
A 3D porous FeP/rGO modulated separator as a dual-function polysulfide barrier for high-performance lithium sulfur batteries†
Yingge
Zhang‡
ab,
Yange
Wang‡
ab,
Rongjie
Luo‡
ab,
Ya
Yang
ab,
Yang
Lu
 *ab,
Yan
Guo
ab,
Xianming
Liu
c,
Shixun
Cao
d,
Jang-Kyo
Kim
e and
Yongsong
Luo
*ab
*ab,
Yan
Guo
ab,
Xianming
Liu
c,
Shixun
Cao
d,
Jang-Kyo
Kim
e and
Yongsong
Luo
*ab
aKey Laboratory of Microelectronics and Energy of Henan Province, Henan Joint International Research Laboratory of New Energy Storage Technology, Xinyang Normal University, Xinyang 464000, P. R. China. E-mail: ysluo@xynu.edu.cn; luyang.181@163.com; Fax: +86 0376 6390801; Tel: +86 0376 6390801
bSchool of Physics and Electronic Engineering, Xinyang Normal University, Xinyang 464000, P. R. China
cCollege of Chemistry and Chemical Engineering, Luoyang Normal University, Luoyang 471934, P. R. China
dDepartment of Physics, and International Center of Quantum and Molecular Structures, Materials Genome Institute, Shanghai University, Shanghai 200444, China
eDepartment of Mechanical and Aerospace Engineering, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, P. R. China
First published on 11th November 2019
Abstract
Lithium–sulfur batteries (LSBs) have gained considerable attention for their desirable energy densities, high theoretical capacities, low cost and environmentally friendly properties. However, the shuttle effect of polysulfides seriously hinders their future practical applications. Herein, a dual-function cathode structure, consisting of 3D porous FeP/rGO microspheres supported on both aluminum foil and a commercial separator, exhibits excellent performance by providing strong adsorption with respect to Li2Sx (x = 1, 2, 4, 6 and 8) and S8. In this rational design, the iron phosphide (FeP) nanoparticles act as a catalyst to accelerate polysulfide conversion and as the designated sites for adsorption. The 3D rGO porous conductive network can provide enough space for sulfur loading and to physically adsorb the polysulfides. More importantly, density functional theory (DFT) calculations also verified the strong interactions (with adsorption energy values of −4.21 to −1.97 eV) between the FeP(111) surface and the sulfur species. The electrochemical results show that the cell using the dual-function cathode structure delivers a capacity of 925.7 mA h g−1, with capacity degradation of 0.05% per cycle after 500 cycles, at a current density of 0.5C. It is also worth mentioning that the cell with sulfur loading of ∼2.2 mg cm−2 maintained a high capacity of 483 mA h g−1 at 0.5C after 500 cycles. In summary, the above results demonstrate the promising application of the dual-function cathode structure for high-performance LSBs.
New conceptsTransition metal phosphides (TMPs) have been regarded as potential host materials in lithium–sulfur batteries (LSBs). Iron phosphide (FeP) possesses good thermal stability and metallicity, and it is an electrocatalyst that can effectively trap the polar polysulfides and expedite the redox reactions of the polysulfides. Herein, a dual-function cathode structure, consisting of 3D porous FeP/rGO microspheres supported on both Al foil and a commercial separator, is tactfully designed to enhance the electrochemical performance of LSBs. In this rational design, the host material of 3D porous FeP/rGO microspheres is prepared by spray-drying and phosphorization. The iron phosphide (FeP) nanoparticles act as a catalyst to accelerate polysulfide conversion and as designated sites for adsorption. More importantly, density functional theory (DFT) calculations also verified the strong interactions (with adsorption energy values of −4.21 to −1.97 eV) between the FeP(111) surface and the sulfur species. In addition, the 3D rGO porous conductive network not only affords enough space for sulfur loading, but also physically adsorbs polysulfides. Finally, the 3D porous FeP/rGO microsphere modified separator plays a significant role in suppressing the migration of polysulfides through physical and chemical synergistic effect. This work demonstrates that the successful combination of FeP and a 3D structure can effectively improve the electrochemical performance of LSBs. |
Introduction
Lithium–sulfur batteries (LSBs) have provoked significant interest for their desirable energy density (2600 W h kg−1), high theoretical capacity (1675 mA h g−1), low cost and eco-friendliness.1,2 Unfortunately, several critical obstacles still limit the future practical application of these batteries. Firstly, both sulfur (5 × 10−30 S cm−1) and the corresponding discharge products, Li2S and Li2S2, possesses inferior electrical conductivities, which will hinder electron transfer in the cathode, resulting in increased polarization and deficient utilization of the active materials.3 Secondly, the high solubility of lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8) means that they can easily dissolve into the organic electrolyte and migrate between the anode and the cathode during electrochemical reaction processes. This will trigger the shuttle effect, causing rapid capacity fading and unsatisfactory coulombic efficiencies. Thirdly, the huge volumetric variation (∼80%) from sulfur to Li2S generates severe pulverization of the cathode and causes the capacity of the cell to fade.4 Fortunately, many efforts to address these tough problems have been made for decades, such as developing new electrolytes, constructing a carbon-based interlayer, utilizing a multifunctional polar binder interlayer etc.5–7 Among these methods, carbon-based materials (e.g. microporous carbon paper, graphene nanosheets, and so on)8–13 have been widely applied to enhance the electrochemical performance of LSBs for their diverse nanostructures and intrinsic excellent conductivity. However, the weak adsorption of nonpolar carbon-based materials onto the polar lithium polysulfides (LiPSs) is insufficient for suppressing the shuttle effect of polysulfides. Given this, the combination of carbon-based materials with a polar metal compound3 has been used to absorb the polar polysulfides and hinder their dissolution into the electrolyte.14 For example, Sun et al. prepared the 3D graphene/VN composite to resolve the shuttle effect in LSBs.15 Paik et al. employed a BN-carbon separator to trap polysulfides and the cell delivered a high capacity of 702 mA h g−1 after 250 cycles at 4C in LSBs.16 Among these polar metal compounds, transition metal phosphides (TMPs) have generated great interest, for example iron phosphide (FeP) possesses good thermal stability, metallicity and electrocatalytic properties, and so can effectively trap the polar and expedite the redox reactions of polysulfides. These advantages indicate that FeP is a promising cathode candidate in LSBs.17–19In this work, a dual-function cathode structure composed of 3D porous FeP/rGO microspheres, supported on both the Al foil and on the commercial separator, was proposed for application in LSBs. The 3D porous FeP/rGO (FPG) microspheres were prepared by a spray-drying process and phosphorization. Then, they were mixed with sulfur to act as the cathode and were cast on the commercial separator (PP separator) through vacuum filtration to serve as an interlayer. The unique dual-function cathode structure we have designed possesses several advantages: firstly, the FeP nanoparticles act as a catalyst to facilitate polysulfide conversion in the LSB redox reactions and decrease the loss of sulfur. Secondly, FeP present on the rGO layer could provide abundant adsorptive interfaces to accelerate Li2S nucleation and growth, and thus suppress the shuttle of the polysulfides. Thirdly, the introduction of rGO can greatly improve the transport of the electrons within the electrode, enhancing the redox reaction kinetics. Finally, the large voids and unique porous structure can help increase the sulfur loading process. Additionally, DFT calculations also showed the strong attraction of the FeP(111) surface to sulfur species (with an adsorption energy of −4.21 to −1.97 eV). This rational design endows the cell with better rate performance and cycling stability, and the cell exhibited a high capacity of 925.7 mA h g−1 after 500 cycles at 0.5C.
Experimental section
Preparation of the 3D porous Fe3O4/rGO microspheres
All reagents used were of analytical grade. 1.5 g polystyrene (PS) nanospheres (synthesized according to the previous report)20 and 12 g colloidal graphene oxide (GO) (0.94 wt%, prepared using a modified Hummers’ method) were uniformly dispersed into 250 mL DI water21 and sonicated for 1 h. Then, 1.5 g iron(III) chloride hexahydrate was added into the above solution and stirred for about 5 h. Then the uniform solution was spray-dried at a temperature of 120 °C and a flow rate of 600 mL h−1. Finally, the collected sample was heated at 450 °C for 3 h accompanied by a heating rate of 3 °C min−1 under argon protection, in order to obtain the 3D porous Fe3O4/GO (FOG) microspheres.Synthesis of the 3D porous FeP/rGO microspheres
The as-prepared FOG microspheres and sodium hypophosphite (NaH2PO2) were put into two separate porcelain crucibles. The porcelain crucible with NaH2PO2 was placed at the upstream part and FOG was placed in the middle part of the furnace (w/w, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), respectively. Subsequently, the product was calcined under 400 °C for 2 h with a ramping rate of 5 °C min−1 in a static argon atmosphere. Finally, the 3D FeP/rGO (FPG) microspheres were collected after completely being cooled.22 The FeP composite was prepared according to the same procedure without adding GO. The rGO was synthesized without adding iron(III) chloride hexahydrate.
:1), respectively. Subsequently, the product was calcined under 400 °C for 2 h with a ramping rate of 5 °C min−1 in a static argon atmosphere. Finally, the 3D FeP/rGO (FPG) microspheres were collected after completely being cooled.22 The FeP composite was prepared according to the same procedure without adding GO. The rGO was synthesized without adding iron(III) chloride hexahydrate.
Synthesis of the 3D porous FeP/rGO–S composite
The FeP/rGO–S (FPGS) composite was constructed by a melt-diffusion method. In summary, the host material FPG and S powder (w/w, 20:80) were homogeneously mixed, followed by sealing them in a vacuum glass bottle and heating at 155 °C for 10 h to assist the uniform distribution of S powder in the host material.
Material characterization
X-ray diffraction (XRD) spectra were conducted on a D2 PHASER (Bruker) with Cu-Kα (λ = 1.5418 Å) radiation at 30 kV and 10 mA. X-ray photoelectron spectrometry (XPS, K-ALPHA 0.5 EV) was performed to analyze the chemical composition of the surface of the sample. Field emission scanning electron microscopy (SEM, S-4800) and field emission transmission electron microscopy (TEM, JEOL JEM-2010) were used to determine the morphologies and structures of the sample. Raman spectra were obtained on an INVIA Raman microprobe (Renishaw Instruments, laser source: 532 nm). The thermogravimetric analysis (TGA) measurements were performed on a STA499f5 analyzer. The nitrogen adsorption/desorption isotherms and the determination of the Brunauer–Emmett–Teller (BET) surface area were conducted on an ASAP2460 2MP at 77 K. UV-vis absorption spectra (U-3900H UV/VIS SPECTROPHOTOMETER. 2J2-0034) were obtained to observe the adsorptions to the sulfur species after adding 10 mg FPG microspheres into the Li2S6 solutions.Cell assembly and electrochemical measurements
:20) were uniformly mixed into an N-methyl-2-pyrrolidinone (NMP) solution and, after stirring for about 2 h, the slurry was cast on Al foil, and then dried at 80 °C for about 12 h. Next, the cast foil was punched to obtain 1.6 × 1.6 cm disks and the loading level of each electrode was ∼1 mg cm−2. CR2032 coin cells were assembled in a glove box using an FPG electrode (acting as both cathode and anode), a Celgard 2500 separator and 50 μL electrolyte (consisting of the mixture of DOL/DME (v/v, 1:1), 1 M bis(trifluoroethanesulfonyl) imide lithium (LiTFSI) and 0.2 M Li2S6). Cyclic voltammetry (CV) tests were performed on a VMP3 Multichannel electrochemical workstation (Bio-Logic, Claix, France) at a scan rate of 3 mV s−1.
:2:1) were dissolved into 10 mL NMP solvent. 5 hours later, the mixture was coated onto Al foil before drying at 60 °C for 12 h. The cathode electrodes were obtained by cutting the foil into 1.6 × 1.6 cm sheets. The cells were assembled with a cathode electrode, separator/the coated separator and Li foil. The electrolyte was 1 M LiTFSI in DOL/DME (1:1 v/v) with 2 wt% 0.1 M lithium nitrate (LiNO3) solution as an additive. CV was tested on a VMP3 Multichannel electrochemical workstation over the potential range of 1.7–2.8 V at different scan rates. The electrochemical impedance spectroscopy (EIS) was also conducted on VMP3 from 100 kHz to 10 mHz with an AC voltage amplitude of 5 mV. The Galvanostatic charge–discharge (GCD) tests were carried out between 1.7 and 2.8 V on a Neware battery-testing system (5 mA and 50 mA, 5 V) based on different current densities.
Visualized adsorption of polysulfides
Adding Li2S and sulfur (molar ratio: 1:5) into a mixture of 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) (v/v, 1:1), then the above solution was stirred at 60 °C to obtain 2 mmol L−1 Li2S6 solution. 10 mg FPG microspheres were soaked into 2 mL of the above solution, followed by stirring and resting for ∼4 h. The above operation was handled in an Ar-filled glove box.
Permeation experiments
Permeation experiments were conducted using a PP separator and FPGI. Suitable amounts of polysulfide solution and the blank electrolyte were put into the inner bottle and outer beaker, respectively. The PP separator and FPGI were placed on the cap of the inner bottle. Then, the optical images of the solutions were recorded using a digital camera. The above operation was handled in an Ar-filled glove box.Density functional theory calculations
The first-principles calculations were carried out based on the framework of DFT23 using the Vienna ab initio simulation package (VASP) program.24 The exchange correlation was evaluated by the Perdew–Burke–Ernzerhof (PBE) functionals of the generalized gradient approximation functional (GGA).25 The cut-off energy was 600 eV and the Brillouin zone was integrated by a 3 × 4 × 1 Γ-centered k mesh. The van der Waals (vdW) interaction was calculated by the DFT-D2 correction method.26 For geometry optimization, the energy and force of the convergence criteria were 10−5 eV and 0.01 eV Å−1 per atom for the total energy, respectively. A 2 × 2 supercell containing 28 atoms (Fe14P14) was used for the model of the FeP(111) substrate. The top two layers of the four-layer slab were free. A vacuum layer of 15 Å was provided to avert the interaction between the layer and its images. The adsorption energy (Ea) was defined as: Ea = Etotal − Eslab − ELi–S, where Etotal, Eslab and ELi–S are the total energy of the FeP slab after adsorbing Li–S, the FeP slab, and Li–S systems, respectively. The energies of the Li–S molecules were calculated in a box of dimensions 20 Å × 20 Å × 20 Å.Results and discussion
The preparation process of the 3D porous FPGS composite is described in Fig. 1. In the first step, the 3D porous FPG microspheres were synthesized via spray-drying and phosphorization (Fig. 1a). In this process, PS nanospheres can help with the dispersion of GO sheets in the spray solution through their hydrophobicity. In addition, the PS nanospheres serve as a sacrificial template, producing a ∼300 ± 50 nm void after calcination, and the TEM image shows a thin layer of rGO after removing the PS nanospheres. The infusion of sulfur into the FPG host and the simple lithiation/delithiation process of the FPGS electrode are shown in Fig. 1b.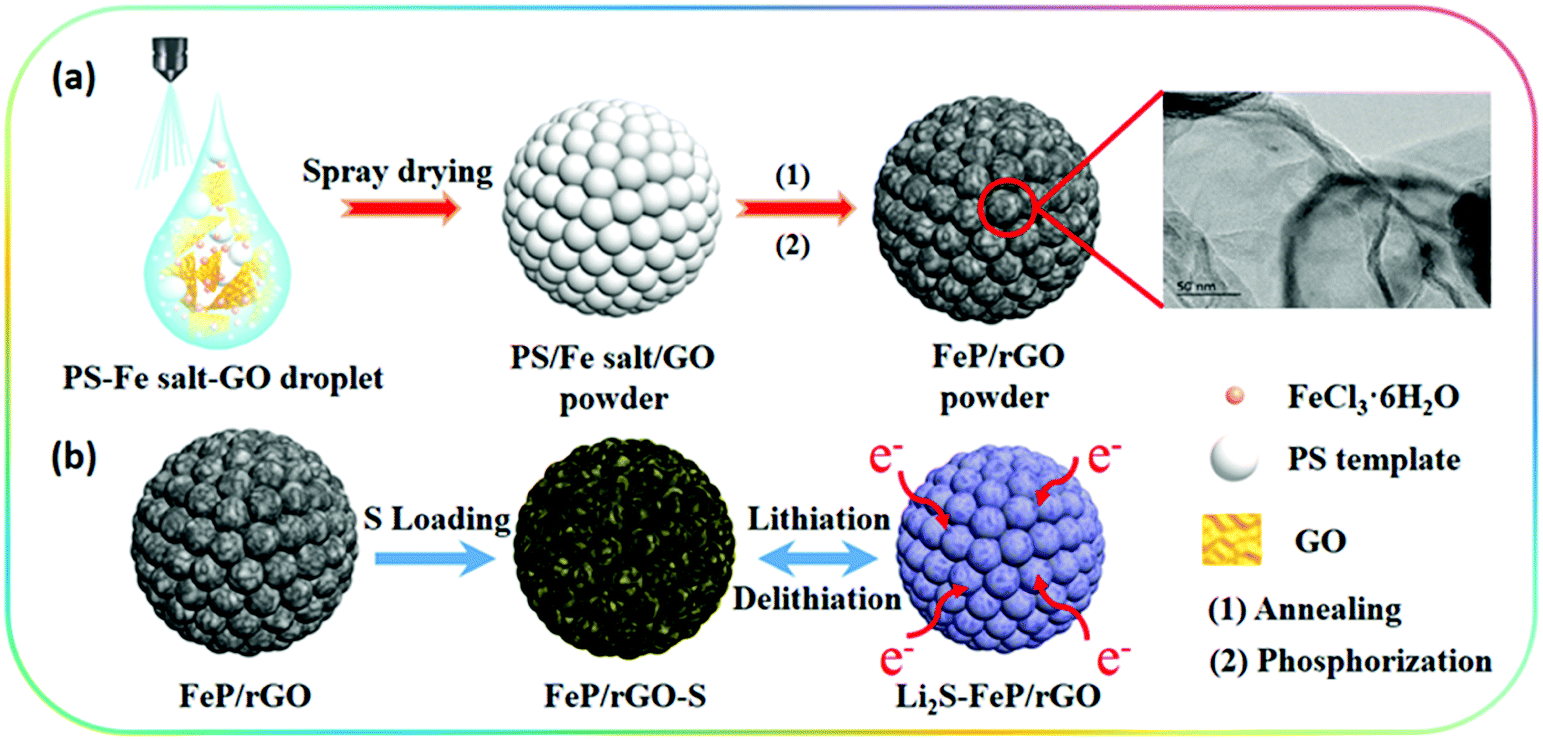 | ||
| Fig. 1 (a) A schematic diagram of the fabrication of the FeP/rGO composite by spray-drying and phosphorization. (b) The preparation of FeP/rGO–S and the lithiation/delithiation process of the composite. | ||
The SEM images of the 3D porous FOG microspheres are shown in Fig. S1a and d (ESI†), and it can be observed that there are many large voids in the whole microspheres. In addition, the morphology of the 3D porous FPG microspheres remains intact after phosphorization (as shown in Fig. S1b and e, ESI†). In Fig. S1c and f (ESI†), the 3D porous FPGS composite was achieved by integrating sulfur into the 3D FPG microspheres. The following TEM results further confirmed the above analysis. In Fig. 2a and Fig. S2 (ESI†), the 3D porous structure of FPG can be clearly seen and the size of each pore is approximately ∼300 ± 50 nm. From the partially enlarged image of FPG (in Fig. 2b), the thin rGO layers can be observed. The high resolution image in Fig. 2c depicts FPG with interplanar distances of 0.187, 0.241, and 0.251 nm, corresponding to the planes of (211), (111) and (102), respectively.27 The high resolution result was also confirmed by XRD analysis (Fig. S3a, ESI†), which revealed the successful transformation of Fe3O4 into FeP (JCPDS No. 89-2746). Additionally, the corresponding selected-area electron diffraction (SAED) pattern of FPG in Fig. 2d is also in accord with the high-resolution analysis. The concentric rings composed of bright discrete diffraction spots in the pattern, indexed to the (211), (111) and (102) planes of orthorhombic FeP, proves that FeP is polycrystalline.22 In Fig. 2e, the morphology of FPGS remains unbroken after the infusion of sulfur. Moreover, in the scanning TEM (STEM) image (Fig. 2f), the energy-dispersive X-ray spectroscopy (EDS) line scanning across FPGS discloses the presence and concentration of S,28 and the energy-dispersive X-ray (EDX) elemental mapping of FPGS in Fig. 2g–k further demonstrates the uniform distribution of S. In addition, the content of sulfur in 3D porous FPGS was 74.5%, which was calculated by the TG curve in Fig. S3c (ESI†).3 Subsequently, Raman spectroscopy was introduced to investigate the carbon in the composite (Fig. S3b, ESI†). The two clear bands at 1344 and 1590 cm−1 are D and G bands, respectively.29 After introducing sulfur into the 3D porous structure, the D band shifts from 1344 to 1350 cm−1 and the G band changes from 1590 to 1596 cm−1. Moreover, the peak intensity ratio between the D and G bands (ID/IG) of FPGS (0.64) is lower than that of FPG (0.86). This indicates that the FPGS composite is more disordered after the encapsulation of sulfur.30 Moreover, the Fe–P bands cannot be observed in the Raman spectrum, demonstrating that the FeP nanoparticles are covered by amorphous carbon.31 The existence and content of carbon were demonstrated by XRD patterns (shown in Fig. S3a, ESI†) and TG curves (shown in Fig. S3c, ESI†). The carbon content is calculated to be 24.7% from the TG curves (the detailed calculation process is depicted in the ESI†).3,22,32 Then, the textural properties of FPG and FPGS were investigated by N2 adsorption/desorption isotherm tests. The curve shown in Fig. S3d (ESI†) is a typical IV with a H3 type hysteresis loop, indicating that there are mesopores within the host material, and this is beneficial to the combination with sulfur. The BET surface areas of the FPG microspheres and FPGS are 81 m2 g−1 and 7.4 m2 g−1, respectively. The inset picture in Fig. S3d (ESI†) shows that the average pore diameter of FPG is 14.7 nm, while for FPGS almost no mesopores appear. The above phenomenon indicates that the impregnation of S will cause a decreased surface area and the mesopores to almost all disappear.33,34
 | ||
| Fig. 2 (a and b) TEM images, (c) a HRTEM image, and (d) the SAED pattern of the FPG composite. (e) TEM images of the FPGS composite. (f) A STEM image and EDS line scan of FPGS: the yellow signals are S along the yellow line. (g) A TEM image of the FPGS composite. (h–k) Elemental mapping images of the Fe, P, S and C elements in the FPGS composite. | ||
XPS measurements were used to probe the surface electronic states and the chemical composition of the samples. In Fig. 3a, the Fe 2p spectrum shows two peaks at 707.1 (Fe–P) and 719.8 eV, corresponding to 2p3/2 and 2p1/2 of FeP, respectively. It is clear that the area ratio between Fe 2p1/2 and Fe 2p3/2 is 0.5, and that the FWHM of them is ∼1.7. The other two peaks that appeared at 712.3 and 725.9 eV can be ascribed to the oxidized form of Fe, due to the inevitable surface oxidation.22,35,36 After infusing with S, FPGS shows higher binding energy (712.6, 720.7, 707.7 and 726.3 eV) in Fig. 3d. In addition, as seen in Fig. 3g, the binding energies of S (163.9 and 165.0 eV) of FPGS exhibited a −0.3 eV and −0.4 eV shift compared with the XPS spectrum of pure S (164.2 and 165.4 eV), shown in Fig. S4 (ESI†).37 The above analysis clearly suggests that S and the host material can strongly interact with each other.28,38,39 The high-resolution XPS spectra of P 2p of the two samples (presented in Fig. 3b and e) showed two peaks (130.1 and 129.5 eV for FPG, and 130.2 and 129.3 eV for FPGS), which correspond to P–C and P–Fe, respectively. The third peaks (134.2 eV for FPG and 133.9 for FPGS) can be ascribed to the P–O oxidized species of phosphide. The P 2p spectrum showed a minor decrease in the P–O component, indicating a slight reduction of the oxidized P species on the surface. In addition, the relative intensity of the P–C bond changed in the fitting curves of FPGS, indicating the slight loss of P content during the heat treatment of S, and this was mainly due to the partial decomposition of the P–O group.35,40,41Fig. 3c presents the XPS spectrum of C 1s of FPG, which can be divided into four peaks. The peak centered at 284.7 eV corresponds to C–C, the peak appearing at 284.9 eV can be assigned to the P–C bond, and the peaks at 285.9 and 289.6 eV represent C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.35,36 The corresponding peaks of C 1s for FPGS are (Fig. 3f) 284.6 eV (C–C), 285.2 eV (P–C), 286.2 eV (C–O) and 289.9 eV (O–CO). The weak peak shift of C–C in FPGS could be attributed to the change in conductivity of rGO by the introduction of S.42
O, respectively.35,36 The corresponding peaks of C 1s for FPGS are (Fig. 3f) 284.6 eV (C–C), 285.2 eV (P–C), 286.2 eV (C–O) and 289.9 eV (O–CO). The weak peak shift of C–C in FPGS could be attributed to the change in conductivity of rGO by the introduction of S.42
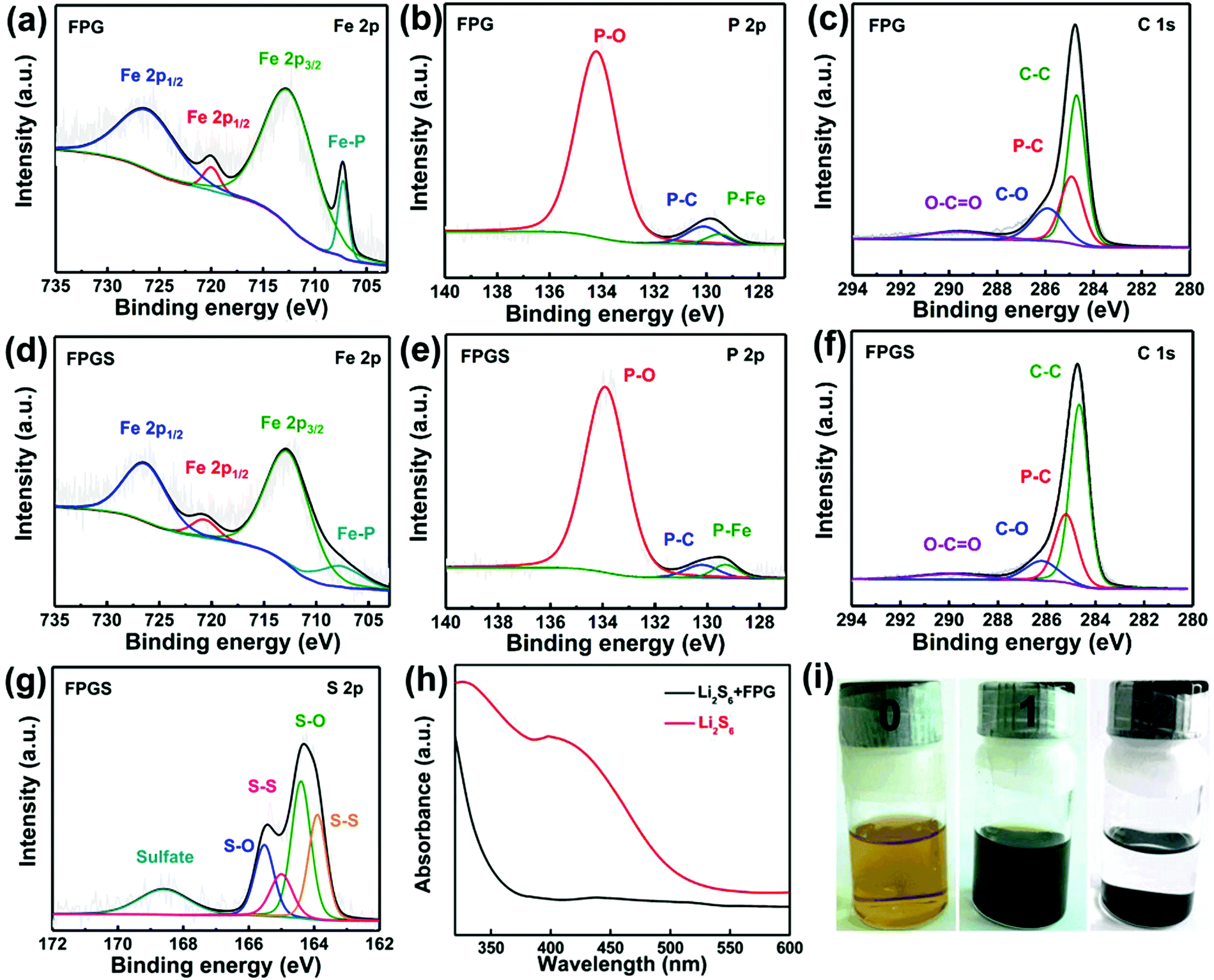 | ||
| Fig. 3 High-resolution (a) Fe 2p, (b) P 2p and (c) C 1s XPS spectra of the FPG composite. High-resolution (d) Fe 2p, (e) P 2p, (f) C 1s and (g) S 2p XPS spectra of the FPGS composite. (h) UV-vis absorption spectra of a Li2S6 solution before and after the addition of FPG. (i) Photographs of the Li2S6 solution before and after the addition of FPG. | ||
UV-vis absorption spectra were measured to investigate the adsorptions to the sulfur species. After adding 10 mg FPG into the Li2S6 solutions, the golden solution became almost transparent after standing for 4 h (Fig. 3i). In contrast, the solution of 10 mg rGO was almost unchanged (as seen in Fig. S5 of the ESI†). It can be suggested that FPG exhibits strong polysulfide adsorption ability. In addition, the UV-vis absorption spectra in Fig. 3h show that the characteristic peaks of S62− almost disappear after the adsorption of Li2S6 by FPG,43 indicating its higher ability to suppress polysulfides.
Fig. 4a shows the fabrication process of the FPG coated separator (with FPGI acting as an interlayer), which is synthesized by vacuum filtration. The optical photographs of the two sides of the coated separator are shown in Fig. 4c and d. The coated FPG microspheres maintain excellent adhesion to the separator and do not flake or peel off after folding (as depicted in Fig. 4e–i and in Video 1, ESI†), indicating its good mechanical strength and flexibility. In Fig. 4j it can be clearly seen that the pristine separator surface possesses sub-micrometer pores, through which polysulfides can migrate to the anode. In contrast, after coating with the FPG microspheres (Fig. 4k), the large area of the pores is covered by the loose FPG microspheres, and this is conducive to the transportation of Li+ and the effective diffusion of the electrolyte.44 Moreover, from the cross-section image of FPGI in Fig. 4l, we can determine that the FPG microspheres are tightly anchored on the surface of the separator (thickness: ∼12.6 ± 1 μm, mass loading: ∼0.75 mg cm−2).45 Finally, as shown in Fig. 4b, FPGI was used to trap polysulfides by cooperative chemical adsorption and as a physical barrier.
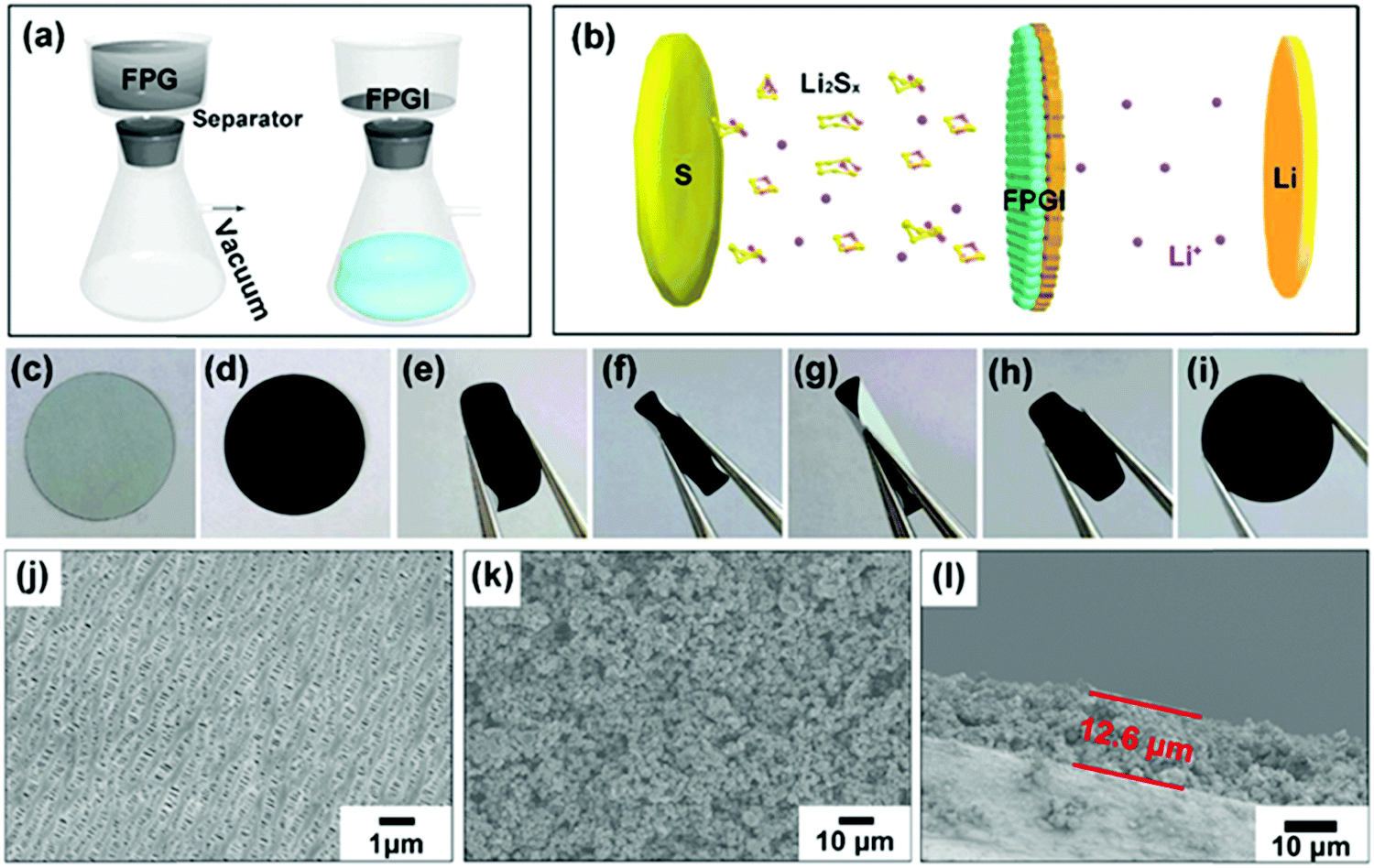 | ||
| Fig. 4 (a) A schematic diagram of the fabrication of FPGI. (b) A schematic diagram of the lithium–sulfur battery with FPGI. (c and d) Digital images of the two sides of FPGI. (e–i) Digital images of folded and recovered FPGI. (j) An SEM image of the raw separator. (k) An SEM image of FPGI. (l) An SEM image of the FPGI cross-section. | ||
Contact angle tests were performed in order to gain further insight into the electrolyte wettability on various separators. It is known from a previous report that deionized water has the similar polarity as the electrolyte of LSBs,46 while the organic electrolyte of LSBs is easily volatile. As seen in Fig. S6 (ESI†), the contact angle of the water on the FeP/rGO coated separator coupling layer is lowest (30.8°) after 0.1 s, compared with that for the other two separators, i.e. 126.5° on the coupling layer of the commercial separator, and 46.3° on the rGO coated separator coupling layer. After 1.2 s, the water droplets almost disappeared on the coupling layer of the FeP/rGO coated separator, while they are still visible on the commercial separator and on the rGO coated separator. The results signified the high wettability of the FeP/rGO modified separator coupling layer, which can accelerate the electron/ion transfer and help the penetration of the electrolyte, resulting in enhanced electrochemical performance.47
The CV curves of FPGS-FPGI (the scan rate is 0.1 mV s−1 and the potential ranges from 1.7–2.8 V versus Li/Li+) are shown in Fig. 5a. In the first cycle, there are two reduction peaks at 2.27 V and 2.05 V, and these belong to the reduction of S8 to lithium polysulfides (Li2Sx, x ≥ 4) and further reduction to solid Li2S2 and Li2S, respectively. In the subsequent anodic scans, only one oxidation peak appeared at 2.38 V, and this can be attributed to the coupled conversion of Li2S2/Li2S to Li2S8/S.48–51 The redox peaks overlap well from the 2nd to the 5th cycles and show no obvious difference in either the peak intensity or potential changes, demonstrating high electrochemical stability. GCD tests of the cell with FPGS-FPGI, FPGS and S were conducted at a current density of 0.1C between 1.7–2.8 V vs. Li/Li+. As seen in Fig. 5d, the cell with FPGS-FPGI presented an initial discharge capacity of 1238.8 mA h g−1, while the cells with FPGS (Fig. 5c) and S electrodes (Fig. 5b) exhibited lower initial discharge capacities of 1132.5 mA h g−1 and 634.7 mA h g−1, respectively. In addition, the discharge capacity of FPGS-FPGI remained at 1071.3 mA h g−1 in comparison with the other two materials (426.5 mA h g−1 for the S electrode and 871.2 mA h g−1 for the FPG electrode) after 100 cycles. Evidently the cell with the dual-function cathode structure exhibited high lithium ion conductivity, as well as quick transfer of lithium ion to the separator and lithium ion diffusion, thus leading to better electrochemical performance.6 Additionally, the lowest ΔE value (half of the charge/discharge capacity) of 0.27 V was observed for FPGS-FPGI, compared with 0.31 V for FPGS and 0.32 V for S, indicating higher facile redox reaction kinetics and lower polarization for the cell with FPGS-FPGI. Furthermore, it can be clearly noted that the voltage plateaus of FPGS-FPGI are longer than those of S and FPGS,4,52,53 demonstrating better conversion and re-utilization of sulfur. In order to testify the catalytic effect of FPG on facilitating polysulfides, symmetrical Li2S6–Li2S6 cells were tested at −1 to 1 V. From the polarization profiles (Fig. 5e), it can be concluded that the major contribution of the redox current comes from Li2S6, and only a fraction of capacitive current is provided by the cell without the Li2S6 electrolyte. The profile for the FPG electrode (areal loading: ∼0.66 ± 0.02 mg cm−2) in the Li2S6 electrolyte showed four distinct redox peaks at 0.56 V, 0.12 V, −0.12 V and −0.56 V, indicating high reversibility of the electrode.54 In addition, Fig. 5f shows that the current density of the FPG electrode is larger than that of the rGO electrode, and this can be attributed to FeP reducing the amount of polarization inside the cell, and effectively accelerating the redox reaction of the polysulfides in the liquid phase (Li2S8 ↔ Li2S6 ↔ Li2S4). Moreover, from the EIS plot in Fig. 5g, we can determine that the FPG symmetric cell shows a smaller high frequency semicircle in the Nyquist plots,55 demonstrating the smallest charge transfer resistance.
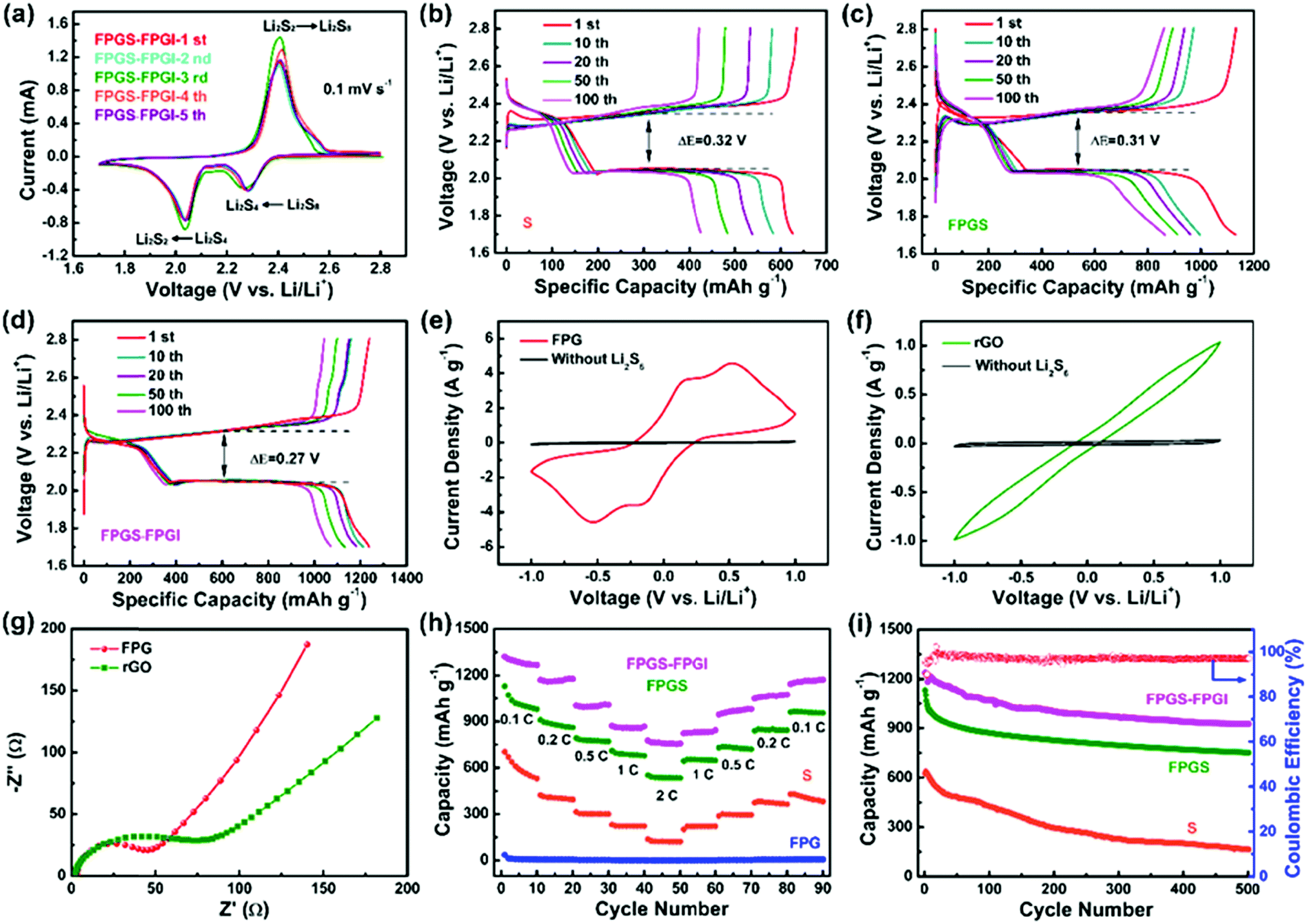 | ||
| Fig. 5 (a) CV profiles of the FPGS-FPGI cell. (b–d) Galvanostatic charge–discharge profiles of cells with S, FPGS and FPGS-FPGI at 0.1C. Cyclic voltammograms of the symmetric cells with identical electrodes of (e) FPG and (f) rGO, in electrolytes with and without 0.2 M Li2S6 at a scan rate of 3 mV s−1 within a voltage window of −1 to 1 V. (g) Electrochemical impedance spectra of FPG and rGO of the symmetric cells. (h) Rate performances of the FPGS-FPGI, FPGS, FPG and S electrodes at various current densities. (i) Cycling stabilities of the FPGS-FPGI, FPGS and S cells at 0.5C. | ||
To further elucidate the enhanced electrochemical performance, the cells with S, FPG, FPGS and FPGS-FPGI (sulfur loading: ∼1 mg cm−2) were tested at discharge current densities of 0.1C to 2C (Fig. 5h). The cell with FPGS-FPGI delivered a specific capacity of 1267.7, 1176.8, 1008.1, 867.5 and 756.5 mA h g−1 at 0.1, 0.2, 0.5, 1 and 2C, respectively. When the current density returned to 0.1C, the capacities still remained at 1171.6 mA h g−1. In contrast, the cells with S and FPGS displayed capacities of 530.7, 394.4, 299.6, 223.2 and 122.3 mA h g−1, and 979.9, 860.8, 771.2, 678.3 and 533.2 mA h g−1 at the same current density, respectively. Then the capacities remained at 382.9 mA h g−1 for S and 954.7 mA h g−1 for FPGS when the current density returned to 0.1C. However, compared with the cells of S, FPGS and FPGS-FPGI, the cell of FPG shows almost no capacity, indicating that the capacity of the cell mainly comes from the active sulfur. The above analysis also manifests that the FPGS-FPGI cell can significantly suppress the shuttle effect through strong chemical and physical adsorption to polysulfides.56 In addition, the results of the permeation tests, shown in Fig. S7 (ESI†), demonstrated that the electrolyte in the bottle containing the PP separator turned light yellow with an increase in time. In contrast, the color of the electrolyte in the bottle with FPGI appears slightly changed after 6 h. This phenomenon directly implies that FPGI plays an important role in confining the diffusion of polysulfides.16
The cycling test results of the different cells are shown in Fig. 5i, where the cell of FPGS-FPGI delivers a capacity of 925.7 mA h g−1 and has a coulombic efficiency of 99.03% after 500 cycles at 0.5C. In contrast, capacities of only 164.1 and 752.0 mA h g−1 for the S and FPGS electrodes were maintained, respectively, verifying that the FPGS-FPGI electrode is conducive to improving LSB performance via its catalytic ability and the strong chemisorption of FeP to polysulfides. In addition, considering the weight of the current collectors, the sulfur content in the FPGS electrode (sulfur loading: 1 mg cm−2) was 13.8 wt% of the whole cathode.57 Moreover, FPGS-FPGI enables the cell to deliver a gravimetric energy density of 1379.6 W h kg−1, based on the content of sulfur in the cathode. Our FPGS-FPGI electrode demonstrates satisfactory electrochemical performance, which is far superior to the previous studies reported so far (Table S1, ESI†).
The role of FPGI in LSBs was further investigated by EIS measurements, shown in Fig. S8 (ESI†). The corresponding equivalent circuits are presented in Fig. S8b (ESI†) and the relevant parameters after fitting are listed in Table S2 (ESI†). In the Nyquist curves, the high-frequency semicircles refer to the interfacial charge-transfer resistance (Rct) and the low-frequency sloping lines represent the Li+ diffusion resistance within the electrodes. In the corresponding equivalent circuits, Rsf stands for the SEI film and/or the contact resistance. The electrolyte resistance is Re, and CM and Cdl represent the double layer capacitance. Zw is the Warburg impedance value, which stands for the Li+ diffusion coefficient.58 As seen in Fig. S8a (ESI†), the cell with FPGS-FPGI shows the lowest Rct value (3.6 Ω) compared to the values for the S (89.5 Ω) and FPGS (25 Ω) electrodes after 500 cycles (Table S2, ESI†). This suggests that FPG has great potential in modifying the separator for improved charge transfer rate.44 Moreover, the electrolyte resistance (Re) of the FPGS cell (as shown in Table S2, ESI†) is slightly lower than that of the FPGS-FPGI cell, i.e. 2.4 vs. 6 Ω, demonstrating that the introduction of FPGI introduces a marginal increase in the internal resistance.
As shown in Fig. 6, the CV curves of S, FPG, FPGS-FPGI were conducted at various scan rates to further study the Li+ diffusion coefficient within the cells based on the Randles–Sevcik equation (eqn (1)). The CV curves of the samples show two cathodic peaks (denoted as C and B) and one anodic peak (denoted as A).
| Ip = (2.69 × 105)n1.5Area DLi+0.5Conc.Liv0.5 | (1) |
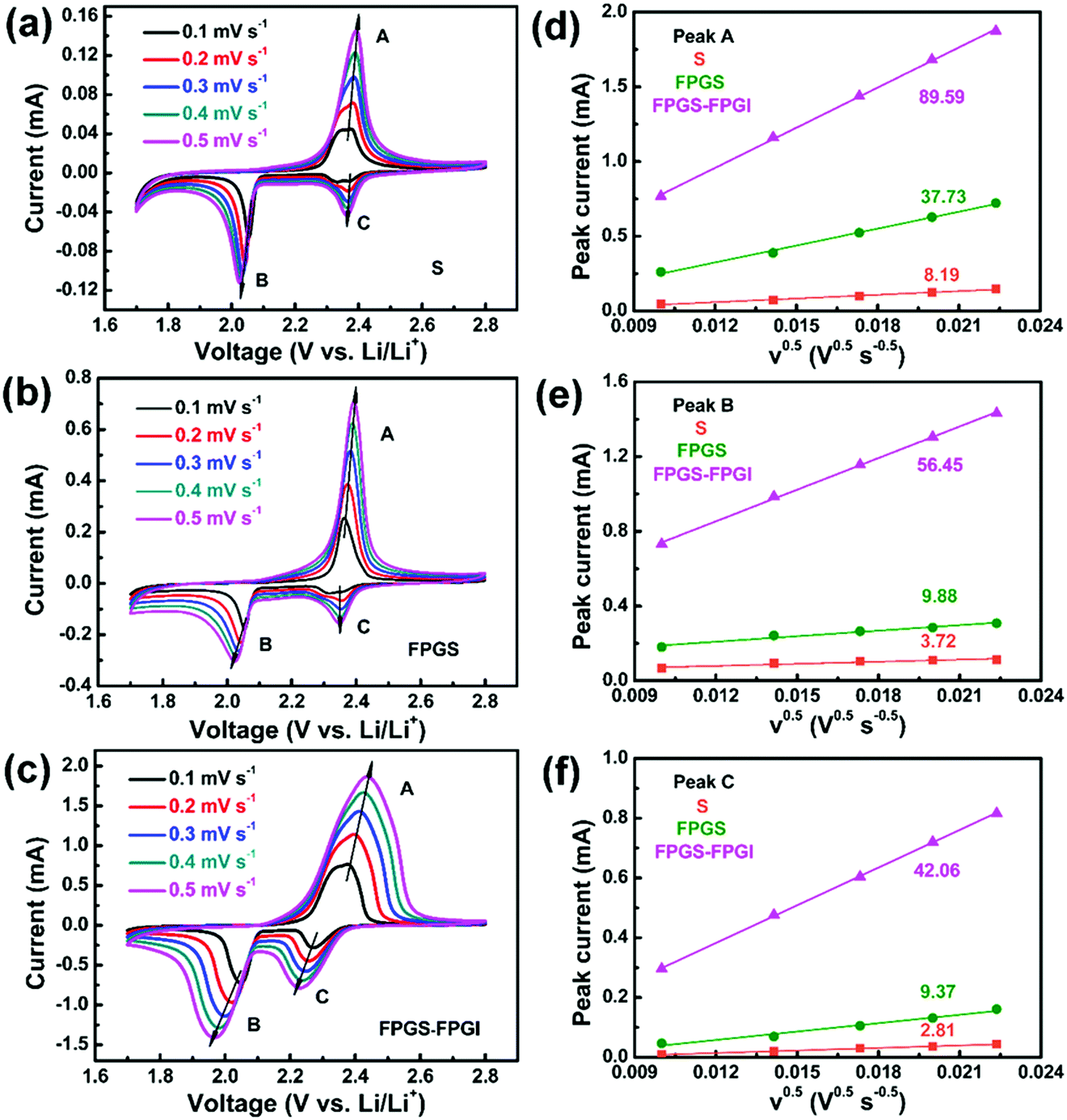 | ||
| Fig. 6 (a–c) CV curves of batteries with S, FPGS and FPGS-FPGI at various scan rates. (d–f) The corresponding linear fits of the peak currents for the cells. | ||
I p refers to the current (A) of the peak, n is the number of electrons that participate in the reaction (2 in LSBs), Area represents the active area of the electrode (2 cm2), DLi+ (cm2 s−1) refers to the Li ion diffusion coefficient, Conc.Li is the Li+ concentration in the electrolyte (0.55 mol L−1) and v is the scan rate (V s−1).59 We can conclude that both the cathodic and anodic peak currents have a linear relationship with v0.5, since the slope of Ipvs. v0.5 corresponds to DLi+ when n, Area and Conc.Li remain unchanged. It is clear that the slopes of FPGS-FPGI, of peaks A, B and C (as seen in Fig. 6d–f), are higher than those of S and FPG. Moreover, in Table S3 (ESI†), the cell with FPGS-FPGI show higher DLi+ values in peaks A, B and C compared with those with S and FPGS. Given this, a conclusion can be reached that the introduction of FPGI does not remarkably reduce DLi+, due to the enhanced electrode conductivity and polysulfide redox reaction by FPG.13,44,60
To gain further insight into the suppressing effect of the FeP nanocrystals on polysulfides, the binding energies between FeP and the five most common polysulfide species were calculated using DFT methods. As presented in Fig. 7a–f, the Ea values of S8 and Li2Sx (x = 1, 2, 4, 6 and 8) on FeP(111) are −2.833, −2.566, −4.21, −1.975, −3.000 and −3.071 eV, respectively. In contrast, taking Li2S4 for example, the Ea value of graphene and Li2S is only −0.47 eV,61 which is weaker than the value of Li2S on FeP(111). This result signifies that the strong interaction between FeP and the sulfur species is chemical adsorption, which mainly consists of two parts: one is the chemical bonding between Li and P, and the other part is between the terminal S and Fe, while the adsorption between graphene and the sulfur species is only through van der Waals interactions.3,61 Overall, FeP exerts a significant influence on the trapping of the polysulfides mainly through strong chemical adsorption.
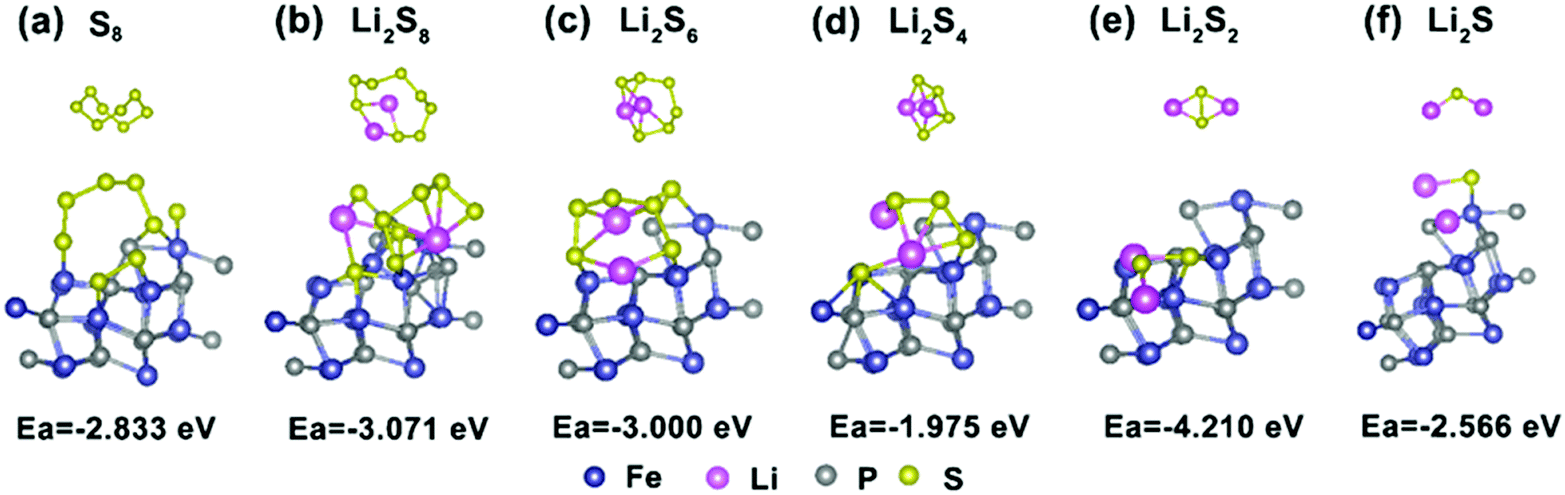 | ||
| Fig. 7 (a–f) The optimized configurations of lithium polysulfide Li2Sx (x = 1, 2, 4, 6, and 8) and S8 clusters adsorbed onto the FeP(111) surface. | ||
Generally, when increasing the mass loading of sulfur in the electrode, the utilization of the electrode material will decrease due to the poor electronic conductivity of S. However, the cells equipped with FPGI with a S loading of ∼1.3 mg cm−2 and ∼2.2 mg cm−2 can still deliver high capacities of 759 mA h g−1 and 483 mA h g−1, respectively, after 500 cycles at 0.5C, as shown in Fig. S9 (ESI†). This suggests that FPGI restricts the shuttle effect by an effective physical barrier and by chemical adsorption to polysulfides, resulting in an excellent electrochemical performance.
Finally, the cells of FPGS-FPGI, FPGS and S were disassembled over 500 cycles to observe the SEM images of the separator with respect to the anode and the cathode. The SEM images of the separator surfaces of the cathode in the cell of FPGS-FPGI before and after the cycles are shown in Fig. S10e (ESI†). Fig. S10f (ESI†) shows that there are almost no polysulfide species on the surface of the separator with respect to the anode. However, for the S cell (Fig. S10a and b, ESI†) and the FPGS cell (Fig. S10c and d, ESI†), both of the surfaces of the separator with respect to the cathode/anode showed some polysulfide species, implying that FPGI plays a remarkable role in capturing the dissolved polysulfides.59
Conclusions
Herein, we have demonstrated the important role of a dual-function cathode structure. It has been confirmed that the 3D FPG/rGO coated separator can confine the migration of polysulfides through its physical barrier and by chemical adsorption. The rGO in this rational structure can provide an additional electrically conductive scaffold and can physically adsorb the polysulfides. The FeP nanoparticles not only show strong adsorption to the sulfur species in the LSB redox reactions, but they also act as a catalyst to speed up polysulfide conversion. More importantly, DFT calculations also certified the strong attraction between the FeP(111) surface and the sulfur species. Therefore, the cell with a dual-function cathode structure delivered a high reversible capacity of 925.7 mA h g−1 after 500 cycles at 0.5C. In summary, this work provides a feasible strategy for the effective suppression of polysulfide migration in high-performance LSBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 61574122 and 61874093) and the Zhongyuan Thousand Talents Plan-Science & Technology Innovation Leading Talents Project (No. 194200510009). This work was also supported by the Xinyang Normal University Analysis & Testing Center.References
- Y. S. Luo, J. S. Luo, J. Jiang, W. W. Zhou, H. P. Yang, X. Y. Qi, H. Zhang, H. J. Fan, D. Y. W. Yu, C. M. Li and T. Yu, Energy Environ. Sci., 2012, 5, 6559–6566 RSC.
- R. J. Luo, Q. H. Yu, Y. Lu, M. J. Zhang, T. Peng, H. L. Yan, X. M. Liu, J. K. Kim and Y. S. Luo, Nanoscale Horiz., 2019, 4, 531–539 RSC.
- S. Z. Huang, Y. V. Lim, X. M. Zhang, Y. Wang, Y. Zheng, D. Z. Kong, M. Ding, S. Y. A. Yang and H. Y. Yang, Nano Energy, 2018, 51, 340–348 CrossRef CAS.
- S. H. Chung, P. Han, R. Singhal, V. Kalra and A. Manthiram, Adv. Energy Mater., 2015, 5, 1500738 CrossRef.
- W. Chen, T. Qian, J. Xiong, N. Xu, X. J. Liu, J. Liu, J. Q. Zhou, X. W. Shen, T. Z. Yang, Y. Chen and C. L. Yan, Adv. Mater., 2017, 29, 1605160 CrossRef.
- Z. A. Ghazi, X. He, A. M. Khattak, N. A. Khan, B. Liang, A. Iqbal, J. X. Wang, H. Sin, L. S. Li and Z. Y. Tang, Adv. Mater., 2017, 29, 1606817 CrossRef.
- J. Q. Huang, Q. Zhang and F. Wei, Energy Storage Mater., 2015, 1, 127–145 CrossRef.
- S. H. Chung and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef.
- G. M. Zhou, S. F. Pei, L. Li, D. W. Wang, S. G. Wang, K. Huang, L. C. Yin, F. Li and H. M. Cheng, Adv. Mater., 2014, 26, 625–631 CrossRef CAS.
- S. H. Chung and A. Manthiram, ChemSusChem, 2014, 7, 1655–1661 CrossRef CAS.
- Z. Li, J. T. Zhang and X. W. (David) Lou, Angew. Chem., Int. Ed., 2015, 54, 12886–12890 CrossRef CAS.
- X. F. Wang, Z. X. Wang and L. Q. Chen, J. Power Sources, 2013, 242, 65–69 CrossRef CAS.
- J. Q. Huang, T. Z. Zhuang, Q. Zhang, H. J. Peng, C. M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS.
- H. D. Yuan, X. L. Chen, G. M. Zhou, W. K. Zhang, J. M. Luo, H. Huang, Y. P. Gan, C. Liang, Y. Xia, J. Zhang, J. G. Wang and X. Y. Tao, ACS Energy Lett., 2017, 2, 1711–1719 CrossRef CAS.
- Z. H. Sun, J. Q. Zhang, L. C. Yin, G. J. Hu, R. P. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef.
- P. J. H. Kim, J. Seo, K. Fu, J. Choi, Z. M. Liu, J. Kwon, L. B. Hu and U. Paik, NPG Asia Mater., 2017, 9, 375 CrossRef.
- Y. M. Shia and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- P. Jiang, Q. Liu, Y. H. Liang, J. Q. Tian, A. M. Asiri and X. P. Sun, Angew. Chem., Int. Ed., 2014, 53, 12855–12859 CrossRef CAS.
- Y. Yan, L. Thia, B. Y. Xia, X. M. Ge, Z. L. Liu, A. Fisher and X. Wang, Adv. Sci., 2015, 2, 1500120 CrossRef.
- Y. G. Zhang, Y. Guo, Y. G. Wang, T. Peng, Y. Lu, R. J. Luo, Y. B. Wang, Y. S. Luo, J. K. Kim and X. M. Liu, Nanoscale Res. Lett., 2018, 13, 389 CrossRef.
- J. W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- X. J. Wang, K. Chen, G. Wang, X. J. Liu and H. Wang, ACS Nano, 2017, 11, 11602–11616 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- J. Jiang, C. D. Wang, J. W. Liang, J. Zuo and Q. Yang, Dalton Trans., 2015, 44, 10297–10303 RSC.
- S. Z. Huang, L. L. Zhang, J. Y. Wang, J. L. Zhu and P. K. Shen, Nano Res., 2018, 11, 1731–1743 CrossRef CAS.
- X. H. Zhu, M. J. Liu, Y. Liu, R. W. Chen, Z. Nie, J. H. Li and S. Z. Yao, J. Mater. Chem. A, 2016, 4, 8974–8977 RSC.
- Z. W. Zhang, Z. Q. Li, F. B. Hao, X. K. Wang, Q. Li, Y. X. Qi, R. H. Fan and L. W. Yin, Adv. Funct. Mater., 2014, 24, 2500–2509 CrossRef CAS.
- Q. R. Yang, W. J. Li, S. L. Chou, J. Z. Wang and H. K. Liu, RSC Adv., 2015, 5, 80536–80541 RSC.
- Q. Wang, B. Y. Wang, Z. Zhang, Y. Zhang, P. Jing, Y. Zhang and H. Wu, Inorg. Chem. Front., 2018, 5, 2605–2614 RSC.
- Y. Guo, Y. G. Zhang, Y. G. Wang, D. Y. Zhang, Y. Lu, R. J. Luo, Y. B. Wang, X. M. Liu, J. K. Kim and Y. S. Luo, Electrochim. Acta, 2019, 296, 989–998 CrossRef CAS.
- G. S. Jiang, F. Xu, S. H. Yang, J. P. Wu, B. Q. Wei and H. Q. Wang, J. Power Sources, 2018, 395, 77–84 CrossRef CAS.
- P. P. Zhu, Z. Zhang, S. J. Hao, B. W. Zhang, P. F. Zhao, J. Yu, J. X. Cai, Y. Z. Huang and Z. Y. Yang, Carbon, 2018, 139, 477–485 CrossRef CAS.
- Y. Yu, Z. Peng, M. Asif, H. T. Wang, W. Wang, Z. X. Wu, Z. Y. Wang, X. Y. Qiu, H. Tan and H. F. Liu, ACS Sustainable Chem. Eng., 2018, 6, 11587–11594 CrossRef CAS.
- G. X. Li, J. H. Sun, W. P. Hou, S. D. Jiang, Y. Huang and J. X. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS.
- W. J. Ren, L. Q. Xu, L. Zhu, X. Y. Wang, X. J. Ma and D. B. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 11642–11651 CrossRef CAS.
- K. Xi, D. Q. He, C. Harris, Y. K. Wang, C. Lai, H. L. Li, P. R. Coxon, S. J. Ding, C. Wang and R. V. Kumar, Adv. Sci., 2019, 1800815 CrossRef PubMed.
- Y. R. Zhong, L. C. Yin, P. He, W. Liu, Z. S. Wu and H. L. Wang, J. Am. Chem. Soc., 2018, 140, 1455–1459 CrossRef CAS.
- J. J. Cai, C. Wu, Y. Zhu, K. L. Zhang and P. K. Shen, J. Power Sources, 2017, 341, 165–174 CrossRef CAS.
- M. A. Baghchesaraa, H. R. Azimib, A. G. Shiravizadehc, M. A. M. Teridid and R. Yousefi, Appl. Surf. Sci., 2019, 466, 401–410 CrossRef.
- L. Yao, X. W. Dong, C. R. Zhang, N. T. Hu and Y. F. Zhang, J. Mater. Chem. A, 2018, 6, 11260–11269 RSC.
- M. X. Wang, L. S. Fan, Y. Qiu, D. D. Chen, X. Wu, C. Y. Zhao, J. H. Cheng, Y. Wang, N. Q. Zhang and K. N. Sun, J. Mater. Chem. A, 2018, 6, 11694–11699 RSC.
- W. L. Cai, G. R. Li, K. L. Zhang, G. N. Xiao, C. Wang, K. F. Ye, Z. W. Chen, Y. C. Zhu and Y. T. Qian, Adv. Funct. Mater., 2018, 28, 1704865 CrossRef.
- W. H. Sun, X. G. Sun, Q. F. Peng, H. Y. Wang, Y. L. Ge, N. Akhtar, Y. Q. Huang and K. Wang, Nanoscale Adv., 2019, 1, 1589–1597 RSC.
- S. S. Yao, J. Cui, J. Q. Huang, Z. H. Lu, Y. Deng, W. G. Chong, J. X. Wu, M. I. U. Haq, F. Ciucci and J. K. Kim, Adv. Energy Mater., 2018, 8, 1800710 CrossRef.
- S. H. Chung and A. Manthiram, Adv. Mater., 2014, 26, 7352–7357 CrossRef CAS PubMed.
- Z. B. Xiao, Z. Yang, L. Wang, H. G. Nie, M. E. Zhong, Q. Q. Lai, X. J. Xu, L. J. Zhang and S. M. Huang, Adv. Mater., 2015, 27, 2891–2898 CrossRef CAS PubMed.
- J. R. He, Y. F. Chen, P. J. Li, F. Fu, Z. G. Wang and W. L. Zhang, J. Mater. Chem. A, 2015, 3, 18605–18610 RSC.
- F. Xu, S. H. Yang, G. S. Jiang, Q. Ye, B. Q. Wei and H. Q. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 37731–37738 CrossRef CAS.
- S. H. Chung, P. Han, R. Singhal, V. Kalra and A. Manthiram, Adv. Energy Mater., 2015, 5, 1614–6840 Search PubMed.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, Adv. Funct. Mater., 2015, 25, 5285–5291 CrossRef CAS.
- Q. H. Yu, Y. Lu, R. J. Luo, X. M. Liu, K. F. Huo, J. K. Kim, J. He and Y. S. Luo, Adv. Funct. Mater., 2018, 28, 1804520 CrossRef.
- H. B. Lin, L. Q. Yang, X. Jiang, G. C. Li, T. R. Zhang, Q. F. Yao, G. Y. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 RSC.
- J. R. He, Y. F. Chen and A. Manthiram, Energy Environ. Sci., 2018, 11, 2560–2568 RSC.
- B. Qi, X. S. Zhao, S. G. Wang, K. Chen, Y. J. Wei, G. Chen, Y. Gao, D. Zhang, Z. H. Sun and F. Li, J. Mater. Chem. A, 2018, 6, 14359–14366 RSC.
- Y. G. Wang, R. J. Luo, Y. G. Zhang, Y. Guo, Y. Lu, X. M. Liu, J. K. Kim and Y. S. Luo, ACS Appl. Energy Mater., 2019, 2, 3314–3322 CrossRef CAS.
- B. Qi, X. S. Zhao, S. G. Wang, K. Chen, Y. J. Wei, G. Chen, Y. Gao, D. Zhang, Z. H. Sun and F. Li, J. Mater. Chem. A, 2018, 6, 14359–14366 RSC.
- Z. A. Ghazi, X. He, A. M. Khattak, N. A. Khan, B. Liang, A. Iqbal, J. X. Wang, H. S. Sin, L. S. Li and Z. Y. Tang, Adv. Mater., 2017, 29, 1606817 CrossRef.
- Q. F. Zhang, Y. P. Wang, Z. W. Seh, Z. H. Fu, R. F. Zhang and Y. Cui, Nano Lett., 2015, 15, 3780–3786 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00532c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |