
Achieving a high-performance Prussian blue analogue cathode with an ultra-stable redox reaction for ammonium ion storage†
Chunyang
Li
 ab,
Wenqi
Yan
b,
Shishuo
Liang
c,
Peng
Wang
b,
Jing
Wang
c,
Lijun
Fu
ac,
Yusong
Zhu
ac,
Yuhui
Chen
*ac,
Yuping
Wu
*abc and
Wei
Huang
a
ab,
Wenqi
Yan
b,
Shishuo
Liang
c,
Peng
Wang
b,
Jing
Wang
c,
Lijun
Fu
ac,
Yusong
Zhu
ac,
Yuhui
Chen
*ac,
Yuping
Wu
*abc and
Wei
Huang
a
aState Key Laboratory of Materials-oriented Chemical Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: cheny@njtech.edu.cn; wuyp@fudan.edu.cn
bInstitute of Advanced Materials (IAM), Nanjing Tech University, Nanjing 210009, China
cSchool of Energy Science and Engineering, Nanjing Tech University, Nanjing 211816, China
First published on 4th March 2019
Abstract
Aqueous rechargeable batteries with advantages of safety, low cost, and environmental kindness have displayed high feasibility of practical applications for large-scale energy storage. Developing high-performance electrode materials is a necessary gateway to commercially available batteries. Here, we demonstrate the controlled synthesis of sodium iron hexacyanoferrates, NaFeIIIFeII(CN)6 (Na-FeHCFs). Ball-cutting Na-FeHCF nanocubes are first synthesized and used as a cathode material for aqueous ammonium-ion batteries. Due to fast charge transfer and diffusion, the ball-cutting Na-FeHCF nanocubes exhibit a high discharge capacity of 62 mA h g−1 at 0.25 A g−1 and 77.4% capacity retention at 2 A g−1. Such excellent capacity and rate performance are superior to those of other Na-FeHCFs and the reported ammonium-ion intercalation cathodes. Furthermore, they present unparalleled cycling stability with no capacity loss over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, thanks to the highly stable redox reaction of the high-spin nitrogen-coordinated FeII/FeIII (FeH) couple. This work supplies a new view to design high-performance cathode materials for ammonium ion storage.
000 cycles, thanks to the highly stable redox reaction of the high-spin nitrogen-coordinated FeII/FeIII (FeH) couple. This work supplies a new view to design high-performance cathode materials for ammonium ion storage.
Conceptual insightsNanostructured electrode materials for batteries with excellent cycling and rate performance are very difficult to prepare, and most reported methods are very complicated. This is the main reason that nanotechnologies are rarely practically applied in the battery field. Here we demonstrated a simple way, i.e. by adjusting the stirring rate of a solution reaction, to achieve a rarely reported morphology, edge-cutting nanocubes for sodium iron hexacyanoferrates [NaFeIIIFeII(CN)6]. Their superlong cycling life for reversible ammonium-ion intercalation/deintercalation in aqueous solution with no evident capacity fading after 50000 cycles paves the way for the possibility of safe and economic energy storage on a large-scale through some simple nanoengineering.
|
1. Introduction
Advanced electrochemical energy storage (EES) devices are essential to the sustainable employment of clean and renewable energy sources, along with the mounting consumption of fossil energy.1,2 Organic rechargeable batteries based on the bulk (de)intercalation of monovalent metal ions, such as lithium-ion batteries, sodium-ion batteries, and potassium-ion batteries, have received extensive attention and in-depth research on account of their high energy densities.3–5 In addition, some batteries utilizing cost-efficient multivalence metal ions (such as Zn2+, Mg2+, Ca2+, and Al3+) as charge carriers have presented wonderful promise for satisfying future energy storage demands.6–9 Yet flammable and venomous organic electrolytes with high price result in safety concerns as well as limited lifespan and power density.10 Aqueous rechargeable batteries (ARBs) have emerged as promising EES devices at the right moment in view of their low cost, safety, and high power density derived from aqueous electrolytes with high ionic conductivity.11,12The first aqueous rechargeable lithium battery (ARLB) was designed and constructed with a LiMn2O4 cathode, VO2(B) anode, and 5 M LiNO3 electrolyte in 1994,13 unlocking the prologue to the development of cost-effective ARBs with high safety. Subsequently, a chain of intercalation-type compounds has been discovered for building a variety of ARBs, such as LiCoO2 (Li+), Na0.35MnO2 (Na+), KCuFe(CN)6 (K+), MnO2 (Zn2+), TiO2 (Al3+), and so forth.14–18 Among various insertion-type materials, Prussian blue analogues (PBAs) with nanostructured open frameworks and large interstitial sites can reversibly intercalate all forementioned metal ions, and have great promise as inexpensive electrode materials for ARBs.19–22 The general formula of PBAs can be expressed as AxM[M′(CN)6]y·mH2O, where A is an alkali metal (e.g., Na and K), M is a transition metal (e.g., Mn, Fe, Co, Ni, Cu, Zn and In), and M′ is usually Mn or Fe, with 0 ≤ x ≤ 2, y < 1.23,24 After Cui et al. testified the reversible intercalation of K+ ions in K0.71Cu[Fe(CN)6]0.72·3.7H2O (CuHCF), CuHCF and other PBAs synthesized by simple co-precipitation were extensively developed and applied in ARBs.25,26
Apart from conventional metal-ion intercalation, the topochemistry of the NH4+ ion in PBAs has been disclosed in recent years, providing a new paradigm in ARBs. Cui et al. explored the insertion chemistry of the NH4+ ion in open framework hexacyanoferrates (CuHCF and NiHCF) compared with the reversible (de)intercalation of Li+, Na+, and K+ ions for the first time.21 Recently, Ji et al. reported the original “rocking-chair” ammonium-ion battery comprising (NH4)1.47Ni[Fe(CN)6]0.88 as the cathode and aromatic compound (PTCDI) as the anode, and systematically investigated the topochemistry of the NH4+ ion in ammonium-source PBA and PTCDI solid.27 However, these PBA cathode materials offer unremarkable capacities and rate capability as well as inferior cycling stability compared to traditional metal-ion intercalation. Hence, it is highly significant and challenging to develop high-performance PBA cathode materials for ammonium-ion batteries in consideration of the rich sources for ammonium on earth.
In this work, we successfully control the morphologies and sizes of sodium iron hexacyanoferrates, NaFeIIIFeII(CN)6 (Na-FeHCFs) by using various stirring speeds and prepare ball-cutting Na-FeHCF nanocubes for the first time. Based on fast charge transfer and diffusion, the ball-cutting Na-FeHCF nanocubes present the highest capacity of 62 mA h g−1 and best rate capability among different cubic Na-FeHCFs and the reported ammonium-ion intercalation cathodes. Moreover, the ball-cutting Na-FeHCF nanocubes deliver an unprecedented lifetime with no capacity fading over 50000 cycles due to the highly stable redox reaction of the FeII/FeIII (FeH) couple. We also systematically investigate the energy storage mechanism and structural evolution of the ball-cutting Na-FeHCF nanocubes during the reversible (de)intercalation of the NH4+ ion.
2. Experimental
2.1 Synthesis of ball-cutting sodium iron hexacyanoferrate (Na-FeHCF) nanocubes
Ball-cutting Na-FeHCF nanocubes were prepared by a modified single-source precipitation method.28 Firstly, 1 g polyvinylpyrrolidone (PVP K30, Sinopharm) and 2 mL hydrochloric acid (37%, Sinopharm) were mixed with 80 mL deionized (DI) water to form solution A under vigorous stirring. Next, 0.58 g sodium ferrocyanide decahydrate (Aladdin) was dissolved in 40 mL DI water by stirring to form solution B. After that, solution B was slowly added to solution A at room temperature under vigorous stirring for 0.5 h. Subsequently, the mixed solution was heated at 80 °C for 12 h under stirring with a speed of 1200 rpm. Finally, the blue precipitate collected by filtration was washed several times with DI water and ethanol, and then dried in a vacuum oven at 80 °C overnight. For comparison, cubic Na-FeHCFs were synthesized using the same procedure but with different stirring speeds of 0, 400, and 800 rpm, respectively (marked as Na-FeHCF-0, Na-FeHCF-400, and Na-FeHCF-800).2.2 Material characterization
The crystal structures and phases of Na-FeHCFs were measured by an X-ray diffractometer (XRD, Rigaku SmartLab TM, 3 KW) with Cu Kα radiation. The morphology and structure of the ball-cutting Na-FeHCF nanocubes were examined by field-emission scanning electron microscope (FESEM, JEOL, JSM-7800F) and transmission electron microscope (TEM, JEOL, JEM-2100F) equipped with an energy dispersive X-ray spectrometer (EDS). SEM images of Na-FeHCF-0, Na-FeHCF-400, and Na-FeHCF-800 were collected by a Phenom ProX microscope. The themogravimetric analysis (TGA, Netzsch STA 449 F3) was recorded form 25 °C to 500 °C in a N2 atmosphere at a heating rate of 10 °C min−1. Fourier transform infrared (FTIR) spectra were recorded on a Bruker ALPHA spectrometer. The valence states of the surface elements were investigated through the X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe) technique. To investigate the energy storage mechanism and structural changes, electrode materials during different charge/discharge states were washed with DI water and ethanol, and dried at 60 °C. Subsequently electrode materials were examined using FTIR and ex situ XRD experiments.2.3 Electrochemical measurements
The working electrodes were prepared by pressing a film composed of active material, acetylene black and poly(tetrafluoroethylene) (PTFE) at a weight ratio of 7:2:1 onto graphite rod (the diameter is 8 mm, the capacity contribution is negligible, Fig. S1, ESI†). The cyclic voltammetry (CV) measurement was conducted by a three-electrode setup on a CHI 760E electrochemical workstation (Chenhua) at room temperature. The three-electrode structure consists of graphite rod as the counter electrode, saturated calomel electrode (SCE) as the reference electrode, and 1 M (NH4)2SO4 solution (pH ≈ 5.4) as the electrolyte. The electrochemical impedance spectra (EIS) were measured in the frequency range from 0.01 Hz to 100 kHz. The cycling and rate tests were carried out on a CT2001A cell system (Land) at room temperature. The mass of active materials in the electrodes was about 1 mg.
3. Results and discussion
In general, it is difficult to control the morphologies and sizes of PBAs during the synthesis process in aqueous solution.28,29 To attain a uniform product, we prepared ball-cutting Na-FeHCF nanocubes by a facile single-source precipitation procedure at a high stirring speed of 1200 rpm and polyvinylpyrrolidone (PVP) as a surfactant (Fig. 1). Well-dispersed ball-cutting Na-FeHCF nanocubes with an edge length of ≈500 nm were observed from the field-emission scanning electron microscopy (FESEM) image in Fig. 2a. The magnified FESEM image indicates unique ball-cutting nanocubes with six smooth circular faces, which can be regarded as nanocubes without eight corners because of high-speed stirring (Fig. 2b). The transmission electron microscopy (TEM) images at various observation angles further demonstrate that ball-cutting Na-FeHCF nanocubes possess a distinctive sphere-like nanostructure and maintain the faces of a cube (Fig. 2c and d), which is consistent with the FESEM results. The dark-field scanning TEM (STEM) image and the corresponding energy dispersive X-ray spectroscopy (EDS) elemental mapping of the ball-cutting Na-FeHCF nanocubes show that N, C, Fe, O, and Na atoms are evenly distributed over the ball-cutting nanocubes (Fig. 2e).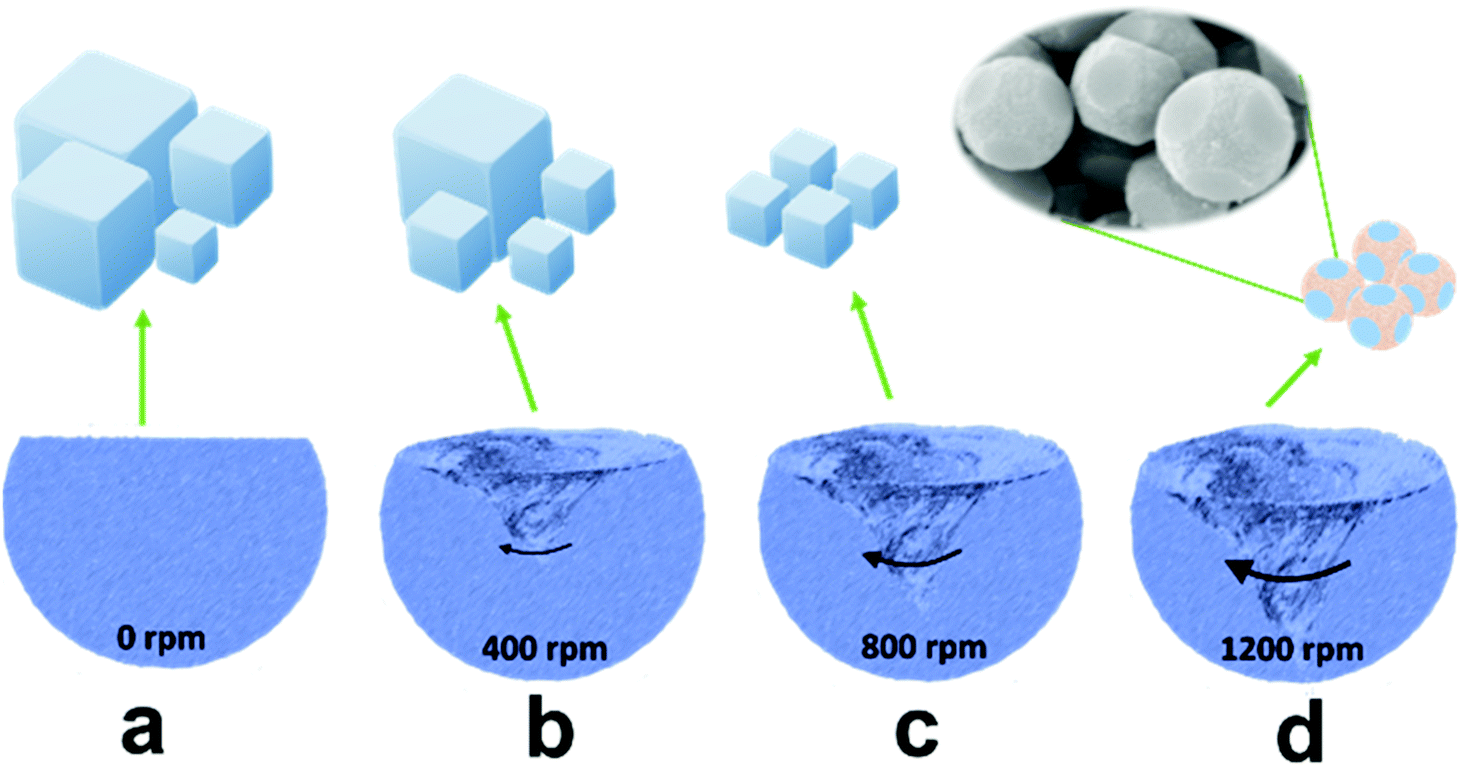 | ||
| Fig. 1 Schematic illustration of the synthesis of Na-FeHCFs with tailored morphology by changing the stirring speed. | ||
 | ||
| Fig. 2 Physical characterization of ball-cutting Na-FeHCF nanocubes: (a and b) FESEM images at different magnifications. (c and d) TEM images at various observation angles. (e) STEM image and EDS elemental mappings. | ||
For comparison, we also prepared Na-FeHCFs by the same procedure but with various stirring speeds of 0, 400, and 800 rpm (Fig. 1a–c). Cubic Na-FeHCF with uneven size distribution (300 nm–4 μm) was obtained by aging for 12 h at 80 °C without stirring (Fig. S2a and b, ESI†). At low stirring speed of 400 rpm, the uniformity of the cubic Na-FeHCF was improved, with large yields of nanoparticles (Fig. S2c and d, ESI†). When the stirring speed was up to 800 rpm, the as-prepared Na-FeHCF exhibits a homogeneous nanocube structure with an average size of 500 nm (Fig. S2e and f, ESI†). Consequently, the morphologies and sizes of Na-FeHCFs are tailorable by controlling the stirring rates during the preparation process. The corresponding growth mechanism can also be explained by Kossel's model: growth units including atoms, molecules, and ions are adsorbed on the crystal surface, migrating and nucleating on the (100) face to form an ordered and active monolayer, eventually growing layer-by-layer into a cube under ideal conditions. The adjustment of the stirring rates is equivalent to the control of the nucleation rates, leading to a sphere-like morphology.24 The X-ray diffraction (XRD) patterns of Na-FeHCFs at various stirring speeds are well indexed to the face-centered cubic (FCC) structure with typical characteristic peaks of (200), (220), (400), (420), (422), (440), (600) and (620) crystal planes (Fig. S3, ESI†).
The Fourier transform infrared (FTIR) spectra of the ball-cutting Na-FeHCF nanocubes show two peaks at 3625 and 1610 cm−1 corresponding to the stretching and bending vibrations of O–H in H2O, respectively (Fig. 3a).30 From the thermogravimetric analysis (TGA) of the ball-cutting Na-FeHCF nanocubes (Fig. 3b), the crystal water content of about 16.5% was detected. The elemental compositions of the ball-cutting Na-FeHCF nanocubes were further determined by X-ray photoelectron spectroscopy (XPS) analysis. The XPS spectra of the ball-cutting Na-FeHCF nanocubes indicate the existence of C, N, O, and Fe (Fig. 3c). The Fe2p spectra of the ball-cutting Na-FeHCF nanocubes displays three peaks at 708.5, 712.6, and 721.4 eV, corresponding to the binding energies of Fe2p3/2 of [Fe(CN)6]4−, Fe2p3/2 and Fe2p1/2 of Fe3+, respectively, which confirms the presence of Fe2+ and Fe3+ (Fig. 3d).31 Therefore the formula of the ball-cutting Na-FeHCF nanocubes was determined to be NaFeIII[FeII(CN)6]·2.7H2O.
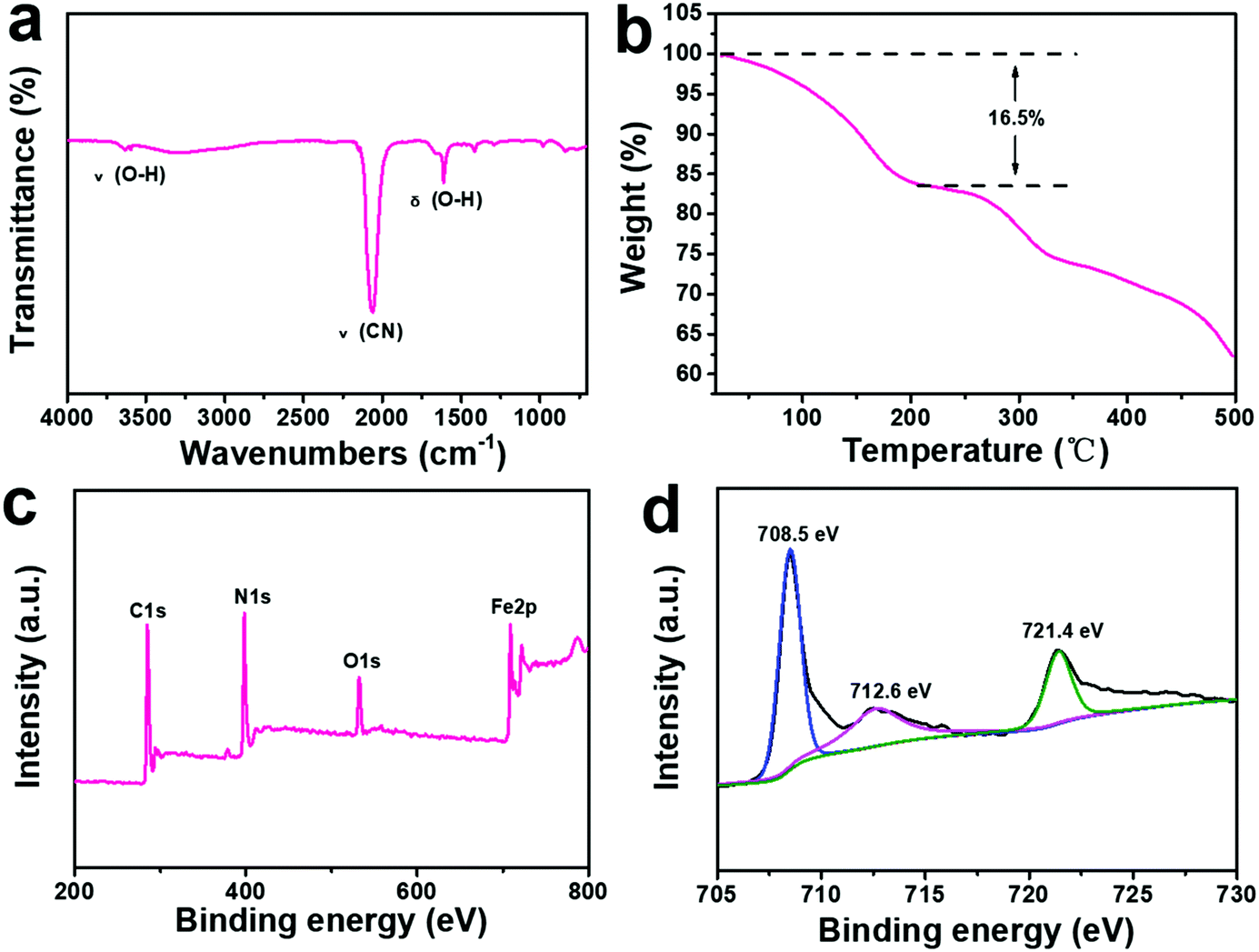 | ||
| Fig. 3 Physical characterization of the ball-cutting Na-FeHCF nanocubes: (a) FTIR analysis. (b) Thermogravimetric analysis. (c) XPS spectra. (d) Fe2p spectra. | ||
The electrochemical performance of the ball-cutting Na-FeHCF nanocubes in 1 M (NH4)2SO4 electrolyte solution (pH ≈ 5.4) was systematically evaluated by cyclic voltammetry (CV) and galvanostatic charge/discharge (GCD) tests in a three-electrode setup with graphite rod as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. Fig. 4a shows the first five CV curves of the ball-cutting Na-FeHCF electrode from −0.1 to 0.8 V at 5 mV s−1, demonstrating two oxidation peaks at 0.31 and 0.59 V (vs. SCE), and only one reduction peak at 0.16 V. Such novel CV results are similar to the (de)intercalation behavior of NH4+ in (NH4)1.47Ni[Fe(CN)6]0.88, which should be assigned to the reversible redox reaction of the high-spin nitrogen-coordinated FeII/FeIII (FeH) couple.27,32 The electrochemical reaction mechanism can be represented as the following eqn (1):
| NaFeIII[FeII(CN)6]·2.7H2O + NH4+ + e− ↔ Na(NH4)FeIIFeII(CN)6·2.7H2O | (1) |
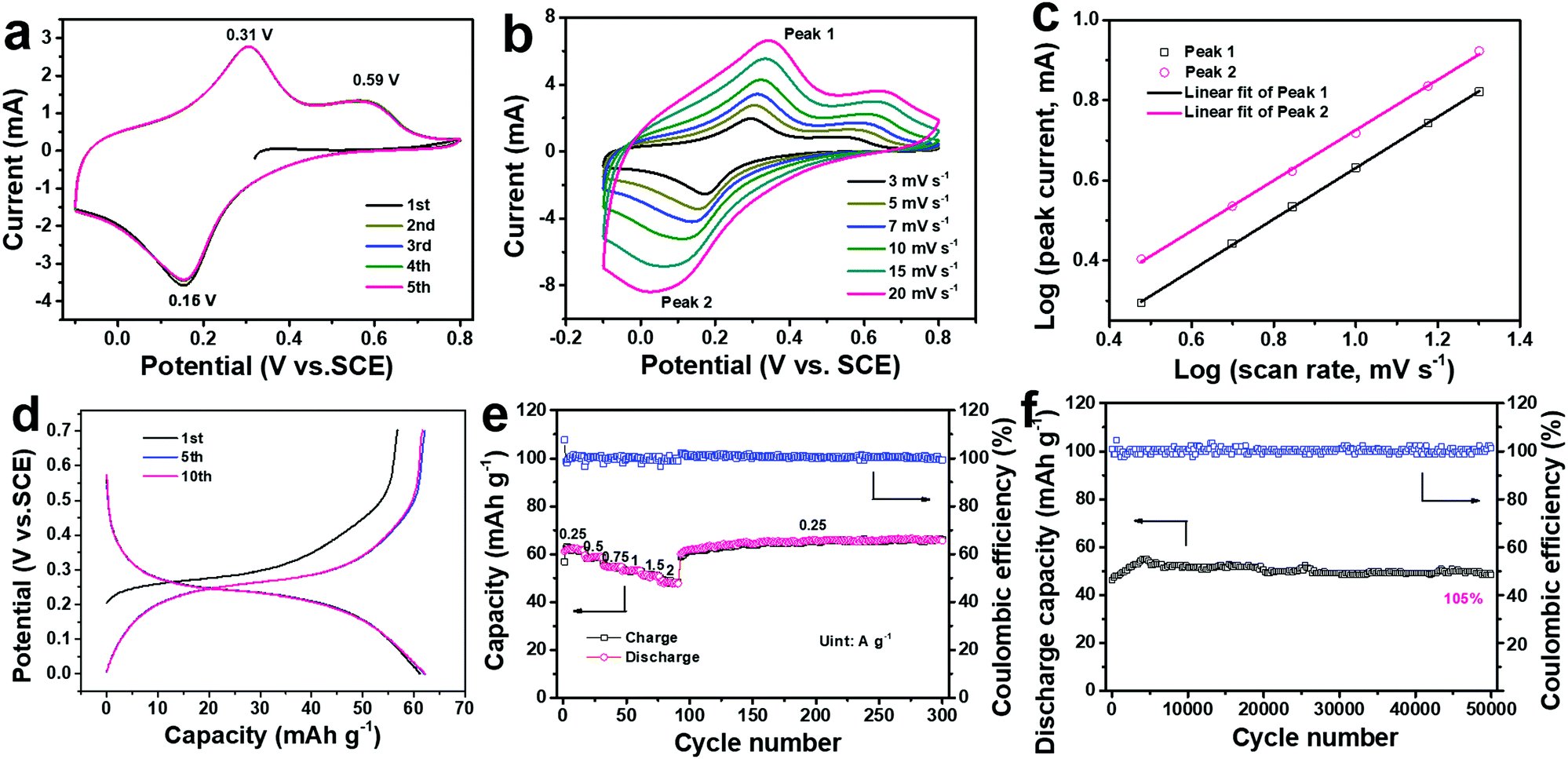 | ||
| Fig. 4 Electrochemical performance of a ball-cutting Na-FeHCF electrode: (a) the first five CV curves at 5 mV s−1. (b) CV curves at various scan rates. (c) Log (peak current) vs. log (scan rate) plots of Peak 1 and Peak 2. (d) GCD profiles at 0.25 A g−1. (e) Rate performance. (f) Long-term cycling performance at 2 A g−1. | ||
To further understand the electrochemistry of the ball-cutting Na-FeHCF nanocubes, the analysis of sweep data was performed by CV measurements at different scan rates (3–20 mV s−1) in Fig. 4b. The total stored charge originates from a capacitive process, involving pseudo-capacitive and electrical double-layer capacitive contributions, and a diffusion-controlled Faradaic intercalation process.33 Generally, the measured peak currents (i) in the CV curves and the corresponding scan rates (v) obey the following equation:
| i = avb | (2) |
| log(i) = log(a) + blog(v) | (3) |
Fig. 4d shows the GCD profiles of the ball-cutting Na-FeHCF electrode from 0 to 0.7 V at a current density of 0.25 A g−1, in which an obvious charge/discharge plateau at 0.27/0.23 V and a slight charge plateau at 0.55 V can be observed, consistent with the unique redox behavior in the CV results. The initial discharge capacity of the ball-cutting Na-FeHCF electrode after the CV test is 61.2 mA h g−1, indicating 66.5% of its theoretical capacity for one electron transfer. The discharge capacity is higher than previously reported ones for CuHCF (ca. 57 mA h g−1 at 0.25 A g−1), NiHCF (ca. 50 mA h g−1 at 0.25 A g−1), and (NH4)1.47Ni[Fe(CN)6]0.88 (ca. 52 mA h g−1 at 0.25 A g−1).21,27Fig. 4e and Fig. S4 (ESI†) illustrate the rate performance of the ball-cutting Na-FeHCF electrode and the associated GCD profiles, respectively. The discharge capacities are approximately 62, 59, 55, 53, 51, and 48 mA h g−1 at current densities of 0.25, 0.5, 0.75, 1, 1.5, 2 A g−1 (77.4% capacity retention), respectively. The high rate capability is superior to the previously reported ammonium-ion intercalation cathodes, which could be ascribed to the short diffusion path from the unique nanocube structure. When the current density goes back to 0.25 g−1, a remarkable capacity recovery can be found with no capacity loss after 300 cycles.
During long-term cycling at a current density of 2 A g−1, the discharge capacity gradually increases and tends to be stable (Fig. 4f). After 50000 cycles, 109.7% of its initial capacity with an average Coulombic efficiency of 100% can be obtained, indicating an unparalleled cycling stability. Such excellent cycling performance over previous literature should be assigned to the stable crystal structure of the ball-cutting Na-FeHCF nanocubes. The SEM and TEM images after 50000 cycles demonstrate that the ball-cutting Na-FeHCF nanocubes still keep a stable framework without obvious structural collapse (Fig. S5, ESI†). Table S1 (ESI†) shows the electrochemical properties of the ball-cutting Na-FeHCF nanocubes compared with some reported insertion-type cathodes for aqueous batteries.
The successful and stable intercalation of NH4+ ions in the ball-cutting Na-FeHCF nanocubes can be verified by the FTIR test of the electrodes. Two new peaks at 3250 and 1375 cm−1 can be observed in the FTIR spectra of the electrodes before and after cycling, which are indexed to the stretching and bending modes of N–H bonds, respectively (Fig. S6, ESI†). The emergence of the N–H signal proves the successful insertion of NH4+ ions into the lattice of the ball-cutting Na-FeHCF nanocubes. To understand the structural change of the ball-cutting Na-FeHCF nanocubes during the (de)intercalation of NH4+ ions, ex situ XRD patterns were recorded at different charge or discharge states. A consistent FCC structure can be found from all XRD patterns at selected potentials, indicating that the (de)intercalation of NH4+ ions is a solid solution reaction (Fig. 5).37 At the first cycle, the charge process is negligible, which is in accordance with the first CV result. Compared with the initial electrode, all characteristic peaks of the electrode at 0.7 V (point a) do not change. When the electrode is discharged to 0 V (point d), these peaks move gradually to lower angles, indicating a volume expansion (NH4+ ion insertion). At the second cycle, all characteristic peaks shift gradually to higher angles during the charge process, suggesting a volume shrinkage due to the extraction of NH4+ ions. During the discharge process (NH4+ ion insertion), these peaks return to the angles of point d along with a volume expansion. This reversible structural evolution reveals the high reversibility of an NH4+ ion in the ball-cutting Na-FeHCF nanocubes.
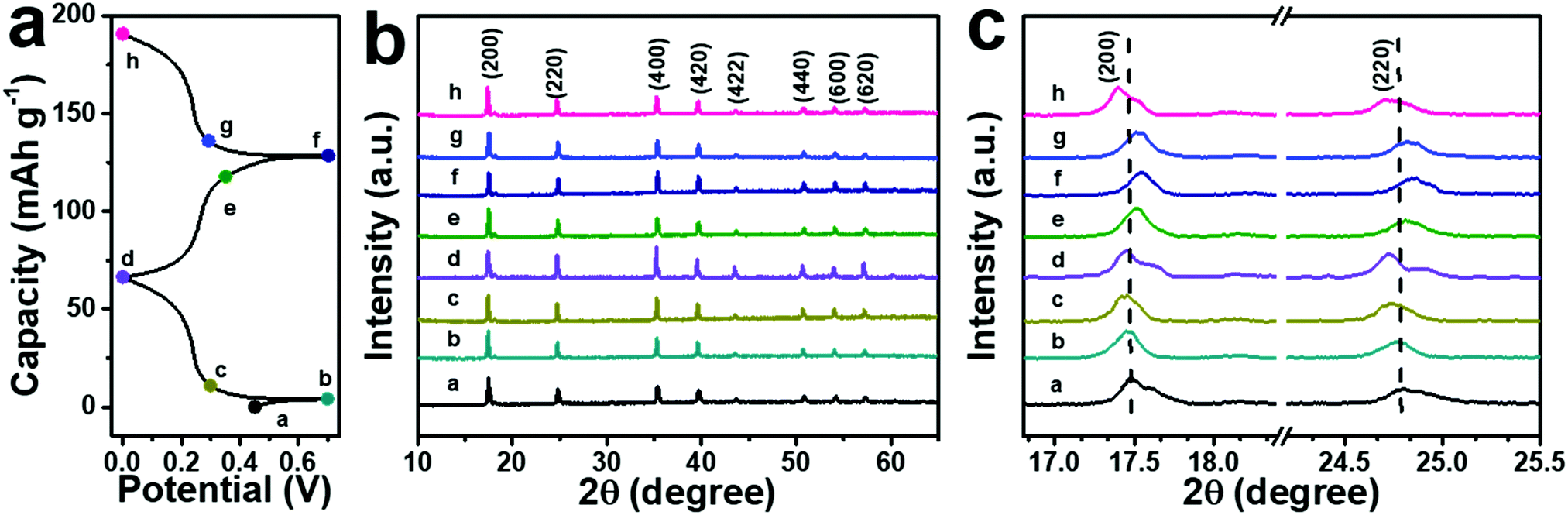 | ||
| Fig. 5 (a) Typical GCD profiles of the electrode. (b and c) Corresponding ex situ XRD patterns at selected potentials. | ||
In theory, two pairs of well-separated redox peaks should emerge with the intercalation/de-intercalation of cations in cubic Na-FeHCF, which are attributed to the valence changes of the FeH couple and low-spin FeII/FeIII![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (FeL) couple.38,39 Therefore, we also evaluate the electrochemical properties of the ball-cutting Na-FeHCF nanocubes for two electrons transferred (the theoretical capacity is 184 mA h g−1). The CV curve of the ball-cutting Na-FeHCF electrode between −0.1 and 1.2 V at 5 mV s−1 presents three oxidation peaks and two reduction peaks (Fig. S7a, ESI†), which is similar to the (de)intercalation behavior of NH4+ in the Berlin green framework.40 At a current density of 0.25 A g−1, the ball-cutting Na-FeHCF electrode delivers a discharge capacity of 73.2 mA h g−1 from 0 to 1.1 V (Fig. S7b, ESI†), and 39.8% of its theoretical capacity for two electrons transferred was achieved, which is worse than the value based on the redox reaction of the FeH couple. Low capacity contribution from the redox reaction of the FeL couple should be responsible for the above result. A discharge capacity of 48 mA h g−1 at 2 A g−1 is gained with 65.5% capacity retention (Fig. S7c, ESI†), demonstrating relatively low rate capability compared with ball-cutting Na-FeHCF nanocubes with limited redox reaction. Moreover, ball-cutting Na-FeHCF nanocubes for two electrons transferred exhibit poor cycling performance with 52.6% capacity retention after 2000 cycles at a current density of 1 A g−1 (Fig. S7d, ESI†). Such bad stability should be ascribed to the structural collapse and dissolution of ball-cutting Na-FeHCF nanocubes originating from the redox reaction of the FeL couple (see the SEM image after cycling, Fig. S8, ESI†). Therefore, restricting the redox reaction of a low-spin couple in PBAs may be an efficient strategy for maintaining the structural stability during a continuous charge/discharge process.
C (FeL) couple.38,39 Therefore, we also evaluate the electrochemical properties of the ball-cutting Na-FeHCF nanocubes for two electrons transferred (the theoretical capacity is 184 mA h g−1). The CV curve of the ball-cutting Na-FeHCF electrode between −0.1 and 1.2 V at 5 mV s−1 presents three oxidation peaks and two reduction peaks (Fig. S7a, ESI†), which is similar to the (de)intercalation behavior of NH4+ in the Berlin green framework.40 At a current density of 0.25 A g−1, the ball-cutting Na-FeHCF electrode delivers a discharge capacity of 73.2 mA h g−1 from 0 to 1.1 V (Fig. S7b, ESI†), and 39.8% of its theoretical capacity for two electrons transferred was achieved, which is worse than the value based on the redox reaction of the FeH couple. Low capacity contribution from the redox reaction of the FeL couple should be responsible for the above result. A discharge capacity of 48 mA h g−1 at 2 A g−1 is gained with 65.5% capacity retention (Fig. S7c, ESI†), demonstrating relatively low rate capability compared with ball-cutting Na-FeHCF nanocubes with limited redox reaction. Moreover, ball-cutting Na-FeHCF nanocubes for two electrons transferred exhibit poor cycling performance with 52.6% capacity retention after 2000 cycles at a current density of 1 A g−1 (Fig. S7d, ESI†). Such bad stability should be ascribed to the structural collapse and dissolution of ball-cutting Na-FeHCF nanocubes originating from the redox reaction of the FeL couple (see the SEM image after cycling, Fig. S8, ESI†). Therefore, restricting the redox reaction of a low-spin couple in PBAs may be an efficient strategy for maintaining the structural stability during a continuous charge/discharge process.
For comparison, the electrochemical performance of Na-FeHCF-0, Na-FeHCF-400, and Na-FeHCF-800 was also investigated in 1 M (NH4)2SO4 electrolyte solution. The same redox behaviors were observed among the three samples and ball-cutting Na-FeHCF nanocubes, while their charge/discharge plateaus are different (Fig. S9a and b, ESI†). The discharge capacities of the three samples are 14.5, 33.4, and 50.8 mA h g−1 for Na-FeHCF-0, Na-FeHCF-400, and Na-FeHCF-800 at a current density of 0.25 A g−1, respectively, which are inferior to that of ball-cutting Na-FeHCF nanocubes. 53.8%, 69.8%, and 72.8% of the initial capacity for Na-FeHCF-0, Na-FeHCF-400, and Na-FeHCF-800 can be retained at a high current density of 2 A g−1 (Fig. S9c, ESI†), respectively, indicating their relatively poor rate capability compared with ball-cutting Na-FeHCF nanocubes. From Na-FeHCF-0 to ball-cutting Na-FeHCF nanocubes, the gradually enhancing capacity and rate performance should be attributed to the boosted morphological homogeneity and particle size of cubic Na-FeHCF. Moreover, all three samples exhibit exceptional stable cycling performance with no capacity loss after 10000 cycles (Fig. S9d, ESI†), which could be attributed to the highly stable redox reaction of the FeH couple.
To better understand the increasing capacity and rate performance from Na-FeHCF-0 to the ball-cutting Na-FeHCF nanocubes, electrochemical impedance spectra (EIS) measurements were conducted to analyze their charge transfer process and diffusion-limited process. Fig. 6a reveals the experimental and fitting EIS curves of a ball-cutting Na-FeHCF electrode. The equivalent series resistance (Rs) and charge transfer resistance (RCT) are 1.99 and 5.02 Ω, respectively, which are the smallest relative to those of Na-FeHCF-0 (Rs: 33.91 Ω; RCT: 26.04 Ω), Na-FeHCF-400 (Rs: 3.35 Ω; RCT: 7.11 Ω), and Na-FeHCF-800 (Rs: 2.29 Ω; RCT: 6.71 Ω) electrodes (Fig. S10a–c, ESI†). Clearly, the charge transfer capability from Na-FeHCF-0 to the ball-cutting Na-FeHCF electrodes is gradually enhanced, which confirms the increasing capacity and rate performance. To the best of our knowledge, the relationship between the real part (Z′) and angular frequency (ω) could be expressed as the following eqn (4),41,42
| Z′ = Rs + RSEI + RCT + σω−1/2 | (4) |
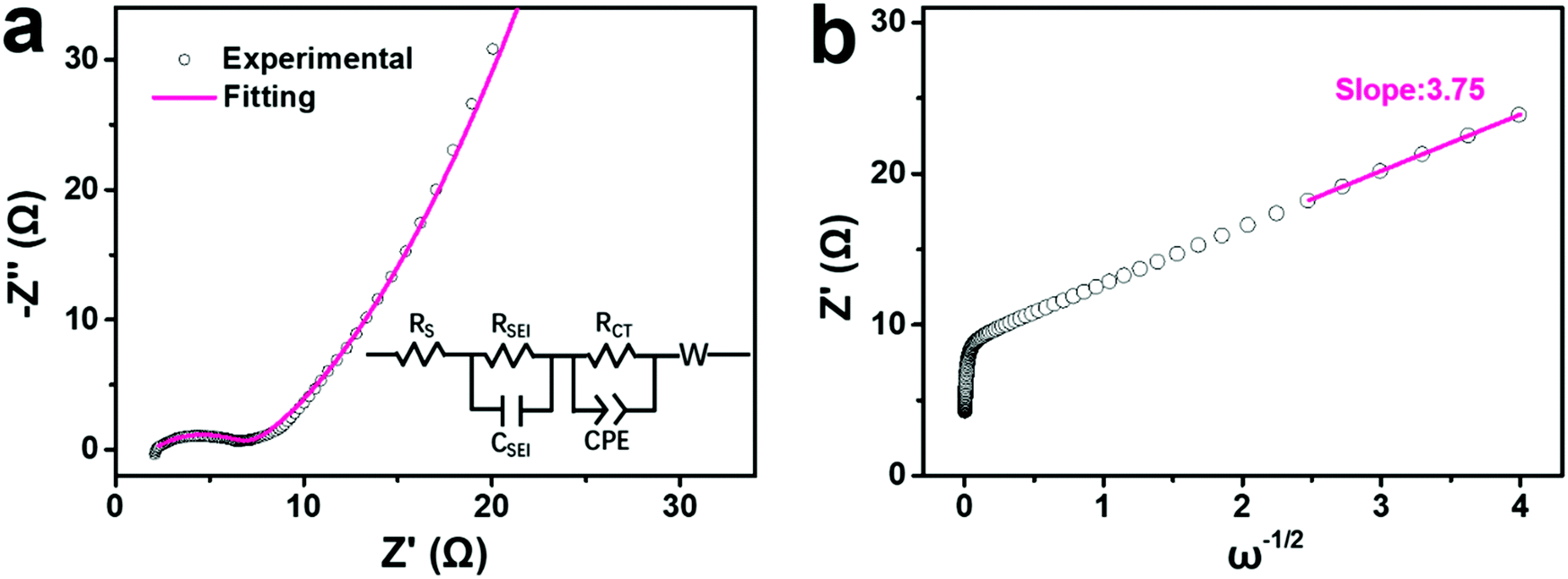 | ||
| Fig. 6 EIS analysis of a ball-cutting Na-FeHCF electrode: (a) Nyquist and fitting plots (inset is equivalent electric circuit). (b) Relationship between Z′ and frequency. | ||
4. Conclusions
In summary, cubic Na-FeHCFs with different morphologies and sizes were synthesized by controlling the stirring speed under the same procedure. Among the different Na-FeHCFs, the ball-cutting Na-FeHCF nanocubes present the highest capacity and best rate capability, which are superior to those of the previously reported studies about NH4+-ion (de)intercalation. Fast charge transfer and diffusion should be responsible for such excellent capacity and rate properties. Compared with the redox reaction of the FeL couple, the FeH couple is highly stable and reversible, which leads to an exceptional cycling stability for the ball-cutting Na-FeHCF nanocubes with limited redox reaction. This study provides a new avenue for developing high-performance cathode materials for aqueous ammonium-ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Project of MOST (2018YFB0104301), the National Natural Science Foundation Committee of China (Distinguished Youth Scientists Project of 51425301, U1601214, 51573013, 51773092 and 51772147), the 1000 Youth Talents Plan of the National Natural Science Foundation of China (51773092), the Research Foundation of State Key Lab (ZK201717), and the Jiangsu Distinguished Professorship Program (2016).Notes and references
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed.
- K. Share, A. P. Cohn, R. Carter, B. Rogers and C. L. Pint, ACS Nano, 2016, 10, 9738–9744 CrossRef CAS PubMed.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- F. Wang, X. Wu, C. Li, Y. Zhu, L. Fu, Y. Wu and X. Liu, Energy Environ. Sci., 2016, 9, 3570–3611 RSC.
- P. Senguttuvan, S.-D. Han, S. Kim, A. L. Lipson, S. Tepavcevic, T. T. Fister, I. D. Bloom, A. K. Burrell and C. S. Johnson, Adv. Energy Mater., 2016, 6, 1600826 CrossRef.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- A. Ponrouch, C. Frontera, F. Barde and M. R. Palacin, Nat. Mater., 2016, 15, 169–172 CrossRef CAS PubMed.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 325–328 CrossRef PubMed.
- W. Tang, Y. Zhu, Y. Hou, L. Liu, Y. Wu, K. P. Loh, H. Zhang and K. Zhu, Energy Environ. Sci., 2013, 6, 2093 RSC.
- C. Liu, X. Wang, W. Deng, C. Li, J. Chen, M. Xue, R. Li and F. Pan, Angew. Chem., Int. Ed., 2018, 57, 7046–7050 CrossRef CAS PubMed.
- M. Pasta, C. D. Wessells, N. Liu, J. Nelson, M. T. McDowell, R. A. Huggins, M. F. Toney and Y. Cui, Nat. Commun., 2014, 5, 3007 CrossRef PubMed.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS PubMed.
- F. Cheng, J. Chen, X. Gou and P. Shen, Adv. Mater., 2005, 17, 2753–2756 CrossRef CAS.
- G. Wang, L. Fu, N. Zhao, L. Yang, Y. Wu and H. Wu, Angew. Chem., 2007, 119, 299–301 CrossRef.
- Y. Liu, B. Zhang, S. Xiao, L. Liu, Z. Wen and Y. Wu, Electrochim. Acta, 2014, 116, 512–517 CrossRef CAS.
- Y. J. He, J. F. Peng, W. Chu, Y. Z. Li and D. G. Tong, J. Mater. Chem. A, 2014, 2, 1721–1731 RSC.
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef PubMed.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2015, 3, 959–962 RSC.
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 159, A98–A103 CrossRef.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed.
- D. Su, A. McDonagh, S. Qiao and G. Wang, Adv. Mater., 2017, 29, 1064007 Search PubMed.
- Q. Wang, Y. Han, X. Wang, N. Bahlawane, G. Pan, M. Yan and Z. Jiang, Science, 2018, 3, 110–133 Search PubMed.
- A. Paolella, C. Faure, V. Timoshevskii, S. Marras, G. Bertoni, A. Guerfi, A. Vijh, M. Armand and K. Zaghib, J. Mater. Chem. A, 2017, 5, 18919–18932 RSC.
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef PubMed.
- X. Wu, Y. Qi, J. J. Hong, Z. Li, A. S. Hernandez and X. Ji, Angew. Chem., Int. Ed., 2017, 56, 13026–13030 CrossRef CAS PubMed.
- L. Zhou, M. Zhang, Y. Wang, Y. Zhu, L. Fu, X. Liu, Y. Wu and W. Huang, Electrochim. Acta, 2017, 232, 106–113 CrossRef CAS.
- M. Hu, S. Ishihara, K. Ariga, M. Imura and Y. Yamauchi, Chem. – Eur. J., 2013, 19, 1882–1885 CrossRef CAS PubMed.
- J. W. Boclair, P. S. Braterman, B. D. Brister, Z. Wang and F. Yarberry, J. Solid State Chem., 2001, 161, 249–258 CrossRef CAS.
- M. Sookhakian, W. J. Basirun, M. A. M. Teridi, M. R. Mahmoudian, M. Azarang, E. Zalnezhad, G. H. Yoon and Y. Alias, Electrochim. Acta, 2017, 230, 316–323 CrossRef CAS.
- M. Morant-Giner, R. Sanchis-Gual, J. Romero, A. Alberola, L. García-Cruz, S. Agouram, M. Galbiati, N. M. Padial, J. C. Waerenborgh, C. Martí-Gastaldo, S. Tatay, A. Forment-Aliaga and E. Coronado, Adv. Funct. Mater., 2018, 28, 1706125 CrossRef.
- Q. Jiang, N. Kurra, M. Alhabeb, Y. Gogotsi and H. N. Alshareef, Adv. Energy Mater., 2018, 8, 1703043 CrossRef.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed.
- Y. Liu, Y. Qiao, W. Zhang, Z. Li, X. Ji, L. Miao, L. Yuan, X. Hu and Y. Huang, Nano Energy, 2015, 12, 386–393 CrossRef CAS.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- W.-J. Li, S.-L. Chou, J.-Z. Wang, Y.-M. Kang, J.-L. Wang, Y. Liu, Q.-F. Gu, H.-K. Liu and S.-X. Dou, Chem. Mater., 2015, 27, 1997–2003 CrossRef CAS.
- X. Wu, Y. Xu, H. Jiang, Z. Wei, J. J. Hong, A. S. Hernandez, F. Du and X. Ji, ACS Appl. Energy Mater., 2018, 1, 3077–3083 CrossRef CAS.
- Y. Wang, B. Chen, Y. Zhang, L. Fu, Y. Zhu, L. Zhang and Y. Wu, Electrochim. Acta, 2016, 213, 260–269 CrossRef CAS.
- H. Li, X. Yu, Y. Bai, F. Wu, C. Wu, L.-Y. Liu and X.-Q. Yang, J. Mater. Chem. A, 2015, 3, 9578–9586 RSC.
- Y. Xu, F. Bahmani, M. Zhou, Y. Li, C. Zhang, F. Liang, S. H. Kazemi, U. Kaiser, G. Meng and Y. Lei, Nanoscale Horiz., 2019, 4, 202–207 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00484f |
| This journal is © The Royal Society of Chemistry 2019 |