
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D defective graphenes with subnanometric porosity obtained by soft-templating following zeolite procedures†
Lu
Peng
a,
Antonio
Doménech-Carbó
 b,
Ana
Primo
*a and
Hermenegildo
García
*a
b,
Ana
Primo
*a and
Hermenegildo
García
*a
aInstituto Universitario de Tecnología Química CSIC-UPV, Universitat Politècnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es
bDepartament de Química Analítica, Universitat de València, Dr Moliner, 50, 46100 Burjassot, València, Spain
First published on 21st October 2019
Abstract
By applying the well-known templating mechanism employed for the synthesis of mesoporous silicas to the structuration of sodium alginate, a novel defective 3D tubular graphene material (graphenolite) with hierarchical macro/meso/micro-porous structure, very high powder specific surface area (1820 m2 g−1) and regular micropore size (0.6 nm) has been obtained. The key feature of the process is the filmogenic property of alginate that is able to replicate the liquid crystal rods formed by the CTAC template in the aqueous phase. The 3D graphene exhibits 2.5 times higher capacitance using Li+ electrolyte compared to K+, indicating that Li+ can ingress to the ultramicropores which, in contrast, are not accessible to K+. Electrochemical impedance measurements also indicate much lower resistance for Li+ in comparison to K+ electrolyte, confirming the benefits of controlled microporosity of 3D graphene granting selective access to Li+, but not to K+. The present report opens the door for the synthesis of a wide range of 3D graphene materials that could be prepared following similar strategies to those employed for the preparation of zeolites and periodic mesoporous aluminosilicates.
Introduction
3D graphene materials are currently attracting considerable attention since these powders allow combination of the unique physical properties of graphenes in terms of electrical and thermal conductivity with large surface area and porosity in a small footprint. In this way, 3D graphenes having mechanical strength can be applied to the development of highly efficient electrocatalysts1 and as active materials in lithium ion batteries and supercapacitors.2–4 In most of these cases, the ideal graphene material requires the combination of high surface area, accessible porosity and electrical conductivity, exceeding in this way the performance of any carbon material employed so far for these applications.2,4,5Procedures for the preparation of 3D graphenes have been recently reviewed.6–9 Basically, the two alternative methods to prepare 3D graphene are based either on hydrothermal synthesis from graphene oxide or the use of hard templates such as silica spheres or metal sponges.8,10–13 In the latter method, graphenes can be prepared by CVD, starting from methane or other gaseous precursors, or by infiltration of graphene oxide into the voids of hard templates followed by subsequent reconstitution of the graphene sheet.8,9 Besides expanding the precursors and procedures for 3D graphene preparation, what is clearly missing in this area is the preparation of 3D graphene materials with uniform pore sizes below 50 nm and, particularly in the micropore range of a few nanometers. In the large majority of the cases, the 3D graphenes reported so far have well defined pores above 100 nm.8 To achieve microporosity and large surface area, an original preparation procedure is needed.
In this context, the use of soft templates in the preparation of 3D graphenes has been, as far as we know, not yet reported. Soft templates are typically organic molecules that self-assemble in a liquid phase forming micelles, rods or other soft objects of nanometric dimensions that can serve to control the morphology and growth of rigid particles.12,14 Soft templates are widely used for synthesis of zeolites, mesoporous aluminosilicates, porous phosphates and other inorganic materials.15–17 Therefore, it could be in principle expected that this methodology should also be applicable to the synthesis of 3D graphene provided a suitable precursor that could undergo templating by these amphiphilic molecules can be found.
Although saccharides, such as sucrose and others, have been employed in the preparation of carbon replicas using hard templates,18–20 procedures based on the use of soft templates to prepare structured carbon materials have not yet been reported.
Herein, we report a novel procedure for the preparation of a 3D graphene material with remarkable uniform size distribution in the (ultra) micropore range based on the use of cetyltrimethylammonium chloride (CTAC) as the soft template and natural sodium alginate (SA) as the precursor. It is proposed that the key factor responsible for the construction of novel 3D graphitic materials is the known filmogenic ability of alginate fibrils21 which can replicate nanometric CTAC micelles and rods.
Results and discussion
In precedents to the present 3D graphene material (3D G–SA), it has been shown that some natural polysaccharides have a filmogenic ability to reproduce conformal flat surfaces and form defect-free subnanometric films.22,23 Subsequent pyrolysis of these polysaccharide films gives rise to the formation of large area, continuous films of single-layer or few-layer defective graphenes.22 We speculate that the filmogenic ability of polysaccharide could also serve to replicate nanometric soft CTAC rods. As it will be shown below, the novel 3D graphene materials prepared by this methodology exhibit a remarkably high specific surface area ranking at the top of 3D graphene solids reported so far.8 Also, the porosity of these novel 3D graphene solids is hierarchical and uniform at different length scales from macro to ultra microporosity, with most of the pore volume corresponding to the subnanometric range, much below the dimensions of the related 3D graphene materials reported until now and reaching the 0.6 nm target value considered optimal for Li-ion batteries.5Scheme 1 illustrates the process of formation of 3D G–SA followed in the present study. Basically the process consists in mixing solutions of appropriate weights of SA and CTAC that are subsequently submitted to hydrothermal treatment in an autoclave at 100 °C for 24 h to produce SA structuration. The templated polysaccharide precursor containing CTAC (CTAC–SA) was recovered by water evaporation. 3D graphene was finally obtained by pyrolysis of CTAC–SA constituted by templated SA embedding CTAC. Pyrolysis was carried out under an Ar flow at 900 °C for 2 h. During the pyrolysis, CTAC decomposes and the byproducts evaporate, while SA transforms into few-layer graphene (see below the thermogravimetric profile of the CTAC–SA material). It has been reported that SA is converted into defective graphene upon pyrolysis at temperatures of 900 °C.24
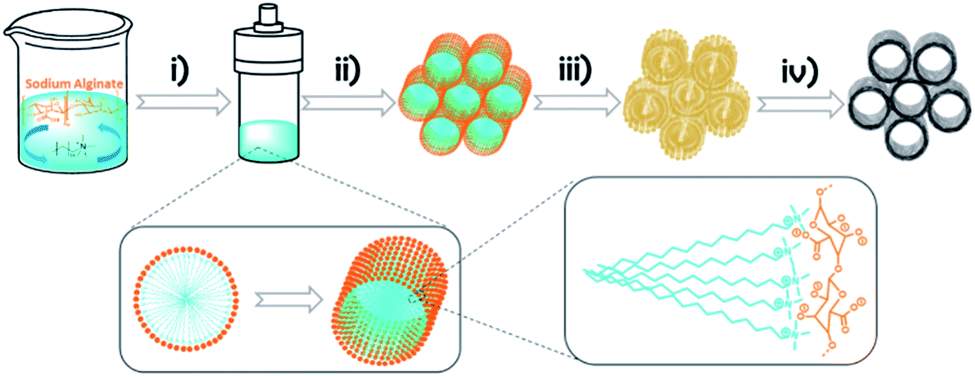 | ||
| Scheme 1 Illustration of the formation of 3D G–SA based on the use of CTAC as the soft template: (i) hydrothermal method; (ii) solvent evaporation; (iii) polysaccharide replication of nanometric soft CTAC rods; (iv) pyrolysis in an argon flow at 900 °C for 2 h. | ||
Formation of defective graphene in the process was ascertained by Raman spectroscopy (see the ESI†). Thus, the Raman spectrum of 3D G–SA exhibits the characteristic 2D, G and D peaks, appearing at 2700, 1590 and 1350 cm−1, respectively, as previously reported for the defective graphene obtained from the pyrolysis of SA.10,24 The G to D peak intensity ratio of about 1.15 is also in agreement with the reported value of this type of graphene.24
X-ray photoelectron spectroscopy (XPS) shows the presence of C and O which is in agreement with the expected composition of defective graphenes derived from SA as the precursor (see Fig. 1). However, small amounts of N, presumably coming from CTAC decomposition, were also detected in the composition of 3D G–SA. The atomic proportion at the surface of the 3D G–SA sample determined by XPS was 85.19, 14.33 and 0.48 at% for C, O and N, respectively. This corresponds to a weight proportion of 81.26, 18.21 and 0.53 wt% for C, O and N, respectively, which agrees well with the combustion elemental analysis data measured independently.
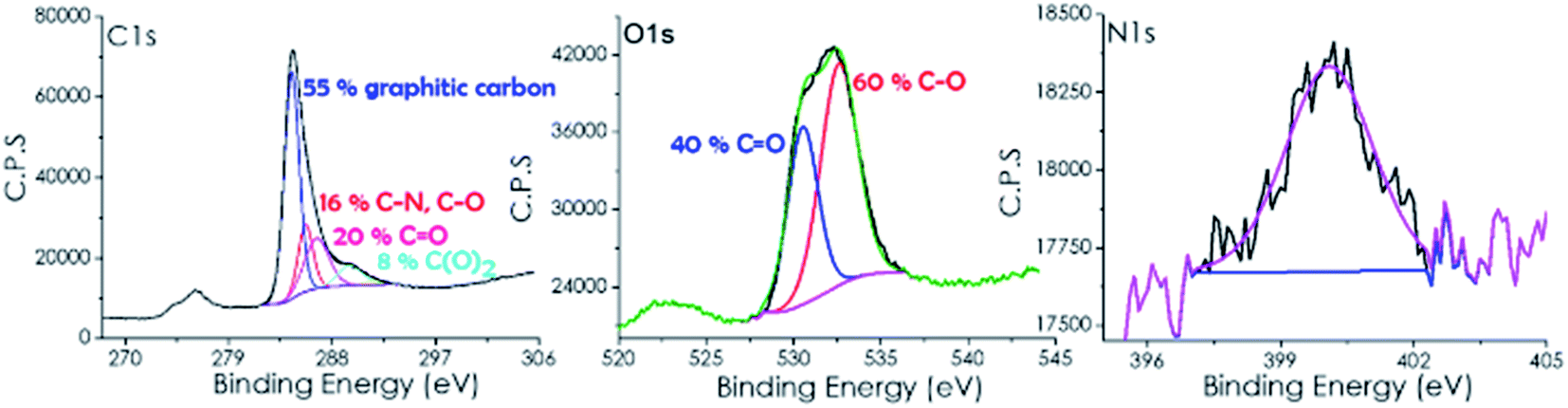 | ||
| Fig. 1 High resolution XPS C 1s, O 1s and N 1s peaks and the best fit to individual components recorded for 3D G–SA. | ||
High-resolution XPS of the C 1s peak shows that it can be deconvoluted into three main components corresponding to graphitic carbon (284.45 eV, 55%), carbon bonded to nitrogen or oxygen through a single bond (285.7 eV, 16%) and carbon bonded by a double bond to oxygen (286.7 eV, 20%) and carboxylic groups (289.6 eV, 8%). These binding energy values and proportions are similar to those previously reported for other defective graphene samples derived from SA as a precursor.25
Analysis of the O 1s peak indicates the presence of two different types of oxygen atoms appearing at binding energy values of 530.56 (40%) and 532.6 (60%) eV, which are attributable to two oxygen families bonded to carbon with double and single bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O).
O and C–O).
As commented, XPS 3D G–SA also exhibits the presence of some nitrogen atoms appearing at about 400 eV with a low atomic percentage of 0.48%.
The above characterization indicates that, as expected in view of the numerous precedents on the conversion of SA into defective graphene with a residual oxygen content,26,27 spectroscopic data of 3D G–SA confirm this transformation in the present case with the occurrence of a low N doping. However, the previous data do not provide any insight on the role of CTAC in the templating and formation of a 3D structure. To address the issue of the morphology and structure of 3D G–SA, the material was further characterized by gas adsorption and scanning and transmission electron microscopy.
Thus, the specific surface area of the 3D G–SA obtained by pyrolysis at 900 °C was initially measured by isothermal N2 adsorption at 77 K, obtaining a value of 1185 m2 g−1 (Fig. 2). This high specific surface area value ranks the 3D G–SA material obtained at 900 °C among the powdered graphene samples with the highest specific surface area ever measured by gas adsorption. It should be noted that gas adsorption measurements on solid samples of powdered graphene typically give much lower specific surface area values in the range of 100 m2 g−1 due to the stacking of the layers and reduced micro/mesoporosity.28 On the other hand, Ar adsorption determines the specific pore volume of 3D G–SA to be 0.61 cm3 g−1, most of the pore system having a diameter below 2 nm with a maximum at 0.6 nm (see Fig. 2).
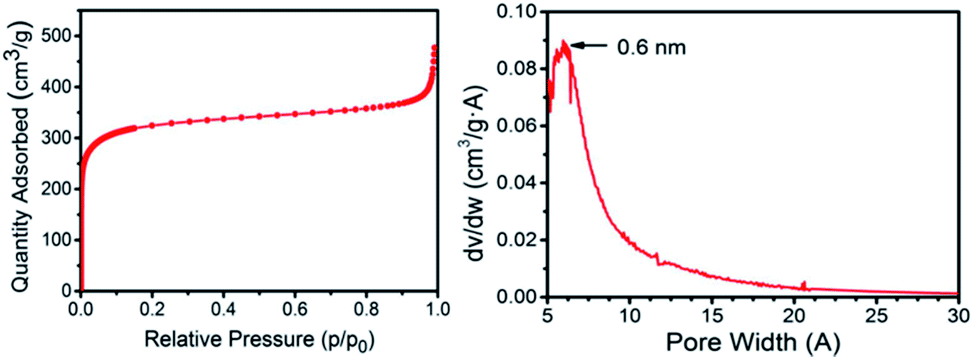 | ||
| Fig. 2 N2 adsorption isotherm at 77 K and pore size distribution measured by Ar for the 3D G–SA sample obtained by pyrolysis at 900 °C. | ||
Considering the small pore size determined by Ar adsorption, it was of interest to determine the possible presence of ultra microporosity by measuring CO2 adsorption at 273 K (see the ESI†). Measurements by CO2 adsorption give a specific surface area value of 1820 m2 g−1. This discrepancy between the specific surface area values determined with N2 at 77 K and CO2 at 273 K can be ascribed to the presence of ultra micropores that are accessible to CO2, but not N2.29,30 Therefore, another feature of 3D G–SA besides high surface area that makes this structured 3D graphene unique is the presence of ultra microporosity with a quite narrow pore size distribution centered at 0.6 nm, never reported in this type of materials. Note that isothermal gas adsorption cannot be routinely used to determine pore dimensions much below this value. Based on the theoretical maximum surface area of graphene of about 2630 m2 g−1, it can be concluded that the average wall configuration in 3D G–SA should be between one and two graphene layers. This estimation was confirmed by transmission electron microscopy measurements (see the ESI†).
In addition, the CO2 adsorption capacity of 3D G–SA at 298 K and 100 kPa was 3.44 mmol of CO2 per g (151 mg CO2 per g), which is a value very similar to other reported values for SWCNTs,31 in line with the above estimation about the average wall thickness in 3D G–SA. The isosteric heat of adsorption of CO2 slightly decreases from 32 to 25 kJ mol−1 with the increase of CO2 adsorption (see the ESI†). This value is considered in the area as a good compromise to achieve high adsorption capacity and adequate regenerability.
Besides micro- and ultra microporosity with a defined subnanometric pore size and high surface area, the templating effect of CTAC on SA was clearly evidenced by recording a set of scanning electron microscopy (SEM) images upon treatment of the CTAC–SA solid at increasing temperatures in the range from 300 to 900 °C. Note that previous studies have determined that pyrolysis of SA at temperatures of 1200 °C results in the volatilization of all the material.32 These images reveal that the CTAC–SA material was constituted by spheres, coated by thin films. By adjusting the CTAC–SA mass ratio to an optimal value (weight ratio about 1), the solid was constituted mainly by spheres of about 1.7 μm diameter and the amount of residual film coating the spheres notably decreased. Heating this CTAC–SA precursor in the range from 300 to 600 °C results in the progressive opening of the spheres, showing that their interior is constituted by long tubes in accordance with the templating mechanism expected for CTAC as indicated in Scheme 1. In this temperature range, some of these tubes, but not all, exhibit an opening at the center.
SEM images upon treatment of CTAC–SA at 900 °C show that at this temperature, the spheres are completely opened and the array of internal long quasi-parallel tubes have fully developed. The 3D G–SA material obtained at this temperature due to SA graphitization exhibits tubes of about 200 nm diameter and about 1 μm length and all of them show at this stage a central opening of about 40 nm that we propose is formed due to the escape of CTAC and those gases formed in the pyrolysis. Fig. 3 provides a set of SEM images to illustrate the morphological changes that occur upon thermal treatment of the as-formed CTAC–SA spheres. Fig. S4† in the ESI† provides additional SEM images of the resulting 3D G–SA formed at 900 °C. SEM images also show the presence of some broken tubes clearly revealing that they are empty along the long axis direction of the tube and a minor proportion of debris generated by some damage of the material. A closer look at the walls reveals some apparent roughness. It would be commented later, based on TEM images, that this apparent roughness can be attributed to the parallel arrangement of inner tubes with smaller diameters and thin walls.
 | ||
| Fig. 3 SEM images of as-prepared CTAC–SA (a) and that treated at 300 (b) or 900 °C (c and d). Images (c) and (d) correspond, therefore, to the 3D G–SA sample referred in the text. | ||
These morphological changes of CTAC–SA with temperature can be interpreted correlating the evolution of the SEM images with thermogravimetry (TG, Fig. 4). It should be noted that according to the TG profile, CTAC–SA loses about 63 wt% upon heating at 300 °C. This weight loss has been attributed to the decomposition of the CTAC template that should, therefore, correspond to more than one half of the CTAC–SA weight. Heating at higher temperatures up to 900 °C produces an additional gradual weight loss of about 15 wt% that corresponds to graphitization of the SA fibrils as has been previously observed in the pyrolysis of SA in the absence of CTAC.32 Therefore, upon pyrolysis, the initial weight of CTAC–SA is decreased to about 14% of the original value. Once SA has been converted into defective graphene, the resulting material does not undergo significant weight loss in the temperature range from ambient to 400 °C.
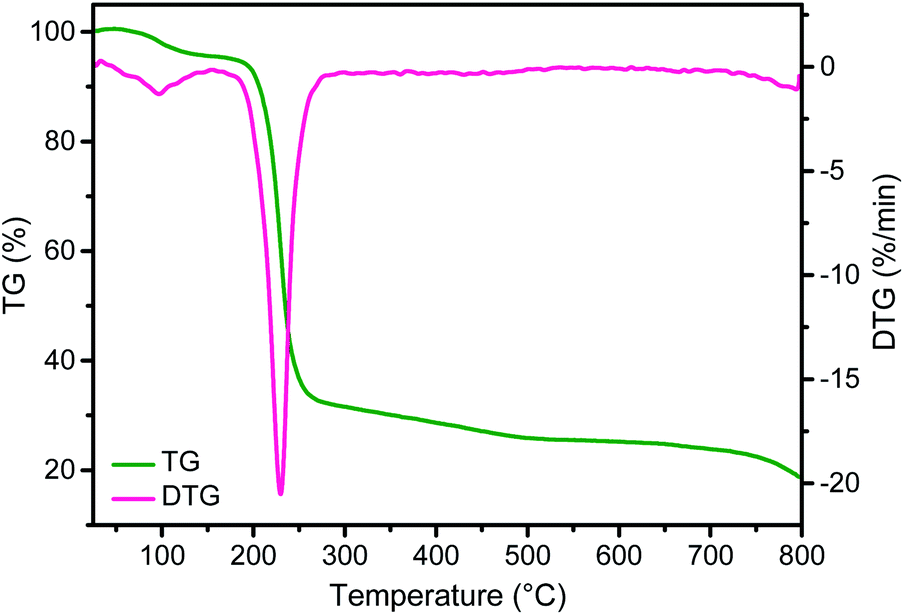 | ||
| Fig. 4 TG profile and differential scanning calorimetry of the CTAC–SA sample. | ||
The structure of these tubes can be further studied by TEM (Fig. 5). It is shown by TEM that these long tubes seen in the SEM images are constituted by an array of parallel tubes of about 20 nm diameter. These 20 nm tubes have been marked in the image and still contain other inner parallel tubes. High-resolution TEM images of 3D G–SA pyrolyzed at 900 °C show the high crystallinity of the very thin walls of about 1 nm thickness corresponding to single or few-layer graphene, as already determined by Raman and XP spectroscopy and in accordance with surface area measurements. It is proposed that this array of long tubular objects correspond to the precise coating by filmogenic SA fibrils of the CTAC rods. Of note is that the ultramicroporosity determined by isothermal Ar adsorption with an average diameter of 0.6 nm cannot be observed in TEM due to the lack of the necessary resolution at this dimension, particularly for materials constituted by light elements like carbon.
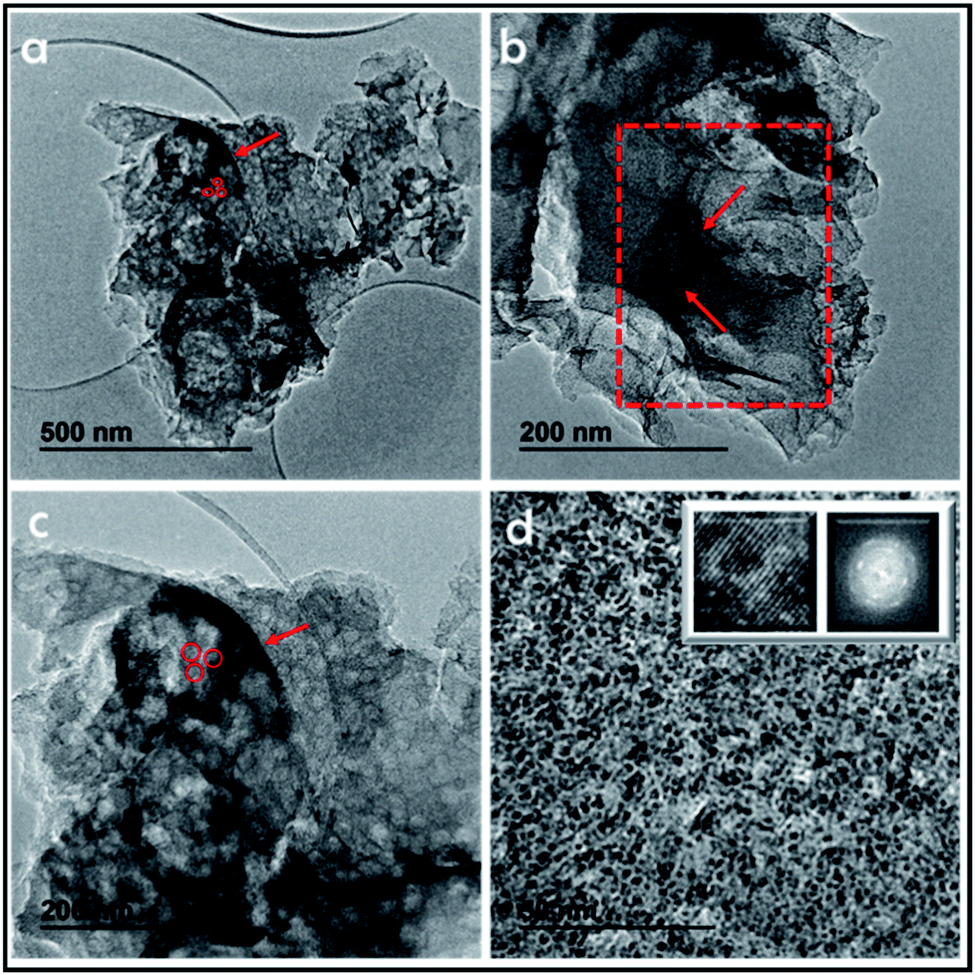 | ||
| Fig. 5 TEM images of 3D G–SA pyrolyzed at 900 °C after ultrasonication at increasing magnifications. The presence of wider tubes between 100 and 200 nm can be seen in images (a–c). These wide tubes contain narrower tubes of about 20 nm (marked by red arrows in frames (a–c)). Higher magnification in image (d) shows even narrower tubes in the nanometric range. Insets of frame (d) show a high resolution image showing the structural atomic arrangement, while selected area electron diffraction shows the polycrystalline nature of the material. | ||
This proposal is in accordance with the mechanism indicated in Scheme 1, which is based on the accepted mechanism for the formation of MCM-41 by CTAC templating of porous silica.33–35Scheme 2 presents a pictorial illustration of the hierarchical structure and the morphological changes undergone by CTAC–SA upon heating at increasing temperatures. Structuration of CTAC by self-assembly and coating by SA of the liquid crystal assembled CTAC nanorods make possible the observation of rods at the lowest temperature for which SA decomposition occurs, i.e. 300 °C, first stage in Scheme 2. Higher temperatures cause the opening of the tubes due to the escape of gases generated by decomposition of the organic molecules, second stage in Scheme 2. Note that pyrolysis of CTAC-SA is carried out in one step and that Scheme 2 indicates two stages that have been identified in the evolution of the material depending on the temperature.
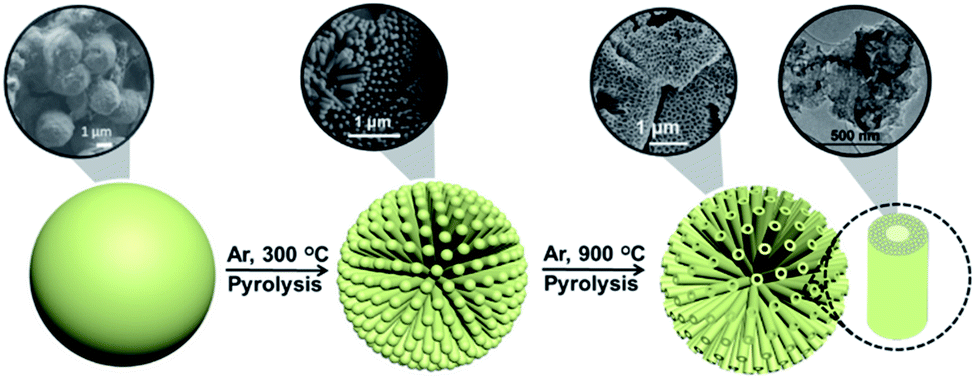 | ||
| Scheme 2 Illustration of the hierarchical structure and morphological changes undergone by CTAC–SA with temperature. | ||
As commented in the Introduction, one of the main applications of 3D graphenes is in the field of electrocatalysis and Li-ion batteries. It has been proposed that the ideal carbon electrode should, in addition to high surface area, exhibit a microporosity of about 0.7 nm to establish strong interactions between the Li+ ions and the graphene walls.5 In this regard, the present 3D G–SA should be an ideal material. Preliminary electrical conductivity measurements of 0.5 mm thick compressed wafers of 3D G–SA powder give a low electrical resistivity value of 11.75 Ω cm, indicating the suitability of 3D G–SA as an electrocatalytic material. Therefore, it was of interest to test the electrochemical performance of 3D G–SA with respect to Li+ ion insertion. Aimed at determining the influence of Li+ ions on the electrochemical response, cyclic voltammetry of 3D G–SA was recorded in 0.1 M KH2PO4 aqueous electrolyte at pH 7 observing the expected rectangular-type profile corresponding to a capacitative electrochemical response with no observable faradaic peaks (Fig. 6). This electrochemical behavior is characteristic of carbon materials.5,36 However, addition of 0.02 M LiClO4 to the electrolyte causes a significant change in the electrochemical response (Fig. 6), not only increasing the area of the rectangular capacitative background by a factor of about 2.5 times, but also allowing the observation of an anodic (0.4 V, marked as Ag in Fig. 6) and a cathodic (−1.2 V, marked as Cg in Fig. 6) peak that can be attributed to the oxidation and reduction of some oxygenated functional groups present in the defective graphene sheets constituting 3D G–SA, as indicated in eqn (1) and (2). In these equations G indicates the graphene sheet, while the redox pairs have been denoted as C–OH (hydroxyl) and CO (carbonyl).
| {G–C–OH} → {G–CO} + H+ (aq) + e− | (1) |
| {G–CO} + Li+ (aq) + e− → {G–O− ⋯ Li+} | (2) |
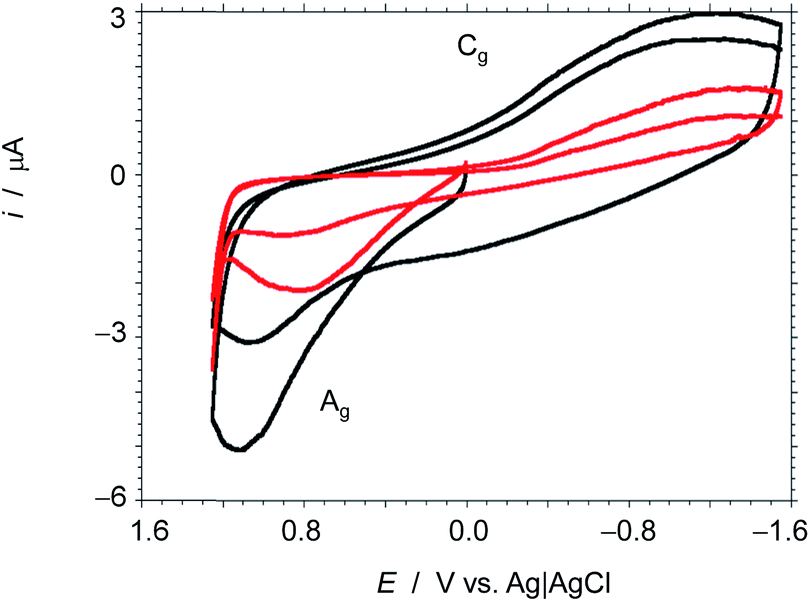 | ||
| Fig. 6 Cyclic voltammograms at glassy carbon electrodes modified with 3D G–SA prepared by pyrolysis at 900 °C immersed into 0.10 M KH2PO4 buffer at pH 7.0 (red), and the same plus 0.02 M LiClO4 (black). Potential scan rate 50 mV s−1. | ||
The voltammograms do not show typical features characterizing adsorption-mediated processes and the presence of Li+ in the electrolyte enhances significantly the cathodic wave for the reduction of dissolved oxygen (see Fig. S5 in the ESI†). Observation of these changes in the electrochemical response by the presence of Li+ in the electrolyte can be attributed to the ability of hydrated Li+ ions, due to their small radii (diameter 0.416 nm),37 to ingress into the ultra micropores (average diameter 0.6 nm) that are not easily accessible to K+ ions (diameter 0.558 nm) due to their larger size.
The remarkable influence of Li+vs. K+ ion on the electrochemical performance of 3D G–SA can also be inferred from electrochemical impedance spectroscopy, by comparing the response of electrodes of this material in contact with air-saturated 5 × 10−2 M aqueous electrolyte of LiCl or KCl salts in the frequency range from 105 to 10−1 Hz upon application of a bias potential sufficiently negative to promote the oxygen reduction reaction (ORR). Under these conditions the ORR process acts as a redox probe to test the electron transfer ability of the modified electrode. The corresponding Nyquist plot is presented in Fig. 7 showing an apparently unique capacitive depressed loop with clearly much higher impedance in the case of the KCl electrolyte than for the LiCl one at all tested frequencies. The spectra do not show linear features characterizing important diffusive effects. Interestingly, the Bode plot of the (minus) phase angle vs. logarithm of frequency (Fig. 7 b) shows clearly higher (minus) phase angles, i.e., larger capacitive effects, for the impedance measurement in KCl electrolyte. This feature cannot be associated with double layer or diffusive effects in the electrolyte as capacitance effects can be mainly associated with graphene surface oxidation and functionalization.38 Then, the observation of large differences in the capacitive effects in the presence of Li+ and K+ ions is consistent with the idea of Li+ intercalation.
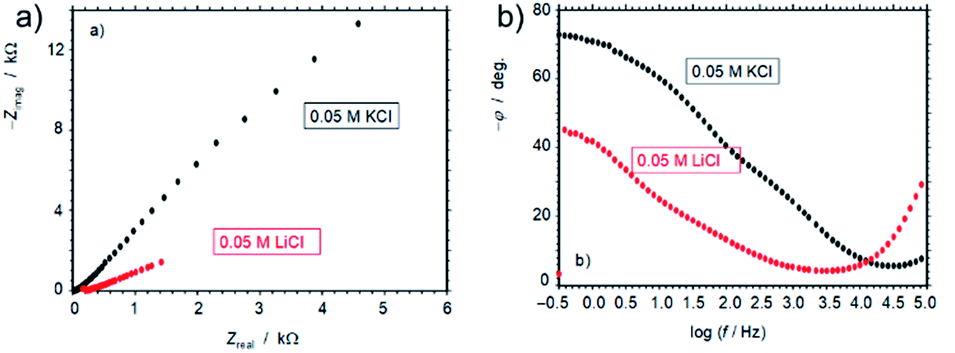 | ||
| Fig. 7 (a) Nyquist and (b) Bode plots for electrochemical impedance spectroscopy of films of 3D G–SA prepared by pyrolysis at 900 °C on glassy carbon electrodes at air-saturated 0.05 M KCl and 0.05 M LiCl aqueous solutions. Bias potential of −0.65 V vs. Ag/AgCl. | ||
Experimental
Sample preparation
Commercially available reagents were purchased from Aldrich and used without further purification.In a general preparation, 100 mg SA from Aldrich and 0.4 mL 25 wt% CTAC aqueous solution were dissolved in 80 mL and 20 mL milliQ H2O, respectively. Different materials were similarly prepared varying SA and CTAC weights. After 2 h magnetic stirring at room temperature, CTAC aqueous solution was slowly added into SA aqueous solution under magnetic stirring. The mixed solution was further stirred for 6 h at room temperature. Then, the solution was transferred to a Teflon-lined autoclave and heated at 100 °C under autogenous pressure for 24 h. The CTAC–SA solid was obtained by water evaporation. To obtain 3D G–SA, the CTAC–SA was pyrolyzed under an Ar flow (200 mL min−1), increasing the temperature at a rate of 5 °C min−1 up to 150 °C then 1 °C min−1 up to 900 °C and holding for 2 h.
Sample characterization
Raman spectra were collected with a Horiba Jobin Yvon-Labram HR UV-visible-NIR (200−1600 nm) Raman Microscope Spectrometer using a 512 nm laser. The spectrum was collected by averaging 10 scans at a resolution of 2 cm−1. Scanning electron microscopy (SEM) images were acquired by using a JEOL JSM 6300 apparatus. HRTEM images were recorded in a JEOL JEM 2100F under an accelerating voltage of 200 kV. Samples were prepared by applying one drop of the suspended material in ethanol onto a carbon-coated nickel TEM grid and allowing them to dry at room temperature.XP spectra were measured on a SPECS spectrometer equipped with a Phoibos 150 9MCD detector using a nonmonochromatic X-ray source (Al and Mg) operating at 200 W. The samples were evacuated in the prechamber of the spectrometer at 1 × 10−9 mbar. The measured intensity ratios of the components were obtained from the area of the corresponding peaks after nonlinear Shirley-type background subtraction and corrected by the transition function of the spectrometer.
The chemical composition of the samples was determined by combustion chemical analysis by using a CHNS FISONS elemental analyser. The micropore volume and specific surface area of the solids were measured by N2 or Ar adsorption isotherms at −196 or −186 °C, respectively, by using a Micromeritics ASAP 2010 instrument. The CO2 adsorption isotherms in the low pressure range were measured by using a Micromeritics ASAP 2010 instrument with approximately 200 mg of solid placed in a sample holder, which was immersed into a liquid circulation thermostatic bath for precise temperature control. Before each measurement, the sample was treated overnight at 400 °C under vacuum, CO2 adsorption isotherms were then acquired at 0, 10, 20, 30 and 40 °C.
Electrochemical measurements
Electrochemical measurements were performed with a CH 660I potentiostat using 3D G–SA films deposited on a glassy carbon electrode (BAS MF 2012, geometrical footprint area 0.071 cm2) as working electrodes. These electrodes were prepared by evaporation of 50 μL of a suspension (1 mg mL−1) of 3D G–SA in a 0.5% solution of Paraloid B52 in acetone on the surface of a glassy carbon electrode. The three-electrode cell was completed with an Ag/AgCl (3 M NaCl) reference electrode and a Pt wire auxiliary electrode.Electrochemistry was studied in air-saturated 0.10 M aqueous potassium phosphate buffer at pH 7.0 adding different concentrations of LiClO4. Optionally the solutions were deaerated by bubbling Ar for at least 10 min. Electrochemical impedance spectroscopy measurements were performed at the open circuit potential with a bias potential of −0.65 V vs. Ag/AgCl in 0.05 M LiCl and KCl air-saturated aqueous solutions using a sinusoidal potential modulation of ±5 mV amplitude in the 105–10−1 Hz range. The bias potential was selected to allow the reduction of dissolved oxygen, acting as a redox probe, as already described.39 In these experiments a Pt-wire was used as a pseudo-reference electrode, the potentials being calibrated after addition of 0.5 mM ferrocene as internal standard.
Conclusions
In conclusion, CTAC as a soft template in aqueous solutions is able to template SA fibrils forming spheres containing hierarchiral tubular structures. Upon pyrolysis at 900 °C, a hierarchical 3D graphene material with a very large surface area (1820 m2 g−1) and ultramicropores (0.6 nm) is formed. The tubes are constituted by few-layer graphene walls of nanometric thickness. The uniqueness of this material is that the micropore size is in the range predicted for maximum interaction with Li+ ions, a fact that is relevant in Li+ ion batteries and supercapacitors. Electrochemical measurements confirm that the material can discriminate between Li+ and K+ ions, the capacitance being larger and the resistance lower for Li+ ions. It is proposed that the present procedure based on soft templating similar to that employed for the synthesis of zeolites and periodic aluminosilicates can be expanded to the preparation of a broad family of 3D structured porous graphenes with tunable structures (graphenolite).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Spanish Ministry of Economy and Competitiveness (Severo Ochoa and RTI2018-98237-CO2-R1) and Generalitat Valenciana (Prometeo 2017-083) is gratefully acknowledged. A.P. thanks the Spanish Ministry of Science and Education for a Ramon y Cajal research associate contract. We are indebted to Dr Miguel Palomino for CO2 adsorption measurements and Dr Pedro Atienzar for measurement of the electrical conductivity.Notes and references
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- X. Cao, Y. Shi, W. Shi, G. Lu, X. Huang, Q. Yan, Q. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- X.-C. Dong, H. Xu, X.-W. Wang, Y.-X. Huang, M. B. Chan-Park, H. Zhang, L.-H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS PubMed.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- A. Eftekhari, Mater. Today Chem., 2018, 7, 1–4 CrossRef CAS.
- Q. Ke and J. Wang, J Materiomics, 2016, 2, 37–54 CrossRef.
- Z.-S. Wu, Y. Sun, Y.-Z. Tan, S. Yang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 19532–19535 CrossRef CAS PubMed.
- S. Mao, G. Lu and J. Chen, Nanoscale, 2015, 7, 6924–6943 RSC.
- Y. Ito, Y. Tanabe, K. Sugawara, M. Koshino, T. Takahashi, K. Tanigaki, H. Aoki and M. Chen, Phys. Chem. Chem. Phys., 2018, 20, 6024–6033 RSC.
- J. Niu, A. Domenech-Carbó, A. Primo and H. Garcia, RSC Adv., 2019, 9, 99–106 RSC.
- A. Rendón-Patiño, J. Niu, A. Doménech-Carbó, H. García and A. Primo, Nanomaterials, 2019, 9, 101 CrossRef PubMed.
- J. Liu, T. Yang, D.-W. Wang, G. Q. M. Lu, D. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798 CrossRef.
- M.-P. Pileni, Nat. Mater., 2003, 2, 145 CrossRef CAS PubMed.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- D. W. Lewis, D. J. Willock, C. R. A. Catlow, J. M. Thomas and G. J. Hutchings, Nature, 1996, 382, 604 CrossRef CAS.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 CrossRef CAS PubMed.
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- H. Yang and D. Zhao, J. Mater. Chem., 2005, 15, 1217–1231 CAS.
- Y. Sakamoto, T. W. Kim, R. Ryoo and O. Terasaki, Angew. Chem., Int. Ed., 2004, 43, 5231–5234 CrossRef CAS PubMed.
- R. Muzzarelli, S. Aiba, Y. Fujiwara, T. Hideshima, C. Hwang, M. Kakizaki, M. Izume, N. Minoura, C. Rha and T. Shouij, in Chitin in Nature and Technology, Springer, 1986, pp. 389–402 Search PubMed.
- A. Primo, P. Atienzar, E. Sanchez, J. M. Delgado and H. García, Chem. Commun., 2012, 48, 9254–9256 RSC.
- M. Latorre-Sánchez, A. Primo, P. Atienzar, A. Forneli and H. García, Small, 2015, 11, 970–975 CrossRef PubMed.
- M. M. Trandafir, M. Florea, F. Neaţu, A. Primo, V. I. Parvulescu and H. García, ChemSusChem, 2016, 9, 1565–1569 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. Primo, P. Concepcion, M. Alvaro and H. Garcia, Chem.–Eur. J., 2013, 19, 7547–7554 CrossRef CAS PubMed.
- M. Latorre-Sánchez, A. Primo and H. García, Angew. Chem., Int. Ed., 2013, 52, 11813–11816 CrossRef PubMed.
- A. Primo, A. Forneli, A. Corma and H. García, ChemSusChem, 2012, 5, 2207–2214 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- J. Jagiello and M. Thommes, Carbon, 2004, 42, 1227–1232 CrossRef CAS.
- D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1233–1242 CrossRef.
- M. Cinke, J. Li, C. W. Bauschlicher, A. Ricca and M. Meyyappan, Chem. Phys. Lett., 2003, 376, 761–766 CrossRef CAS.
- J. He, A. Anouar, A. Primo and H. Garcia, Nanomaterials, 2019, 9, 895 CrossRef PubMed.
- C.-Y. Chen, S. L. Burkett, H.-X. Li and M. E. Davis, Microporous Mater., 1993, 2, 27–34 CrossRef CAS.
- C. Kresge, M. Leonowicz, W. J. Roth, J. Vartuli and J. Beck, nature, 1992, 359, 710 CrossRef CAS.
- C.-Y. Chen, H.-X. Li and M. E. Davis, Microporous Mater., 1993, 2, 17–26 CrossRef CAS.
- A. Balducci, R. Dugas, P.-L. Taberna, P. Simon, D. Plee, M. Mastragostino and S. Passerini, J. Power Sources, 2007, 165, 922–927 CrossRef CAS.
- E. Casero, A. Parra-Alfambra, M. Petit-Domínguez, F. Pariente, E. Lorenzo and C. Alonso, Electrochem. Commun., 2012, 20, 63–66 CrossRef CAS.
- L. Osmieri, A. H. M. Videla and S. Specchia, J. Solid State Electrochem., 2016, 20, 3507–3523 CrossRef CAS.
- R. Ballesteros-Garrido, M. de Miguel, A. Doménech-Carbó, M. Alvaro and H. Garcia, Chem. Commun., 2013, 49, 3236–3238 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00554d |
| This journal is © The Royal Society of Chemistry 2019 |