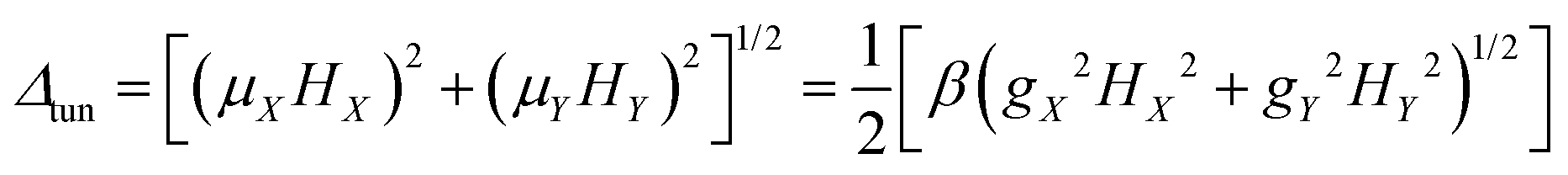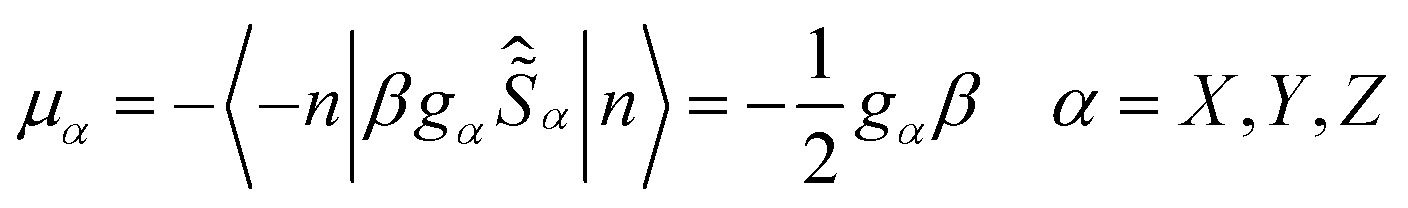DOI:
10.1039/C8NJ04019B
(Paper)
New J. Chem., 2019,
43, 454-462
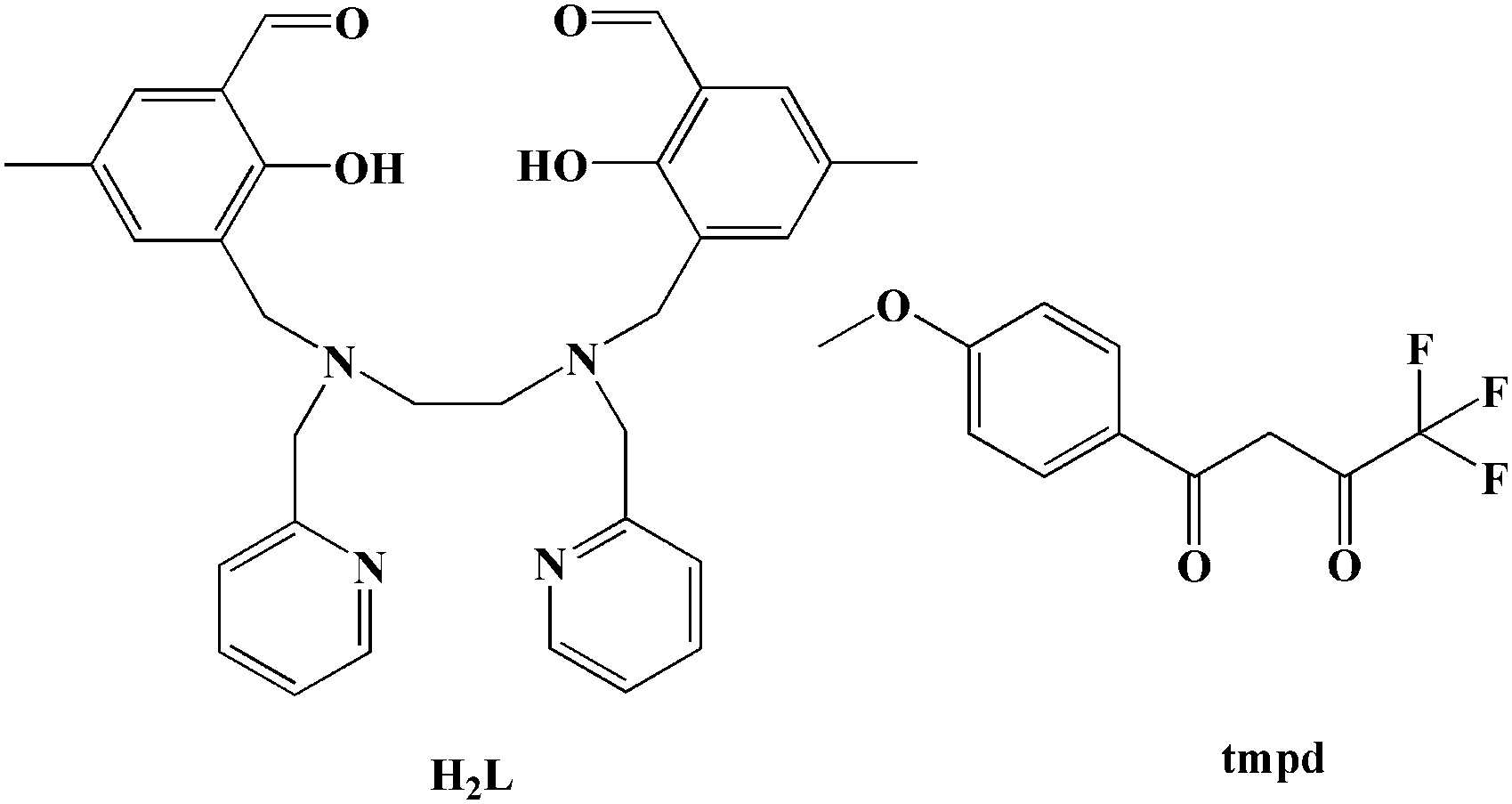
Designing a mononuclear DyIII single-molecule magnet (SMM) by using a N,O,N,O-based multichelating Schiff base ligand and a β-diketonate ligand†
Received
8th August 2018
, Accepted 9th November 2018
First published on 21st November 2018
Abstract
Two mononuclear LnIII compounds, in which each LnIII is eight-coordinated, namely [Ln(L)(tmpd)] (Ln = Dy (1) or Er (2)), have been prepared using a multichelating Schiff base ligand N,N′-bis(2-hydroxy-5-methyl-3-formylbenzyl)-N,N′-bis-(pyridin-2-ylmethyl)ethylenediamine (H2L) and a bidentate chelating β-diketonate ligand (tmpd). The local geometry of the LnIII ions in 1 and 2 is close to the D2d symmetry. Notably, magnetic studies reveal that compound 2 displays no slow relaxation of magnetization while compound 1 exhibits single-molecule magnet (SMM) behaviour in the absence of a static magnetic field, with an effective energy barrier (Ueff) of 66.81 cm−1 (96.63 K). To deeply understand their different magnetic behaviours, the magnetic anisotropy of 1 and 2 is systematically studied by ab initio calculations. There was an obvious difference between the first excited state (KD1) and the experimental Ueff in 1 because the QTM within KD0 is not completely prohibited and the residual QTM could lead to a large discrepancy between experimentally-fitted Ueff and ab initio calculated crystal field splitting. No temperature/frequency dependence is recorded in the ac susceptibility of 2 because the ab initio magnetic easy axis of 2 does not lie close to any atom of the first sphere since it tries to avoid any strong electrostatic repulsion. Moreover, the gXY value of KD0 of 2 is larger than that of 1 by three orders of magnitude leading to the probability of stronger QTM within KD0 of 2.
Introduction
Single-molecule magnets (SMMs), which are molecules that show slow relaxation, have been a hot topic of research in the fields of chemical and materials sciences due to their potential applications in high-density information storage, quantum computing and molecular spintronics.1–4 The pursuing high performance SMMs and understanding relaxation mechanisms deeply has propelled this field forward enormously over the past three decades. Currently, the study of lanthanide (Ln) based SMMs, especially for LnIII mononuclear SMMs, is the most eye-catching field because of their large unquenched orbital angular momentum resulting in significant anisotropy.5–10 Some lanthanide ions, such as TbIII, DyIII, HoIII, and ErIII ions, have become attractive candidates for SMMs. The mononuclear LnIII SMMs with a relatively simple magnetic system are not only conducive to investigating the magnetic anisotropy and spin dynamics so as to deeply understand the magneto-structural correlations but can also show excellent SMM properties.11–17 Within the context of lanthanide SMMs, well-known organometallic ligands, β-diketonate ligands, phthalocyanine, polyoxometalate, Schiff base ligands and so on, have been used to influence the properties of dysprosium and erbium-containing SMMs.18 Certainly, theoretical studies also have provided detailed insight into how the properties of ligands impact upon the electronic structure of the LnIII cation, leading to striking increases in the magnetic blocking temperature (TB) and magnetic reversal barrier (Ueff). Most recently, a remarkable Ueff of 1277 cm−1 as well as a magnetic hysteresis around 60 K have been found in a mononuclear DyIII metallocene compound [Dy(Cpttt)2][B(C6F5)4].16,17 Additionally, a series of seven-coordinated mononuclear SMMs with outstanding magnetic properties confirm the great potential of Ln-SMMs and fuel the exploration of mononuclear LnIII SMMs with more enhanced properties.11–16 Among various strategies to design high-performance LnIII SMMs, the strategy of enhancing the coordination symmetries of the lanthanide ions such as D2d, D4d, D5h, and D6d is very efficient.18–25 Herein, the selection of a Schiff base ligand, namely N,N′-bis(2-hydroxy-5-methyl-3-formylbenzyl)-N,N′-bis-(pyridin-2-ylmethyl)ethylenediamine (H2L), formed by reaction between N,N′-bis(pyridin-2-ylmethyl)ethylenediamine and 2-hydroxy-3-(chloromethyl)-5-methyl-benzaldehyde, is especially useful for constructing mononuclear LnIII compounds because of the following considerations: (1) the ligand provides N,O,N,O-based multichelating sites forming a coordination pocket, which is especially favorable for accommodating lanthanide ions to generate mononuclear compounds;26 (2) similar ligands have successfully synthesized several structurally and magnetically outstanding mononuclear dysprosium SMMs with different configurations and coordination numbers, such as N,N′-bis(2-hydroxybenzyl)-N,N′-bis((2-methylpyridyl)ethylenediamine) (bbpen),14N,N′,N′′-trimethyl-N,N′′-bis(2-hydroxy-3-methoxy-5-methylbenzyl) diethylene triamine (H2L′)27 and the derivative of bbpen with an electron-donating group (CH3-bbpen).15 Additionally, a type of β-diketonate ligand, 4,4,4-trifluoro-1-(4-methoxyphenyl)-1,3-butanedione (tmpd), was introduced into the mononuclear system because its simple chelating coordination model would be conducive to obtaining a constitutionally stable mononuclear compound and the β-diketonate ligand could provide a negative charge to reducing counter cations. Fortunately, two mononuclear compounds were obtained using H2L, tmpd and different rare-earth salts, namely [Dy(L)(tmpd)] (1) and [Er(L)(tmpd)] (2) (see Scheme 1). Their structures were determined by single-crystal X-ray diffraction, showing a trigonal dodecahedron configuration (D2d) for DyIII and ErIII ions. Moreover, the uniaxial magnetic anisotropies and magnetostructural correlations, as well as relaxation mechanism, were investigated by magnetic studies, ab initio calculations and electrostatic analyses.
 |
| | Scheme 1 H2L ligand and the tmpd ligand. | |
Experimental
Materials and instructions
All the materials and reagents were obtained commercially without further purification. The FT-IR spectra were recorded in the range of 400–4000 cm−1 using KBr pellets on an EQUINOX55 FT/IR spectrophotometer. Elemental analysis (C, H, and N) was implemented using a Perkin-Elmer 2400 CHN elemental analyzer. The phase purity of the bulk or polycrystalline samples was confirmed by powder X-ray diffraction (PXRD) measurements performed using a Rigaku RU200 diffractometer at 60 kV, 300 mA, and Cu Kα radiation (λ = 1.5406 Å), with a scan speed of 5° min−1 and a step size of 0.02° in 2θ. The diffuse reflectance spectrum was obtained by a U-41000 spectrophotometer and applying BaSO4 powder as a 100% reflectance reference. Magnetic measurements were performed in the temperature range of 1.8–300 K with an applied field of 1000 Oe, using a Quantum Design MPMS-XL-7 SQUID magnetometer on polycrystalline samples. The diamagnetic corrections for the compounds were estimated using Pascal's constants. Alternating current (ac) susceptibility experiments were performed using an oscillating ac field of 2.0 Oe at ac frequencies ranging from 1 to 1000 Hz. The magnetization was measured in the field range of 0–7 T.
X-ray crystallography
X-ray single-crystal diffraction analysis: the single crystal X-ray experiment was performed using a Rigaku SCX mini CCD diffractometer equipped with graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å) using ω and φ scan mode. Data integration and reduction were processed with SAINT software. Absorption correction based on multiscan was performed using the SADABS program.28 The structures were solved by the direct method using SHELXTL and refined by means of full-matrix least-squares procedures on F2 with the SHELXL-97 program.29 All non-hydrogen atoms were refined anisotropically. Other details of crystal data, data collection parameters, and refinement statistics are given in Table 1. The selected bond lengths and angles are listed in Tables S1 and S2 (ESI†).
Table 1 Crystal data and structure refinement details for 1 and 2
| Compound |
1
|
2
|
|
R
1 = ∑‖Fo| − |Fc‖/∑|Fo|.
wR2 = [∑(Fo2 − Fc2)/∑w(Fo)2]1/2, w = [σc2(Fo2) + (xP)2 + yP]−1, P = (Fo2 + 2Fc2)/3.
|
| Empirical formula |
C43H40DyF3N4O7 |
C43H40ErF3N4O7 |
| CCDC |
1851377
|
1851378
|
| Formula weight |
944.290 |
949.050 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/c |
|
a/Å |
10.9674(8) |
10.9617(6) |
|
b/Å |
11.0167(15) |
11.0136(8) |
|
c/Å |
33.215(3) |
33.9591(17) |
|
α/° |
90.000 |
90 |
|
β/° |
95.602(9) |
103.177(5) |
|
γ/° |
90.000 |
90 |
|
V/Å3 |
3994.0(7) |
3991.9(4) |
|
Z
|
4 |
4 |
|
ρ
calc./g cm−3 |
1.570 |
1.579 |
|
μ/mm−1 |
10.607 |
10.010 |
|
F(000) |
1900 |
2460 |
|
θ
min–max/° |
4.1330 to 67.6390 |
4.1280 to 66.9870 |
| Goodness-of-fit on F2 |
1.044 |
1.000 |
|
R
1
/wR2b [I > 2σ(I)] |
0.0748/0.1002 |
0.0584/0.1002 |
|
R
1
/wR2b [all data] |
0.0845/0.1585 |
0.1106/0.1206 |
Theoretical methods and computational details
Multiconfigurational ab initio calculations, including spin–orbit coupling (SOC), were performed on the experimental structures of 1 and 2 to explore their SMM properties. This type of calculation includes two steps:30 (1) a set of spin eigenstates are obtained by the state-averaged (SA) CASSCF method;31 (2) the low-lying SOC states, i.e., Kramers doublets (KD) herein, are obtained by state interaction which is the diagonalization of the SOC matrix in the space spanned by the spin eigenstates from the first step. In the CASSCF step, the active space consisted of 9 or 11 electrons in 7 orbitals for 1 or 2 respectively. The spin eigenstates of 21 sextets or 35 quartets were included for 1 or 2 respectively. Due to the hardware limitation, other highly excited spin states were not considered. The step of state interaction was performed by the RASSI-SO module32 with the SOC integrals from the AMFI method.33 The ANO-RCC basis sets,34–36 including VTZP for Dy/Er, VDZ for C and H and VDZP for other atoms, were used. All the calculations were carried out with the MOLCAS@UU, a version of MOLCAS 8.037,38 which is freely distributed for academic users. The SINGLE_ANISO module,39,40 developed by Chibotaru et al., was used to obtain g-tensors, transition magnetic moments and other parameters characterizing the magnetic anisotropy.
Synthesis
Synthesis of [Dy(L)(tmpd)] (1).
Methanol solution (10 ml) of Dy(ClO4)3·6H2O (0.0461 g, 0.1 mmol) and methanol solution (10 ml) of H2L (0.0510 g, 0.1 mmol) and tmpd (0.0246 g, 0.1 mmol) were mixed at room temperature, to which CH3OK (0.0210 g, 0.3 mmol) in methanol (5 ml) was added. The mixture was stirred at the room temperature for 4 h to afford a yellow precipitate. The resultant solution was filtered and allowed to stand undisturbed at room temperature for three weeks. Finally, orange-yellow single crystals suitable for X-ray analysis were obtained in 41% yield (based on the DyIII salts). Anal. calcd for C43H40DyF3N4O7: C, 54.64; H, 4.24; N, 5.93. Found: C, 54.56; H, 4.31; N, 5.82. IR (KBr, cm−1): 3432 (m), 1587 (s), 1455 (s), 1371 (w), 1288 (s), 1270 (w), 1166 (m), 1022 (w), 916 (m), 784 (w), 759 (w), 551 (m).
Synthesis of [Er(L)(tmpd)] (2).
A similar synthetic procedure as that for 1 was used to synthesize 2 except that Dy(ClO4)3·6H2O (0.0461 g, 0.1 mmol) was replaced by Er(ClO4)3·6H2O (0.0574 g, 0.3 mmol). Yield: 53% (based on the ErIII salts). Finally, yellow crystals were obtained after several days. Anal. calcd for C43H40ErF3N4O7: C, 54.37; H, 4.21; N, 5.90. Found: C, 54.27; H, 4.32; N, 5.81. (KBr, cm−1): 3424 (m), 1589 (s), 1453 (s), 1355 (w), 1283 (s), 1268 (w), 1174 (m), 1011 (w), 929 (m), 782 (w), 760 (w), 555 (m).
Results and discussion
Description of the structures
The crystal structures of both compounds are shown in Fig. 1 and 2. Compounds 1 and 2 crystallize in the monoclinic space group P21/c and the monoclinic space group P21/n, respectively. Each LnIII ion is surrounded by one negative L2− ligand and one negative β-diketonate ligand. The LnIII ions display an LnN4O4 octa-coordinate environment. The average lengths of the Dy–O and Dy–N bonds are 2.288 Å and 2.616 Å, respectively. The average lengths of the Er–O and Er–N bonds are 2.245 Å and 2.573 Å, respectively. By using the SHAPE 2.1 software,41 the configurations of LnIII ions in 1 and 2 were calculated (Table S3, ESI†), indicating that compounds 1 and 2 belong to an approximately trigonal dodecahedron (D2d) configuration. The closest distances of LnIII⋯LnIII ions are 9.733 Å for 1 and 9.700 Å for 2, respectively. In [Dy(H2L)(NO3)3]·2CH3OH,27 the DyIII ion exhibits a rather unsymmetrical DyO9 coordination environment. The H2L′ ligand surrounds the DyIII ion. Three bidentate nitrate anions provide six coordinated oxygen atoms and three negative charges to finishing charge balance. The DyIII⋯DyIII distances are 9.476 and 9.780 Å. For [Dy(bbpen)X]14 and [Dy(bbpen-CH3)X] (X = Cl or Br),15 the DyIII sites in them possess seven-coordinate geometry with a distorted pentagonal bipyramidal coordination sphere occupied by one X− ion, two O, and four N atoms from a single divalent bbpen ligand. Two phenol O atoms from the ligand are negatively charged and axially coordinated to the DyIII ion. One Br−(Cl−) and four N atoms in the equatorial plane are all weakly coordinated with the DyIII ion. In our compounds, the ligand H2L changes into the L2− pocketing LnIII ion in 1 (Dy) and 2 (Er). Meanwhile, one negative β-diketonate ligand coordinates the DyIII ion with the bidentate chelating coordination mode to form an eight-coordinated configuration.
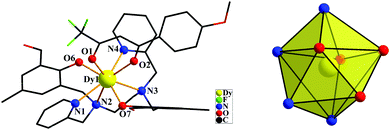 |
| | Fig. 1 The crystal structures of compound 1 (left) showing general ligand configurations. Local coordination geometry of the DyIII ion for compound 1 (right) (hydrogen atoms are omitted for clarity). | |
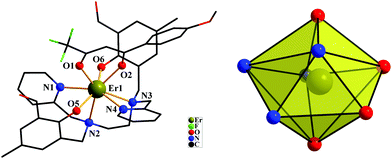 |
| | Fig. 2 The crystal structures of compound 2 (left) showing general ligand configurations. Local coordination geometry of the ErIII ion for compound 2 (right) (hydrogen atoms are omitted for clarity). | |
Magnetic properties
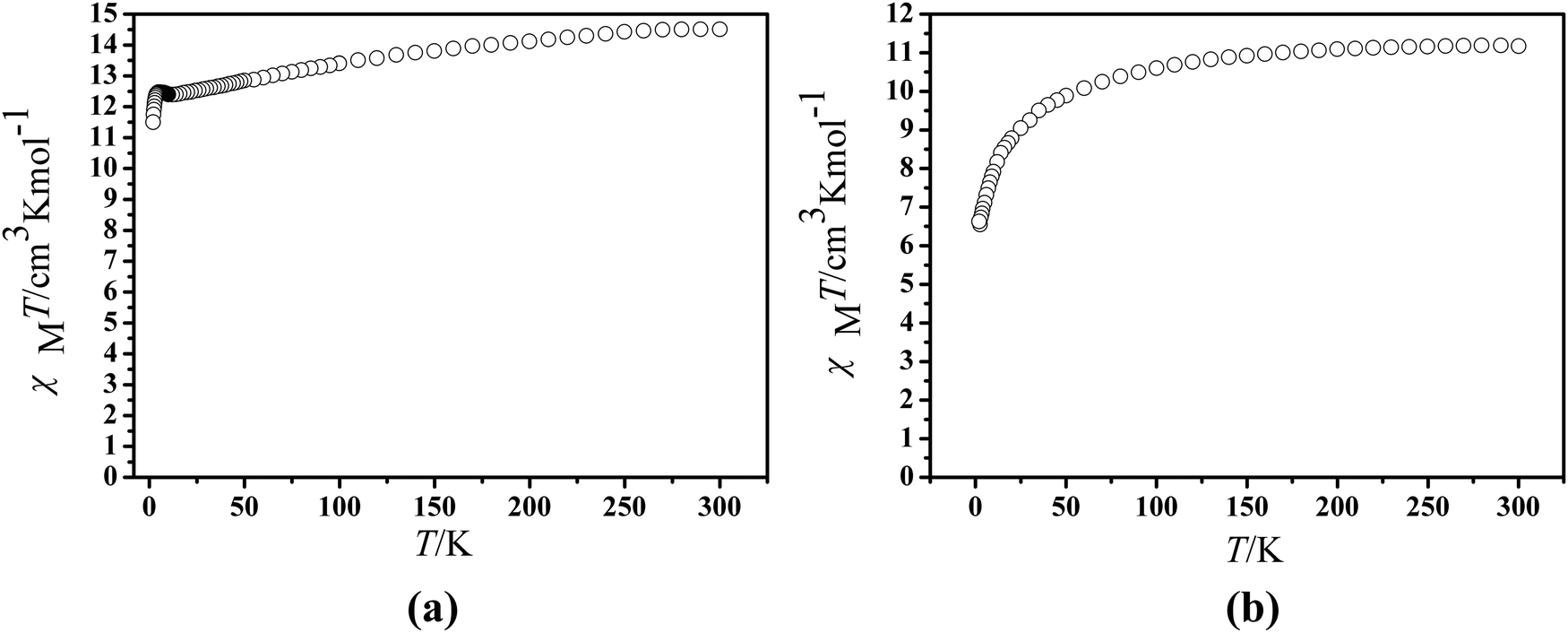
The magnetic experiments of 1 and 2 were performed on polycrystalline samples. The PXRD results of 1 and 2 support the pure state of the bulk materials (Fig. S1, ESI†). The temperature-dependent magnetic direct-current susceptibility measurements were performed on 1 and 2 under an applied field of 1 kOe from 2–300 K (Fig. 3). The room temperature χMT value is 14.51 cm3 mol−1 K, which is in good agreement with the expected value of 14.17 cm3 mol−1 K for a single DyIII ion (6H15/2, S = 5/2, L = 5, J = 15/2, and g = 4/3).42 Upon cooling, the χMT product decreases gradually and more rapidly below 100 K, which is likely due to crystal-field effects (i.e. thermal depopulation of the LnIII Stark sublevels) and/or the possible antiferromagnetic dipole–dipole interaction between the molecules.42 On the other hand, a plateau of the χMT value around 12.48 cm3 K mol−1 at a low temperature is observed, suggesting negligible magnetic interactions. Finally, the χMT products decrease to a minimum of 11.50 cm3 mol−1 K in 1 at 2.0 K. The room temperature χMT value of 2 is determined to be 11.17 cm3 mol−1 K, in good agreement with the expected paramagnetic value of 11.48 cm3 mol−1 K for the ErIII ion.43 When cooled, the χMT curves for compound 2 reduce tardily in the range of 300 to 50 K. Subsequently, the χMT products decrease sharply below 50 K to a minimum of 6.63 cm3 mol−1 K at 2.0 K. The downturn of each set of the χMT value on cooling can be explained by depopulation of the Stark level split by the ligand field, suggesting the presence of significant magnetic anisotropy.43
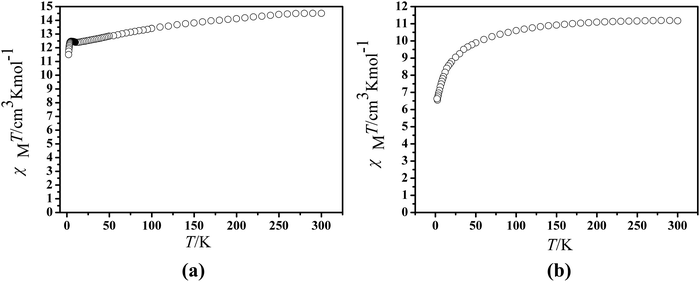 |
| | Fig. 3 Temperature dependence of the χMT product at 1000 Oe for compounds 1 (a) and 2 (b). | |
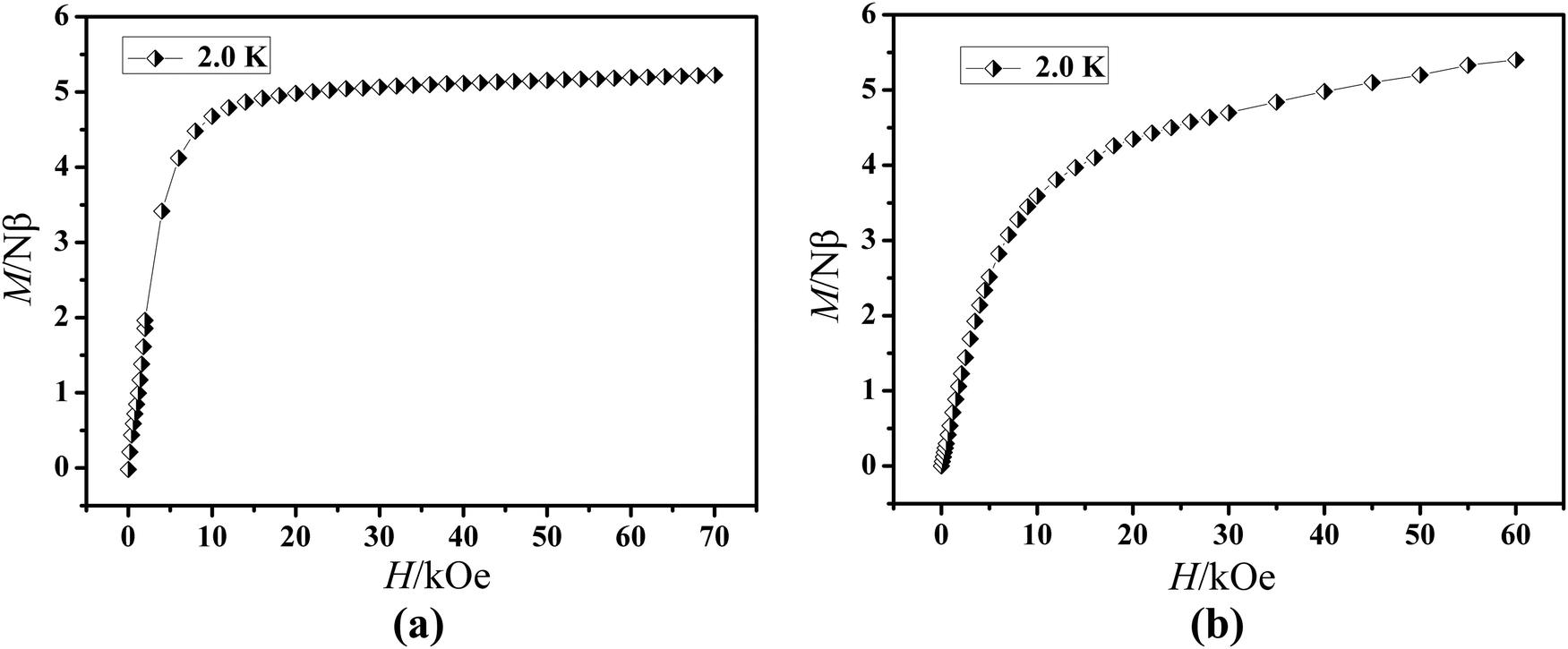
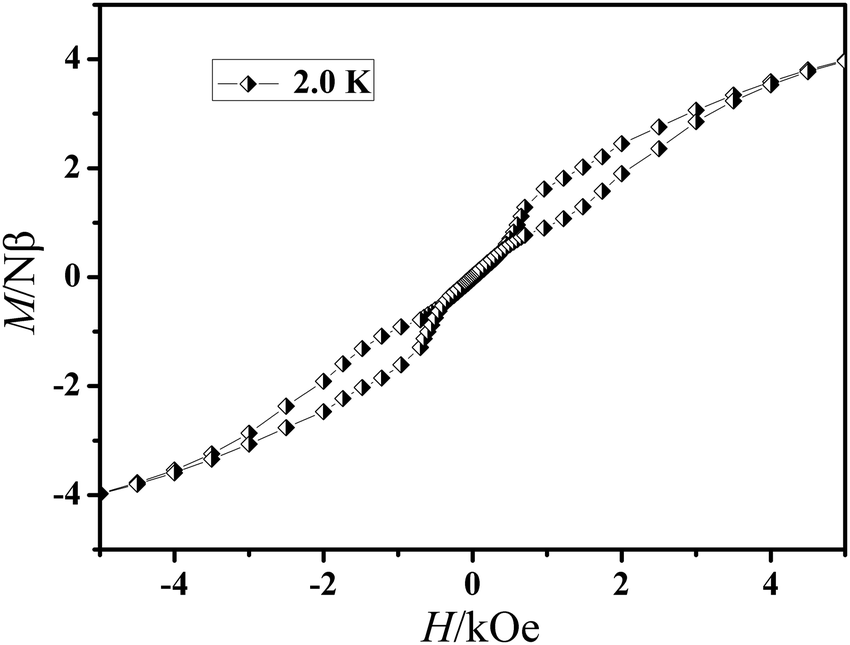
The field dependence of magnetization was determined at 2.0 K in the field range of 0–7 T for 1 and 0–6 T for 2 (Fig. 4). On increasing the static field up to 7 T for 1 and 6 T for 2 at 2.0 K, the magnetizations for DyIII and ErIII reach values of 5.22 for 1 and 5.40 Nβ for 2, respectively and deviate much from theoretical saturation values of 10 and 9 Nβ, which can be attributed to the ligand-field-induced splitting of the Stark level as well as magnetic anisotropy.42–45 As another significant characteristic of the magnetic bistability of a magnet, the magnetic hysteresis loop was examined for 1 (Fig. 5). The M versus H data exhibit obvious butterfly-shaped hysteresis loops at 2 K for 1, indicating fast zero-field relaxation between the two ground states.
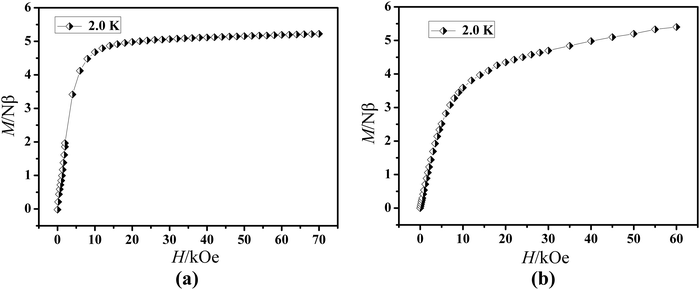 |
| | Fig. 4
M vs. H plots at 2.0 K for compounds 1 (a) and 2 (b). | |
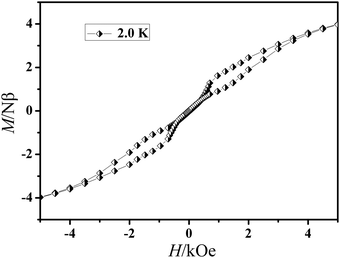 |
| | Fig. 5 The magnetic hysteresis of 1 at 2.0 K. | |
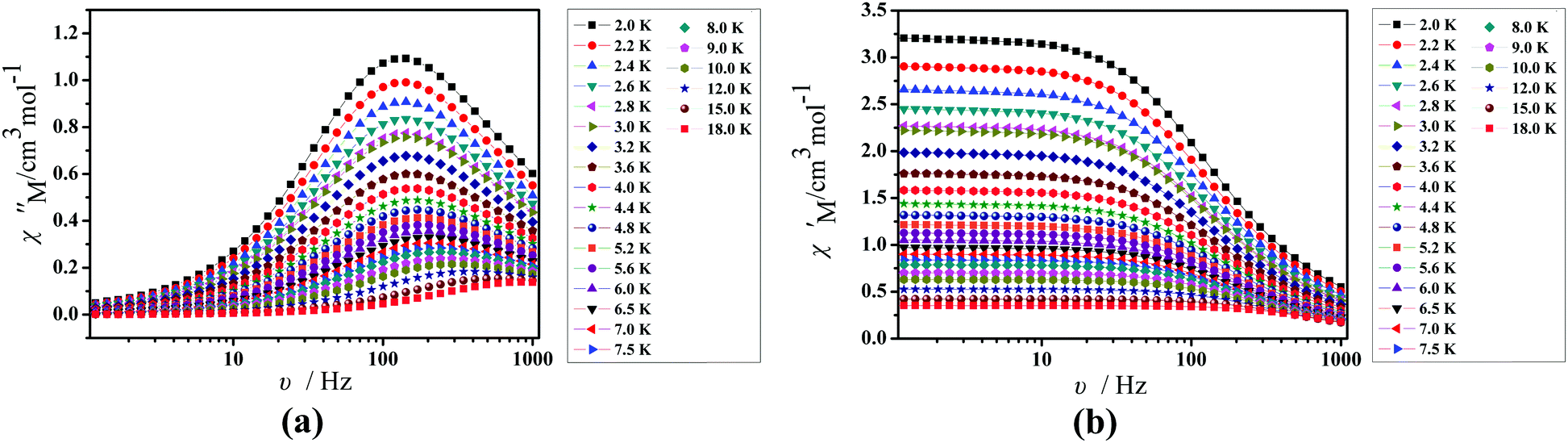
Under an oscillating field of 2.0 Oe, zero-field AC susceptibility experiments were performed in the range of 2.0–25.0 K and at frequencies 1, 10, 100, 499 and 999 Hz in 1. Compound 1 exhibits weak out-of-phase (χ′′) signals (Fig. 6). No signals in out-of-phase (χ′′) susceptibilities can be observed in 2 (Fig. S2, ESI†).
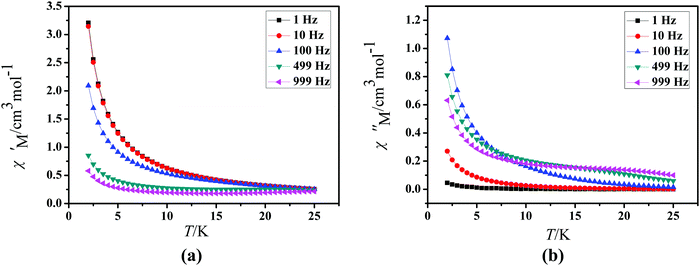 |
| | Fig. 6 Temperature dependence of the in-of-phase (χ′, a) and out-of-phase (χ′′, b), respectively, of the ac susceptibility for 1 under a zero applied dc field. | |
To further understand the dynamics of the magnetization of 1, the frequency dependencies of the alternating-current (ac) susceptibility were measured under a zero dc field. From frequency dependencies of ac susceptibility (Fig. 7), the in-phase (χ′) and out-of-phase (χ′′) signals of 1 show frequency dependencies, which also reveal the presence of slow relaxation of the magnetization in 1. Obviously, with increasing temperature, the peaks of the out-of-phase (χ′′) gradually shift from the middle frequency to the high frequency for 1. The Cole–Cole plots of 1 based on frequency-dependent ac susceptibility data present semi-circular shapes (Fig. 8). On fitting the corresponding data with a generalized Debye model,45,46 the parameter (α) could be obtained in the range of 0.262–0.356 for 1 (Table S4, ESI†), indicating a narrow distribution of relaxation times for a single relaxation process. Furthermore, a combination of multiple relaxation mechanisms, that is, Raman, Orbach and QTM mechanisms, is taken into account so as to clarify the ln(τ) vs. 1/T behaviors of 1 by using eqn (1).16,45
| | τ−1 = τQTM−1 + CTn + τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ueff/kT) exp(−Ueff/kT) | (1) |
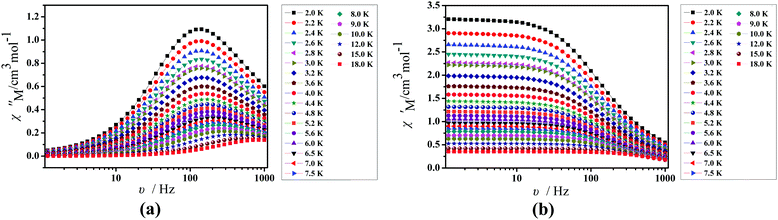 |
| | Fig. 7 Frequency dependence of the in-phase (χ′, a) and out-of-phase (χ′′, b) of the AC susceptibility for 1 under a zero applied dc field. | |
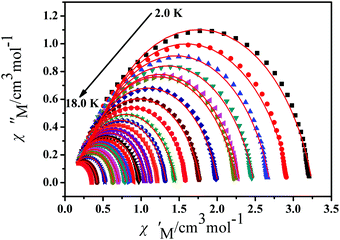 |
| | Fig. 8 Cole–Cole plots for 1 from 2.0 K to 18.0 K using the AC susceptibility data under a zero applied dc field. The solid lines are the best fits for the generalized Debye model between 2.0 K and 18.0 K. | |
The best fits are obtained giving τQTM = 1.15 × 10−3 s, n = 1.71, C = 21.97 s−1 K−1.71, τ0 = 3.38 × 10−6 s, and an effective energy barrier of Ueff = 66.81 cm−1 (96.63 K) for 1 (Fig. 9).
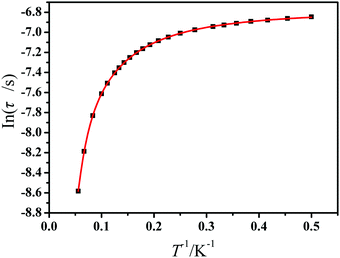 |
| | Fig. 9 Magnetization relaxation time (ln(τ)) versus T−1 plots under a zero applied dc field for 1. The solid red line represents the best fitting to the multiple relaxation process (eqn (1), see text for parameters). | |
Theoretical analysis
In principle, the magnetic relaxation of Ln-SIM is a collective result of several processes including Orbach, Raman, and direct as well as quantum tunneling of magnetization (QTM) simultaneously.47–51 Thus, the necessary condition for slow relaxation, i.e., SMM behavior, in mononuclear Ln compounds should be the effective suppression of all the fast relaxation processes.48,49
Usually QTM within the ground state is the most effective fast relaxation process and thus it is the first target of suppression. In principle, the probability of QTM scales as the square of the so-called tunnel splitting Δtun47–49,52 and, for Kramers systems, Δtun is provided by the Zeeman interaction (eqn (1a)) between the transversal fields (HX and HY) and the corresponding components of the magnetic moments ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) of the KD (μX and μY).47–49,52,53
of the KD (μX and μY).47–49,52,53
| | 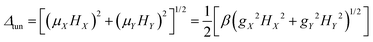 | (1a) |
| | 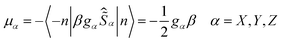 | (1b) |
| | gα = 2〈−n|![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) α|n〉/β α = X, Y, Z α|n〉/β α = X, Y, Z | (1c) |
Clearly, small values of the transversal gX and gY (eqn (1c)) of the ground KD, i.e., KD0, will lead to a low magnitude of the transversal magnetic moment μX,Y as well as a low magnitude of Δtun. Thus, as indicated by Ruiz et al.,54 zero field mononuclear SMM behavior could exist if the gXY value (eqn (2a)) of KD0 is smaller than 0.015 for mononuclear DyIII systems. Besides the principal g values of each KD, ab initio calculations also provide the averaged absolute value of the transversal magnetic moments, μQTM (eqn (2b)), which could be used to measure the strength of QTM too.
| | 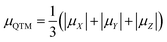 | (2b) |
In the case of 1, the gXY value of KD0 is 0.2795 × 10−2 (Table 2) which is remarkably smaller than 0.015. Thus 1 has good axiality47,48,52,53 and zero-field slow magnetic relaxation should be observed.54 This theoretical prediction is consistent with the experimental results of the ac susceptibility measurements. However, the Ueff, fitted from the experimental data, is only 96.63 K (67.16 cm−1) which is significantly lower than the theoretical energy of the first excited KD, i.e., KD1 (226.64 cm−1). As a phenomenological parameter, the Ueff could be close to the energy of a certain excited KD only if the relaxation is dominated by the thermally-activated Orbach process. Although effectively suppressed, the QTM within KD0 of 1 is not completely prohibited since gXY values are not exactly zero. Thus the residual QTM should be the main reason for the large discrepancy between experimentally-fitted Ueff and ab initio calculated crystal field splitting. Besides QTM, the Raman process, which is not of thermally-activated type neither, could also lead to such a discrepancy.
Table 2
Ab initio computed relative energies (in cm−1), principal values of the g-tensors and averaged transition magnetic moment μQTM (in β) of the four lowest KDs of the compounds studied in this work
|
|
1
|
2
|
|
Experimental Ueff is shown in parenthesis.
|
| KD0 |
E
|
0.000 |
0.000 |
|
g
Z
|
19.9260 |
14.7872 |
|
g
X
|
0.1602 × 10−2 |
0.4896 × 10 |
|
g
Y
|
0.2291 × 10−2 |
0.2763 × 101 |
|
g
XY
|
0.2795 × 10−2 |
0.2806 × 101 |
|
g
XY
/gZ |
0.1403 × 10−3 |
0.1897 × 10 |
|
μ
QTM
|
0.6488 × 10−3 |
0.5420 × 10 |
|
|
| KD1 |
E
|
226.642 (66.81)a |
26.826 |
|
g
Z
|
17.0609 |
13.3277 |
|
g
X
|
0.5155 × 10−1 |
0.1164 × 101 |
|
g
Y
|
0.5775 × 10−1 |
0.1985 × 101 |
|
g
XY
|
0.7741 × 10−1 |
0.2301 × 101 |
|
g
XY
/gZ |
0.4537 × 10−2 |
0.1726 × 10 |
|
μ
QTM
|
0.1822 × 10−1 |
0.5248 × 10 |
|
|
| KD2 |
E
|
441.093 |
90.249 |
|
g
Z
|
13.9322 |
10.1792 |
|
g
X
|
0.5107 × 10 |
0.2973 × 101 |
|
g
Y
|
0.6334 × 10 |
0.6342 × 101 |
|
g
XY
|
0.8137 × 10 |
0.7004 × 101 |
|
g
XY
/gZ |
0.5840 × 10−1 |
0.6881 × 10 |
|
μ
QTM
|
0.1907 × 10 |
0.1552 × 101 |
|
|
| KD3 |
E
|
561.000 |
129.161 |
|
g
Z
|
12.4784 |
11.5135 |
|
g
X
|
0.3778 × 101 |
0.9007 × 10 |
|
g
Y
|
0.5246 × 101 |
0.3810 × 101 |
|
g
XY
|
0.6465 × 101 |
0.3915 × 101 |
|
g
XY
/gZ |
0.5181 × 10 |
0.3400 × 10 |
|
μ
QTM
|
0.1504 × 101 |
0.7850 × 10 |
Due to the oblate electron density of the ground multiplet of the DyIII ion,55,56 the electronic structure, suitable for promising mononuclear SMM properties, could be rationalized via a simple route,49,57 which is that the electrostatic repulsion along a given (axial) direction should be stronger than other (transversal) directions as much as possible. The effectiveness of this route has been verified in our previous studies on various DyIII-SIM systems.15,42
As shown in Fig. 10, the ab initio magnetic easy axis lies close to the bonds of Dy–O4 and Dy–O5. Clearly, the two atoms O4 and O5 have the largest amounts of negative charges within the first coordination sphere (Table 3). Therefore, a large axial electrostatic repulsion does exist in 1 and thus it leads to the zero-field mononuclear SMM property with moderate Ueff.
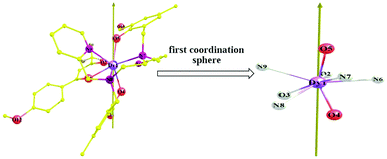 |
| | Fig. 10 Direction of the ab initio magnetic easy axis of the ground KDs of compound 1. | |
Table 3 The negative charges (in |e|) from ab initio calculations and the related Dy–O/N bond lengths (in Å) of the atoms in the first sphere
|
1
|
O4 |
O5 |
O2 |
O3 |
| Charge |
0.9063 |
0.9315 |
0.7137 |
0.7017 |
| Dy–O/N |
2.2590 |
2.2404 |
2.3687 |
2.2828 |
| N6 |
N7 |
N8 |
N9 |
| 0.3430 |
0.2950 |
0.2912 |
0.3473 |
| 2.5673 |
2.6340 |
2.6740 |
2.5875 |
|
2
|
O4 |
O5 |
O2 |
O3 |
| Charge |
0.8844 |
0.8829 |
0.7087 |
0.7071 |
| Dy-O/N |
2.2313 |
2.2195 |
2.3208 |
2.2984 |
| N6 |
N7 |
N8 |
N9 |
| 0.3387 |
0.2983 |
0.2892 |
0.3421 |
| 2.5170 |
2.6184 |
2.6177 |
2.5376 |
Being opposite the DyIII ion, ErIII is of probate electron density of the ground state.56,57 Thus, from an electrostatic viewpoint,15,42,49,57 a coordination environment might disfavor Er-SIM if it favors Dy-SIM. The gXY value of KD0 of 2 is 0.2806 × 10, larger than that of 1 by three orders of magnitude. According to eqn (1a), the probability of QTM within KD0 of 2 could be as large as 103 times of that of 1. Clearly, it should be impossible for the observation of the mononuclear SMM property of 2. This theoretical prediction is consistent with the fact that no temperature/frequency dependence is recorded in the ac susceptibility of 2. As shown in Fig. 11, the ab initio magnetic easy axis of 2 does not lie close to any atom of the first sphere since it tries to avoid any strong electrostatic repulsion. Commonly, a strongly axial CF means that the axial CF parameters B20 considerably exceed the nonaxial CF parameters Bkq with q ≠ 0 (Table S5, ESI†). Therefore, the CF experienced by DyIII in 1 is more axial than that in 2.
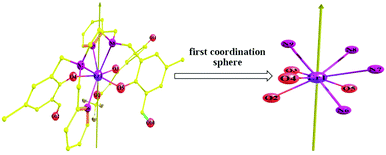 |
| | Fig. 11 Direction of the ab initio magnetic easy axis of the ground KDs of compound 2. | |
It needs to be stated that the electrostatic route used here is preliminary and it might be simple as recent theoretical work has indicated the importance of covalency in the ligand/crystal field felt by the central LnIII ion.58 Thus the expansion of the usage of this route should be carefully examined.
Conclusions
Two mononuclear LnIII compounds, 1 and 2, have been designed and successfully synthesized by selecting the appropriate Schiff base ligand and the auxiliary ligand, and realizing a DyN4O4 eight-coordinated configuration with the D2d symmetry. An experimental and theoretical investigation demonstrates that 1 exhibits SMM behaviour under a zero dc field with the energies of the first excited state (KD1) of 226.64 cm−1 and an experimental Ueff of 67.16 cm−1. There is a big difference between the two values because the QTM within KD0 of 1 is not completely prohibited since gXY values are not exactly zero and the residual QTM could lead to a large discrepancy between experimentally-fitted Ueff and ab initio calculated crystal field splitting. The gXY value of KD0 of 2 is larger than that of 1 by three orders of magnitude leading to the probability of stronger QTM within KD0 of 2. Thus no temperature/frequency dependence can be observed in the ac susceptibility of 2.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 21703002, 21701168, 21103137, 21727805 and 21673180), the 61th China Postdoctoral Science Foundation Funded Project (2017M610646), the Doctoral Scientific Research Starting Foundation of Baoji University of Arts and Science (No. ZK2017024) and the Fund of Northwest University (334100048).
References
- S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015–1018 CrossRef CAS.
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 2002, 115, 1804–1816 CrossRef.
-
(a) S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC;
(b) P. Zhang, Y.-N. Guo and J.-K. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef CAS;
(c) L. Ungur, S.-Y. Lin, J.-K. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC;
(d) Z.-H. Zhu, M. Guo, X.-L. Li and J.-K. Tang, Coord. Chem. Rev., 2019, 378, 350–364 CrossRef CAS.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS.
- A. J. Hutchings, F. Habib, R. J. Holmberg, I. Korobkov and M. Murugesu, Inorg. Chem., 2014, 53, 2102–2112 CrossRef CAS.
- E. M. Pineda and N. F. Chilton, Nat. Commun., 2014, 5, 5243 CrossRef.
-
(a) L. Zhang, J. Jung, P. Zhang, M. Guo, L. Zhao, J.-K. Tang and B. L. Guennic, Chem. – Eur. J., 2016, 22, 1392–1398 CrossRef CAS;
(b) Y. Bi, Y.-N. Guo, L. Zhao, Y.-N. Guo, S.-Y. Lin, S.-D. Jiang, J.-K. Tang, B.-W. Wang and S. Gao, Chem. – Eur. J., 2011, 17, 12476–12481 CrossRef CAS;
(c) J.-F. Wu, J.-L. Jung, P. Zhang, H.-X. Zhang, J.-K. Tang and B. L. Guennic, Chem. Sci., 2016, 7, 3632–3639 RSC.
-
(a) X.-Y. Chu, H.-X. Zhang, Y.-X. Chang, Y.-Y. Nie, J.-Z. Cui and H.-L. Gao, New J. Chem., 2018, 42, 5688–5697 RSC;
(b) J.-F. Wu, O. Cador, X.-L. Li, L. Zhao, B. L. Guennic and J.-K. Tang, Inorg. Chem., 2017, 56, 11211–11219 CrossRef CAS.
- X. Yao, P.-F. Yan, G.-H. An, C. Shi, Y.-X. Li and G.-M. Li, New J. Chem., 2018, 42, 8438–8444 RSC.
-
(a) J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC;
(b) Y.-N. Guo, G.-F. Xu, Y. Guo and J.-K. Tang, Dalton Trans., 2011, 40, 9953–9963 RSC;
(c) P. Zhang, L. Zhang and J.-K. Tang, Dalton Trans., 2015, 44, 3923–3929 RSC;
(d) J.-J. Lu, M. Guo and J.-K. Tang, Chem. – Asian J., 2017, 12, 2772–2779 CrossRef CAS.
- J.-L. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS.
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS.
- S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Sci., 2016, 7, 5181–5191 RSC.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y.-H. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- Z.-J. Jiang, L. Sun, Q. Yang, B. Yin, H.-S. Ke, J. Han, Q. Wei, G. Xie and S.-P. Chen, J. Mater. Chem. C, 2018, 6, 4273–4280 RSC.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- A. Yamashita, A. Watanabe, S. Akine, T. Nabeshima, M. Nakano, T. Yamamura and T. Kajiwara, Angew. Chem., Int. Ed., 2011, 50, 4016–4019 CrossRef CAS.
- P. Zhang, L. Zhang, S.-F. Xue, S.-Y. Lin and J.-K. Tang, Chin. Sci. Bull., 2012, 57, 2517–2524 CrossRef CAS.
- N. Ishikawa, Y. Mizuno, S. Takamatsu, T. Ishikawa and S. Y. Koshihara, Inorg. Chem., 2008, 47, 10217–10219 CrossRef CAS.
- K. Bernot, J. Luzon, L. Bogani, M. Etienne, C. Sangregorio, M. Shanmugam, A. Caneschi, R. Sessoli and D. Gatteschi, J. Am. Chem. Soc., 2009, 131, 5573–5579 CrossRef CAS.
- X.-L. Wang, L.-C. Li and D.-Z. Liao, Inorg. Chem., 2010, 49, 4735–4737 CrossRef CAS.
- S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Arino, A. Camon, M. Evangelisti, F. Luis, M. J. Martinez-Perez and J. Sese, J. Am. Chem. Soc., 2012, 134, 14982–14990 CrossRef CAS.
- J. Zhu, C. Wang, F. Luan, T. Liu, P. Yan and G. Li, Inorg. Chem., 2014, 53, 8895–8901 CrossRef CAS.
- C.-M. Liu, D.-Q. Zhang and D.-B. Zhu, Inorg. Chem., 2013, 52, 8933–8940 CrossRef CAS PubMed.
- H.-X. Zhang, S.-Y. Lin, S.-F. Xue, C. Wang and J.-K. Tang, Dalton Trans., 2014, 43, 6262–6268 RSC.
- J. Ruiz, A. J. Mota, A. Rodríguez-Diéguez, S. Titos, J. M. Herrera, E. Ruiz, E. Cremades, J. P. Costesc and E. Colacio, Chem. Commun., 2012, 48, 7916–7918 RSC.
-
G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
-
G. M. Sheldrick, SHELXTL, Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed.
- J. Luzon and R. Sessoli, Dalton Trans., 2012, 41, 13556–13567 RSC.
- B. O. Roos, P.
R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- P. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- B. A. Hess, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef CAS.
- B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov and P. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov and P. Widmark, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef CAS.
- B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov, P. Widmark and A. C. Borin, J. Phys. Chem. A, 2008, 112, 11431–11435 CrossRef CAS PubMed.
- F. Aquilante, L. De Vico, N. Ferre, G. Ghigo, P. A. Malmqvist, P. Neogrady, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andres, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fdez. Galvan, N. Ferre, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. A. Malmqvist, T. Muller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Marti, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS.
- L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112 CrossRef CAS.
- L. F. Chibotaru, Adv. Chem. Phys., 2013, 153, 397–519 CrossRef CAS.
-
M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE, v2.1, Universitat de Barcelona, Barcelona, 2013 Search PubMed.
- S. Zhang, H.-P. Wu, L. Sun, H.-S. Ke, S.-P. Chen, B. Yin, Q. Wei, D.-S. Yang and S.-L. Gao, J. Mater. Chem. C, 2017, 5, 1369–1382 RSC.
- M. Ramos Silva, P. Martín-Ramos, J. T. Coutinho, L. C. J. Pereira, V. Lavín, I. R. Martín, P. S. Pereira Silva, J. Martín-Gil, S.-D. Jiang, S.-S. Liu, L.-N. Zhou, B.-W. Wang, Z.-M. Wang and S. Gao, Inorg. Chem., 2012, 51, 3079–3087 CrossRef.
- C.-M. Liu, D.-Q. Zhang, X. Hao and D.-B. Zhu, Sci. China: Chem., 2017, 60, 358–365 CrossRef CAS.
- G.-Z. Huang, Z.-Y. Ruan, J.-Y. Zheng, J.-Y. Wu, Y.-C. Chen, Q.-W. Li, M. Akhtar, J.-L. Liu and M.-L. Tong, Sci. China: Chem., 2018, 61, 1399–1404 CrossRef CAS.
- X.-J. Zhang, V. Vieru, X.-W. Feng, J.-L. Liu, Z.-J. Zhang, B. Na, W. Shi, B.-W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef CAS PubMed.
- L. F. Chibotaru, Struct. Bonding, 2015, 164, 185–230 CrossRef CAS.
- L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016, 55, 10043–10056 CrossRef CAS.
- M. Li, H.-P. Wu, Q. Yang, H.-S. Ke, B. Yin, Q. Shi, W.-Y. Wang, Q. Wei, G. Xie and S.-P. Chen, Chem. – Eur. J., 2017, 23, 17775–17787 CrossRef CAS.
- S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC.
-
B. Elena, A. Ana, L. Javier, B. Juan and B. Fernando, In Handbook of Magnetic Materials, Elsevier B.V., 2017, ch. 1, vol. 26 Search PubMed.
- L. Ungur and L. F. Chibotaru, Phys. Chem. Chem. Phys., 2011, 13, 20086–20090 RSC.
-
L. Ungur and L. F. Chibotaru, Computational Modelling of the Magnetic Properties of Lanthanide Compounds, in Lanthanides and Actinides in Molecular Magnetism, ed. R. A. Layfield and M. Murugesu, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2015, ch. 6, pp. 153–184 Search PubMed.
- D. Aravena and E. Ruiz, Inorg. Chem., 2013, 52, 13770–13778 CrossRef CAS.
- Z. Sievers, Z. Phys. B: Condens. Matter, 1982, 45, 289–296 CrossRef.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- M. Guo, J.-F. Wu, O. Cador, J.-J. Lu, Y. Bing, B. L. Guennic and J.-K. Tang, Inorg. Chem., 2018, 57, 4534–4542 CrossRef CAS.
- L. Ungur and L. F. Chibotaru, Chem. – Eur. J., 2017, 23, 3708–3718 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1851377 and 1851378. For the ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj04019b |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Wenjiao
Mo
a,
Bing
Yin
*ab,
Wenjiao
Mo
a,
Bing
Yin