13) indicating that the iodonium ion (I+) was possibly driving the reaction. Recently, we found that NCS could be used for decarboxylative oxidation of tetrahydro-β-carbolines so it was planned to investigate its utility for oxidative cyclisation.24a Interestingly, NCS was able to provide the desired product albeit in low yield (32%) along with an unidentified polar impurity (Table 1, entry 14). The use of DMP, NH4Cl and transition metal catalysts like FeCl3, CuI, Cu(OTf)2 and La(OTf)2 failed to deliver the desired product (Table 1, entries 15–20). Eventually, the optimization investigations led to the inference that DMSO was the best solvent and Cs2CO3 was a suitable base for the oxidative cyclization of 1aA in a short duration with 85% yield (Table 1, entries 5 and 6).
Next, it was considered worthwhile to develop a one-pot procedure for this transformation (Scheme 1). Accordingly, condensation of 1a with A and successive oxidative cyclisation was attempted in MeOH using Cs2CO3 and I2 at 70 °C but the formation of β-carboline substituted 1,3,4-oxadiazole 2aA required more than 24 h due to the precipitation of hydrazone 1aA from the reaction mixture and the desired product was obtained in 25% yield only. Then the one-pot assembly of reactants was executed in DMSO but condensation of 1a and A was not completed even after 12 h. It was realised that MeOH was a suitable solvent for hydrazone synthesis 1aA while DMSO was suited for oxidative cyclisation. Therefore, an alternate procedure was adopted where first hydrazone synthesis was achieved in MeOH within 1 h, then MeOH was evaporated and the hydrazone was re-dissolved in DMSO and subjected to oxidative cyclisation with 1.5 equiv. of iodine and 3.0 equiv. of Cs2CO3 at 90 °C for 45 min. To our delight, a clean reaction was observed and no column chromatographic separation was required.
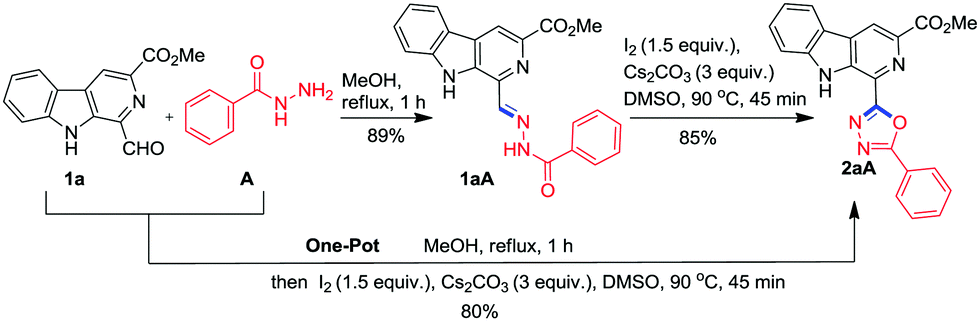 | ||
| Scheme 1 Synthesis of β-carboline tethered 1,3,4-oxadiazole 2aA from 1-formyl β-carboline (1a) and benzoic hydrazide (A). | ||
With established conditions in hand, the scope of this one-pot procedure was investigated for condensation and oxidative cyclisation of 1-formyl β-carbolines24 (1a, 1i and 1k) with substituted acylhydrazides (A–L) to afford the desired β-carboline substituted 1,3,4-oxadiazoles as depicted in Scheme 2. It was pleasing to see that the methodology worked well and smoothly yielded the anticipated products 2aA–aK, 2iA, 2kA, 2kG, 2kI and 2kLvia oxidative cyclisation (C–O bond formation) within 1.75–5 h. The strategy was found compatible with a broad range of acylhydrazides (A–K) bearing electron withdrawing as well as electron donating groups. It was analysed that the acylhydrazides with an electron-donating group (F) reacted faster and afforded higher yield (87%) in comparisons to those bearing electron-withdrawing groups (B–D) (46–76%). The heteroaryl acylhydrazides (G–I) also reacted efficiently and furnished the corresponding products in good to excellent yields (74–92%). Furthermore, this method was extended to aliphatic hydrazides, such as acetylhydrazide K which generated the respective products 2aK in 50% yield.
 | ||
| Scheme 2 Synthesis of β-carboline C-1 tethered 1,3,4-oxadiazole derivatives with the scope of variation in substituted acylhydrazides. | ||
Encouraged by these results, we further extended our study by employing N-9 substituted 1-formyl β-carboline derivatives (1b–h and 1j) for oxidative cyclisation with benzohydrazide A. To our delight, the reactions under optimum conditions easily yielded the corresponding β-carboline C1 substituted 1,3,4-oxadiazole derivatives 2bA–gA and 2jA within 1.5–3 h in good to excellent yield ranging from 73 to 87% (Scheme 3). It is important to mention that N-alkylated derivatives (except N-propargyl, 1f) furnished the desired product in better yield and took less time for completion as compared to the free N–H derivative of β-carboline 1a. Surprisingly, when 1h was subjected to annulation with A under optimal reaction conditions, cleavage of the 2-nitrobenzyl group from the N-9 position of the β-carboline ring was observed and 2aA was obtained as the product instead of 2hA. It is valuable to mention that the analytically pure products (2bA–gA and 2jA) could be obtained by washing the crude product with diethyl ether and column chromatographic separation was not required.
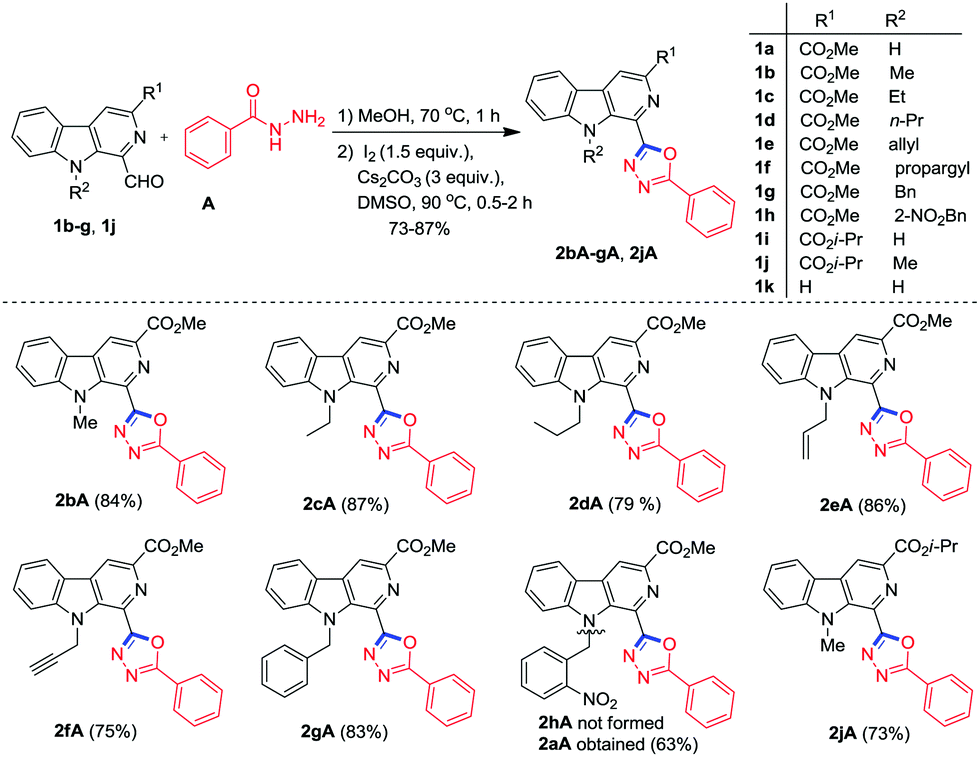 | ||
| Scheme 3 Synthesis of N-alkylated β-carboline substituted 1,3,4-oxadiazole derivatives. | ||
Delighted by the successful synthesis of β-carboline C1 tethered 1,3,4-oxadiazoles, we examined the scope of this methodology for the synthesis of β-carboline tethered bis-oxadiazoles by employing β-carboline 1,3-dicarbaldehyde 4 as the precursor prepared from 3.25 The reaction of dialdehyde 4 with benzohydrazide A in the presence of I2 and Cs2CO3 at 90 °C smoothly afforded the desired product 5A in 43% yield (Scheme 4) after a silica gel column chromatographic separation.
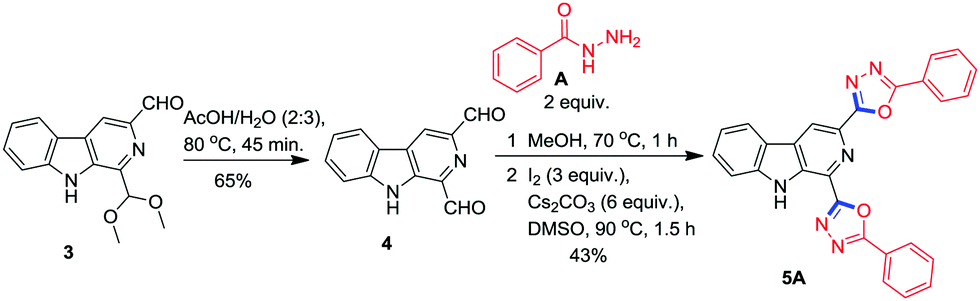 | ||
| Scheme 4 Synthesis of β-carboline tethered bis-1,3,4-oxadiazole. | ||
Surprisingly, when formic hydrazide L was used for oxidative annulation with 1k (Scheme 5), the 1-cyano β-carboline 6 was obtained as the sole product instead of 2kL as confirmed on the basis of 1H-, 13C-NMR and HRMS data.
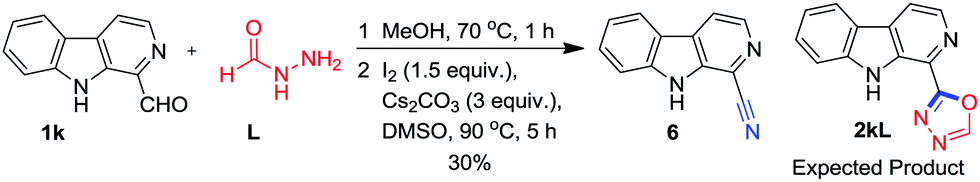 | ||
| Scheme 5 Synthesis of 1-cyano β-carboline. | ||
Next, it was envisaged to further extend the scope of this practical methodology to prepare β-carboline containing 2-amino-1,3,4-oxadiazole derivatives by replacing benzohydrazide A with semicarbazide hydrochloride M. However, Kumujian C 1k failed to react with semicarbazide hydrochloride M in the presence of I2 and Cs2CO3 in DMSO. The screening of various solvents indicated that the desired product 2kM could be isolated in 73% yield in 1,4-dioxane at 90 °C, as depicted in Scheme 6. In order to demonstrate the applicability of this protocol for industrial application, a one gram scale reaction was also conducted using 1a and A as substrates and to our pleasure, the expected product 2aA was obtained in an analytically pure form (77%) without column chromatographic purification (Scheme 7).
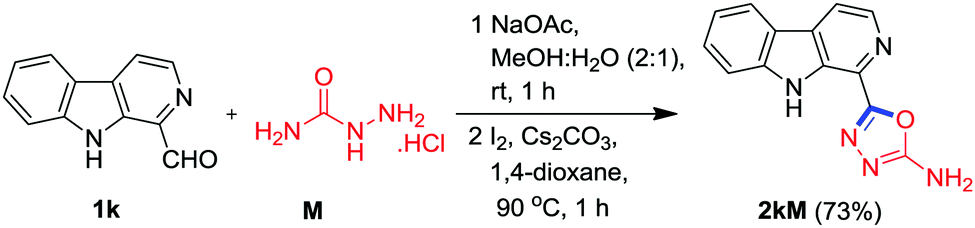 | ||
| Scheme 6 Synthesis of β-carboline C1 tethered 2-amino 1,3,4-oxadiazole from semicarbazide. | ||
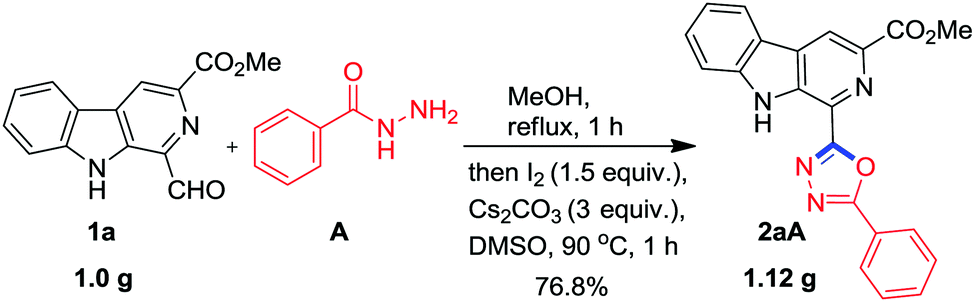 | ||
| Scheme 7 Gram scale synthesis of 2aA. | ||
To probe the reaction mechanism, two control experiments were conducted where the isolated benzohydrazone 1bA was separately reacted with iodine monochloride (source of I+) and KI (source of I−) in the presence of Cs2CO3 in DMSO under optimised conditions. It was found that ICl readily afforded the desired β-carboline substituted 1,3,4-oxadiazole, 2bA within 2h while KI failed to execute the reaction (Scheme 8). These experiments indicated that the iodonium ion played a key role in initiation of this oxidative annulation process.
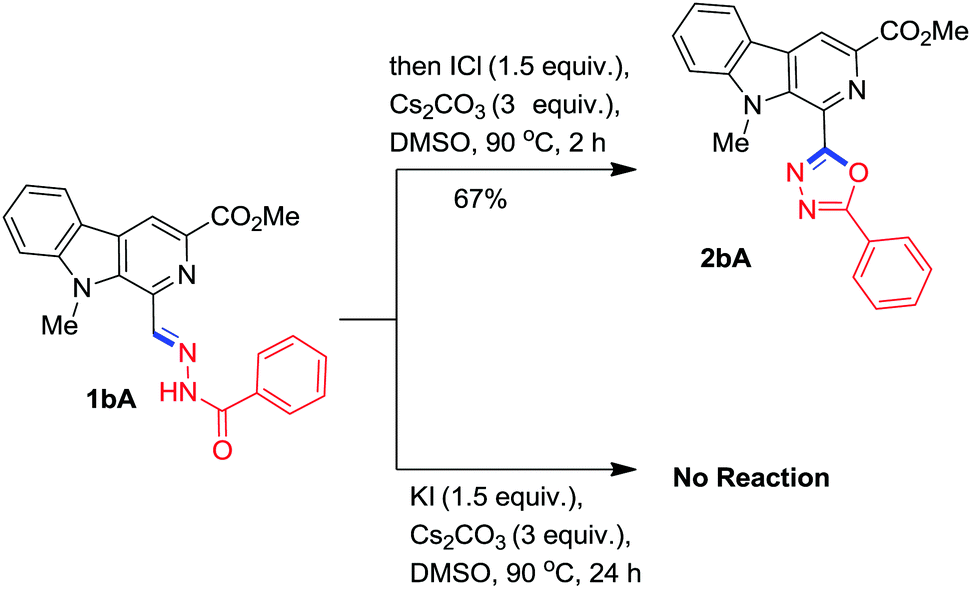 | ||
| Scheme 8 Control experiments. | ||
A plausible mechanism for the formation of β-carboline substituted 1,3,4-oxadiazoles is presented in Fig. 3 based on the results of the study. Initially, the condensation of 1-formyl β-carboline 1a and benzohydrazide (A) resulted in the formation of isolable benzoylhydrazone 1aA. Then, it is proposed that benzoylhydrazone 1aA may undergo Cs2CO3 mediated oxidative C-iodination via route-I to generate the intermediate 6. The successive formation of a new C–O bond via a SN2-type intramolecular cyclization of 7 may result in the formation of 1,3,4-oxadiazole framework 2aA. Alternatively, benzoylhydrazone 1aA may undergo N-iodination via route-II to generate an N-iodo intermediate 8 which may follow base/heat mediated annulation with loss of 1 mole of HI to afford the 1,3,4-oxadiazole framework 2aA as depicted in Fig. 2. The desired product could not be produced in the presence of Lewis acids (FeCl3, Cu(OTf)2 and La(OTf)2) which are likely to follow route-II during annulation; therefore, there is good probability that the reaction is following route-I. The generation of HI during the course of reaction was confirmed by the pH study of a reaction medium which was performed in the absence of base (SI). It was observed that after completion of the reaction under base free conditions, the pH of the reaction medium was 2.61.
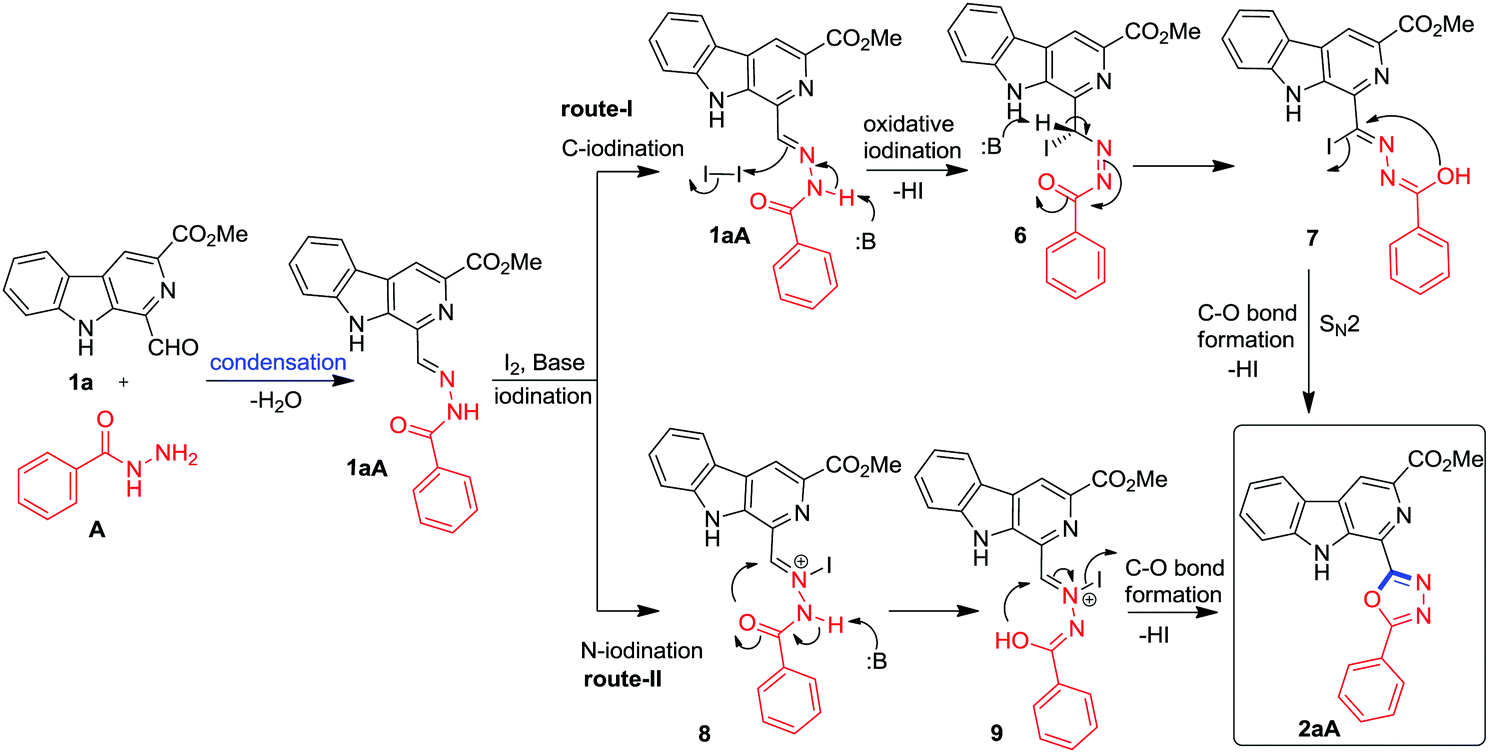 | ||
| Fig. 3 The proposed mechanism for the construction of β-carboline substituted 1,3,4-oxadiazole. | ||
Conclusions
In summary, we have developed a transition metal-free approach for the synthesis of β-carboline C1 tethered 1,3,4-oxadiazoles as well as 2-amino 1,3,4-oxadiazoles via I2 mediated oxidative cyclocondensation. This sequential one-pot synthetic procedure was found to be compatible for the annulation of diversely substituted hydrazides and 1-formyl β-carboline derivatives and desired 1,3,4-oxadiazoles could be isolated at a gram scale without column chromatographic purification. This methodology provides easy access to heterocycles with potential pharmaceutical applications. The investigation of the application of β-carboline linked 1,3,4-oxadiazoles as a ligand for complexation with metals and their development as analytical sensors is currently in progress in our laboratory.Experimental section
General methods
The reagents and chemicals were procured from Sigma Aldrich and Spectrochem Ltd and used as such without additional purification. The anhydrous solvents (glacial AcOH, DCM, DMSO, DMF, and MeOH) were used as available commercially. TLC was examined on precoated aluminum plates (E. Merck; silica gel 60 PF254, 0.25 mm). The column chromatographic separation was conducted on silica gel (SRL; 60–120 mesh). The melting points were recorded in open-ended capillary tubes on a Precision Digital melting-point apparatus (LABCO make) that contained silicon oil and are uncorrected. The IR spectra were recorded on an Agilent FTIR spectrophotometer. 1H and 13C NMR spectra were recorded on an Avance III Bruker spectrometer at operating frequencies of 400 MHz or 100 MHz (13C), as mentioned in the individual spectrum, by using TMS as a standard. The HRMS spectra were recorded on Xevo G2-SQ TOF (Water; USA) or Thermo Finnigan LCQ Advantage, Ion-Trap Mass Spectrometer. Elemental analysis was performed on a Carlo–Erba 108 or an Elementar Vario EL III microanalyzer. During the study, room temperature varied between 25 and 40 °C. The multiplicity in the 1H NMR spectra is mentioned as follows: s for singlet, d for doublet, t for triplet, q for quartet, dd for doublet of doublet, dt for doublet of triplet and m for multiplet.General procedure (one-pot) for the synthesis of compounds 2aA–aK, 2iA, 2kA, 2kG, 2kI, 2kL, 2bA–hA and 2jA as exemplified for compound 2aA
The stirred solution of 1-formyl β-carboline 1a (0.200 g, 0.787 mmol) and benzohydrazide A (0.112 g, 0.826 mmol) in MeOH (10 mL) was refluxed for 1 h. After the condensation was completed as monitored by TLC, the solvent was evaporated under reduced pressure. The resulting benzohydrazone 1aA was re-dissolved in DMSO (5 mL), followed by addition of cesium carbonate (0.770 g, 2.362 mmol) and molecular iodine (0.300 g, 1.181 mmol) in sequence and the reaction mixture was stirred at 90 °C for 45 min; the conversion was monitored by the TLC technique. The reaction mixture was cooled to room temperature and then the content was poured into ice-cold water followed by treatment with 5% aq. Na2S2O3 (20 mL), and light brown precipitates were obtained which were filtered and sintered under high vacuum to get a crude product which was triturated and washed with diethyl ether (twice) to afford the analytically pure product, β-carboline C1 tethered 1,3,4-oxadiazole 2aA in 80% yield.Methyl 1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b] indole-3-carboxylate (2aA)
Yield: 80% (0.234 g from 0.200 g) as a light brown solid; m.p. 199–201 °C; Rf = 0.35 (hexane/EtOAc, 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v); IR (neat): νmax (cm−1) = 3426 (NH), 1710 (CO2CH3), 1626 (C
:30, v/v); IR (neat): νmax (cm−1) = 3426 (NH), 1710 (CO2CH3), 1626 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1261 (C–O), 1064 (C–O–C); 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ = 4.02 (s, 3 H, CO2CH3), 7.31 (t, J = 7.5 Hz, 1 H, ArH), 7.46–7.52 (m, 3 H, ArH), 7.57 (t, J = 7.6 Hz, 1 H, ArH), 7.69 (d, J = 8.2 Hz, 1 H, ArH), 8.14 (d, J = 7.9 Hz, 1 H, ArH), 8.21 (d, J = 7.9 Hz, 2 H, ArH), 8.93 (s, 1 H, ArH), 10.87 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ = 52.8, 112.9, 119.6, 121.2, 121.6, 121.9, 123.1, 125.3, 127.5, 129.0, 129.8, 130.9, 132.2, 135.6, 137.4, 141.1, 163.1, 165.0, 165.8 ppm; HRMS (ESI) m/z: calcd for C21H14N4O3 [M + Na]: 393.0964, found: 393.1002.
N), 1261 (C–O), 1064 (C–O–C); 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ = 4.02 (s, 3 H, CO2CH3), 7.31 (t, J = 7.5 Hz, 1 H, ArH), 7.46–7.52 (m, 3 H, ArH), 7.57 (t, J = 7.6 Hz, 1 H, ArH), 7.69 (d, J = 8.2 Hz, 1 H, ArH), 8.14 (d, J = 7.9 Hz, 1 H, ArH), 8.21 (d, J = 7.9 Hz, 2 H, ArH), 8.93 (s, 1 H, ArH), 10.87 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ = 52.8, 112.9, 119.6, 121.2, 121.6, 121.9, 123.1, 125.3, 127.5, 129.0, 129.8, 130.9, 132.2, 135.6, 137.4, 141.1, 163.1, 165.0, 165.8 ppm; HRMS (ESI) m/z: calcd for C21H14N4O3 [M + Na]: 393.0964, found: 393.1002.
Methyl 1-(5-(2-fluorophenyl)-1,3,4-oxadiazol-2-yl)-9H-pyrido [3,4-b]indole-3-carboxylate (2aB)
Yield: 75% (0.230 g from 0.200 g) as a light brown solid; m.p. 204–206 °C; Rf = 0.30 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 3565 (NH), 1701 (CO2CH3), 1620 (CN), 1265 (C–O), 1058 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.10 (s, 3 H, CO2CH3), 7.28–7.43 (m, 3 H, ArH), 7.59–7.67 (m, 3 H, ArH), 8.23 (dd, J1 = 15.5 Hz, J2 = 7.7 Hz, 2 H, ArH), 9.00 (s, 1 H, ArH), 10.48 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 53.0, 111.9 (d, J = 11 Hz), 112.7, 117.1 (d, J = 21 Hz), 119.8, 121.4, 122.0 (d, J = 34 Hz), 124.7, 124.8, 125.2, 130.2 (d, J = 20 Hz), 131.1, 134.1, 134.2, 135.9, 138.0, 141.2, 159.2, 162.5 (d, J = 143 Hz), 166.0 ppm; HRMS (ESI) m/z: calcd for C21H13FN4O3 [M + H+]: 389.1050, found: 389.1077.
Methyl 1-(5-(4-bromophenyl)-1,3,4-oxadiazol-2-yl)-9H-pyrido [3,4-b]indole-3-carboxylate (2aC)
Yield: 46% (0.163 g from 0.200 g) as a pale yellow solid; m.p. > 260 °C; Rf = 0.45 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 3483 (NH), 1711 (CO2CH3), 1629 (CN), 1277 (C–O), 1064 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.12 (s, 3 H, CO2CH3), 7.43 (dt, J1 = 8.0 Hz, J2 = 4.1 Hz, 1 H, ArH), 7.68 (d, J = 3.8 Hz, 2 H, ArH), 7.70–7.72 (m, 2 H, ArH), 8.15–8.19 (m, 2 H, ArH), 8.24 (dd, J1 = 7.9 Hz, J2 = 0.7 Hz, 1 H, ArH), 9.02 (d, J = 0.5 Hz, 1 H, ArH), 10.45 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 53.1, 112.7, 120.0, 121.6, 122.0, 122.2, 122.4, 125.3, 127.3, 129.1, 130.3, 131.2, 132.6, 136.0, 138.1, 141.3, 163.5, 164.6, 166.0 ppm; HRMS (ESI) m/z: calcd for C21H13BrN4O3 [M + H+]: 449.0249, found: 449.0194.
Methyl 1-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)-9H-pyrido [3,4-b]indole-3-carboxylate (2aD)
Yield: 76% (0.242 g from 0.200 g) as an off-white solid; m.p. 257–259 °C; Rf = 0.40 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 3512 (NH), 1707 (CO2CH3), 1626 (CN), 1269 (C–O), 1067 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.12 (s, 3 H, CO2CH3), 7.40–7.44 (m, 1 H, ArH), 7.54 (d, J = 8.3 Hz, 2 H, ArH), 7.68 (d, J = 3.9 Hz, 2 H, ArH), 8.23 (d, J = 8.3 Hz, 3 H, ArH), 9.01 (s, 1 H, ArH), 10.44 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 53.1, 112.7, 120.0, 121.6, 121.7, 122.0, 122.4, 125.3, 129.0, 129.6, 130.2, 131.2, 136.0, 138.0, 138.8, 141.2, 163.5, 164.5, 166.0 ppm; HRMS (ESI) m/z: calcd for C21H13ClN4O3 [M + H+]: 405.0754, found: 405.0702.
Methyl 1-(5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b]indole-3-carboxylate (2aE)
Yield: 51% (0.161 g from 0.200 g) as an off-white solid; m.p. 254–256 °C; Rf = 0.40 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 3450 (NH), 1704 (CO2CH3), 1623 (CN), 1266 (C–O), 1060 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 3.92 (s, 3 H, OCH3), 4.13 (s, 3 H, CO2CH3), 7.07 (d, J = 8.9 Hz, 2 H, ArH), 7.40–7.45 (m, 1 H, ArH), 7.68 (d, J = 3.3 Hz, 2 H, ArH), 8.23–8.27 (m, 3 H, ArH), 9.03 (s, 1 H, ArH), 10.51 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 53.1, 55.6, 112.6, 114.6, 115.7, 119.7, 121.6, 121.9, 122.3, 125.7, 129.6, 130.1, 131.0, 135.9, 137.9, 141.2, 162.9, 163.0, 165.3, 166.1 ppm; HRMS (ESI) m/z: calcd for C22H16N4O4 [M + H+]: 401.1250, found: 401.1254.
Methyl 1-(5-(p-tolyl)-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b] indole-3-carboxylate (2aF)
Yield: 87% (0.263 g from 0.200 g) as a pale yellow solid; m.p. 233–235 °C; Rf = 0.37 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 3412 (NH), 1709 (CO2CH3), 1618 (CN), 1266 (C–O), 1126 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 2.47 (s, 3 H, ArCH3), 4.13 (s, 3 H, CO2CH3), 7.39 (d, J = 7.9 Hz, 2 H, ArH), 7.43 (dd, J1 = 7.9 Hz, J2 = 4.5 Hz, 1 H, ArH), 7.70 (d, J = 3.4 Hz, 2 H, ArH), 8.22 (d, J = 8.0 Hz, 2 H, ArH), 8.26 (d, J = 7.9 Hz, 1 H, ArH), 9.05 (s, 1 H, ArH), 10.52 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 21.7, 53.1, 100.1, 112.9, 119.8, 120.5, 121.6, 121.9, 122.3, 127.7, 127.8, 129.9, 130.1, 135.9, 138.0, 141.3, 143.1, 163.1, 165.5, 166.1 ppm; HRMS (ESI) m/z: calcd for C22H16N4O3 [M + H+]: 385.1301, found: 385.1289.
Methyl 1-(5-(pyridin-3-yl)-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b]indole-3-carboxylate (2aG)
Yield: 80% (0.235 g from 0.200 g) as a light brown solid; m.p. > 260 °C; Rf = 0.30 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 3392 (NH), 1714 (CO2CH3), 1595 (CN), 1261 (C–O), 1107 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.12 (s, 3 H, CO2CH3), 7.42–7.46 (m, 1 H, ArH), 7.52–7.55 (m, 1 H, ArH), 7.70 (d, J = 4.0 Hz, 2 H, ArH), 8.25 (d, J = 7.9 Hz, 1 H, ArH), 8.58 (dt, J1 = 8.1 Hz, J2 = 1.9 Hz, 1 H, ArH), 8.84 (dd, J1 = 4.8 Hz, J2 = 1.5 Hz, 1 H, ArH), 9.04 (s, 1 H, ArH), 9.55 (d, J = 1.9 Hz, 1 H, ArH), 10.47 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 53.2, 112.7, 119.9, 120.1, 121.5, 122.2, 122.4, 124.0, 125.1, 130.3, 131.3, 134.9, 136.0, 138.1, 141.3, 148.7, 153.0, 163.3, 163.8, 165.9 ppm; HRMS (ESI) m/z: calcd for C20H13N5O3 [M + H+]: 372.1097, found: 372.1078.
Methyl 1-(5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b]indole-3-carboxylate (2aH)
Yield: 83% (0.243 g from 0.200 g) as a pale yellow solid; m.p. > 260 °C; Rf = 0.35 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 3399 (NH), 1719 (CO2CH3), 1623 (CN), 1260 (C–O), 1107 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.12 (s, 3 H, CO2CH3), 7.41–7.46 (m, 1 H, ArH), 7.70 (dd, J1 = 5.9 Hz, J2 = 1.0 Hz, 2 H, ArH), 8.16 (dd, J1 = 4.5 Hz, J2 = 1.6 Hz, 2 H, ArH), 8.24 (d, J = 8.0 Hz, 1 H, ArH), 8.88 (dd, J1 = 4.5 Hz, J2 = 1.6 Hz, 2 H, ArH), 9.04 (s, 1 H, ArH), 10.50 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 53.2, 112.8, 120.3, 121.0, 121.5, 122.1, 122.4, 124.9, 130.4, 130.5, 131.5, 136.1, 138.1, 141.3, 151.1, 163.5, 164.2, 165.9 ppm; HRMS (ESI) m/z: calcd for C20H13N5O3 [M + H+]: 372.1097, found: 372.1114.
Methyl 1-(5-(furan-2-yl)-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b]indole-3-carboxylate (2aI)
Yield: 92% (0.261 g from 0.200 g) as a pale yellow solid; m.p. 255–257 °C; Rf = 0.45 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 3393 (NH), 1712 (CO2CH3), 1624 (CN), 1262 (C–O), 1128 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.11 (s, 3 H, CO2CH3), 6.67 (dd, J1 = 3.5 Hz, J2 = 1.7 Hz, 1 H, ArH), 7.39–7.42 (m, 2 H, ArH), 7.65–7.73 (m, 3 H, ArH), 8.23 (d, J = 7.8 Hz, 1 H, ArH), 9.01 (s, 1 H, ArH), 10.49 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 53.1, 112.6, 112.7, 116.0, 120.0, 121.5, 122.0, 122.3, 125.2, 130.2, 131.2, 136.0, 138.0, 139.1, 141.3, 146.6, 158.0, 162.6, 166.0 ppm; HRMS (ESI) m/z: calcd for C19H12N4O4 [M + H+]: 361.0937, found: 361.0874.
Methyl 1-(5-methyl-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b] indole-3-carboxylate (2aK)
Yield: 50% (0.122 g from 0.200 g) as a light brown solid; m.p. 235–237 °C; Rf = 0.30 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 3391 (NH), 1712 (CO2CH3), 1587 (CN), 1260 (C–O), 1086 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 2.74 (s, 3 H, CH3), 4.09 (s, 3 H, CO2CH3), 7.39–7.43 (m, 1 H, ArH), 7.66 (d, J = 3.8 Hz, 2 H, ArH), 8.22 (d, J = 7.9 Hz, 1 H, ArH), 8.99 (s, 1 H, ArH), 10.40 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 11.5, 53.2, 112.6, 119.8, 121.5, 121.9, 122.3, 125.5, 130.2, 131.1, 136.7, 137.8, 141.2, 163.6, 164.7, 166.0 ppm; HRMS (ESI) m/z: calcd for C16H12N4O3 [M + H+]: 309.0988, found: 309.0999.
Isopropyl 1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b] indole-3-carboxylate (2iA)
Yield: 90% (0.255 g from 0.200 g) as a light brown solid; m.p. 197–199 °C; Rf = 0.50 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 3419 (NH), 1711 (CO2i-Pr), 1598 (CN), 1234 (C–O), 1108 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 1.53 (d, J = 6.3 Hz, 6 H, CH(CH3)2), 5.41–5.47 (m, 1 H, CH(CH3)2), 7.40–7.44 (m, 1 H, ArH), 7.58 (d, J = 7.7 Hz, 3 H, ArH), 7.68 (dd, J1 = 3.7 Hz, J2 = 1.5 Hz, 2 H, ArH), 8.26 (d, J = 7.9 Hz, 1 H, ArH), 8.31 (dd, J1 = 7.8 Hz, J2 = 1.6 Hz, 2 H, ArH), 8.97 (s, 1 H, ArH), 10.49 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 22.2, 69.8, 112.7, 119.7, 121.6, 121.8, 122.3, 123.4, 125.7, 127.7, 129.2, 130.1, 131.0, 132.4, 135.9, 138.7, 141.3, 163.5, 164.9, 165.3 ppm; HRMS (ESI) m/z: calcd for C23H18N4O3 [M + H+]: 399.1457, found: 399.1443.
2-Phenyl-5-(9H-pyrido[3,4-b]indol-1-yl)-1,3,4-oxadiazole (2kA)
Yield: 77% (0.245 g from 0.200 g) as a brown solid; m.p. 209–211 °C; Rf = 0.50 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 3465 (NH), 1630 (CN), 1249 (C–O), 1088 (C–O–C); 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ = 7.32 (t, J = 7.5 Hz, 1 H, ArH), 7.57–7.62 (m, 4 H, ArH), 7.80 (d, J = 8.2 Hz, 1 H, ArH), 8.14–8.18 (m, 2 H, ArH), 8.27 (d, J = 7.2 Hz, 2 H, ArH), 8.62 (d, J = 5.1 Hz, 1 H, ArH), 10.96 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ = 112.3, 116.6, 120.0, 120.2, 121.1, 122.9, 125.1, 126.7, 128.5, 128.7, 130.0, 131.5, 133.9, 138.3, 140.8, 163.1, 163.9 ppm; HRMS (ESI) m/z: calcd for C19H12N4O [M + H+]: 313.1089, found: 313.1067.
2-(Pyridin-3-yl)-5-(9H-pyrido[3,4-b]indol-1-yl)-1,3,4-oxadiazole (2kG)
Yield: 74% (0.236 g from 0.200 g) as a brown solid; m.p. 213–215 °C; Rf = 0.35 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 1628 (CN), 1225 (C–O), 1122 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 7.34–7.38 (m, 1 H, ArH), 7.51 (dd, J1 = 7.9 Hz, J2 = 4.9 Hz, 1 H, ArH), 7.64 (d, J = 3.9 Hz, 2 H, ArH), 8.14 (d, J = 5.0 Hz, 1 H, ArH), 8.18 (d, J = 7.8 Hz, 1 H, ArH), 8.54 (d, J = 8.0 Hz, 1 H, ArH), 8.66 (d, J = 5.1 Hz, 1 H, ArH), 8.82 (d, J = 4.4 Hz, 1 H, ArH), 9.51 (s, 1 H, ArH), 10.18 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 112.3, 117.6, 120.1, 121.0, 121.1, 122.1, 123.9, 125.2, 129.7, 130.9, 134.6, 134.8, 139.4, 140.9, 148.4, 152.8, 162.8, 164.3 ppm; HRMS (ESI) m/z: calcd for C18H11N5O [M + H+]: 314.1042, found: 314.1042.
2-(Furan-2-yl)-5-(9H-pyrido[3,4-b]indol-1-yl)-1,3,4-oxadiazole (2kI)
Yield: 85% (0.262 g from 0.200 g) as a yellow solid; m.p. > 260 °C; Rf = 0.60 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 3409 (NH), 1640 (CN), 1271 (C–O), 1118 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 6.67 (dd, J1 = 3.4 Hz, J2 = 1.6 Hz, 1 H, ArH), 7.34–7.38 (m, 1 H, ArH), 7.42 (d, J = 3.5 Hz, 1 H, ArH), 7.62–7.66 (m, 2 H, ArH), 7.72 (d, J = 0.7 Hz, 1 H, ArH), 8.14 (d, J = 5.1 Hz, 1 H, ArH), 8.19 (d, J = 7.9 Hz, 1 H, ArH), 8.66 (d, J = 5.1 Hz, 1 H, ArH), 10.18 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 112.3, 112.5, 115.4, 117.5, 121.1, 121.3, 122.1, 125.5, 129.7, 130.8, 134.9, 139.3, 139.6, 141.0, 146.4, 157.7, 163.2 ppm; HRMS (ESI) m/z: calcd for C17H10N4O2 [M + H+]: 303.0882, found: 303.0870.
Methyl 9-methyl-1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9H-pyrido [3,4-b]indole-3-carboxylate (2bA)
Yield: 84% (0.240 g from 0.200 g) as an off-white solid; m.p. 228–230 °C; Rf = 0.45 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 1711 (CO2CH3), 1588 (CN), 1267 (C–O), 1129 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.09 (s, 3 H, NCH3), 4.10 (s, 3 H, CO2CH3), 7.45 (t, J = 7.5 Hz, 1 H, ArH), 7.58 (t, J = 7.6 Hz, 4 H, ArH), 7.74 (d, J = 7.5 Hz, 1 H, ArH), 8.25–8.28 (m, 3 H, ArH), 9.06 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 33.5, 53.2, 110.6, 119.4, 121.0, 121.8, 122.0, 123.7, 126.4, 127.6, 129.2, 130.2, 132.3, 132.4, 137.3, 143.6, 162.0, 165.9, 166.0 ppm; HRMS (ESI) m/z: calcd for C22H16N4O3 [M + Na]: 407.1120, found: 407.1070.
Methyl 9-ethyl-1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9H-pyrido [3,4-b]indole-3-carboxylate (2cA)
Yield: 87% (0.246 g from 0.200 g) as an off-white solid; m.p. 204–206 °C; Rf = 0.55 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 1714 (CO2CH3), 1586 (CN), 1269 (C–O), 1119 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 1.28 (t, J = 7.2 Hz, 3 H, NCH2CH3), 4.09 (s, 3 H, CO2CH3), 4.80 (q, J = 7.2 Hz, 2 H, NCH2CH3), 7.43 (t, J = 7.5 Hz, 1 H, ArH), 7.54–7.62 (m, 4 H, ArH), 7.69–7.73 (m, 1 H, ArH), 8.23–8.29 (m, 3 H, ArH), 9.06 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 14.3, 40.2, 53.1, 110.7, 119.4, 121.3, 121.7, 122.0, 123.6, 126.2, 127.5, 129.2, 130.1, 132.3, 132.7, 136.1, 137.1, 142.6, 162.1, 165.9 ppm; HRMS (ESI) m/z: calcd for C23H18N4O3 [M + H+]: 399.1457, found: 399.1457.
Methyl 1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9-propyl-9H-pyrido [3,4-b]indole-3-carboxylate (2dA)
Yield: 79% (0.110 g from 0.100 g) as an off-white solid; m.p. 201–202 °C; Rf = 0.65 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 1713 (CO2CH3), 1590 (CN), 1261 (C–O), 1123 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 0.73 (t, J = 7.1 Hz, 3 H, NCH2CH2CH3), 1.62-1.67 (m, 2 H, NCH2CH2CH3), 4.09 (s, 3 H, CO2CH3), 4.71 (t, J = 7.1 Hz, 2 H, NCH2CH2CH3), 7.41 (t, J = 7.2 Hz, 1 H, ArH), 7.53–7.60 (m, 4 H, ArH), 7.67–7.71 (m, 1 H, ArH), 8.25 (d, J = 5.0 Hz, 3 H, ArH), 9.05 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 11.2, 22.6, 46.6, 53.1, 110.9, 119.3, 121.1, 121.5, 121.6, 121.9, 123.6, 126.3, 127.5, 129.2, 130.0, 132.2, 132.5, 136.3, 137.1, 143.0, 162.0, 165.9 ppm; HRMS (ESI) m/z: calcd for C24H20N4O3 [M + H+]: 413.1614, found: 413.1569.
Methyl 9-allyl-1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9H-pyrido [3,4-b]indole-3-carboxylate (2eA)
Yield: 86% (0.240 g from 0.200 g) as a light brown solid; m.p. 160–162 °C; Rf = 0.45 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 1706 (CO2CH3), 1598 (CN), 1297 (C–O), 1089 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.10 (s, 3 H, CO2CH3), 4.57 (d, J = 16.7 Hz, 1 H, =CHH), 4.90 (dd, J1 = 10.5 Hz, J2 = 0.7 Hz, 1 H, =CHH), 5.48–5.51 (m, 2 H, NCH2), 5.68–5.75 (m, 1 H, NCH2CH), 7.45 (t, J = 7.6 Hz, 1 H, ArH), 7.55–7.60 (m, 4 H, ArH), 7.71 (t, J = 7.2 Hz, 1 H, ArH), 8.24 (dd, J1 = 8.0 Hz, J2 = 1.5 Hz, 2 H, ArH), 8.29 (d, J = 7.8 Hz, 1 H, ArH), 9.09 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 47.2, 53.2, 110.9, 116.7, 119.4, 121.2, 121.9, 122.0, 123.7, 126.8, 127.6, 129.2, 130.2, 132.1, 132.2, 132.8, 136.3, 137.7, 143.1, 161.9, 165.8, 165.9 ppm; HRMS (ESI) m/z: calcd for C24H18N4O3 [M + H+]: 411.1457, found: 411.1491.
Methyl 1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9-(prop-2-yn-1-yl)-9H-pyrido[3,4-b]indole-3-carboxylate (2fA)
Yield: 75% (0.105 g from 0.100 g) as a light brown solid; m.p. 205–207 °C; Rf = 0.50 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 1710 (CO2CH3), 1622 (CN), 1275 (C–O), 1127 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 2.00 (t, J = 2.4 Hz, 1 H, CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 4.11 (s, 3 H, CO2CH3), 5.74 (d, J = 2.4 Hz, 2 H, NCH2), 7.47 (t, J = 7.5 Hz, 1 H, ArH), 7.55–7.60 (m, 3 H, ArH), 7.64 (d, J = 8.4 Hz, 1 H, ArH), 7.74 (t, J = 7.4 Hz, 1 H, ArH), 8.27 (dd, J1 = 7.9 Hz, J2 = 1.7 Hz, 3 H, ArH), 9.07 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 35.3, 53.2, 73.3, 77.0, 110.7, 119.3, 121.5, 122.1, 122.3, 123.7, 127.0, 127.6, 129.2, 130.4, 132.3, 133.4, 135.8, 138.1, 142.6, 162.1, 165.8, 166.0 ppm; HRMS (ESI) m/z: calcd for C24H16N4O3 [M + H+]: 409.1301, found: 409.1350.
CH), 4.11 (s, 3 H, CO2CH3), 5.74 (d, J = 2.4 Hz, 2 H, NCH2), 7.47 (t, J = 7.5 Hz, 1 H, ArH), 7.55–7.60 (m, 3 H, ArH), 7.64 (d, J = 8.4 Hz, 1 H, ArH), 7.74 (t, J = 7.4 Hz, 1 H, ArH), 8.27 (dd, J1 = 7.9 Hz, J2 = 1.7 Hz, 3 H, ArH), 9.07 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 35.3, 53.2, 73.3, 77.0, 110.7, 119.3, 121.5, 122.1, 122.3, 123.7, 127.0, 127.6, 129.2, 130.4, 132.3, 133.4, 135.8, 138.1, 142.6, 162.1, 165.8, 166.0 ppm; HRMS (ESI) m/z: calcd for C24H16N4O3 [M + H+]: 409.1301, found: 409.1350.
Methyl 9-benzyl-1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9H-pyrido [3,4-b]indole-3-carboxylate (2gA)
Yield: 83% (0.222 g from 0.200 g) as a light brown solid; m.p. 144–145 °C; Rf = 0.65 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 1714 (CO2CH3), 1628 (CN), 1257 (C–O), 1149 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 4.06 (s, 3 H, CO2CH3), 6.01 (s, 2 H, NCH2), 6.53 (d, J = 7.3 Hz, 2 H, ArH), 6.90 (t, J = 7.3 Hz, 2 H, ArH), 6.95 (t, J = 7.1 Hz, 1 H, ArH), 7.48–7.51 (m, 3 H, ArH), 7.55 (d, J = 7.3 Hz, 1 H, ArH), 7.65 (d, J = 8.4 Hz, 1 H, ArH), 7.71–7.75 (m, 1 H, ArH), 8.00 (d, J = 7.1 Hz, 2 H, ArH), 8.35 (d, J = 7.8 Hz, 1 H, ArH), 9.12 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 48.1, 53.2, 110.8, 119.4, 121.1, 122.1, 122.2, 123.6, 125.8, 127.2, 127.5, 128.7, 129.0, 130.4, 132.1, 132.9, 135.4, 136.1, 137.7, 143.8, 161.3, 165.8, 165.9 ppm; HRMS (ESI) m/z: calcd for C28H20N4O3 [M + H+]: 461.1614, found: 461.1661.
Isopropyl 9-methyl-1-(5-phenyl-1,3,4-oxadiazol-2-yl)-9H-pyrido[3,4-b]indole-3-carboxylate (2jA)
Yield: 73% (0.203 g from 0.200 g) as a pale yellow solid; m.p. 185–187 °C; Rf = 0.60 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 1707 (CO2i-Pr), 1594 (CN), 1255 (C–O), 1121 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 1.50 (d, J = 6.2 Hz, 6 H, CH(CH3)2), 4.10 (s, 3 H, NCH3), 5.38–5.45 (m, 1 H, CH(CH3)2), 7.43 (t, J = 7.2 Hz, 1 H, ArH), 7.56–7.59 (m, 4 H, ArH), 7.70–7.74 (m, 1 H, ArH), 8.25 (d, J = 1.8 Hz, 1 H, ArH), 8.27 (d, J = 7.8 Hz, 2 H, ArH), 8.98 (s, 1 H, ArH) ppm; 13C NMR (100 MHz, CDCl3) δ = 22.2, 33.5, 69.7, 110.6, 119.0, 121.1, 121.6, 121.9, 123.7, 126.5, 127.5, 129.2, 130.0, 132.2, 137.1, 137.9, 143.6, 162.2, 164.7, 165.8 ppm; HRMS (ESI) m/z: calcd for C24H20N4O3 [M + H+]: 413.1614, found: 413.1637.
9H-Pyrido[3,4-b]indole-1-carbonitrile (6)
Yield: 30% (0.030 g from 0.100 g) as a light brown solid; m.p. 201–203 °C; Rf = 0.40 (hexane/EtOAc, 50:50, v/v); IR (neat): νmax = 2239 (CN), 3404 (NH); 1H NMR (400 MHz, CDCl3) δ = 7.38 (t, J = 7.4 Hz, 1 H, ArH), 7.61–7.69 (m, 2 H, ArH), 8.16 (dd, J1 = 6.3 Hz, J2 = 4.3 Hz, 2 H, ArH), 8.57 (d, J = 5.1 Hz, 1 H, ArH), 9.10 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 112.2, 116.1, 116.7, 118.4, 121.1, 121.6, 122.4, 130.4, 131.0, 138.8, 140.6 ppm; HRMS (ESI) m/z: calcd for C12H7N3 [M + H+]: 194.0718, found: 194.0780.
One pot procedure for the synthesis of 5A
A stirred solution of β-carboline 1,3-dicarbaldehyde 4 (0.150 g, 0.670 mmol) and benzohydrazide A (0.182 g, 1.339 mmol) in MeOH (10 mL) was refluxed for 1 h. After the condensation was completed as monitored by TLC, the solvent was evaporated under reduced pressure. The resulting benzohydrazone 4A was re-dissolved in DMSO (5 mL), followed by addition of cesium carbonate (1.309 g, 4.017 mmol) and molecular iodine (0.510 g, 2.008 mmol) in sequence and the reaction mixture was stirred at 90 °C for 1.5 h; the conversion was monitored by the TLC technique. The reaction mixture was cooled to room temperature and then the content was poured into ice-cold water followed by treatment of 5% aq. Na2S2O3 (20 mL), and light brown precipitates were obtained which were filtered and sintered under high vacuum to get a crude product which was purified through a short silica gel column chromatography using EtOAc:hexane (30:70, v/v) as an eluent to afford the analytically pure product, β-carboline tethered bis-1,3,4-oxadiazole 5A in 43% yield.
5,5′-(9H-Pyrido[3,4-b]indole-1,3-diyl)bis(2-phenyl-1,3,4-oxadiazole) (5A)
Yield: 43% (0.131 g from 0.150 g) as a yellow solid; m.p. > 260 °C; Rf = 0.70 (hexane/EtOAc, 70:30, v/v); IR (neat): νmax = 3393 (NH), 1565 (CN), 1275 (C–O), 1139 (C–O–C); 1H NMR (400 MHz, CDCl3) δ = 7.38–7.42 (m, 1 H, ArH), 7.57–7.61 (m, 6 H, ArH), 7.64–7.67 (m, 2 H, ArH), 8.22 (d, J = 7.9 Hz, 1 H, ArH), 8.27–8.31 (m, 4 H, ArH), 9.06 (s, 1 H, ArH), 10.45 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 112.6, 117.5, 121.3, 121.9, 122.4, 123.4, 124.1, 127.4, 127.7, 129.2, 129.3, 130.4, 131.3, 132.0, 132.5, 133.5, 135.4, 141.3, 163.4, 164.6, 165.3, 166.5 ppm; HRMS (ESI) m/z: calcd for C27H16N6O2 [M + H+]: 457.1413, found: 457.1454.
General procedure for the synthesis of compound 2kM
To a stirred solution of semicarbazide hydrochloride M (0.085 g, 0.765 mmol) and sodium acetate (0.063 g, 0.765 mmol) in MeOH (7 mL), 1k (0.150 g, 0.765 mmol) was added and stirring continued at room temperature for 1 h. After the condensation was completed as monitored by TLC, the solvent was evaporated under reduced pressure. The resulting residue was re-dissolved in 1,4-dioxane (5 mL), followed by addition of cesium carbonate (0.748 g, 2.295 mmol) and iodine (0.291 g, 1.147 mmol) in sequence and the reaction mixture was stirred at 90 °C for 1 h; the conversion was monitored by the TLC technique. The reaction mixture was cooled to room temperature and then, the reaction content was poured into ice-cold water followed by treatment with 5% aq. Na2S2O3 (20 mL), and yellow precipitates were obtained which were filtered and sintered to get a crude product which was triturated and washed with diethyl ether (twice) to afford the analytically pure product 2kM in 73% yield.5-(9H-Pyrido[3,4-b]indol-1-yl)-1,3,4-oxadiazol-2-amine (2kM)
Yield: 73% (0.14 g from 0.150 g) as a pale yellow solid; m.p. > 260 °C; Rf = 0.30 (hexane/EtOAc, 20:80, v/v); 1H NMR (400 MHz, DMSO-d6) δ = 7.29 (d, J = 7.4 Hz, 1 H, ArH), 7.55 (s, 2 H, NH2), 7.59 (d, J = 7.5 Hz, 1 H, ArH), 7.89 (d, J = 8.2 Hz, 1 H, ArH), 8.26 (dd, J1 = 8.9 Hz, J2 = 6.7 Hz, 2 H, ArH), 8.44 (d, J = 5.1 Hz, 1 H, ArH), 11.57 (s, 1 H, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 113.4, 116.2, 120.0, 120.4, 121.7, 126.8, 128.7, 129.4, 132.5, 138.0, 141.3, 157.1, 164.1 ppm; HRMS (ESI) m/z: calcd for C13H9N5O [M + H+]: 252.0885, found: 252.0849.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. S. acknowledges the Council of Scientific & Industrial Research New Delhi (India), for a Senior Research Fellowship. V. S. gratefully acknowledges the financial support in the form of a research grant from the Science and Engineering Research Board, New Delhi (EMR/2017/000155 and SB/FT/CS-188/2011) and the Council of Scientific & Industrial Research ((02)0202/14/EMR-II) New Delhi (India) and TEQIP-III (NIT Jalandhar). The central instrumentation facility, Guru Nanak Dev University Amritsar and Tezpur University, Assam is gratefully acknowledged for recording some of the spectroscopic data reported in this paper.Notes and references
- (a) R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479 CrossRef CAS; (b) H. Huang, Y. Yao, Z. He, T. Yang, J. Ma, X. Tian, Y. Li, C. Huang, X. Chen, W. Li, S. Zhang, C. Zhang and J. Ju, J. Nat. Prod., 2011, 74, 2122 CrossRef CAS PubMed; (c) P.-C. Kuo, L.-S. Shi, A. G. Damu, C.-R. Su, C.-H. Huang, C.-H. Ke, J.-B. Wu, A.-J. Lin, K. F. Bastow, K.-H. Lee and T.-S. Wu, J. Nat. Prod., 2003, 66, 1324 CrossRef CAS PubMed; (d) K. K. H. Ang, M. J. Holmes, T. Higa, M. T. Hamann and U. A. K. Kara, Antimicrob. Agents Chemother., 2000, 44, 1645 CrossRef CAS; (e) K. G. Brahmbhatt, N. Ahmed, S. Sabde, D. Mitra, I. P. Singh and K. K. Bhutani, Bioorg. Med. Chem. Lett., 2010, 20, 4416 CrossRef CAS; (f) Y.-H. Wang, J.-G. Tang, R.-R. Wang, L.-M. Yang, Z.-J. Dong, L. Du, X. Shen, J.-K. Liu and Y.-T. Zheng, Biochem. Biophys. Res. Commun., 2007, 355, 1091 CrossRef CAS; (g) Y. Liu, H. Song, Y. Huang, J. Li, S. Zhao, Y. Song, P. Yang, Z. Xiao, Y. Liu, Y. Li, H. Shang and Q. Wang, J. Agric. Food Chem., 2014, 62, 9987 CrossRef CAS; (h) L. C. Meurer, R. N. Guthikonda, J. L. Huber and F. DiNinno, Bioorg. Med. Chem. Lett., 1995, 5, 767 CrossRef CAS.
- (a) W. Horton, A. Sood, S. Peerannawar, N. Kugyela, A. Kulkarni, R. Tulsan, C. D. Tran, J. Soule, H. LeVine, B. Török and M. Török, Bioorg. Med. Chem. Lett., 2017, 27, 232 CrossRef CAS; (b) R. Otto, R. Penzis, F. Gaube, T. Winckler, D. Appenroth, C. Fleck, C. Tränkle, J. Lehmann and C. Enzensperger, Eur. J. Med. Chem., 2014, 87, 6 CrossRef; (c) Y. Rook, K. U. Schmidtke, F. Gaube, D. Schepmann, B. Wünsch, J. Heilmann, J. Lehmann and T. Winckler, J. Med. Chem., 2010, 53, 3611 CrossRef CAS.
- H. Guan, H. Chen, W. Peng, Y. Ma, R. Cao, X. Liu and A. Xu, Eur. J. Med. Chem., 2006, 41, 1167 CrossRef CAS.
- Y. Song, D. Kesuma, J. Wang, Y. Deng, J. Duan, J. H. Wang and R. Z. Qi, Biochem. Biophys. Res. Commun., 2004, 317, 128 CrossRef CAS.
- Y. Funayama, K. Nishio, K. Wakabayashi, M. Nagao, K. Shimoi, T. Ohira, S. Hasegawa and N. Saijo, Mutat. Res., 1996, 349, 183 CrossRef.
- A. C. Castro, L. C. Dang, F. Soucy, L. Grenier, H. Mazdiyasni, M. Hottelet, L. Parent, C. Pien, V. Palombella and J. Adams, Bioorg. Med. Chem. Lett., 2003, 13, 2419 CrossRef CAS.
- (a) L. A. Sorbera, L. Martin, P. A. Leeson and J. Castaner, Drugs Future, 2001, 26, 15 CrossRef CAS; (b) L. Turski, D. N. Stephens, L. H. Jensen, E. N. Peterson, B. S. Mel-drum, S. Patel, J. B. Hansen, W. Loscher, H. H. Schneider and R. Schmiechen, J. Pharmacol. Exp. Ther., 1990, 253, 344 CAS.
- (a) S. Sharma, P. K. Sharma, N. Kumar and R. Dudhe, Der Pharma Chem., 2010, 2, 253 CAS; (b) S. Bhatia and M. Gupta, J. Chem. Pharm. Res., 2011, 3, 137 CAS; (c) Z. Li, P. Zhan and X. Liu, Mini-Rev. Med. Chem., 2011, 11, 1130 CrossRef CAS; (d) V. K. R. Sahu, A. K. Singh and D. Yadav, Int. J. ChemTech Res., 2011, 3, 1362 CAS; (e) A. K. Singh, V. K. R. Sahu and D. Yadav, Int. J. Pharm. Sci. Res., 2011, 2, 135 CAS; (f) C. S. de Oliveira, B. F. Lira, J. M. Barbosa-Filho, J. G. Lorenzo and P. F. de Athayde-Filho, Molecules, 2012, 17, 10192 CrossRef PubMed; (g) H. Khalilullah, M. J. Ahsan, M. Hedaitullah, S. Khan and B. Ahmed, Mini-Rev. Med. Chem., 2012, 12, 789 CrossRef CAS.
- S. Singh, L. K. Sharma, A. Sarswat, I. R. Siddiqui, H. K. Kheri and R. K. P. Singh, RSC Adv., 2013, 3, 4237 RSC.
- (a) U. Mitschke and P. Bauerle, J. Mater. Chem., 2000, 10, 1471 RSC; (b) N. Rehmann, C. Ulbricht, A. Kohnen, P. Zacharias, M. C. Gather, D. Hertel, E. Holder, K. Meerholz and U. S. Schubert, Adv. Mater., 2008, 20, 129 CrossRef CAS; (c) M. Guan, Z. Q. Bian, Y. F. Zhou, F. Y. Li, Z. J. Lib and C. H. Huang, Chem. Commun., 2003, 2708 RSC.
- A. Savarino, Expert Opin. Invest. Drugs, 2006, 15, 1507 CrossRef CAS.
- N. D. James and J. W. Growcott, Drugs Future, 2009, 34, 624 CrossRef CAS.
- H. Brandenberger and R. A. A. Maes, Clinical Biochemistry: Analytical Toxicology for Clinical, Forensic and Pharmaceutical Chemists, Walter de Gruyter, Berlin, 1997 Search PubMed.
- G. W. Adelstein, C. H. Yen, E. Z. Dajani and R. G. Bianchi, J. Med. Chem., 1976, 19, 1221 CrossRef CAS.
- (a) H. Ichiro, K. Yasuhiko and U. Ryuzo, Nippon Kagaku Zassi, 1967, 88, 574 CrossRef; (b) M. Ogata, H. Atobe, H. Kushida and K. J. Yamamoto, Antibiotics, 1971, 24, 443 CrossRef CAS.
- L. Guo, W. K. Hagmann, S. He, Z. Lai, J. Liu, R. P. Nargund, S. K. Shah and Q. T. Truong, US2014-8754099, Chem. Abst., 2016, 1920231 Search PubMed.
- (a) V. K. Tandon and R. B. Chhor, Synth. Commun., 2001, 31, 1727 CrossRef CAS; (b) S. Cesarini, N. Colombo, M. Pulici, E. R. Felder and W. K.-D. Brill, Tetrahedron, 2006, 62, 10223 CrossRef CAS; (c) P. Stabile, A. Lamonica, A. Ribecai, D. Castoldia, G. Guercio and O. Curcuruto, Tetrahedron Lett., 2010, 51, 4801 CrossRef CAS.
- (a) C. Dobrota, C. C. Paraschivescu, I. Dumitru, L. Lavinia, I. Baciu and L. L. Ruta, Tetrahedron Lett., 2009, 50, 1886 CrossRef CAS; (b) P. Gao and Y.-Y. Wei, Heterocycl. Commun., 2013, 19, 113 CAS; (c) P. Gao and Y.-Y. Wei, J. Chem. Res., 2013, 506 CrossRef CAS; (d) W. Yu, G. Huang, Y. Zhang, H. Liu, L. Dong, X. Yu, Y. Li and J. Chang, J. Org. Chem., 2013, 78, 10337 CrossRef CAS; (e) G. Majji, S. K. Rout, S. Guin, A. Gogoi and B. K. Patel, RSC Adv., 2014, 4, 5357 RSC.
- (a) T. Kawano, T. Yoshizumi, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 3072 CrossRef CAS; (b) M. Nishino, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 4457 CrossRef CAS PubMed; (c) X.-Y. Fan, X. Jiang, Y. Zhang, Z.-B. Chen and Y.-M. Zhu, Org. Biomol. Chem., 2015, 13, 10402 RSC.
- T. Fang, Q. Tan, Z. Ding, B. Liu and B. Xu, Org. Lett., 2014, 16, 2342 CrossRef CAS.
- (a) M. Dabiria, P. Salehib, M. Baghbanzadeha and M. Bahramnejada, Tetrahedron Lett., 2006, 47, 6983 CrossRef; (b) N. Montazeri and K. Rad-Moghadam, Chin. Chem. Lett., 2008, 19, 1143 CrossRef CAS; (c) Q. Gao, S. Liu, X. Wu, J. Zhang and A. Wu, Org. Lett., 2015, 17, 2960 CrossRef CAS PubMed; (d) C. Xu, F.-C. Jia, Q. Cai, D.-K. Li, Z.-W. Zhou and A.-X. Wu, Chem. Commun., 2015, 51, 6629 RSC.
- W. Yu, G. Huang, Y. Zhang, H. Liu, L. Dong, X. Yu, Y. Li and J. Chang, J. Org. Chem., 2013, 78, 10337 CrossRef CAS PubMed.
- S. Guin, T. Ghosh, S. K. Rout, A. Banerjee and B. K. Patel, Org. Lett., 2011, 13, 5976 CrossRef CAS PubMed.
- (a) D. Singh, V. Kumar, N. Devi, C. C. Malakar, R. Shankar and V. Singh, Adv. Synth. Catal., 2017, 359, 1213 CrossRef CAS; (b) D. Singh, P. Sharma, R. Kumar, S. K. Pandey, C. C. Malakar and V. Singh, Asian J. Org. Chem., 2018, 7, 383 CrossRef CAS; (c) N. Devi, S. Kumar, S. K. Pandey and V. Singh, Asian J. Org. Chem., 2018, 7, 6 CrossRef CAS; (d) V. Singh and S. Batra, Curr. Org. Synth., 2012, 9, 513 CrossRef CAS.
- (a) D. Singh, C. K. Hazra, C. C. Malakar, S. K. Pandey, B. S. Kaith and V. Singh, ChemistrySelect, 2018, 3, 4859 CrossRef CAS; (b) N. Devi, D. Singh, G. Kaur, S. Mor, V. P. R. K. Putta, S. Polina, C. C. Malakar and V. Singh, New J. Chem., 2017, 41, 1082 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj04294b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |