
Pt nanozyme for O2 self-sufficient, tumor-specific oxidative damage and drug resistance reversal†
Zhaoyu
Ma
a,
Long
Wu
 b,
Kai
Han
*b and
Heyou
Han
*ab
b,
Kai
Han
*b and
Heyou
Han
*ab
aState Key Laboratory of Agricultural Microbiology, College of Life Science and Technology, Huazhong Agricultural University, Wuhan 430070, China. E-mail: hyhan@mail.hzau.edu.cn
bState Key Laboratory of Agricultural Microbiology, College of Science, Huazhong Agricultural University, Wuhan 430070, China. E-mail: hank@mail.hzau.edu.cn
First published on 12th April 2019
Abstract
Herein, an ultrathin Pt nanozyme has been developed. The Pt nanozyme exhibited both oxidase and peroxidase-mimetic properties, leading to relieved hypoxia and specific oxidative damage in tumor cells in a cascade manner. Meanwhile, hydrophilic Pt2+ was released sustainably under tumor acidity. These properties endow the Pt nanozyme with great potential in killing drug resistant tumor cells while not damaging normal cells.
New conceptsDrug resistance always leads to the failure of clinical chemotherapy due to the increased efflux pump effect of hydrophobic drugs and hypoxia in the tumor. Although various nanocarriers have been developed, their efficacy is still unsatisfactory. Herein, an ultrathin platinum (Pt) nanozyme, PtAu@SiO2, was designed for tumor acidity-enhanced oxidative damage and subsequent drug resistance reversal. The ultrathin Pt shell in PtAu@SiO2 could suffer from tumor acidity-dependent etching, releasing cytotoxic Pt ions. Meanwhile, the hydrophilic Pt ions could not be easily pumped-out from the drug resistant tumor cells. Specifically, PtAu@SiO2 exhibited a high catalase-like activity, which could alleviate hypoxia by H2O2-activated oxygen (O2) generation. More interestingly, the O2 self-sufficiency could ensure that PtAu@SiO2 could further exert oxidase-mimetic properties, leading to subsequent reactive oxygen species generation and enhanced oxidative damage to the tumor cells. Different from the previous reactive oxygen species generation systems, neither laser penetration depth nor the O2 content in the tumor needs to be considered in our peroxidase/oxidase mimetic nanozyme system. This nanozyme system should provide a new nanoplatform in drug resistant tumor therapy. |
1. Introduction
Although considerable progress has been achieved in tumor therapy, drug resistance in the tumor after repeated dosage remains one of the greatest challenges at present.1–3 One of the most recognized mechanisms of drug resistance is that the overexpressed ABC transporters in the cell membrane can specifically and actively pump hydrophobic drugs outside the tumor cells, decreasing the drug concentration below the effective therapeutic window.4,5 To address this hurdle, various nanoparticles have been developed to elevate the hydrophobic drug concentration inside the tumor cells through enhanced drug internalization and controlled release.6–8 However, once these hydrophobic drugs are liberated inside the cells to exert toxicity, they will suffer from free diffusion to the cell membrane, be recognized by ABC transporters and subsequently be pumped outside the tumor cells, which compromises the efficacy dramatically. On the other hand, tumor microenvironments, especially hypoxia, not only promote tumor angiogenesis and metastasis, but also are responsible for drug resistance and failure of treatment.9–11 Hypoxia can alter the cellular metabolism to decrease drug cytotoxicity and enhance genetic instability, leading to more rapid development of therapeutic resistance in the tumor.Since tumor tissue usually contains increased levels of H2O2, ranging from 100 μM to 1 mM,12in situ generation of oxygen in the tumor tissue with catalase through the decomposition of H2O2 to O2 has obtained increasing attention.13,14 However, natural enzymes usually have poor cell internalization and unsatisfactory stability and can be inactivated. As an alternative, the Pt nanozyme has exhibited multiple enzyme mimetic activities similar to peroxidase, oxidase and superoxide dismutase (SOD), endowing the Pt nanozyme with great potential to manipulate the tumor hypoxic microenvironment.15,16 However, the application of the Pt nanozyme in tumor therapy is still scarce, partly because of the good stability and non-toxicity of Pt nanoparticles under physiological environments. Very recently, some pioneering works have demonstrated that when the particle size of the Pt nanoparticles is small enough (≪3 nm), most of the Pt atoms will be on the surface of the crystal. As a result, the increased O2 adsorption and water oxidation in the presence of acidity induce surface corrosion, facilitating the release of cytotoxic Pt ions.17,18 Encouraged by these findings, we speculated that when Pt nanoparticles with appropriate size are integrated with the tumor microenvironment including acidity and excess H2O2, tumor-specific, sustained release of hydrophilic Pt ions (avoiding the active pump-up by ABC transporters), as well as hypoxia reversal in the tumor, can be realized simultaneously to overcome drug resistance.
As a proof of concept, we reported a simple and rational PtAu@SiO2 nanozyme for tumor-specific reversal of drug resistance and selective therapy. As shown in Scheme 1, the Pt shell was coated in Au islands. The isolated Au islands could hinder the aggregation of the neighboring Pt nanoparticles. SiO2 nanoparticles were used herein as carriers to avoid the rapid clearance of PtAu due to the small size.19 Tumor mild acidity, but not the physiological environment, would accelerate the corrosion of the Pt shell, making PtAu@SiO2 a continuous drug depot with tumor-triggered Pt ion release and growth inhibition. During the exploration of the enzyme-mimetic activity of PtAu@SiO2, we were surprised to find that PtAu@SiO2 could act as an oxidase to even catalyze O2 to form ROS. And tumor mild acidity could further improve the oxidase activity. Besides, PtAu@SiO2 could also act as a peroxidase to decompose H2O2 to O2 in the tumor, which relieved the tumor hypoxia and inhibited drug resistance. Importantly, this self-supplied O2 further allowed the Pt nanozyme to exert oxidase-like activity, realizing enhanced tumor oxidative damage and reversal of drug resistance. This mild acidity activated Pt ions release, hypoxia alleviation and ROS generation of PtAu@SiO2, which resulted in efficient drug resistance reversal in the tumor cells, while not damaging the normal cells. Different from the previous reactive oxygen species generation systems,20,21 neither the laser penetration depth nor the O2 content in the tumor needs to be considered in our peroxidase/oxidase mimetic nanozyme system.
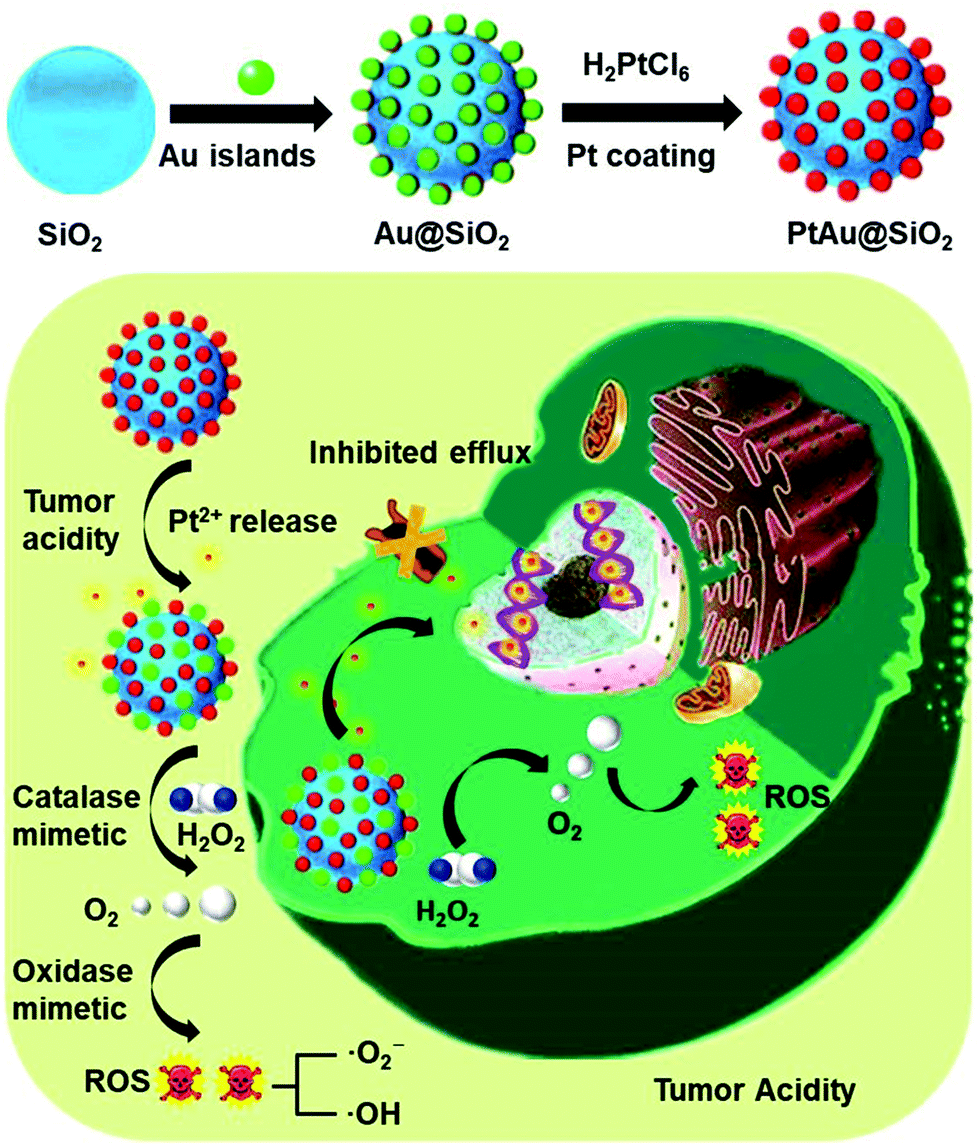 | ||
| Scheme 1 Schematic of the preparation process of PtAu@SiO2 and the mechanism of PtAu@SiO2 in overcoming drug resistance. | ||
2. Results and discussion
Transmission electron microscopy (TEM, Fig. 1a) showed that the Au nanoparticles were homogeneously distributed on the surface of the SiO2 nanospheres and the obtained Au@SiO2 showed an average size of ∼90 nm. In order to form the platinum shell in situ, H2PtCl6 was added to the Au@SiO2 for 20 minutes. Then, most of the platinum nanoparticles would be reduced on the surface of the Au nanoparticles with NaBH4. Fig. 1b demonstrated that the formation of the Pt shell on the Au islands had no effect on the morphology. The dynamic light scattering (DLS) results revealed that the PtAu@SiO2 exhibited good stability in 10% FBS (Fig. 1c). To substantially confirm the successful growth of the Pt shell on the Au islands, energy-dispersive X-ray (EDX) mapping was conducted. As shown in Fig. 1d, a layer of Au and Pt hybrid nanoparticles (∼10 nm) was uniformly attached to the surface of the silicon sphere. Furthermore, the EDX line-scan was performed along a central axis direction of a single sphere, indicating that the Au and Pt hybrid nanoparticles belong to a core–shell structure (Pt: shell, Au: core), and the platinum shell size is very small (Fig. 1e). The amplified EDX surface scan profile of the small Au/Pt hybrid nanoparticles was also performed in Fig. 1f. Meanwhile, the X-ray photoelectron spectroscopy (XPS) of the PtAu@SiO2 was also shown in Fig. 1g, where characteristic peaks of both Pt and Au appeared. The high-resolution spectra of the Pt 4f peaks (Fig. 1h) revealed the existence of two chemical states for the Pt species, where the Pt0 was dominant, and some bivalent Pt also existed.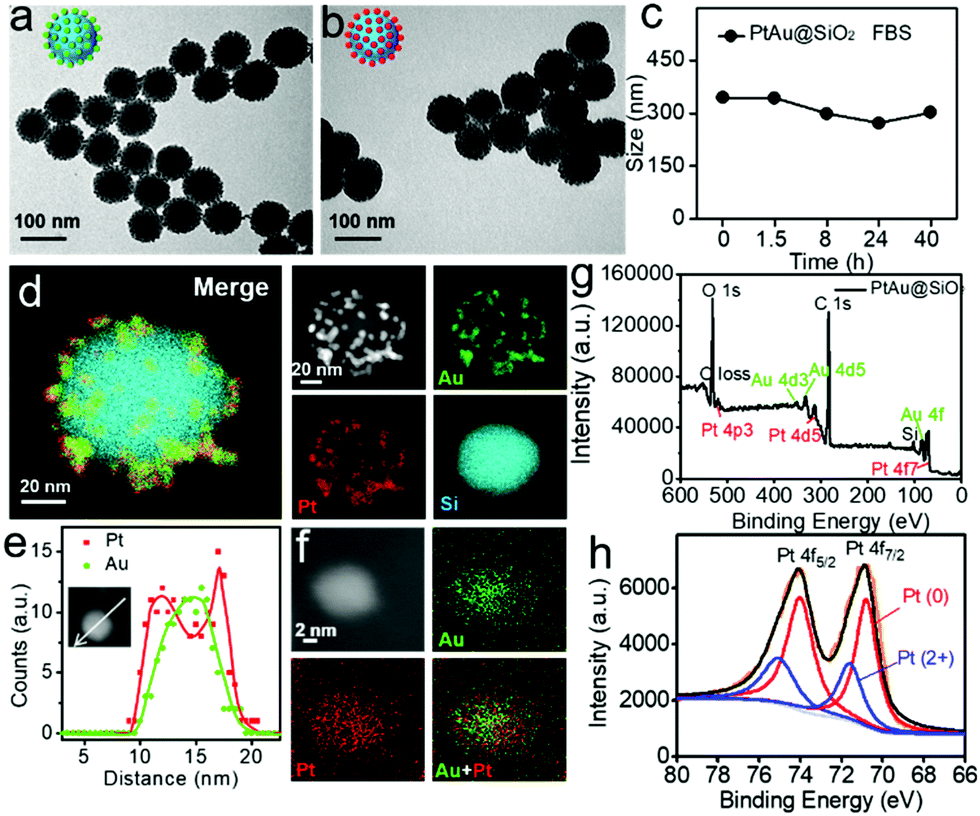 | ||
| Fig. 1 TEM images of (a) Au@SiO2 and (b) PtAu@SiO2. (c) The hydrodynamic sizes of PtAu@SiO2 in 10% FBS with increasing time. (d) The EDX mapping image of an individual PtAu@SiO2 (green: Au, red: Pt, blue: Si). (e) Line-scan EDX spectra of the elemental Au and Pt that were recorded from an individual particle (inset) along a direction as indicated by the white arrow. (f) EDX mapping image of an individual Au/Pt hybrid nanoparticle. (g) XPS spectra of PtAu@SiO2. (h) High-resolution XPS spectra of the Pt 4f region shown in (g). | ||
In order to accurately calculate the thickness of the platinum shell, inductively coupled plasma-mass spectrometry (ICP-MS) analysis was conducted. The mass ratio of Au to Pt was measured to be 0.5326![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Given the size of the Au spherical seeds, and the densities of Au and Pt, the thickness of the Pt shell was quantified to be 1.486 nm (see Fig. S1, ESI† for details), suggesting that the platinum shell is very thin. Since the Pt shell is very thin, lots of Pt atoms will be located on the surface of the crystal, resulting in increased O2 adsorption and water oxidation for surface corrosion, thus facilitating Pt ion release. As shown in Fig. 2a, more Pt ions were released at pH 6.5 than at pH 7.4. This pH-sensitive leaching of Pt ions should endow PtAu@SiO2 with tumor selectivity.
:1. Given the size of the Au spherical seeds, and the densities of Au and Pt, the thickness of the Pt shell was quantified to be 1.486 nm (see Fig. S1, ESI† for details), suggesting that the platinum shell is very thin. Since the Pt shell is very thin, lots of Pt atoms will be located on the surface of the crystal, resulting in increased O2 adsorption and water oxidation for surface corrosion, thus facilitating Pt ion release. As shown in Fig. 2a, more Pt ions were released at pH 6.5 than at pH 7.4. This pH-sensitive leaching of Pt ions should endow PtAu@SiO2 with tumor selectivity.
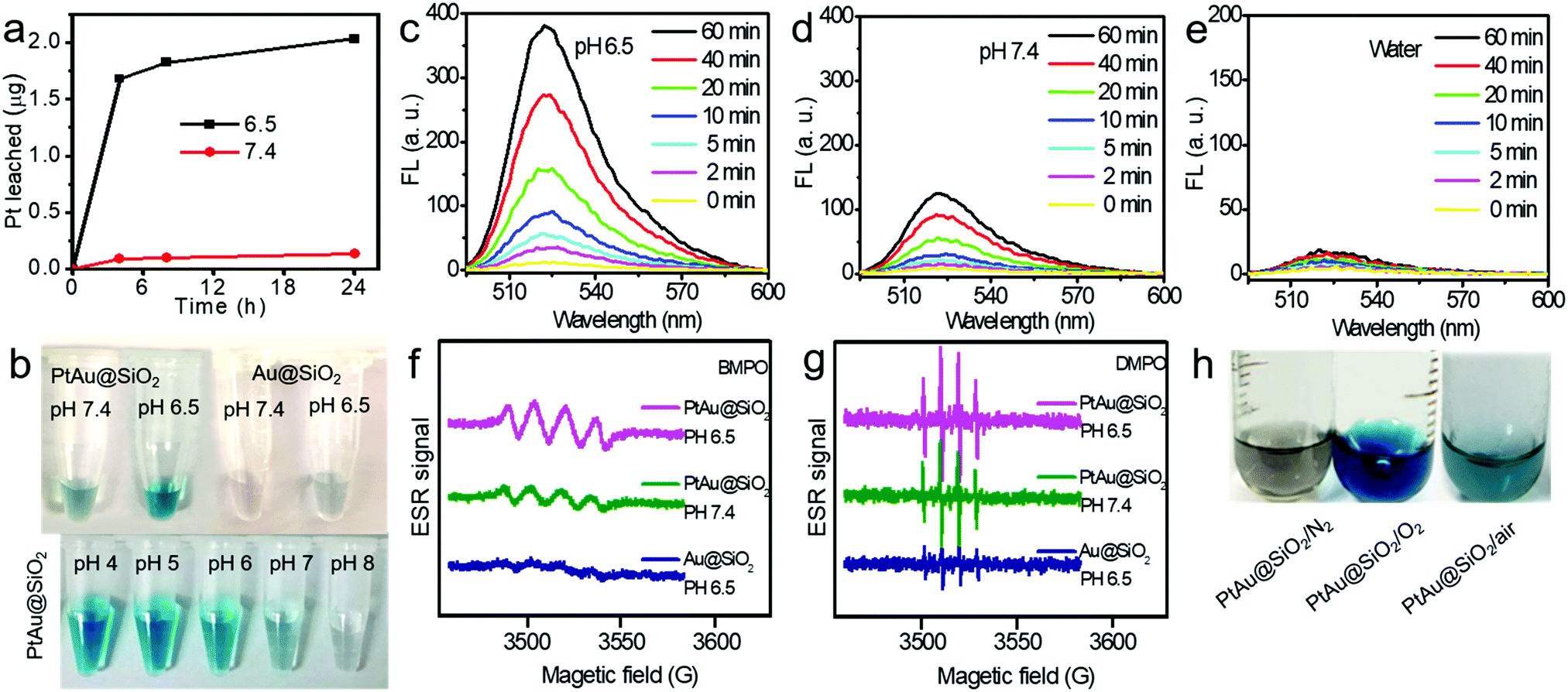 | ||
| Fig. 2 (a) The in vitro cumulative Pt ion release from PtAu@SiO2 at different pHs (pH 7.4 and 6.5). (b) Color evolution of TMB oxidation catalyzed by PtAu@SiO2 and Au@SiO2 at different pHs (pH 7.4 and 6.5) (top); color evolution of TMB oxidation catalyzed by PtAu@SiO2 at different pHs (pH 4, 5, 6, 7 and 8, bottom). The ROS generation of the PtAu@SiO2 over time at (c) pH 6.5 and (d) pH 7.4 using DCFH-DA as the sensor. (e) Fluorescence spectra of DCF over time at pH 6.5 in the absence of PtAu@SiO2. ESR spectra of the PtAu@SiO2 and Au@SiO2 at pH 7.4 and 6.5 using (f) BMPO as spin-trapping agents for detecting ˙O2− and (g) DMPO for detecting ˙OH. (h) The color change of TMB oxidation catalyzed by PtAu@SiO2 at pH 6.5 in different environments (nitrogen-filled, oxygen-rich and air). | ||
Pt-based nanoparticles have multiple enzyme mimetic activities. Herein, the oxidase-like activity of PtAu@SiO2 was tested using tetramethylbenzidine (TMB) oxidation as a chromophoric substrate for the oxidase mimetic studies.22 As shown in Fig. 2b (top panel), PtAu@SiO2 could effectively catalyze TMB oxidation by dissolved O2 in water at pH 6.5, generating blue color for TMB within 5 min in the absence of H2O2. However, this phenomenon had not been observed in a neutral environment (pH 7.4), suggesting that the oxidase-like activity of PtAu@SiO2 is pH dependent. Interestingly, lower acidity would accelerate the color change (Fig. 2b, bottom panel). For comparison, there was no significant color change for Au@SiO2 regardless of the pH change, indicating that the Pt shell played a crucial role in the oxidase-mimetic activity. Similar results were also reported in previous literature.23 However, few rational explanations were provided for the pH dependent oxidase-mimetic activity. Considering the XPS results (Fig. 1h), there should be some PtO coating on the Pt shell, which would significantly retard the oxidation of TMB. And an acidic environment can destroy the surface PtO in the Pt shell and expose the shielded Pt, improving the oxidase activity of the PtAu@SiO2.
Furthermore, although the oxidase-mimetic activity of PtAu@SiO2 was widely reported, the mechanism was unclear to date. Some pioneering works have suggested that Pt nanoparticles could trigger the ROS generation.24,25 It was reasonable to speculate that ROS oxidize the TMB. To confirm this, 2′,7′-dichlorofluorescein (DCFH) was used to assess the generation of ROS.26 PtAu@SiO2 at pH 6.5 (Fig. 2c) induced more ROS generation than that at pH 7.4 (Fig. 2d). While there was low ROS production for the water blank group (Fig. 2e), the Au@SiO2 group at pH 6.5 and PtAu@SiO2 at pH 6.5 in the presence of Vc (Fig. S2, ESI†). As expected, the platinum nanozyme had high pH selectivity for the production of ROS. This pH-dependent ROS generation could also be attributed to the existence of PtO. To identify which kind of ROS played a critical role in the oxidase-mimetic activity, the electron spin resonance (ESR) technique was used to monitor the ROS signals from PtAu@SiO2. 5-tert-Butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO) and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) (spin-trapping agents of superoxide radicals (˙O2−), and hydroxyl radicals (˙OH), respectively), were employed to distinguish the type of ROS.27,28 The ESR spectra of PtAu@SiO2 at pH 6.5 showed a more strong spin trapped signal of ˙O2− and ˙OH than at pH 7.4, while the ESR spectra of Au@SiO2 at pH 6.5 showed weak signal (Fig. 2f and g). These results substantially demonstrated the pH-dependent ROS generation of PtAu@SiO2. Besides, the oxidase-mimetic activity of PtAu@SiO2 is also O2 dependent. As shown in Fig. 2h, in the TMB assay, blue color was not observed in the hypoxia solution (via purging with N2) for PtAu@SiO2. While in a normal solution, PtAu@SiO2 could catalyze TMB to show blue color. And purging with O2 could further accelerate this color change process. The potential generation process of ˙O2− and ˙OH catalyzed by PtAu@SiO2 was performed (Fig. S3, ESI†). Furthermore, the intracellular ROS generation was also detected by a confocal laser scanning microscope using DCFH as a ROS sensor (Fig. 3).
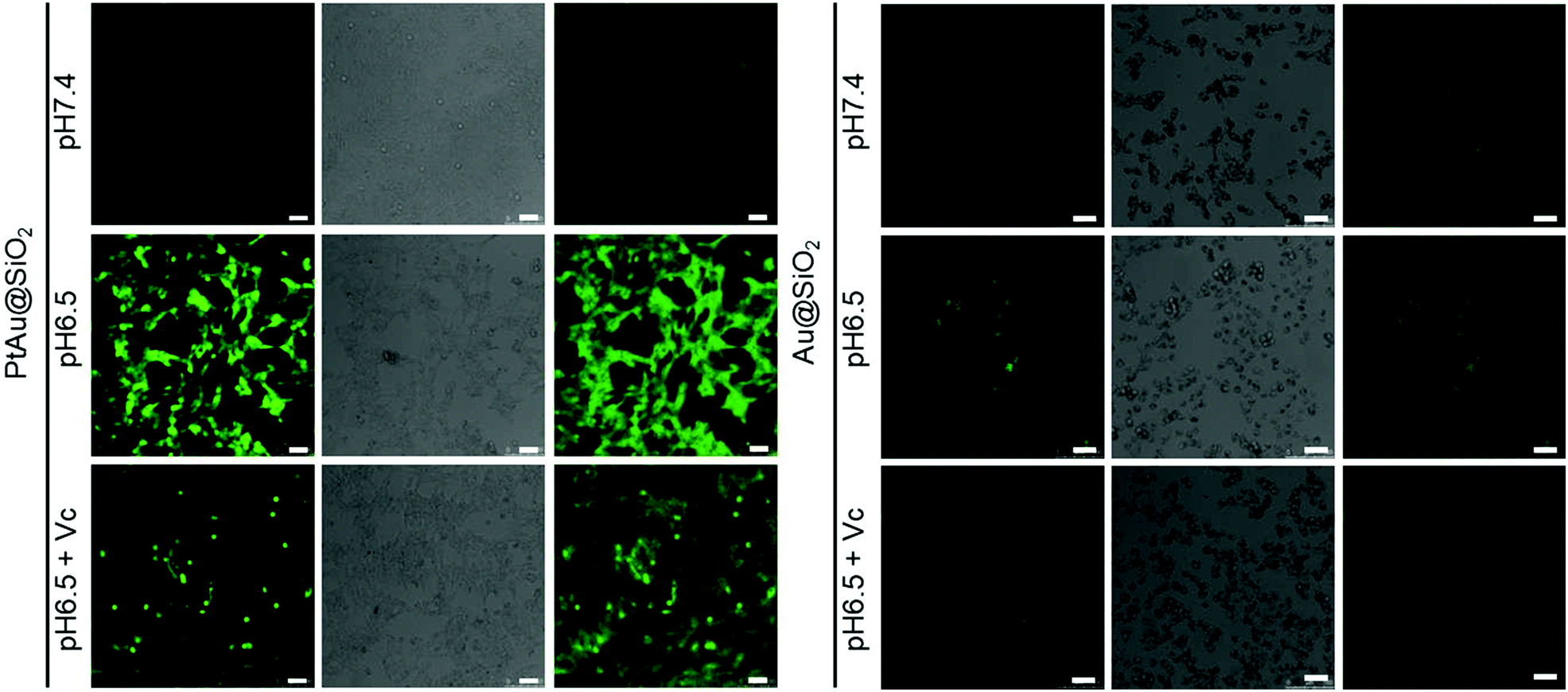 | ||
| Fig. 3 The production of ROS in HeLa cells treated with PtAu@SiO2 and Au@SiO2 under different environments (pH 7.4, pH 6.5 and pH 6.5 + Vc). DCF fluorescence (green) depicted intracellular ROS. The scale bar is 50 μm. | ||
Given the mild acidity triggered Pt ion release as well as ROS generation, PtAu@SiO2 was expected to possess tumor selective damage due to the fact that solid tumors are always slightly acidic. To confirm this, the cell viability of HeLa cells was examined by calcein-AM and propidium iodide (PI) staining.29 As shown in Fig. 4a, when PtAu@SiO2 was incubated in HeLa cells at pH 7.4, only green fluorescence could be observed, suggesting the good biocompatibility of PtAu@SiO2 under normal physiological pH environments. When the cell experiments were performed at pH 6.5, most cells exhibited red fluorescence, suggesting that the PtAu@SiO2 could induce tumor cell death efficiently. Subsequently, Vc (antioxidant) was added in HeLa cells at pH 6.5. Interestingly, the number of HeLa cells with green fluorescence increased but a small amount of cells with red fluorescence still existed. These results demonstrated that both the Pt ions and the ROS triggered the death of HeLa cells. The cytotoxic Pt ions inside the cancer cells can cause DNA damage by binding to DNA, leading to cell death.30 In sharp contrast, all HeLa cells treated with Au@SiO2 in the same environment were alive. Quantitative statistics of the cell life and death were also performed in Fig. 4b. The methylthiazolyl tetrazolium (MTT) assay in Fig. 4c further showed that PtAu@SiO2 possessed greater toxicity at pH 6.5 than at pH 7.4. Once Vc was added, the cytotoxicity of PtAu@SiO2 was significantly reduced. The control group of Au@SiO2 was essentially non-toxic under the same conditions (Fig. 4d). In order to prove that PtAu@SiO2 has good selectivity, the toxicity of PtAu@SiO2-treated normal cells (COS7 cells) in the physiological environment (pH 7.4) for 40 h was provided in Fig. S4, ESI.† There was no toxicity after PtAu@SiO2 treatment, even at high concentrations (>10 mg L−1). All these results substantially indicated the tumor-selective damage of PtAu@SiO2.
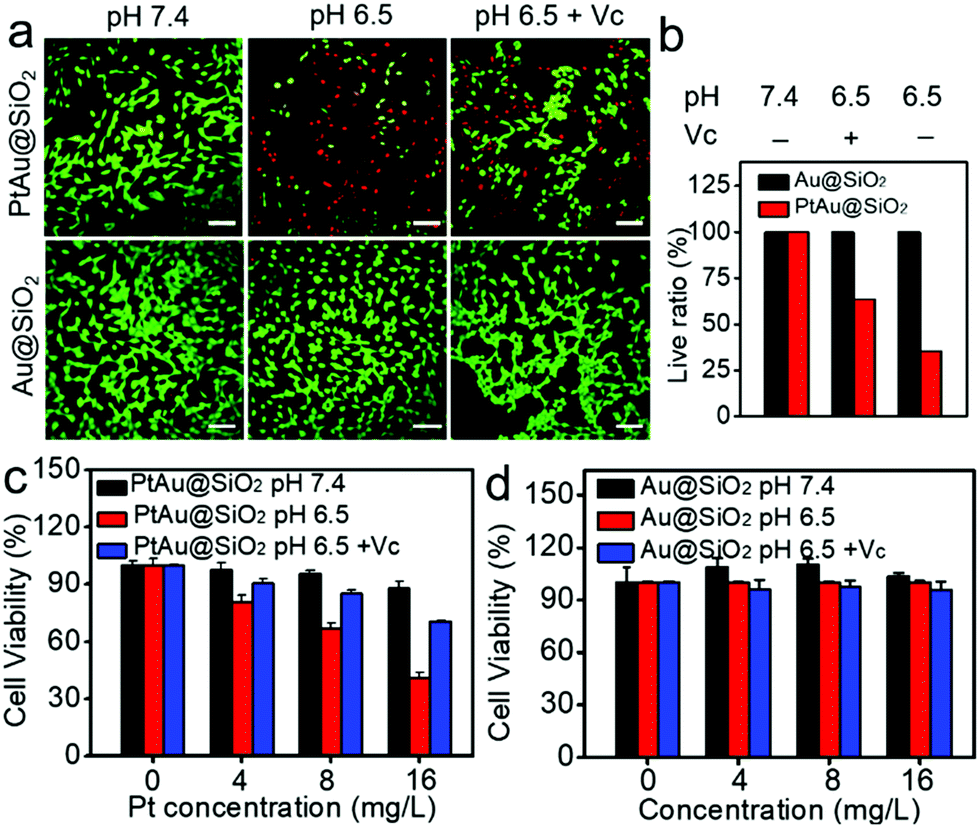 | ||
| Fig. 4 (a) Calcein AM/PI staining of HeLa cells treated with PtAu@SiO2 or Au@SiO2 under different conditions (pH 7.4, 6.5 and 6.5 + Vc), calcein AM: green signal, PI: red signal. The scale bar is 80 μm. (b) Live cell ratio (live ratio = number of stained green cells/number of total cells). (c) The cell viability of PtAu@SiO2 (d) and Au@SiO2 against HeLa cells at different conditions (pH 7.4, 6.5 and 6.5 + Vc). | ||
It was recognized that repeated administration of hydrophobic drugs would evoke drug resistance in the tumor cells and finally cause therapeutic failure. Promoted by efficient inhibition of HeLa cells by PtAu@SiO2, the potential ability of PtAu@SiO2 in overcoming drug resistance was further investigated. A549/DDP cells (cis-Pt resistant lung cancer A549) were chosen as a cell model. The MTT assay revealed that PtAu@SiO2 had the highest toxicity at pH 6.5 and the addition of Vc at pH 6.5 would decrease the toxicity (Fig. 5a). These results were similar to that in HeLa cells, suggesting the universality of PtAu@SiO2 in killing tumor cells at mild acidity. CLSM imaging (Fig. S5, ESI†) demonstrated that PtAu@SiO2 could also elevate the ROS content in A549/DDP cells. Besides, cisplatin could not kill A549/DDP cells at the low concentration gradient. In contrast, PtAu@SiO2 exhibited concentration-dependent cell toxicity (Fig. 5b). Clearly, A549/DDP cells could transport intercellular hydrophobic cisplatin outside the cells, elevating the cell viability dramatically. However, for PtAu@SiO2, it underwent different cell inhibition mechanisms. PtAu@SiO2 mediated ROS could kill the drug resistant tumor cells. More importantly, the substrate of ABC transporters in drug resistant cells should be hydrophobic molecules, and the sustained released hydrophilic Pt ions would not be pumped out, ensuring the high content of therapeutic dosage.
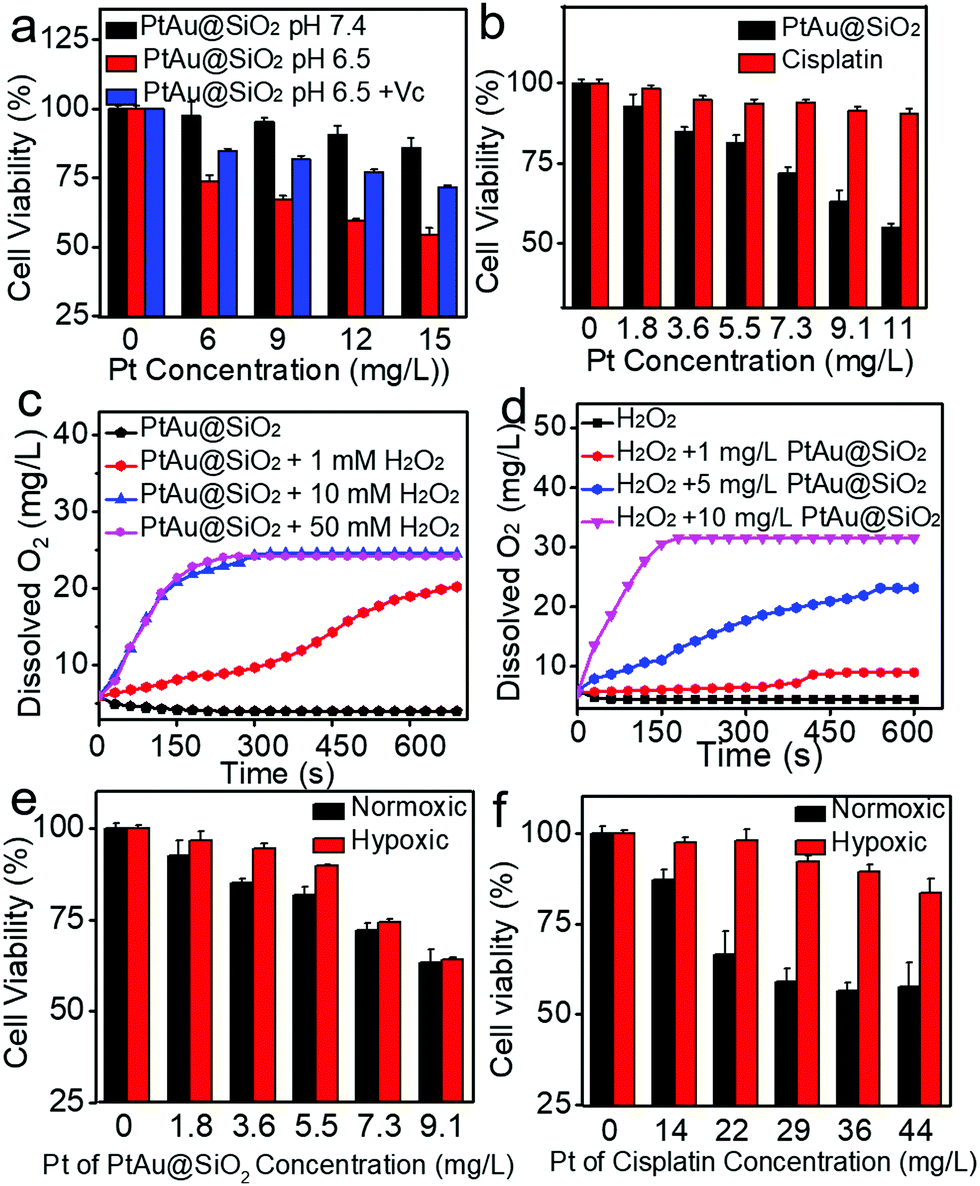 | ||
| Fig. 5 (a) The cell viability of PtAu@SiO2 against A549/DDP cells under different conditions (pH 7.4, 6.5 and 6.5 + Vc) under normoxic conditions. (b) The cell viability of PtAu@SiO2 and cisplatin against A549/DDP cells at pH 6.5 under hypoxic conditions. (c) Quantitative oxygen generation of PtAu@SiO2 at pH 6.5 after reaction with increasing concentrations of H2O2: 0, 1 mM, 10 mM, 50 mM; PtAu@SiO2: 10 mg L−1. (d) Quantitative oxygen generation of PtAu@SiO2 at pH 6.5 after reaction with increasing concentrations of PtAu@SiO2: 0, 1 mg L−1, 5 mg L−1, 10 mg L−1; H2O2: 10 mM. The cell viability of PtAu@SiO2 (e) or cisplatin (f) against A549/DDP cells at pH 6.5 under normoxic and hypoxic conditions. | ||
Except for the transporter-mediated drug efflux, tumor hypoxia has also been considered to render the solid tumors with drug resistance.31,32 Relieving the hypoxia is believed to reverse the drug resistance. PtAu@SiO2 possesses peroxidase mimetic activity, and could catalyze the abundant H2O2 to generate O2. To simulate the decomposition of H2O2 catalyzed by PtAu@SiO2, PtAu@SiO2 was incubated with H2O2 and the O2 production increases, when either the concentration of PtAu@SiO2 or the concentration of H2O2 increases (Fig. 5c and d). Meanwhile, gas bubbles formed in the tubes, suggesting the generation of O2 (Fig. S6a and b, ESI†). Considering excessive H2O2 is one of the basic characteristics of the tumor microenvironment,33–35 we anticipated that PtAu@SiO2 could alleviate hypoxia at the tumor site and finally reverse the drug resistance. To confirm this, A549/DDP cells were treated with PtAu@SiO2 or cisplatin under normoxia (21% O2) and hypoxia (5% O2) to detect the cell viability. The cell viability of the A549/DDP cells gradually decreased with increased concentration of PtAu@SiO2 in both normoxic and hypoxic environments (Fig. 5e), demonstrating that PtAu@SiO2 effectively alleviated the hypoxic environment and the toxicity was unaffected by hypoxia. In contrast, although cisplatin exhibited great toxicity against A549/DDP cells at a high concentration under normoxic conditions, the toxicity was greatly inhibited in a hypoxic environment (Fig. 5f). Clearly, the peroxidase mimetic activity of PtAu@SiO2 endowed it with better tolerance in hypoxic conditions, when compared with commercial cisplatin.
To intuitively observe the influence of various samples on A549/DDP cells, calcein AM/PI (live/dead staining) was designed. In normoxic conditions, Fig. 6a revealed that the PtAu@SiO2 at pH 6.5 caused the highest death, when compared with PtAu@SiO2 at pH 6.5 with Vc and PtAu@SiO2 at pH 7.4. This result was consistent with the MTT results. Besides, all the A549/DDP cells treated with a high dosage of cisplatin emitted red fluorescence under normoxia, while in the hypoxic condition, most of the A549/DDP cells were stained with calcein AM (Fig. 6b). This dramatic difference convincingly revealed that hypoxia would benefit the drug resistance in the tumor cells. However, the A549/DDP cells treated with PtAu@SiO2 were almost all red in both normoxic and hypoxic conditions. This result was due to the fact that PtAu@SiO2 could catalyze the H2O2 to O2. The relieved hypoxic environment, combined with the subsequent generation of ROS as well as Pt ion release, effectively kills the drug resistant tumor cells.
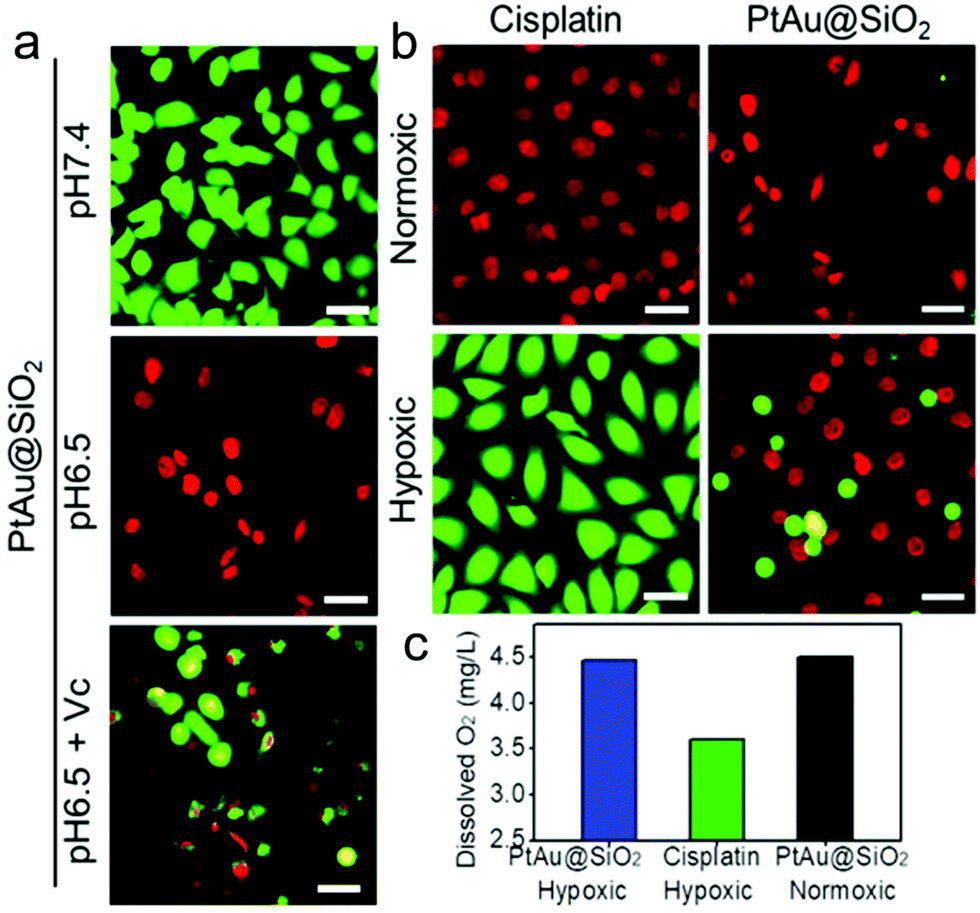 | ||
| Fig. 6 (a) Calcein AM/PI staining of A549/DDP cells treated with PtAu@SiO2 under different conditions (pH 7.4, 6.5 and 6.5 + Vc) under normoxia, AM: green signal, PI: red signal. (b) AM/PI staining of A549/DDP cells treated with PtAu@SiO2 or cisplatin at pH 6.5 under normoxic or hypoxic condition. The scale bar is 50 μm. (c) Quantification of the dissolved oxygen concentration of the culture solution under different conditions (PtAu@SiO2 under hypoxia and normoxia, cisplatin under hypoxia). | ||
Next, a systematic investigation of the cellular internalization mechanism and the pathway for PtAu@SiO2 was carried out. It has been reported that cells can take up nanoparticles by several endocytic processes such as clathrin-mediated endocytosis, caveolae-mediated endocytosis, macropinocytosis, and so on.36,37 The cancer cells were pre-treated with a variety of endocytosis inhibitors such as chlorpromazine, genistein, and amiloride. As shown in Fig. 7a, PtAu@SiO2-FAM was well colocalized with the cancer cells, and mostly in the cytoplasm without any treatment. A significantly decreased fluorescence was observed when the incubation temperature decreases from room temperature to 4 °C, indicating that the intracellular internalization is an energy-consuming process. The cells treated with chlorpromazine showed a significantly decreased fluorescence; this suggested that the internalization of PtAu@SiO2-FAM in cancer cells was clathrin-mediated. However, the genistein and amiloride-treated cells showed fluorescence similar to that of the control; this indicated that the endocytosis pathway of the PtAu@SiO2 was not mediated by caveolae or micropinocytosis. The quantitative results from flow cytometry are shown in Fig. 7b. It can, therefore, be concluded that the intracellular internalization of PtAu@SiO2-FAM is mainly through a clathrin-mediated pathway, whereas the caveolae or micropinocytosis pathways play a less important role. In addition, the intracellular platinum content after treatment with different inhibitors was studied by ICP-MS. In Fig. 7c, the cell internalization percentages decrease to 43% when the incubation temperature decreases from room temperature to 4 °C, as less energy is produced at low temperature due to the decreased enzyme activity. The internalization percentage of PtAu@SiO2 decreases to 78.2%, 96.1% and 96.7% when co-incubated with chlorpromazine, amiloride and genistein, respectively. These results are also consistent with CLSM.
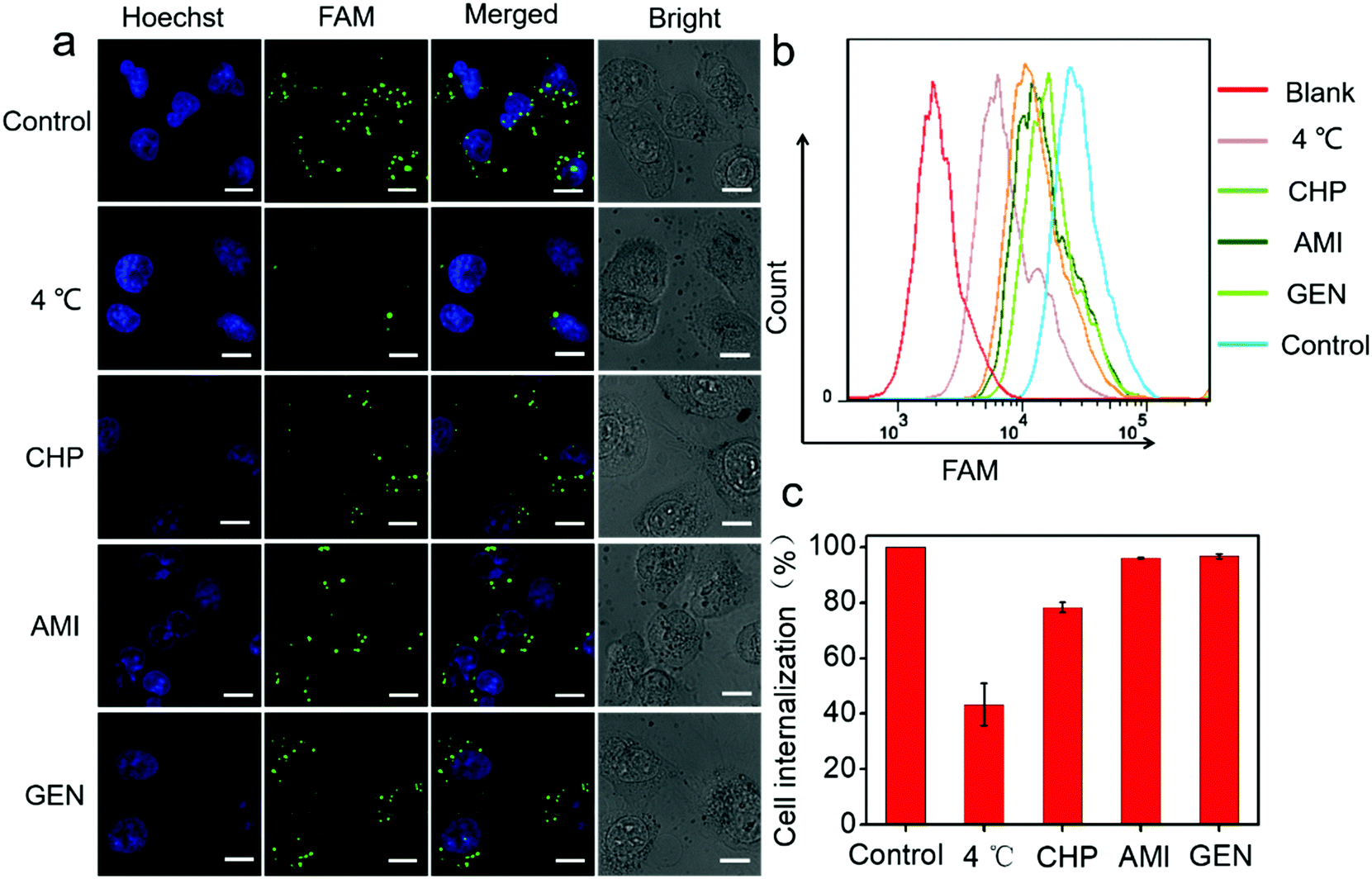 | ||
| Fig. 7 The cell uptake and intracellular distribution of PtAu@SiO2-FAM in the presence of different endocytosis inhibitors. (a) The cellular internalization of FAM-labeled PtAu@SiO2 formulations acquired by CLSM; the nuclei were stained with H33342 (blue), and FAM-labeled PtAu@SiO2 (green). The scale bar was 10 μm. (b) The uptake of FAM-labeled PtAu@SiO2 in the cells was evaluated by flow cytometry. (c) Statistical analysis of the relative uptake of PtAu@SiO2 by ICP-MS. | ||
To substantially verify that PtAu@SiO2 could manipulate the hypoxic environment, the amount of dissolved O2 in the A549/DDP cells was tested. As shown in Fig. 6c, when the cells were treated with PtAu@SiO2, the O2 in the culture medium was rich. The dissolved O2 was 4.47 mg L−1 and 4.5 mg L−1 in hypoxic and normoxic environments, respectively. The slightly decreased O2 in a hypoxic environment should be utilized by the PtAu@SiO2 to exert the oxidase-like activity. As a control, the dissolved O2 decreased to 3.6 mg L−1 when the cells were incubated with cisplatin in a hypoxic environment. Clearly, the PtAu@SiO2 could relieve the hypoxic environment.
The cell cycle of the A549/DDP cells after 24 h of different treatments was recorded by a flow cytometer in Fig. 8a. Compared with the control group, the cells treated with PtAu@SiO2 had a significant reduction in the percentage of S phase, and most cells were arrested in the G1 phase in both normoxic and hypoxic conditions. These findings suggested that PtAu@SiO2 inhibits cell proliferation by inducing cell cycle arrest at the G1 phase. The western blotting analysis in Fig. 8b further revealed that the apoptosis-mediated p21 and p53 proteins were greatly upregulated when the A549/DDP cells were treated with PtAu@SiO2 both in normoxia and hypoxia. While the p21 and p53 expressions in the control groups were weak, suggesting that PtAu@SiO2 mediated significant apoptosis of cancer cells. The quantitative analysis of the western blotting is provided in Fig. S7, ESI.†
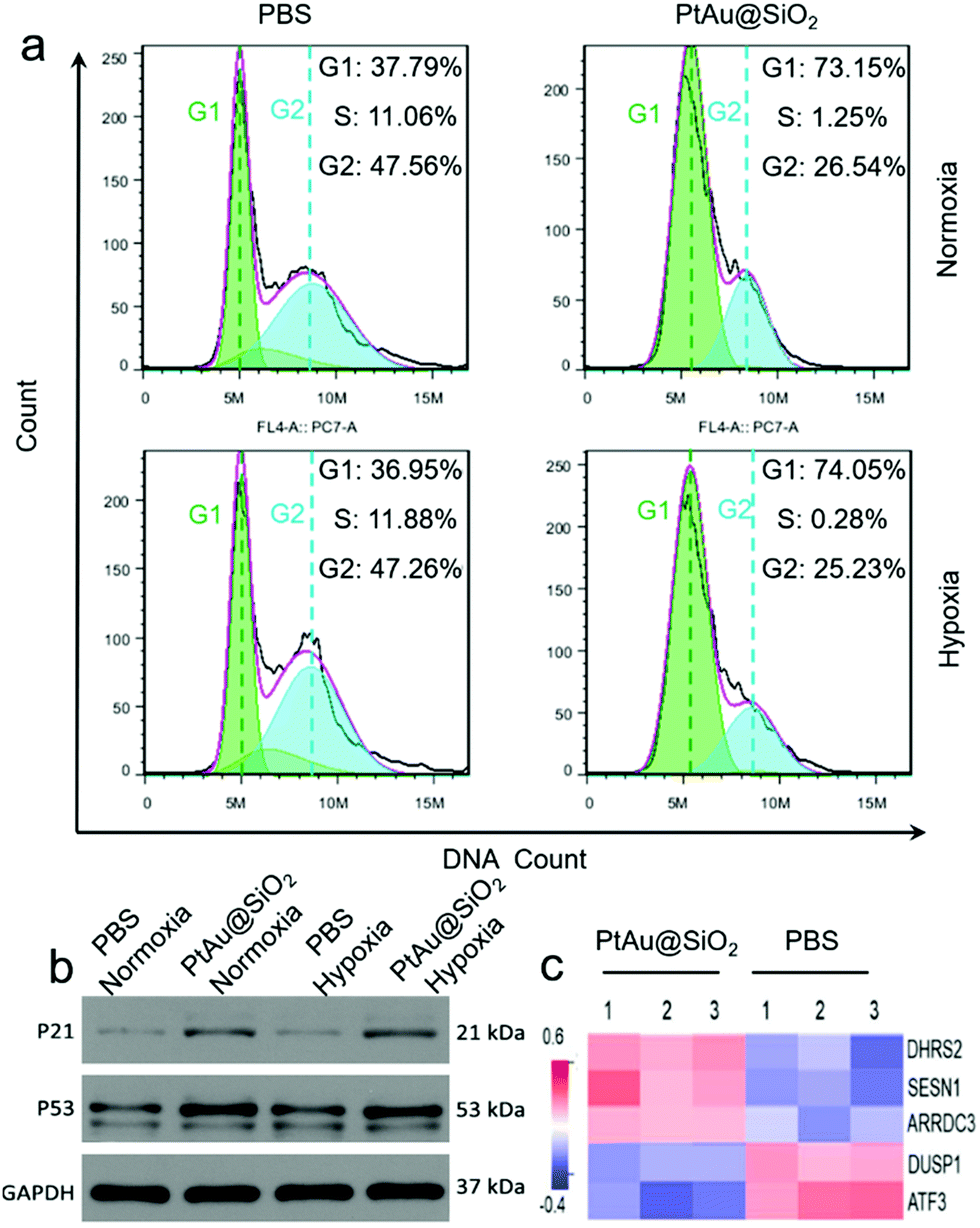 | ||
| Fig. 8 The effect of PtAu@SiO2 on the cell cycle progression of A549/DDP cells. The A549/DDP cells were pretreated with PBS or PtAu@SiO2 under normoxic and hypoxic conditions respectively for 24 hours. (a) The experimental groups were stained with PI followed by flow cytometry analysis. (b) The cellular levels of p53 and p21 were compared by western blotting with GAPDH as a loading control. (c) The heat map analysis of the genes involved in the regulation of apoptosis in A549/DDP cells after PtAu@SiO2 treatment in hypoxia. A red-blue color scale depicts normalized miRNA expression levels in Ct values (red: high, blue: low). | ||
In order to investigate the regulation of A549/DDP cell genes by PtAu@SiO2 treatment, eukaryotic transcriptome sequencing was performed. The data were analyzed on the free online Majorbio I-Sanger Cloud Platform (http://www.i-sanger.com). As shown in Fig. 8c, three genes, i.e., DHRS2, SESN1 and ARRDC3 were significantly upregulated after PtAu@SiO2 treatment, and both the DHRS2 and ARRDC3 genes are associated with the inhibition of tumor progression.38,39 The SESN1 gene is related to the sestrin family, which plays a role in the cellular response to DNA damage and oxidative stress.40 In contrast, DUSP1 and ATF3 were downregulated genes. It is known that DUSP1 is closely associated with tumor resistance,41 while ATF3 downregulated p53 expression and p53 is an important tumor suppressor molecule.42 Clearly, these changes in gene expression further demonstrated that PtAu@SiO2 enhanced the oxidative stress in drug resistant cells, which activated p53 protein, induced DNA damage and finally reversed drug resistance.
3. Conclusions
In summary, we designed a pH-sensitive ultrathin Pt nanozyme for tumor resistance reversal and selective therapy. This Pt nanozyme as a nanodepot showed tumor mild acid responsive Pt ion release and DNA damage due to the ultrathin platinum shell. Specifically, this platinum nanozyme exhibited both catalase and oxidase-like activity. The catalase-like activity could catalyze the decomposition of H2O2 to produce O2 to alleviate drug resistance. The sufficient oxygen environment ensured that the platinum nanozyme with oxidase-like activity could produce ROS, which specifically enhanced the oxidative damage of the tumor cells. Different from photodynamic systems, neither laser penetration depth nor O2 content in the tumor needs to be considered in our nanozyme system. This nanozyme system should open a window in drug resistant tumor therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21778020 and 51603080), the Sci-tech Innovation Foundation of Huazhong Agricultural University (2662017PY042 and 2662018PY024), the Fundamental Research Funds for the Central Universities (2662015QD026), the National Key R&D Program of China (2016YFD0500706) and the Science and Technology Major Project of Guangxi (Gui Ke AA18118046).References
- Q. Q. Ni, F. W. Zhang, Y. L. Zhang, G. Z. Zhu, Z. Wang, Z. G. Teng, C. Y. Wang, B. C. Yung, G. Niu, G. M. Lu, L. J. Zhang and X. Y. Chen, Adv. Mater., 2018, 30, 1705737 CrossRef PubMed.
- M. Chen, X. L. Liang, C. Gao, R. R. Zhao, N. S. Zhang, S. B. Wang, W. Chen, B. Zhao, J. R. Wang and Z. F. Dai, ACS Nano, 2018, 12, 7312 CrossRef CAS PubMed.
- H. Wang, Z. Gao, X. Y. Liu, P. Agarwal, S. Zhao, D. W. Conroy, G. Ji, J. H. Yu, C. P. Jaroniec, Z. G. Liu, X. B. Lu, X. D. Li and X. M. He, Nat. Commun., 2018, 9, 562 CrossRef PubMed.
- R. W. Robey, K. M. Pluchino, M. D. Hall, A. T. Fojo, S. E. Bates and M. M. Gottesman, Nat. Rev. Cancer, 2018, 18, 452 CrossRef CAS PubMed.
- J. Conde, N. Oliva and N. Artzi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1278 CrossRef CAS PubMed.
- M. Z. Ye, Y. X. Han, J. B. Tang, Y. Piao, X. R. Liu, Z. X. Zhou, J. Q. Gao, J. H. Rao and Y. Q. Shen, Adv. Mater., 2017, 29, 1702342 CrossRef PubMed.
- D. Q. Chen, G. Q. Zhang, R. M. Li, M. R. Guan, X. Y. Wang, T. J. Zou, Y. Zhang, C. R. Wang, C. Y. Shu, H. Hong and L. J. Wan, J. Am. Chem. Soc., 2018, 140, 7373 CrossRef CAS PubMed.
- N. H. Park, W. Cheng, F. Lai, C. Yang, P. F. de Sessions, B. Periaswamy, C. W. Chu, S. Bianco, S. Q. Liu, S. Venkataraman, Q. F. Chen, Y. Y. Yang and J. L. Hedrick, J. Am. Chem. Soc., 2018, 140, 4244 CrossRef CAS PubMed.
- F. Lucien, P. P. Pelletier, R. R. Lavoie, J. M. Lacroix, S. Roy, J. L. Parent, D. Arsenault, K. Harper and C. M. Dubois, Nat. Commun., 2017, 8, 15884 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393 CrossRef CAS PubMed.
- C. C. Huang, W. T. Chia, M. F. Chung, K. J. Lin, C. W. Hsiao, C. Jin, W. H. Lim, C. C. Chen and H. W. Sung, J. Am. Chem. Soc., 2016, 138, 5222 CrossRef CAS PubMed.
- S. Y. Li, H. Cheng, B. R. Xie, W. X. Qiu, J. Y. Zeng, C. X. Li, S. S. Wan, L. Zhang, W. L. Liu and X. Z. Zhang, J. Am. Chem. Soc., 2017, 139, 10992 CrossRef PubMed.
- J. Kim, H. R. Cho, H. Jeon, D. Kim, C. Song, N. Lee, S. H. Choi and T. Hyeon, ACS Nano, 2017, 11, 7006 CrossRef PubMed.
- Q. Chen, C. Liang, X. Q. Sun, J. W. Chen, Z. J. Yang, H. Zhao, L. Z. Feng and Z. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5343 CrossRef CAS PubMed.
- Y. Zhang, F. M. Wang, C. Q. Liu, Z. Z. Wang, L. H. Kang, Y. Y. Huang, K. Dong, J. S. Ren and X. G. Qu, ACS Nano, 2018, 12, 651 CrossRef CAS PubMed.
- Y. Z. Chen, Z. U. Wang, H. Wang, J. Lu, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035 CrossRef CAS PubMed.
- B. H. Kim, M. J. Hackett, J. Park and T. Hyeon, Chem. Mater., 2014, 26, 59 CrossRef CAS.
- H. P. Xia, F. Y. Li, W. Park, S. F. Wang, Y. Jang, Y. Du, S. Baik, S. Cho, T. Kang, D. H. Kim, D. S. Ling, K. M. Hui and T. Hyeon, ACS Cent. Sci., 2016, 2, 802 CrossRef CAS PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673 CrossRef CAS PubMed.
- W. T. Zhang, S. H. Li, X. N. Liu, C. Y. Yang, N. Hu, L. N. Dou, B. G. Zhao, Q. Y. Zhang, Y. R. Suo and J. L. Wang, Adv. Funct. Mater., 2018, 28, 1706375 CrossRef.
- J. H. Wang, H. He, X. Xu, X. Wang, Y. B. Chen and L. C. Yin, Biomaterials, 2018, 171, 72 CrossRef CAS PubMed.
- S. T. Zhang, D. X. Zhang, X. H. Zhang, D. H. Shang, Z. H. Xue, D. L. Shan and X. Q. Lu, Anal. Chem., 2017, 89, 3538 CrossRef CAS PubMed.
- L. Wu, W. M. Yin, K. Tang, K. Shao, Q. Li, P. Wang, Y. P. Zuo, X. M. Lei, Z. C. Lu and H. Y. Han, Biosens. Bioelectron., 2016, 82, 177 CrossRef CAS PubMed.
- P. V. Asharani, N. Xinyi, M. P. Hande and S. Valiyaveettil, Nanomedicine, 2010, 5, 51 CrossRef CAS PubMed.
- Z. Z. Wang, Y. Zhang, E. G. Ju, Z. Liu, F. F. Cao, Z. W. Chen, J. S. Ren and X. G. Qu, Nat. Commun., 2018, 9, 3334 CrossRef PubMed.
- K. Han, Z. Y. Ma, X. X. Dai, J. Zhang and H. Y. Han, J. Controlled Release, 2018, 279, 198 CrossRef CAS PubMed.
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599 CrossRef CAS PubMed.
- S. Wang, A. Riedinger, H. Li, C. Fu, H. Liu, L. Li, T. Liu, L. Tan, M. J. Barthel, G. Pugliese, F. De Donato, M. S. D'Abbusco, X. W. Meng, L. Manna, H. Meng and T. Pellegrino, ACS Nano, 2015, 9, 1788 CrossRef CAS PubMed.
- Z. Y. Ma, K. Han, X. X. Dai and H. Y. Han, ACS Nano, 2018, 12, 6252 CrossRef CAS PubMed.
- J. Gao, G. Liang, B. Zhang, Y. Kuang, X. Zhang and B. Xu, J. Am. Chem. Soc., 2007, 129, 1428 CrossRef CAS PubMed.
- M. W. Dewhirst, Y. Cao and B. Moeller, Nat. Rev. Cancer, 2008, 8, 425 CrossRef CAS PubMed.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437 CrossRef CAS PubMed.
- C. Zhang, W. H. Chen, L. H. Liu, W. X. Qiu, W. Y. Yu and X. Z. Zhang, Adv. Funct. Mater., 2017, 27, 1700626 CrossRef.
- C. Yao, W. X. Wang, P. Y. Wang, M. Y. Zhao, X. M. Li and F. Zhang, Adv. Mater., 2018, 30, 1704833 CrossRef PubMed.
- H. C. Chen, J. W. Tian, W. J. He and Z. J. Guo, J. Am. Chem. Soc., 2015, 137, 1539 CrossRef CAS PubMed.
- F. Zhao, Y. Zhao, Y. Liu, X. L. Chang, C. Y. Chen and Y. L. Zhao, Small, 2011, 7, 1322 CrossRef CAS PubMed.
- S. S. Wang, Z. K. Liu, Y. X. Zou, X. F. Lai, D. Ding, L. Chen, L. Q. Zhang, Y. Wu, Z. Chen and W. H. Tan, Analyst, 2016, 141, 3337 RSC.
- Y. Zhou, L. Wang, X. Ban, T. Zeng, Y. Zhu, M. Li, X. Y. Guan and Y. Li, Oncogene, 2018, 37, 1086 CrossRef CAS PubMed.
- X. Shen, X. H. Sun, B. Sun, T. Li, G. L. Wu, Y. T. Li, L. Chen, Q. X. Liu, M. Cui and Z. Z. Zhou, FEBS Lett., 2018, 592, 599 CrossRef CAS PubMed.
- J. H. Lee, A. V. Budanov and M. Karin, Cell Metab., 2013, 18, 792 CrossRef CAS PubMed.
- F. Teng, Z. Y. Xu, J. H. Chen, G. W. Zheng, G. D. Zheng, H. Lv, Y. P. Wang, L. J. Wang and X. D. Cheng, Oncol. Rep., 2018, 40, 1203 CAS.
- X. Wu, B. C. Nguyen, P. Dziunycz, S. Chang, Y. Brooks, K. Lefort, G. F. Hofbauer and G. P. Dotto, Nature, 2010, 465, 368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00088g |
| This journal is © The Royal Society of Chemistry 2019 |