
Selective chemotherapy and imaging of colorectal and breast cancer cells by a modified MUC-1 aptamer conjugated to a poly(ethylene glycol)-dimethacrylate coated Fe3O4–AuNCs nanocomposite†
Roya
Binaymotlagh
ab,
Farid
Hajareh Haghighi
ab,
Fatemeh
Aboutalebi
a,
Seyede Zohreh
Mirahmadi-Zare
 *a,
Hassan
Hadadzadeh
*b and
Mohammad-Hossein
Nasr-Esfahani
a
*a,
Hassan
Hadadzadeh
*b and
Mohammad-Hossein
Nasr-Esfahani
a
aDepartment of Molecular Biotechnology, Cell Science Research Center, Royan Institute for Biotechnology, ACECR, Isfahan 81651-31378, Iran. E-mail: mirahmadi_zare@royaninstitute.org
bDepartment of Chemistry, Isfahan University of Technology, Isfahan 84156-83111, Iran. E-mail: hadad@cc.iut.ac.ir
First published on 14th November 2018
Abstract
The combination of diagnosis and targeted therapy within a single nanoplatform is one of the remarkable advances in molecular medicine. Herein, we synthesized a magnetite–gold nanocluster–aptamer(epirubicin) (Fe3O4–AuNC–Apt(EPI)) nanocomposite with an average particle size of 35 nm for specific detection, fluorescent imaging, and therapy of colorectal (HT-29) and breast (MCF-7) cancer cells. AuNCs were synthesized in situ in (γ-mercaptopropyl) trimethoxysilane (MPS) solution to produce a highly fluorescent nanocomposite. In this study, short-length poly(ethylene glycol)-dimethacrylate (PEGDMA) was used for efficient immobilization of further aptamer on the nanocomposite along with the reduction of the growth rate of the silica network and thickness of the coating, subsequently. A thiol-modified MUC-1 aptamer (Apt) with an increased CG base pair region was conjugated to the surface of the nanocomposite which provides the situation for a high amount of epirubicin (EPI) loading as a chemotherapeutic drug specifically targeted toward the desired cancer cells. Also, the calculated binding free energies (ΔGb) showed that the binding affinity of EPI to the active sequence modified Apt was higher than the traditional one. According to the MTS results, this nanocomposite is able to kill the MUC-1 positive cancer cells (HT-29 and MCF-7) more efficiently than MUC-1 negative cells (HepG2) which shows its selective behavior when compared to the free EPI. Surprisingly, the toxicity threshold of EPI increased by 1.5 times using the nanocomposite. Taking advantage of imaging and chemotherapy, this nanocomposite is able to detect HT-29 and MCF-7 specifically as MUC-1 expressing cancer cells and release EPI inside of them.
Introduction
Despite many advances in the field of cancer treatment such as chemotherapy, radiology, and surgery, it still remains the second leading cause of death in our world.1 The main challenge in cancer therapy is the significant toxicity and unavoidable side effects of chemotherapeutic drugs on healthy tissues due to their non-selective nature.2 So, designing a suitable drug delivery system to overcome these problems is a necessity.3Theranostics in nanotechnology refers to multi-functional nanoparticles with potential use in both diagnostic and therapeutic applications.4–6 Using multi-functional nanoparticles has received significant attention in biomedicine as promising tool for target specific delivery of imaging and therapeutic agents.7 Inorganic nanomaterials with their unique intrinsic properties have attracted a lot of interest in the field of nanomedicine and are applicable in tumor cell imaging,8 targeted drug delivery,9 bioseparation,2 and biosensing.10 Fluorescent and magnetism are the most popular properties of inorganic nanomaterials.11 Among fluorescent nanomaterials, gold nanoclusters (AuNCs) have attracted a lot of interest8 due to their ultrafine size, high photobleaching threshold, large Stokes shift, good biocompatibility, and low environmental hazard.12,13 Fusing of AuNCs and magnetic nanoparticles could lead to the formation of a multi-functional nanocomposite with the enjoyment of properties of both components within a single nanoplatform such as enhanced stabilization, optical, and superparamagnetic properties.14,15
Nanoparticles can be coated with biocompatible polymers such as polyethylene glycol (PEG) to increase their circulation time in the biological environment by evading macrophage-mediated uptake.16 The ability of PEG to hide nanoparticles from the immune system is known as the stealth effect.17 Functionalization of PEG with reactive functional groups provides the situation for conjugation to biologically-relevant targeting moieties such as aptamers and antibodies.
Surface functionalization of nanomaterials with targeting ligands (e.g. antibodies, aptamers, peptides, and small molecules) can overcome several drawbacks of nanoparticles, such as insufficient cell uptake and non-specific accumulation of nanomaterials in normal tissues during the transportation process of drugs and imaging agents to the tumor sites.18 Among targeting ligands, single-stranded oligonucleotides, widely known as aptamers, are of interest.19–21 Ease of synthesis and surface modification, reproducibility, lack of immunogenicity, and high stability are the advantages of aptamers over other technologies.7 To date, variable forms of magnetic and gold nanomaterials have been developed for protein or cell separation, sensing, and catalytic applications. But few reports are devoted to the formation of a single platform of magnetite and gold nanocomposites (smaller than 100 nm) which are functionalized with aptamer or targeting ligands for simultaneous tumor cell imaging, targeting, drug delivery, and tumor therapy. For example, Li and co-workers7 developed a five function aptamer-conjugated nanorose (Fe3O4@Au, 70 nm in diameter) for specific delivery of doxorubicin as a fluorescent imaging agent and chemotherapeutic drug to CCRF-CEM cancer cells. Magnetic resonance imaging and photothermal therapy were the reported applications of the synthesized nanorose. In 2011, Durgadas et al.8 reported the green synthesis of highly blood compatible superparamagnetic quantum clusters with a mean diameter of 10 nm. The surface of the quantum cluster was coated with transferrin protein for selective detection and separation of C6 glioma cells from blood. They fused dopamine anchored magnetic nanoparticles to the BSA capped gold nanoclusters through a carboxyl activating agent. They reported that in the cytotoxicity assay, dopamine conjugated magnetic nanoparticles decreased the cell viability below 75%; while their combination with BSA capped gold nanoclusters showed an ascending trend in the cell viability. Ocsoy and co-workers22 synthesized nanoflowers of Au@MnO (average particle size = 18 nm). The surface of the nanoflowers was functionalized with the sgc8 aptamer to target CCRF-CEM cancer cells. Also, by surface functionalization with an anti-ATP aptamer, the nanoflowers were able to capture and sense adenosine triphosphate (ATP) as the metabolite product of cancer cell lysates using mass spectral analysis. In 2016, Machulkin et al.23 synthesized Fe3O4–Au dumbbell like nanoparticles decorated with a prostate specific membrane antigen (PSMA) aptamer for diagnostics of prostate cancer cells. They reported that the fabricated nanoparticles have the ability for tissue-specific magnetic resonance imaging.
In this study, we synthesize a nanocomposite comprising silica coated magnetite nanoparticles (Fe3O4@SiO2) decorated with gold nanoclusters (AuNCs). (γ-mercaptopropyl) trimethoxysilane (MPS) was employed to synthesize small uniform and highly stable AuNCs. Furthermore, the hydrolysable methoxy silane groups of MPS provide the condition for immediate reaction with silanol groups on the surface of the silica coated Fe3O4 NPs. Coating of the fabricated nanocomposite with poly(ethylene glycol)-dimethacrylate (PEGDMA) provides the condition for efficient conjugation of the thiol-modified MUC-1 aptamer (Apt) via a rapid and simple thiol-en click reaction. Subsequently, functionalization of the Fe3O4–AuNC with the CG-rich region modified MUC-1 Apt enables the nanocomposite to load a high amount of chemotherapeutic drug. MUC-1 is a transmembrane glycoprotein abnormally overexpressed on the surface of a variety of cancer cells including breast, ovarian, lung, and pancreatic cancers.2,24 In the following, the prepared Fe3O4–AuNC–Apt was loaded with epirubicin (EPI) to investigate the ability of selective drug delivery to MCF-7 and HT-29 as MUC-1 expressing cancer cells versus HepG2 as MUC-1 negative cells. Epirubicin belongs to a group of drugs called anthracyclines.25 Anthracyclines (e.g. doxorubicin, daunorubicin, and epirubicin) are red aromatic polyketides used to treat many types of cancers such as breast, colon, lymphoma, and leukemia.26 They consist of three moieties: a pigmented aglycone, an amino sugar, and a lateral chain. They can intercalate to the CG-rich region of the DNA or RNA aptamers.27 Epirubicin is the 4-epimer of doxorubicin and due to fewer side effects and reduced toxicity it is more favorable than doxorubicin (the most popular chemotherapeutic drug).41
Finally, the ability of the synthesized Fe3O4–AuNC–Apt(EPI) for selective detection, imaging, and therapy of the cancer cell lines in vitro was investigated.
Experimental
Materials
Epirubicin hydrochloride, poly(ethylene glycol)-dimethacrylate (PEGDMA, average Mn 250), and sephadex LH-20 were purchased from Sigma-Aldrich. 3-(4,5-dimethyl thiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophynyl)-2H-tetrazolium (MTS, >98%) was purchased from Promega. All solvents and other chemical reagents were purchased from Merck and were used as received without further purification. The DMEM medium and fetal bovine serum (FBS) were obtained from Gibco. The cancer cell lines were purchased from the national center for genetic and biological diversity of Iran. The MUC-1 targeting DNA aptamer modified by a thiol group (denoted as Apt) was purchased from SBS Genetech Co. Ltd (Shanghai, China) with the following sequence: 5′HS-C6 spacer GATCCTTTGGATA GATCCTTTGGATA GATCCTTTGGATA-3′. All the solutions were prepared in doubly-distilled deionized water.Characterization
Steady-state luminescence measurements were performed on a SHIMADZU RF 5301PC spectrofluorophotometer. Fourier transform infrared spectra were recorded on an FT-IR JASCO 680-PLUS spectrometer in the region of 4000–400 cm−1 using KBr pellets. The transmission electron microscopy (TEM) images were obtained using a Zeiss-EM10C-80 kV (Germany). The size distribution of the particles was determined from the TEM images using the SPSS program. Field-emission scanning electron microscopy (FE-SEM) and energy-dispersive X-ray analysis spectroscopy (EDAX) of the nanocomposite were further performed using a MIRA3TESCAN-XMU. X-ray diffraction measurements (XRD) were performed using an X-ray diffractometer (Philips EXPERTMPD) using Cu Kα (λ = 0.154 nm) radiation. The samples were scanned from 20 to 80° of 2θ. The hysteresis loop was measured on a Lake Shore 7410 at room temperature. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was performed using an Optima 7300 DV instrument. The zeta potentials of the freshly prepared NPs were measured with a Malvern Zetasizer (Nano ZS, Malvern Instruments, UK). A vibrating sample magnetometer (VSM, 7400, Lakeshare, USA) was utilized to determine the magnetic properties of nanoparticles in a magnetic field with ±8 kOe at 25 °C. The flow cytometry analysis was done with a BD FACScalibur cytometer. Also, the flow cytometry data were analyzed using Flowing 2.5.1 software (downloaded from http://flowingsoftware.btk.fi/download/).Synthesis of Fe3O4 NPs and silica modification (Fe3O4@SiO2)
Synthesis of Fe3O4 NPs and their further coating with a silica layer were done according to a literature method with some modifications.28,29 Briefly, 258 mg of trisodium citrate, 160 mg of sodium hydroxide, and 17 g of sodium nitrate were stirred into 20 mL of deionized water and heated to 100 °C. Then, 0.45 mg of iron(II) sulfate tetrahydrate solution was added rapidly and allowed to complete the reaction for 1 h at the same condition. Finally, after cooling and magnetic separation, in order to apply the silica coating, the freshly prepared Fe3O4 NPs (0.05 g) were dispersed in 2.5 mL deionized water and sonicated for 60 min. After that, the volume of the resulting dispersion was adjusted to 50 mL and the resultant suspension was poured into 175 mL ethanol containing 6.2 mL NH3 (25 wt%) during mechanical stirring at 40 °C. Then a solution of tetraethyl orthosilicate (TEOS, 1.25 mL) in ethanol (12.5 mL) was added to the dispersion in a dropwise manner under continuous mechanical stirring. The resultant dispersion was stirred mechanically for 14 hours at 25 °C. Finally, the Fe3O4@SiO2 NPs were collected by an external magnet and washed with ethanol and deionized water.Synthesis of AuNC@MPS
The AuNCs were prepared in situ in an ethanolic solution of MPS.30 Briefly, 32 μL of pure MPS was added to 8.0 mL ethanol, followed by the addition of 16 μL of HAuCl4·3H2O aqueous solution (0.6 mg μL−1). Then, we allowed the reaction mixture to stir for about 10 min until the color of the mixture changed from yellow to colorless. After that, 1 mL of a 0.16 M NaBH4 methanolic solution was added to the Au+@MPS intermediate in one portion under vigorous stirring. The mixture was stirred at 25 °C for 20 min to complete the reaction. The final light brown solution was gel filtered over a sephadex LH-20 column to remove the excess MPS or salts.Synthesis of Fe3O4–AuNC–Apt
In order to decorate the AuNCs on the surface of Fe3O4@SiO2 NPs, 1.0 mL of the AuNC sol (187 μg mL−1) was immediately after its preparation added to 1.0 mL of 12.7 μg mL−1 Fe3O4@SiO2 aqueous solution. Then, PEGDMA (170 μL) was added to the above solution and stirred at room temperature for 12 h. The final product was magnetically separated, washed with water, and redispersed in Tris–HCl buffer (1.0 mL, 10 mM, and pH 7.4). After that, the Apt was immobilized on the surface of the nanocomposite, immediately. First, 20 μL of 10 μM of Apt in Tris–HCl (10 mM, pH 7.4) was activated with 100 μL of 1 mg mL−1 DTT by incubating at 30 °C for 30 min. Then, the aptamer solution was mixed with the nanocomposite in the presence of thermal initiator V50 (1 mg) for being assembled on PEGDMA functionalized Fe3O4–AuNC in the incubator at 55 °C for 5 h. The final product was magnetically separated and the supernatant was collected to measure the amount of unreacted Apt by spectrophotometry.Introduction of EPI to the Fe3O4–AuNC–Apt
The absorbance titration method was used to measure the amount of EPI loaded onto the Fe3O4–AuNC–Apt. For this purpose, a fixed concentration of EPI (10 μM) was incubated with increasing concentrations of purified Fe3O4–AuNC–Apt (10–100 nM) for 10 min. The absorbance of the unbound EPI was calculated at 470 nm relative to the calibration curve recorded under the same condition, allowing the drug loading efficiency to be estimated.Cell imaging
MCF-7, HT-29, and HepG2 cells were allowed to adhere to a glass cover slip in a 24-well plate for 24 h (i.e., 2 × 104 cells per well). The cells were then incubated with 60 nM of Fe3O4–AuNC–Apt for 2 h at 37 °C. Then, the culture medium was aspirated and the cells were washed three times with PBS− to remove the excess nanocomposites in advance; finally, they were fixed with 4% (by mass) paraformaldehyde in PBS− for 15 min. All the experiments were performed in triplicate.Flow cytometry analysis
In order to assess the internalization of EPI and Fe3O4–AuNC–Apt(EPI), flow cytometry analysis was performed. MCF-7, HT-29, and HepG2 cells were grown in 12-well plates for 24 h (i.e., 2.5 × 105 cells per well). The cells were then incubated with 75 nM of Fe3O4–AuNC–Apt(EPI) (containing 7.5 μM of the intercalated EPI) for 2 h at 37 °C. After that, the cells were digested with enzyme and centrifuged at 1800 rpm for 7 min. After removing the supernatant, the cells were dispersed in PBS−. The fluorescence of EPI was used to detect the localization of EPI in the cells.Cell culture and cytotoxicity assay
Cell viability was measured by MTS assay. Briefly, the cells were grown to about 70% confluence in 96-well plates. Before incubation with nanocomposites, cells were washed with PBS− and incubated with 150 μL of fresh medium for 30 min. The cells were incubated with either Fe3O4–AuNC–Apt (1–4 nM), or Fe3O4–AuNC–Apt(EPI) (1–4 nM containing 0.1–0.4 μM of the intercalated EPI). Free EPI (7.5 μM) and untreated cells were considered as positive and negative controls, respectively. After 48 h, the supernatant was removed and cells were washed immediately with PBS−. Then, an aliquot of 150 μL DMEM and 15 μL MTS stock solution were subsequently added to each well and incubated for 4 h at 37 °C. Finally, the absorbance was read with a microplate reader at 490 nm.Results and discussion
Synthesis and characterization of the Fe3O4–AuNC nanocomposite
In order to synthesise a highly dispersed and fluorescent nanocomposite, an in situ method was used for the preparation of the AuNCs. Then, the freshly prepared AuNCs were directly conjugated to the silica coated Fe3O4 NPs. PEGDMA was added to the sol–gel mixture to increase the amount of aptamer conjugation along with the biocompatibility without increasing the size. Also, to obtain a nanocomposite with a high drug loading capacity, a thiol-modified MUC-1 Apt with a modified CG base pair region was employed. The sequence of the Apt was reported in the literature,31 but its conjugation to the nanoparticle was used in this study for the first time. The schematic procedure for the preparation of the nanocomposite is shown in Scheme 1. | ||
| Scheme 1 Schematic illustration for the preparation of the Fe3O4–AuNC–Apt(EPI). | ||
In the first step, highly dispersed and water stable Fe3O4 NPs were synthesized in the way reported earlier with some modifications.28 A water-based method was used for the preparation of these NPs. Synthesis of the Fe3O4 NPs in water at room temperature and neutral pH value facilitates the biological application of them in comparison to the hydrophobic Fe3O4 NPs accompanied by toxic chemicals produced in the thermal decomposition method.32
Fig. 1(A and C) shows the TEM image of the synthesized Fe3O4 NPs as well as the size distribution histogram. According to the histogram analysis, the nanoparticles have an average particle size of 20 ± 2.5 nm with a relatively narrow size distribution range. Also the FESEM image (Fig. 1B) shows the morphology of the Fe3O4 NPs. In addition, zeta potential analysis reveals a 23.5 mV negative surface charge which is related to the presence of sodium citrate salt which increases the dispersity of the produced nanoparticles. Also, addition of sodium nitrate to the reaction solution provides a rich ionic strength in the reactive environment which makes the electrical charges around the formed NPs equally distributed.33 The stability of dispersed Fe3O4 NPs is a key point that guarantees uniform coverage in subsequent layers.
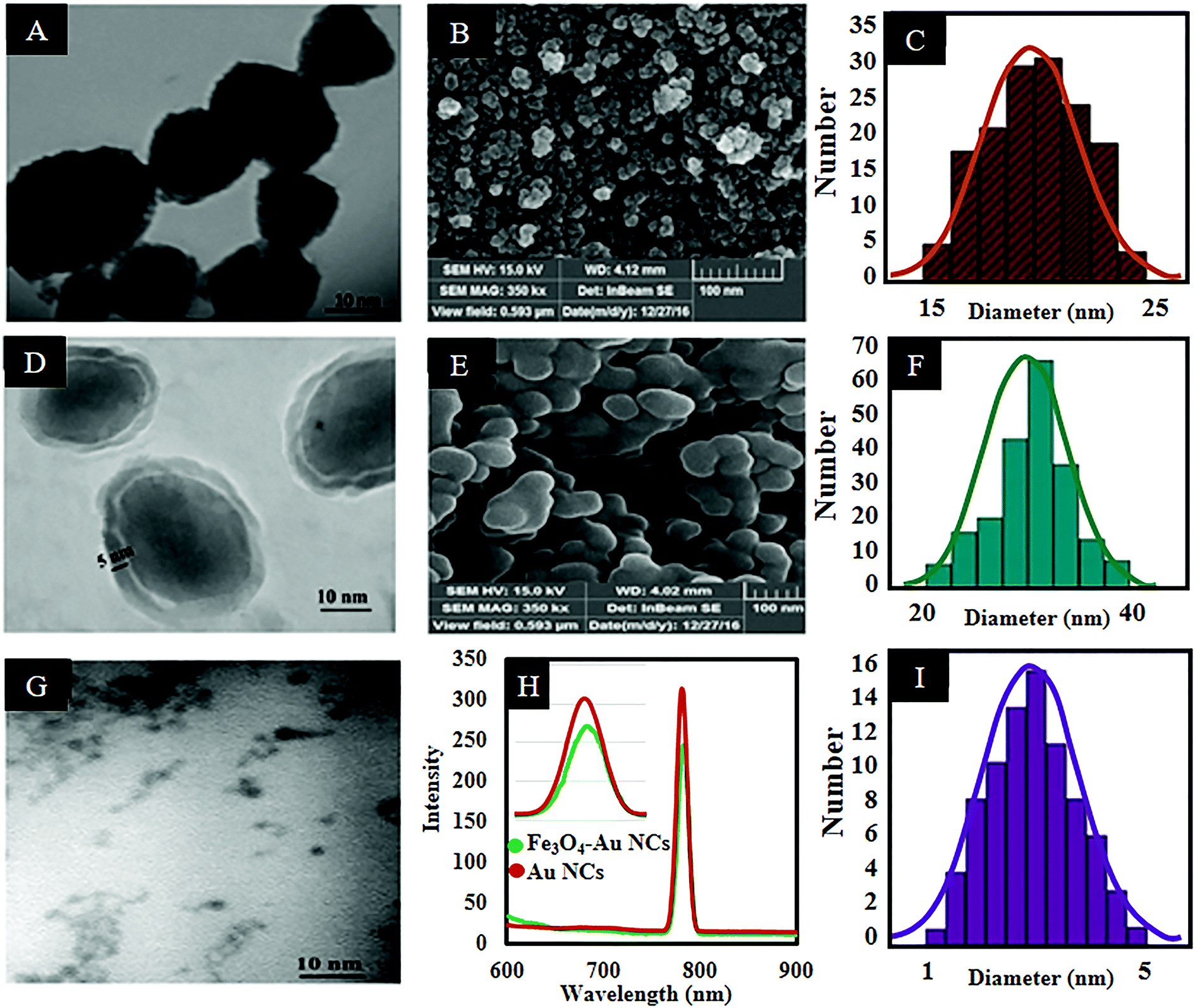 | ||
| Fig. 1 TEM and SEM images of Fe3O4 (A and B) and Fe3O4@SiO2 (D and E) nanoparticles. The corresponding size distribution histogram of Fe3O4 (C) and Fe3O4@SiO2 (F) nanoparticles. TEM and size distribution histogram of AuNCs (G and I). Emission spectra of AuNCs before and after conjugation of Fe3O4 NPs (H). | ||
Silica coated Fe3O4 NPs were synthesized by employing Fe3O4 NPs as the core and TEOS as a silica source via a modified sol–gel method.29Fig. 1(D and E) shows TEM and FESEM images of the Fe3O4@SiO2 NPs. As can be seen in Fig. 1D, the relatively translucent region surrounding the Fe3O4 core is attributed to the silica shell.34 It is well established that a thicker silica shell can reduce the magnetic power of the Fe3O4 NPs due to the diamagnetic effect of the silica shell.35 So, the thickness of the silica shell was adjusted to about 5 ± 1 nm. Also according to the obtained histogram, the average size of the Fe3O4@SiO2 NPs is 30 ± 2.5 nm (Fig. 1F).
Afterward, MPS coated-AuNCs (AuNC@MPS) as a fluorescent component of the nanocomposite were synthesized. The TEM image and the size distribution histogram of the prepared AuNCs are shown in Fig. 1(G and I). As can be seen, the nanoclusters are well dispersed with a narrow size distribution and an average particle size of 2.5 ± 0.5 nm. The fluorescence spectrum (Fig. 1H) of the resultant AuNCs shows a red emission peak located at 785 nm (λex = 520 nm).8,12,36 Under consideration of the large Stokes shift (λem − λex = 785 − 520 = 265 nm) and the narrow emission spectrum, it can be concluded that the prepared AuNCs are highly fluorescent.37 The emission spectra of AuNCs are typically broad, but it is not general. A narrow FWHM (less than 20 or even 10 nm) for AuNCs (with a size of about 2 nm) was reported previously in the literature.38 The narrow peak can be attributed to less distortion in the excited state. The fluorescence quantum yield (QY) of the AuNCs was calculated. Using rhodamine 6G (QY: 0.95 in ethanol) as the standard, the QY of the synthesized AuNCs was found to be 1.5% at room temperature.
The AuNCs were synthesized in situ in the MPS solution as a thiol capping agent. Our previous experiments showed that in situ generation of gold nanoparticles can significantly reduce the size of the produced nanoparticles due to growth inhibition of the newborn nucleus, and thus keep the size dependent fluorescent property.39 Also, the presence of thiol groups in the structure of capping ligands can lead to the formation of AuNCs, owing to the formation of high affinity Au–S covalent bonding (soft–soft interactions).39,40 After addition of HAuCl4 aqueous solution to the ethanolic mixture of the MPS, the colorless Au(I)–S complex forms.30 It may be concluded that a redox reaction occurs to produce Au(I) by oxidizing the thiol group of the MPS to a disulfide.41 Finally, addition of NaBH4 reduces this Au(I)–S complex to form light-brown AuNC@MPS. Also, FT-IR spectroscopy was employed to confirm the presence of MPS on the surface of AuNCs. Fig. S1 (ESI†) shows the spectra of MPS in the absence and presence of the AuNCs. As can be seen in the spectrum of the free MPS, three bands located at 2959, 2840, and 1191 cm−1 are attributed to the methoxysilane groups.30 It is noticeable that the weak band at 2500 cm−1 assigned to the stretching of the thiol group (H–S)42 disappears completely in the spectrum of the AuNC@MPS indicating the chemisorption of MPS on the surface of the AuNCs.30 Also, the band at 954 cm−1 in the spectrum of AuNC@MPS refers to the partial hydrolysis of the methoxysilane groups.30,43
In this work, the freshly prepared AuNCs were seeded directly on the surface of the Fe3O4@SiO2 through a silanization reaction between the alkoxy groups of the MPS and the hydroxyl groups on the surface of the Fe3O4@SiO2. So, it can be concluded that the MPS has two-way potential to form stable covalent bonds with both the AuNCs via formation of a Au–S bond and also with the hydroxyl groups on the surface of the Fe3O4@SiO2 (Si–O–Si bond). Therefore, by continuing the hydrolysis condition, the stabilized AuNCs in a continuous shell of thiol are embedded at the surface of the Fe3O4@SiO2. According to the fluorescence spectra (Fig. 1H), after conjugation of the AuNC@MPS to the Fe3O4@SiO2, the fluorescence intensity of the nanoclusters slightly decreases without any significant shift. However, the thiol and silica shell can insulate the gold core from electron transfer reaction and support the florescent activity of AuNCs. But in this case, the fluorescent decrease of the AuNCs can be attributed to the quenching effect of the Fe3O4 NPs due to some intrinsic properties of the Fe3O4 NPs.2,12 TEM and FESEM were employed to determine the size and morphology of the Fe3O4–AuNC nanocomposite, respectively. According to the TEM image and the size distribution histogram of the Fe3O4–AuNC nanocomposite (Fig. 2(A and C)), the size distribution is narrow and more than 90% of the nanocomposites are distributed in the range of 35 ± 2 nm. The FESEM image of the Fe3O4–AuNC nanocomposite (Fig. 2B) shows that the Au shell consists of a few adjacent AuNCs.44 EDAX (Fig. 2E) of selected areas of the Fe3O4–AuNC nanocomposite further reveals its elemental composition. Fe, Au, O, C, Si, and S can all be easily found in the EDAX graph of the Fe3O4–AuNC nanocomposite. The metal composition of the nanocomposite was quantitatively determined by ICP-OES. The Fe3O4/AuNC molar ratio of the product was estimated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :68. We examined different ratios of Fe3O4@SiO2 to AuNCs. According to our experimental results, the chosen ratio (1:68) is the optimized ratio for our system. Higher than this ratio results in unbound AuNCs and lower than that leads to vacancies on the surface of the Fe3O4@SiO2, due to the lack of AuNCs, which shows a decrease in fluorescence intensity. In addition, XRD analyses were used to check the composition and crystallinity of Fe3O4, Fe3O4@SiO2, and Fe3O4–AuNC nanomaterials. The obtained XRD pattern of Fe3O4 NPs is in accordance with the literature reports (Fig. S2, ESI†).34 In the XRD pattern of the Fe3O4@SiO2, a broad peak from θ = 23.66 to 28.99° can be assigned to amorphous silica and proves the existence of the silica (see Fig. S3, ESI†).34 After conjugation of the AuNCs to the surface of the Fe3O4@SiO2, the peaks of the silica-coated magnetite are unchanged and four new peaks appear, which are assigned to the (111), (200), (220), and (311) planes of Au in the cubic phase, confirming the crystallinity of the Fe3O4–AuNC nanocomposite (Fig. 2E).34
:68. We examined different ratios of Fe3O4@SiO2 to AuNCs. According to our experimental results, the chosen ratio (1:68) is the optimized ratio for our system. Higher than this ratio results in unbound AuNCs and lower than that leads to vacancies on the surface of the Fe3O4@SiO2, due to the lack of AuNCs, which shows a decrease in fluorescence intensity. In addition, XRD analyses were used to check the composition and crystallinity of Fe3O4, Fe3O4@SiO2, and Fe3O4–AuNC nanomaterials. The obtained XRD pattern of Fe3O4 NPs is in accordance with the literature reports (Fig. S2, ESI†).34 In the XRD pattern of the Fe3O4@SiO2, a broad peak from θ = 23.66 to 28.99° can be assigned to amorphous silica and proves the existence of the silica (see Fig. S3, ESI†).34 After conjugation of the AuNCs to the surface of the Fe3O4@SiO2, the peaks of the silica-coated magnetite are unchanged and four new peaks appear, which are assigned to the (111), (200), (220), and (311) planes of Au in the cubic phase, confirming the crystallinity of the Fe3O4–AuNC nanocomposite (Fig. 2E).34
 | ||
| Fig. 2 Characterization of the Fe3O4–AuNC. TEM image (A), FESEM image (B), size distribution histogram (C), XRD spectrum (D), EDAX analysis (E), and VSM curves of (1) Fe3O4, (2) Fe3O4@SiO2 and (3) the Fe3O4–AuNC nanocomposite (F). | ||
The magnetic properties of the synthesized products were investigated using VSM analysis at room temperature. As can be seen in Fig. 2F, the products exhibit superparamagnetic behavior without any hysteresis loops. The superparamagnetic property can prevent agglomeration of the nanoparticles and they can be well dispersed in the solution after removing the magnetic field. This property enables the magnetic nanoparticles to be suitable for biological applications such as bioseparation and biodetection.44 According to Fig. 2F, the magnetic saturation (MS) value of the Fe3O4 NPs is about 38 emu g−1 which is decreased to 30 and 27 emu g−1 after wrapping with silica and AuNC layers, respectively. The reduction of the MS value shows that the Fe3O4 NPs are covered with non-magnetic silica and AuNC layers as confirmed by TEM and FESEM images.2,12
Also, as shown in the inset of Fig. 2F, the produced Fe3O4–AuNC nanocomposite is attracted easily to the walls of the vial in the presence of an external magnet owing to its strong magnetic response.44 The colloidal stability of Fe3O4, Fe3O4@SiO2 and Fe3O4–AuNC nanoparticles in Tris–HCl buffer (1.0 mL, 10 mM, and pH 7.4) was evaluated. As can be seen in Fig. S4 (ESI†), there is no aggregation in the colloidal solution of the freshly prepared nanoparticles (Fig. S4(A), ESI†) and after 10 days (Fig. S4(B), ESI†) at room temperature. Hydrodynamic sizes of the mentioned nanoparticles were measured to confirm their colloidal stability. The sizes of the freshly prepared Fe3O4@SiO2, Fe3O4@SiO2–AuNC, and Fe3O4@SiO2–AuNC–Apt were measured to be 97, 141, and 198 nm, respectively. The long term stability of the nanoparticles was also evaluated by measuring their hydrodynamic sizes after 10 days (see Fig. S4(C), ESI†). The results showed that the sizes of nanoparticles do not change significantly, suggesting their high colloidal stability. Most of the cell functions can be regulated by intracellular pH. Cancer cells are generally associated with higher values of intracellular pH 7.12–7.65 and lower extra cellular pH 6.2–6.9.45–47 Regarding this point, the colloidal stability of the Fe3O4–AuNC nanocomposite was also studied at various pH (6.2, 7.4, and 7.6) using dynamic light scattering (DLS). As can be seen in Fig. S5 (ESI†), the size of the Fe3O4–AuNC nanocomposite does not change significantly after this period of time.
Bioconjugation of the Fe3O4–AuNC nanocomposite
In order to fabricate a nanocomposite with molecule recognizing ability for specific cell detection, an aptamer was conjugated to the surface of the Fe3O4–AuNC. In this regard, PEGDMA was employed to improve the dispersity, biocompatibility, and also facilitate the conjugation of the Apt on to the surface of the nanocomposite. Furthermore, the presence of PEGDMA in the sol–gel mixture reduces the growth rate of the silica network and the thickness of the coating, subsequently. The presence of PEGDMA on the surface of the nanocomposite was confirmed by FT-IR spectroscopy (Fig. S6, ESI†). As shown in the spectra, the strong bands at 1622, 1588, and 955 cm−1 are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, and CC–H of the conjugated PEGDMA, respectively. After conjugation of the Apt to the surface of the Fe3O4–AuNC nanocomposite, a new peak at 625 cm−1 appears which corresponds to the heterocyclic groups of the Apt. Also, the intensity of the CC–H band is decreased significantly. These results confirmed that the surface of the nanocomposite was successfully functionalized by the Apt. Also, elemental analysis of Fe3O4–AuNC–Apt indicates the contents (w/w) of C, H, N, and S are 50.47, 8.18, 8.06, and 3.14%, respectively. The presence of N atoms is more evidence to confirm the conjugation of the Apt to the surface of the nanocomposite.
O, CC, and CC–H of the conjugated PEGDMA, respectively. After conjugation of the Apt to the surface of the Fe3O4–AuNC nanocomposite, a new peak at 625 cm−1 appears which corresponds to the heterocyclic groups of the Apt. Also, the intensity of the CC–H band is decreased significantly. These results confirmed that the surface of the nanocomposite was successfully functionalized by the Apt. Also, elemental analysis of Fe3O4–AuNC–Apt indicates the contents (w/w) of C, H, N, and S are 50.47, 8.18, 8.06, and 3.14%, respectively. The presence of N atoms is more evidence to confirm the conjugation of the Apt to the surface of the nanocomposite.
In the present study, the 5′ end thiol-modified MUC-1 DNA aptamer (Apt) was introduced as a recognizing agent. The active targeting region of the Apt is repeated three times to improve the targeting and drug intercalation/delivery efficacy of the Apt.45 The aim of this study is to calculate the amount of EPI that can intercalate within the CG-rich region when the number of these regions triplicates. Fig. 3A shows the optimized structure of the Apt contains a one (Apt-L1) or three (Apt-L3) loop sequence. The molecular docking analyses of the drug (EPI) with the Apt-L1 or Apt-L3 were performed using AutoDock 4.2 software.48 The binding free energies (ΔGb) of Apt-L1 and Apt-L3 were found to be −4.64 and −5.70 kcal mol−1, respectively. These results show that EPI has higher binding affinity to Apt-L3 as compared with Apt-L1. Fig. 3B demonstrates the molecular docking and the binding sites of the drug interacting with Apt-L1 and Apt-L3. Also, the interaction schemes of the residues of binding sites of the above aptamers with EPI (generated by the LIGPLOT+ software) are shown in Fig. 3C. It can be seen that the dominant interactions between the drug and the residues in the active site of Apt-L1 are hydrophobic; while in the case of Apt-L3 simultaneous hydrophobic and hydrogen interactions stabilize the intercalation of the drug in the active site of the aptamer. We expected that by increasing the number of active regions of Apt-L1, more EPI can be loaded. Previous studies showed that the ratio of EPI to Apt-L1 is 1:2.25 After increasing the number of active regions, this ratio reached 1:1. Also ΔGb showed that the affinity of Apt-L3 increased for EPI.
 | ||
| Fig. 3 The optimized structure of the Apt contains a one (Apt-L1) or three (Apt-L3) loop sequence (A), molecular docking and the binding sites of the drug interacting with Apt-L1 and Apt-L3, respectively (B) and the interaction schemes of the residues of binding sites of the above aptamers with EPI (generated by the LIGPLOT+ software) (C). | ||
Conjugation of EPI to the Apt was evaluated by fluorescence spectroscopy. EPI is a fluorescent chemotherapeutic drug that can be intercalated to the CG-rich region of DNA or RNA aptamers.49 Conjugation of EPI within the Apt, results in the quenching of the EPI fluorescence (Fig. 4).25 According to Fig. 4A, addition of various concentrations of Apt (1–9 μM) to a constant concentration of EPI (10 μM) leads to a reduction in the fluorescence intensity of EPI. The results show that maximum quenching occurs at approximately a 1:1.1 molar ratio of drug to aptamer and complete intercalation is achieved. As reported previously, the maximum quenching of EPI occurs at approximately a 1:2 molar ratio of drug to aptamer,25 while our theoretical calculations show that the repeating sequences of the active targeting region of the Apt provide stronger binding and so a higher amount of drug loading which are in harmony with the florescence results.
 | ||
| Fig. 4 (A) Fluorescence spectra of EPI in the presence of increasing molar ratio of Apt (λex = 488 nm, λem = 588 nm), (B) fluorescence spectra of EPI in the presence of increasing molar ratio of nanocomposite. | ||
Then using a spectrophotometer, increasing concentrations of Fe3O4–AuNC–Apt (0.105–0.135 μM) were incubated with 15 μM of EPI (Fig. 4B). The prepared Fe3O4–AuNC–Apt(EPI) nanocomposite contains approximately 95 EPI molecules per nanocomposite. With consideration of the molar ratio of drug to aptamer, there are also 95 Apt molecules on the surface of the nanocomposite.
Specific cellular targeting with the nanocomposite
The specificity and sensitivity of the fabricated Fe3O4–AuNC–Apt(EPI) nanocomposite as an imaging, drug delivery, and therapy system in vitro were assessed. First of all, the ability of the Fe3O4–AuNC–Apt nanocomposite as a labeling agent in recognizing targeted cancer cells (MCF-7 and HT-29 as MUC-1 positive cells) versus non targeted cancer cells (HepG2 as MUC-1 negative cells) was evaluated using microscopic imaging and flow cytometry techniques. All cell lines were incubated with the Fe3O4–AuNC–Apt conjugate for 2 h at 37 °C. After washing with PBS− to remove unbound nanocomposite, the cells were analyzed by fluorescent microscopy using the inherent fluorescence properties of the AuNCs. The bright field images of Fig. 5A indicate that the morphology of the cells does not change which shows the good biocompatibility of the nanoparticles at the tested doses and time. Also, from the dark field images of the cancer cells it can be observed that the fluorescence intensity of the Fe3O4–AuNC–Apt in the desired cells (MCF-7 and HT-29) is significantly higher than the control cells (HepG2). These results show that the fabricated Fe3O4–AuNC–Apt exhibits cell specificity and can be selectively taken up by the MUC-1 positive cells. In order to determine the quantity of the Fe3O4–AuNC–Apt taken up by the cells, ICP-OES was employed. The results of ICP-OES analysis revealed that the uptake of the Fe3O4–AuNC–Apt conjugate by MCF-7 and HT-29 cells is 9 times higher than that by Hep-G2 (Fig. S7, ESI†). | ||
| Fig. 5 Bright field, fluorescent and overlay microscopic images of HepG2, MCF-7 and HT-29 after incubation with Fe3O4–AuNC–Apt (A), flow cytometry histograms of the mentioned three cells after incubation with free EPI and Fe3O4–AuNC–Apt(EPI) (B). | ||
Flow cytometry analysis was used to determine the binding properties of Fe3O4–AuNC–Apt after intercalation of EPI. The three cell lines were treated with EPI and the Fe3O4–AuNC–Apt(EPI) nanocomposite and the fluorescence of EPI was used to evaluate the nanocomposite uptake by the MUC-1 positive and MUC-1 negative cells. As can be seen in Fig. 5B, for MUC-1 positive MCF-7 and HT-29 cells, the fluorescent signals generated by free EPI or Fe3O4–AuNC–Apt(EPI) are similar; whereas for MUC-1 negative HepG2 cells, the fluorescent signal generated by Fe3O4–AuNC–Apt(EPI) is remarkably lower than that generated by free EPI. The results again suggest that Fe3O4–AuNC–Apt(EPI) could be selectively taken up by the MUC-1 positive cancer cells and the intercalation of EPI within the MUC-1 aptamer does not affect its binding properties.25 Taken together, the results of fluorescent microscopy and flow cytometry analysis show that the Fe3O4–AuNC–Apt(EPI) nanocomposite can recognize MUC-1 positive cancer cells specifically and release EPI inside of them.
In vitro cytotoxicity
MTS analysis was performed to evaluate the specific cell toxicity of the fabricated nanomaterials. For this purpose, Fe3O4–AuNC–Apt and Fe3O4–AuNC–Apt(EPI) nanocomposites, and free EPI were used to treat the three cell types. As can be seen in Fig. 6A, all the cell lines show no appreciable viability loss after treatment with a series of concentrations of Fe3O4–AuNC–Apt. These results show the excellent biocompatibility of the synthesized nanocomposite. Then, the cytotoxicity of the parent EPI and Fe3O4–AuNC–Apt(EPI) nanocomposite was examined. According to the MTS results, non-selective cytotoxicity of parent EPI on the three cell lines indicates its adverse side effect on normal tissues. While, the toxicity of the Fe3O4–AuNC–Apt(EPI) nanocomposite is significantly higher in MCF-7 and HT-29 than HepG2 cells (Fig. 6B). Surprisingly, according to the data obtained from Fig. 6B, the toxicity threshold of EPI increased by 1.5 times using the nanocomposite. These results show the efficiency of the nanocomposite in transporting the chemotherapeutic drug to the desired cancer cells. Also, a drug release experiment (Fig. S8, ESI†) shows that after 1h, about 30% of EPI is released which shows the relatively fast drug release.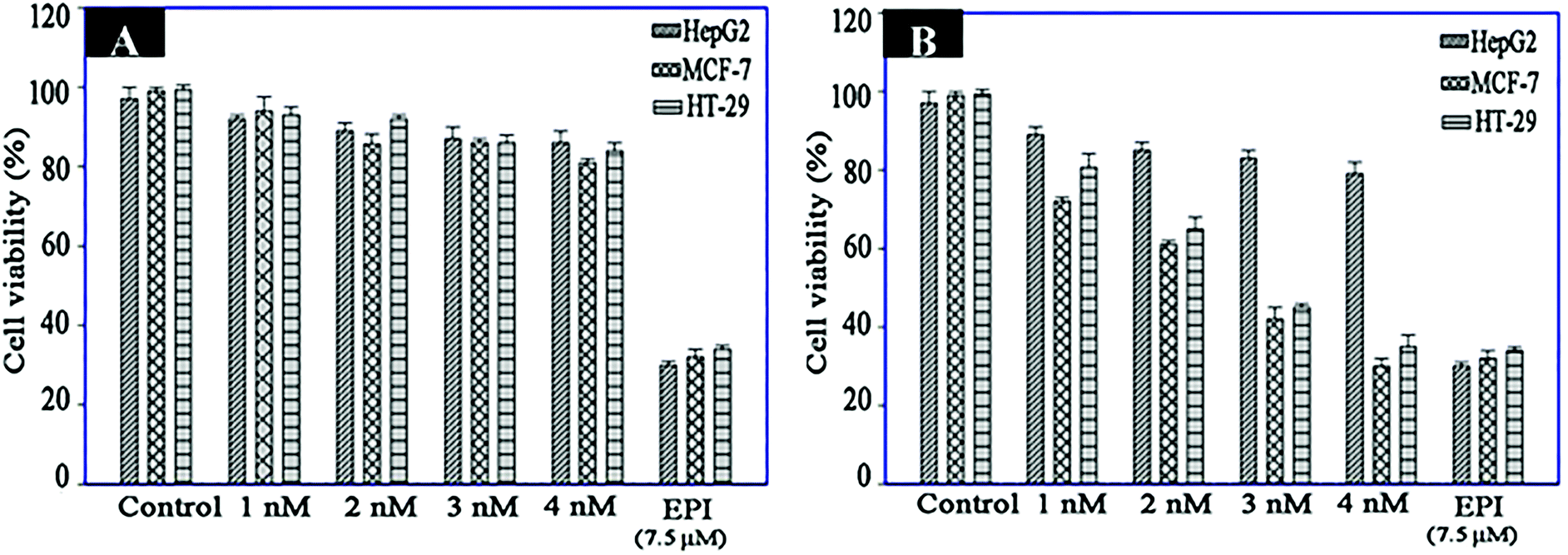 | ||
| Fig. 6 Viability assay of HT-29, MCF-7 (target cells) and HepG2 (control cells) treated with different concentrations of Fe3O4–AuNC–Apt (A) and Fe3O4–AuNC–Apt(EPI) (B) (n = 3). | ||
Conclusions
In this work, we synthesized a multi-functional Fe3O4–AuNC–Apt(EPI) nanocomposite with high drug loading capacity for selective detection, imaging, and therapy of colorectal and breast cancer cells. The presence of in situ generated AuNCs in MPS solution with 2.5 ± 0.5 nm diameter provides the condition for highly fluorescent imaging of the cancer cells. We used PEGDMA to immobilize the Apt on the surface of the nanocomposite. The presence of the PEGDMA on the surface of the nanocomposite provides a rapid and highly efficiency conjugation of the MUC-1 Apt via a thiol-en click reaction and reduces the growth rate of the silica network and the thickness of the coating. Also, in this research, we used a modified MUC-1 Apt with repeating CG-base pair region to increase the efficiency of drug loading. Theoretical calculations showed that the binding affinity of EPI to the modified Apt is higher than the traditional one reported in the literature. The results of MTS assay showed that besides the biocompatibility of the Fe3O4–AuNC–Apt as a nanocarrier, the Fe3O4–AuNC–Apt(EPI) nanocomposite indicates more cytotoxicity toward the targeted cancer cells in comparison to the non-targeted cells. Finally, it is expected that by changing the type of aptamer and chemotherapeutic drug, this nanocomposite has potential to be used for detection and treatment of various cancer cells. In future work, we will investigate the ability of the nanocomposite to be used for MRI of cancer cells. Also, the hyperthermia effect of the magnetic nanocomposite will be studied.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Iran National Science Foundation, (No. 94802693), Royan Institute for Biotechnology, and Isfahan University of Technology.References
- H. Z. Chen, S.-Y. Tsai and G. Leone, Nat. Rev. Cancer, 2009, 9, 785–797 CrossRef CAS PubMed.
- P.-H. Zhang, J.-T. Cao, Q.-H. Min and J.-J. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 7417–7424 CrossRef CAS PubMed.
- S. Kunjachan, R. Pola, F. Gremse, B. Theek, J. Ehling, D. Moeckel, B. Hermanns-Sachweh, M. Pechar, K. Ulbrich and W. E. Hennink, Nano Lett., 2014, 14, 972–981 CrossRef CAS PubMed.
- J. Liao, X. Wei, B. Ran, J. Peng, Y. Qu and Z. Qian, Nanoscale, 2017, 9, 2479–2491 RSC.
- Q. Chen, C. Liang, C. Wang and Z. Liu, Adv. Mater., 2015, 27, 903–910 CrossRef CAS PubMed.
- J. Mou, T. Lin, F. Huang, H. Chen and J. Shi, Biomaterials, 2016, 84, 13–24 CrossRef CAS PubMed.
- F. Zhou, T. Zheng, E. S. Abdel-Halim, L. Jiang and J.-J. Zhu, J. Mater. Chem. B, 2016, 4, 2887–2894 RSC.
- C. Durgadas, C. P. Sharma and K. Sreenivasan, Nanoscale, 2011, 3, 4780–4787 RSC.
- O. Shimoni, A. Postma, Y. Yan, A. M. Scott, J. K. Heath, E. C. Nice, A. N. Zelikin and F. Caruso, ACS Nano, 2012, 6, 1463–1472 CrossRef CAS PubMed.
- P. Yang, Q. Z. Xu, S. Y. Jin, Y. Lu, Y. Zhao and S. H. Yu, Chem. – Eur. J., 2012, 18, 9294–9299 CrossRef CAS PubMed.
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. Wang, B. Xing and Y. Zhao, Adv. Funct. Mater., 2012, 22, 5144–5156 CrossRef CAS.
- C. Wang, Y. Yao and Q. Song, J. Mater. Chem. C, 2015, 3, 5910–5917 RSC.
- L. Zhang and E. Wang, Nano Today, 2014, 9, 132–157 CrossRef CAS.
- J. Gao, H. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097–1107 CrossRef CAS PubMed.
- D. Wang, J. He, N. Rosenzweig and Z. Rosenzweig, Nano Lett., 2004, 4, 409–413 CrossRef CAS.
- H. R. Ramay and M. Zhang, Biomaterials, 2003, 24, 3293–3302 CrossRef CAS PubMed.
- N. H. Abd Ellah and S. A. Abouelmagd, Expert Opin. Drug Delivery, 2017, 14, 201–214 CrossRef CAS PubMed.
- Y. Lu, P. Wu, Y. Yin, H. Zhang and C. Cai, J. Mater. Chem. B, 2014, 2, 3849–3859 RSC.
- P. Wu, Y. Gao, Y. Lu, H. Zhang and C. Cai, Analyst, 2013, 138, 6501–6510 RSC.
- L. Peng, C. S. Wu, M. You, D. Han, Y. Chen, T. Fu, M. Ye and W. Tan, Chem. Sci., 2013, 4, 1928–1938 RSC.
- A. B. Iliuk, L. Hu and W. A. Tao, Anal. Chem., 2011, 83, 4440–4452 CrossRef CAS PubMed.
- I. Ocsoy, B. Gulbakan, M. I. Shukoor, X. Xiong, T. Chen, D. H. Powell and W. Tan, ACS Nano, 2012, 7, 417–427 CrossRef PubMed.
- A. Machulkin, A. Garanina, O. Zhironkina, E. Beloglazkina, N. Zyk, A. Savchenko, V. Kotelyanskii and A. Mazhuga, Bull. Exp. Biol. Med., 2016, 161, 706 CrossRef CAS PubMed.
- H. Xing, N. Y. Wong, Y. Xiang and Y. Lu, Curr. Opin. Chem. Biol., 2012, 16, 429–435 CrossRef CAS PubMed.
- S. H. Jalalian, S. M. Taghdisi, N. S. Hamedani, S. A. M. Kalat, P. Lavaee, M. ZandKarimi, N. Ghows, M. R. Jaafari, S. Naghibi and N. M. Danesh, Eur. J. Pharm. Sci., 2013, 50, 191–197 CrossRef CAS PubMed.
- S. Duan, W. K. Bleibel, R. S. Huang, S. J. Shukla, X. Wu, J. A. Badner and M. E. Dolan, Cancer Res., 2007, 67, 5425–5433 CrossRef CAS PubMed.
- R. A. Forrest, L. P. Swift, B. J. Evison, A. Rephaeli, A. Nudelman, D. R. Phillips and S. M. Cutts, Cancer Chemother. Pharmacol., 2013, 71, 809–816 CrossRef CAS PubMed.
- C. Hui, C. Shen, T. Yang, L. Bao, J. Tian, H. Ding, C. Li and H.-J. Gao, J. Phys. Chem. C, 2008, 112, 11336–11339 CrossRef CAS.
- E. Shoghi, S. Z. Mirahmadi-Zare, R. Ghasemi, M. Asghari, M. Poorebrahim and M. H. Nasr-Esfahani, Microchim. Acta, 2018, 4, 241–252 CrossRef PubMed.
- P. A. Buining, B. M. Humbel, A. P. Philipse and A. J. Verkleij, Langmuir, 1997, 13, 3921–3926 CrossRef CAS.
- L. Tan, K. Gee Neoh, E. T. Kang, W. S. Choe and X. Su, J. Pharm. Sci., 2012, 101, 1672–1677 CrossRef CAS PubMed.
- T. Hyeon, Chem. Commun., 2003, 927–934 RSC.
- K. D. Collins and M. W. Washabaugh, Q. Rev. Biophys., 1985, 18, 323–422 CrossRef CAS PubMed.
- F. Li, Z. Yu, L. Zhao and T. Xue, RSC Adv., 2016, 6, 10352–10357 RSC.
- Y.-S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979–3986 CrossRef CAS.
- F. Aldeek, M. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509–2521 CrossRef CAS PubMed.
- N. Vasimalai and S. A. John, J. Mater. Chem. B, 2013, 1, 5620–5627 RSC.
- C. Yang, G. An and X. Zhao, J. Mater. Sci.: Mater. Electron., 2013, 24, 3490–3495 CrossRef CAS.
- R. Binaymotlagh, H. Hadadzadeh, H. Farrokhpour, F. H. Haghighi, F. Abyar and S. Z. Mirahmadi-Zare, Mater. Chem. Phys., 2016, 177, 360–370 CrossRef CAS.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- X. Qi, W. Li, J. Gu, C. Guo and J. Zhang, RSC Adv., 2016, 6, 105110–105118 RSC.
- G. Andrade, E. Barbosa-Stancioli, A. P. Mansur, W. Vasconcelos and H. Mansur, Biomed. Mater., 2006, 1, 221 CrossRef CAS PubMed.
- W. R. Thompson, M. Cai, M. Ho and J. E. Pemberton, Langmuir, 1997, 13, 2291–2302 CrossRef CAS.
- J. Wang, X. Wu, C. Wang, Z. Rong, H. Ding, H. Li, S. Li, N. Shao, P. Dong and R. Xiao, ACS Appl. Mater. Interfaces, 2016, 8, 19958–19967 CrossRef CAS PubMed.
- M. V. Shirmanova, I. N. Druzhkova and M. M. Lukina, et al. , Biochim. Biophys. Acta, 2015, 1850, 1905–1911 CrossRef CAS PubMed.
- M. Damaghi, J. W. Wojtkowiak and R. J. Gillies, Front. Physiol., 2013, 4, 370–380 Search PubMed.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, Autodock4 and AutoDockTools4: automated docking with selective receptor exibility, J. Comput. Chem., 2009, 16, 2785 CrossRef PubMed.
- A. Erdem and M. Ozsoz, Anal. Chim. Acta, 2001, 437, 107–114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: FT-IR spectra of MPS in the absence (1) and presence (2) of the AuNCs. Fig. S2: XRD spectrum of the Fe3O4 NPs. Fig. S3: XRD spectrum of the Fe3O4@SiO2 NPs. Fig. S4: Colloidal stability of the freshly prepared nanoparticles (A) and after 10 days (B). Hydrodynamic size of the nanoparticles in different storage time periods (C). Fig. S5: Hydrodynamic size of the Fe3O4–AuNC at different pH. Fig. S6: FT-IR spectra of Fe3O4–AuNC before (1) and after (2) conjugation of the MUC-1 aptamer. Fig. S7: The amount of Fe3O4–AuNC–Apt taken up by three cell lines measured by ICP-OES and denoted as percentage of Au uptake. Fig. S8: Profile of EPI release from the Fe3O4–AuNC–Apt(EPI) nanocomposite depending on time (n = 3). See DOI: 10.1039/c8nj04236e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |