
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhage-mimicking antibacterial core–shell nanoparticles†
Juliane
Hopf
a,
Margo
Waters
b,
Veronica
Kalwajtys
c,
Katelyn E.
Carothers
c,
Ryan K.
Roeder
 bd,
Joshua D.
Shrout
a,
Shaun W.
Lee
c and
Prakash D.
Nallathamby
*bde
bd,
Joshua D.
Shrout
a,
Shaun W.
Lee
c and
Prakash D.
Nallathamby
*bde
aDepartment of Civil and Environmental Engineering and Earth Sciences, University of Notre Dame, Notre Dame, IN, USA
bDepartment of Aerospace and Mechanical Engineering, University of Notre Dame, Notre Dame, IN, USA. E-mail: pnallath@nd.edu; Tel: +1 574 631 7868
cDepartment of Biological Sciences, University of Notre Dame, Notre Dame, IN, USA
dCenter for Nanoscience and Technology (NDnano), University of Notre Dame, Notre Dame, IN, USA
eCenter for Advanced Diagnostics and Therapeutics (AD&T), University of Notre Dame, Notre Dame, IN, USA
First published on 7th November 2019
Abstract
The increasing frequency of nosocomial infections caused by antibiotic-resistant microorganisms concurrent with the stagnant discovery of new classes of antibiotics has made the development of new antibacterial agents a critical priority. Our approach is an antibiotic-free strategy drawing inspiration from bacteriophages to combat antibiotic-resistant bacteria. We developed a nanoparticle-based antibacterial system that structurally mimics the protein-turret distribution on the head structure of certain bacteriophages and explored a combination of different materials arranged hierarchically to inhibit bacterial growth and ultimately kill pathogenic bacteria. Here, we describe the synthesis of phage-mimicking antibacterial nanoparticles (ANPs) consisting of silver-coated gold nanospheres distributed randomly on a silica core. The silver-coating was deposited in an anisotropic fashion on the gold nanospheres. Structurally, our nanoparticles mimicked the bacteriophages of the family Microviridae by up to 88%. These phage-mimicking ANPs were tested for bactericidal efficacy against four clinically relevant nosocomial pathogens (Staphylococcus aureus USA300, Pseudomonas aeruginosa FRD1, Enterococcus faecalis, and Corynebacterium striatum) and for biocompatibility with skin cells. Bacterial growth of all four bacteria was inhibited (21% to 90%) as well as delayed (by up to 5 h). The Gram-positive organisms were shown to be more sensitive to the nanoparticle treatment. Importantly, the phage-mimicking ANPs did not show any significant cytotoxic effects against human skin keratinocytes. Our results indicate the potential for phage-mimicking antimicrobial nanoparticles as a highly effective, alternative antibacterial agent, which may be suitable for co-administration with existing available formulations.
1. Introduction
In 2013, the Centers for Disease Control and Prevention (CDC) published a list of bacteria causing severe human infections and currently presenting the highest risk of developing multidrug resistance.1 Most of these bacteria can be found in regular healthcare/hospital settings, spreading via surfaces and human contact. The ESKAPE pathogens Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species are the leading cause of nosocomial infections (hospital-acquired infections, HAI) world-wide2 and increasingly exhibit multidrug resistance to most available classes of antibiotics.3 More than two-thirds of HAI will turn into chronic, skin, or high-density biofilm infections, which are difficult to treat due to their virulent and adaptive nature.4At the current low rate of antibiotic discovery and development,5 we may lose the race to contain antibiotic-resistant bacterial strains. Without urgent action, antibiotic-resistant infections will kill more patients per year by 2050 than all cancers combined.6 Due to the fast emergence and global spread of new resistance, presently treatable infections such as pneumonia, tuberculosis, gonorrhea, and salmonellosis will become harder or near impossible to treat.7 There is a critical need for broad-spectrum antibacterial agents independent of antibiotics to (a) effectively prevent the progression of antibiotic resistance and (b) provide a necessary alternative to fight infectious diseases.
The use of nanomaterials as alternative antibacterial agents has shown promise owing to their unique physical and chemical properties.8–11 Nanoparticles (NPs) are below 100 nm in size and exhibit a high surface-to-volume ratio, which enables close interaction with microbial membranes.12 Specifically, metal and metal oxide NPs have gained attention as a new class of antibacterial agents over the last decade due to their bactericidal properties.13 NPs containing Ag, Au, Zn, Cu, Ti, Mg, Ni, Ce, Se, Al, Cd, Y, Pd, and super-paramagnetic Fe have been studied in detail.8,14 Among them, silver NPs showed the most effective, concentration-dependent bactericidal activity against Escherichia coli and Pseudomonas aeruginosa.15 Metal and metal oxide NPs inhibit the growth of bacteria (e.g. Staphylococcus aureus) by a combination of mechanisms (induction of oxidative stress, cell membrane disruption, and release of metal ions).16 The advantage of utilizing metal NPs over antibiotics is that the probability of development of bacterial resistance to NPs is less likely.8 But some bacteria (Pseudomonas aeruginosa and Bacillus subtilis) have been found to utilize efflux pumps to rid themselves of NPs17 effectively. Additionally, the high toxicity of these elemental NPs to eukaryotic cells further narrows their therapeutic dosage range.14
We hypothesized that the drawbacks associated with antibacterial NPs might be minimized by modularly assembling individually potent antibacterial components into a hierarchical nanostructure accentuating only the desired antibacterial properties of the components while minimizing their toxicity to normal healthy cells. A previous study investigated the shape-dependent antibacterial activity of NPs but used mono-elemental (silver) NPs that were not rationally designed to mimic any naturally occurring antibacterial agents.17 We designed modular NPs that mimic the structure and dimensions of bacteriophages. Phages use structural interactions to infect only bacteria exhibiting the appropriate nanoscale features on the bacterial membrane, and the infectious activity of phages is unaffected by bacterial antibiotic resistance.18,19 97% of bacteriophages infecting ESKAPE organisms belong to the Caudovirales, an order of tailed bacteriophages (45–170 nm).20 We synthesized NPs to mimic tailless bacteriophages as a more scalable and reproducible approach compared to mimicking tailed bacteriophages. Therefore, our phage-mimicking ANPs targeted the evolutionarily conserved bacterial membrane with high specificity by structurally mimicking the architecture and function of tailless phages.
Our phage-mimicking ANPs were composed of a silica core studded with smaller, silver-coated gold nanospheres on its surface, mimicking the protein-turret distribution density and high surface-area of icosahedral phage heads (e.g., ΦX174).21 These phage-mimicking ANPs were designed to (a) electrostatically attract negatively charged bacterial cell membranes with pockets of positive surface charge on the exposed surfaces of the amine-functionalized silica cores, (b) create a high surface area antibacterial silver coating with negligible silver ion leaching compared with current antibacterial formulations, (c) inhibit bacterial growth, (d) be non-toxic to and biocompatible with human cells, and (e) facilitate scalable manufacturing. The synthesis of these phage-mimicking ANPs was confirmed by Transmission Electron Microscopy (TEM) and Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES). Bactericidal efficiency was evaluated against four clinically relevant nosocomial pathogens (Staphylococcus aureus USA300, Pseudomonas aeruginosa FRD1, Corynebacterium striatum, and Enterococcus faecalis). The ethidium homodimer based dead cell detection assay was used to verify that the phage-mimicking ANPs were noncytotoxic to HaCaT keratinocyte cells (human skin).
2. Methods and materials
2.1 Materials
Staphylococcus aureus USA300;22Corynebacterium striatum;23,24Enterococcus faecalis;24,25Pseudomonas aeruginosa FRD1;26 200 proof ethanol (VWR); 20% w/v ammonium hydroxide (NH4OH; BDH); tetraethyl orthosilicate (TEOS; Sigma Aldrich, 99% GC grade); 3-aminopropyltriethoxysilane (APTES; Sigma Aldrich, 99%); sodium hydroxide (NaOH; Sigma Aldrich, >97%); sodium citrate dihydrate (C6H5Na3O7·2H2O; Sigma Aldrich, >99%); tetrakis(hydroxymethyl)phosphonium chloride (80% purity) (THPC; Sigma Aldrich, 80% in water); gold chloride (anhydrous; Sigma Aldrich); silver nitrate (AgNO3; Sigma Aldrich, 99% ACS grade); hydroquinone (Sigma Aldrich, >99% reagent grade); deionized water (DI water); 16% w/v paraformaldehyde (Beantown Chemical); Dulbecco's Modified Eagle's Medium (DMEM; Gibco); heat-inactivated fetal bovine serum (FBS); phosphate buffered saline (pH 7.4; Gibco); ethidium homodimer (Molecular Probes); 0.1% (w/v) saponin (Sigma).2.2 Nanoparticle synthesis
Silica cores were synthesized by a sol–gel method (Stöber process). Alkaline reduction was performed at room temperature to synthesize gold nanospheres.27 In a multistep process derived from established chemistry, the gold nanospheres were immobilized on the silica cores28 and coated with a thin layer of silver.29 To investigate the influence of nanoparticle size on bacterial growth, two sizes of silica cores – small (45 nm) and large (130 nm) – were produced. Additionally, the concentration of silver coating the gold nanospheres was also varied, including a low silver (LS), medium silver (MS), and high silver (HS) concentration. In total, 10 nanoparticle variations were synthesized and characterized: 45 nm SiO2, 45 nm SiO2@Au, 45 nm SiO2@Au@Ag LS, 45 nm SiO2@Au@Ag MS, 45 nm SiO2@Au@Ag HS, 130 nm SiO2, 130 nm SiO2@Au, 130 nm SiO2@Au@Ag LS, 130 nm SiO2@Au@Ag MS, and 130 nm SiO2@Au@Ag HS. The process of synthesizing the phage-mimicking nanoparticles is illustrated in Fig. 1.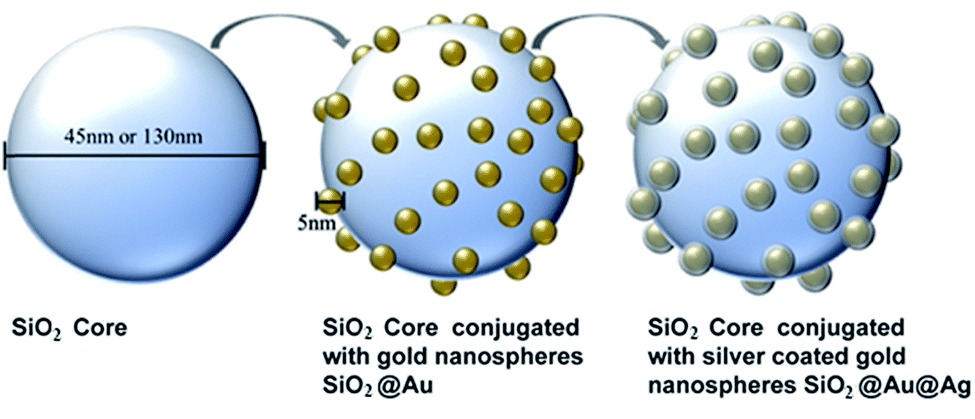 | ||
| Fig. 1 Schematic illustration of the three major steps for synthesizing phage-mimicking silica-based ANPs. First, 45 nm and 130 nm SiO2 nanoparticle cores were synthesized, followed by the conjugation of gold nanospheres to the cores. Finally, three different concentrations of silver were added as a coating over the gold nanosphere surface to produce low silver (LS), medium silver (MS), and high silver (HS) variants. The schematic is not to scale. | ||
To adsorb the gold nanospheres on the SiO2 cores, the solutions of silica cores (45 nm and 130 nm) were amine-functionalized by stirring overnight with additional 15 ml ethanol, 5 ml DI water, and 1 ml APTES. 45 nm and 130 nm SiO2–APTES cores were centrifuged at 9000 rcf for 30 min at room temperature and separated from the supernatant. The amine-functionalized nanoparticle pellets were washed, sonicated (20 s at 40% amplitude), and rinsed with 10 ml ethanol twice by centrifugation at 9000 rcf. After the last rinse, the centrifuged pellet was resuspended in 10 ml DI water and sonicated (20 s at 40% amplitude).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v), the gold nanosphere solution was mixed into the SiO2–APTES core solutions and stirred (minimum of 8 h) in the dark. Afterward, both SiO2@Au nanoparticle solutions were centrifuged at 9000 rcf for 15 min at room temperature, followed by two cycles of washing–sonication–centrifugation in 10 ml of DI water.
:2 (v/v), the gold nanosphere solution was mixed into the SiO2–APTES core solutions and stirred (minimum of 8 h) in the dark. Afterward, both SiO2@Au nanoparticle solutions were centrifuged at 9000 rcf for 15 min at room temperature, followed by two cycles of washing–sonication–centrifugation in 10 ml of DI water.
| Phage-mimicking ANPs | SiO2@Au nanoparticle solution (45 and 130 nm) 1% w/v SiO2 | Silver nitrate solution (10 mM) | Hydroquinone solution (10 mM) |
|---|---|---|---|
| Low silver (LS) | 30 ml | 3 ml | 0.3375 ml |
| Medium silver (MS) | 30 ml | 3 ml | 1.8 ml |
| High silver (HS) | 30 ml | 7.2 ml | 5.76 ml |
After mixing, the solutions were stirred for one hour for the low silver (LS) phage-mimicking ANPs and overnight for the medium silver (MS) and high silver (HS) phage-mimicking ANPs. The silver coating reaction was stopped by centrifugation and discarding of the supernatant. The resulting SiO2@Au@Ag nanoparticle pellets were washed and centrifuged two additional times in 10 ml DI water. All SiO2@Au@Ag nanoparticle solutions were stored in the dark until they were used for characterization and bacterial testing.
2.3 Nanoparticle characterization
2.4 Biocidal activity against planktonic bacteria
All ten synthesized nanoparticle variations, SiO2, SiO2@Au, SiO2@Au@Ag LS (low silver), SiO2@Au@Ag MS (medium silver), and SiO2@Au@Ag HS (high silver) in both sizes (45 and 130 nm), were screened against four clinically relevant nosocomial bacterial strains/pathogens (Staphylococcus aureus USA300, Pseudomonas aeruginosa FRD1, Corynebacterium striatum, and Enterococcus faecalis). Glycerol stocks of all four strains were kept at −80 °C, and each week fresh lysogeny broth agar (LB) plates were streaked with each strain. To adapt the strains to a planktonic lifestyle, a single colony from each LB plate was transferred to a 6 ml LB growth tube and grown at 37 °C under continuous shaking for approximately 12 h to produce a pre-culture before each plate reader run. Additionally, this preparation resulted in very consistent bacterial cell counts (Staphylococcus aureus USA300: 5 × 109 CFU ml−1; Pseudomonas aeruginosa FRD1: 2 × 109 CFU ml−1; Corynebacterium striatum: 2 × 109 CFU ml−1; Enterococcus faecalis: 6 × 108 CFU ml−1) used to inoculate the experimental runs. The biocidal activity experiments were run with at least four replicates for each NP variation, the background (LB), and the control (strain growing in LB only) in a 96 well plate. All single tests were premixed in a small growth tube containing 2 ml with subsequent additions of 10 μl bacterial inoculate from the pre-culture and different concentrations of the three different silver variant phage-mimicking ANPs. These premixed samples were thoroughly mixed, and 200 μl was pipetted into each well of a sterile 96 well plate. With shaking, the 96-well plate containing all biocidal activity tests was incubated at 37 °C for 24 h with OD600 measurements taken every 10 min. The resulting growth curves were corrected with the LB media background measurements.2.5 Dark-field imaging
S. aureus USA300 isolates (methicillin-resistant, community-associated) are continuously evolving their resistance profiles.22 Therefore, the interaction of this pathogen with phage-mimicking ANPs was directly observed by dark-field microscopy. S. aureus USA300 cells were incubated for 24 h with different concentrations of SiO2@Au@Ag HS, aspirated from their individual wells (96 well plate) and collected by centrifugation for 10 min at room temperature. After removal of the supernatant, 4% paraformaldehyde (w/v in PBS) was added to the residual cell pellet. To ensure that all bacterial cells are fixed, the sample was vortexed for 1 min and then incubated for 30 min at room temperature. The fixing agent was removed by centrifugation, discarding the supernatant, and two consecutive rinses with PBS. Sample solutions were stored in PBS at 4 °C until further investigation.Fixed S. aureus USA300 cells were imaged using a dark-field optical microscope (DFOM) equipped with a QIClick CCD camera, a QImaging 01-Rgb-Hm-S-Ir high-resolution color filter, a dark-field condenser (oil 1.43–1.20, Nikon), and a 100× objective (Nikon Plan Fluor 100× oil, iris, SL NA 0.5–1.3, WD 0.20 mm) with a depth of field (focus) of 190 nm. Typically, 10 μl of the samples were added to 1 mm thick glass slides and covered with a #1 glass coverslip. Color images were 12 bit, 2 × 2 binned, and had exposure times between 100 and 200 ms. Images were analysed using ImageJ.
2.6 Biocompatibility testing
For all biocompatibility studies, HaCaT human epithelial keratinocytes were used, maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), and incubated at 37 °C with 5% CO2 in 100 mm culture dishes. The ethidium homodimer cell death assay30 was used to determine the cytotoxicity of all ten synthesized nanoparticle variations (SiO2, SiO2@Au, SiO2@Au@Ag LS (low silver), SiO2@Au@Ag MS (medium silver), and SiO2@Au@Ag HS (high silver), both sizes (45 and 130 nm)). For this assay, HaCaT cells were grown to approximately 75–80% confluency in 24 well tissue culture plates with DMEM. HaCaT cells were between passage numbers 16–25 when cultured from liquid nitrogen storage. Before the nanoparticle treatment, DMEM was aspirated from the wells, and the cells were washed with PBS. Then PBS was aspirated from the wells and the nanoparticle solution (in DMEM) was added in two concentrations (LC, low concentration = approximately 6.99 × 1011 particles per ml of 45 nm phage-mimicking ANPs and 1.09 × 1011 particles per ml of 130 nm phage-mimicking ANPs; HC, high concentration = approximately 1.40 × 1012 particles per ml of 45 nm phage-mimicking ANPs and 2.19 × 1011 particles per ml of 130 nm phage-mimicking ANPs). Cells were then incubated for 16 h at 37 °C with 5% CO2. After incubation, DMEM was aspirated, and cells were washed with PBS to remove the free-floating nanoparticles. 4 μM ethidium homodimer in PBS was added, and cells were incubated for 30 min in the cell culture incubator. The level of fluorescence was determined using a plate reader set to 528 nm excitation and 617 nm emission with a cut-off value of 590 nm. The percentage of dead cells was determined by adding 0.1% (w/v) saponin to each well following the initial reading in order to permeabilize all cells and incubating the plate for an additional 20 min at room temperature followed by shaking the plate on an orbital shaker before reading the plate a second time at the same settings. Percent membrane permeabilization was calculated by dividing the fluorescence values with intact cells by the fluorescence values after cell disruption by saponification. Each treatment condition was performed in triplicate, with the average cell viability (as a percent of the control) plus standard deviation of all conditions plotted together for comparison. Significance was determined by ANOVA and the t-test (one tail and two tail).3. Results and discussion
3.1 Structural similarity of phage-mimicking antibacterial nanoparticles to bacteriophages
For a visual comparison, Fig. 2A and B show the structure of a tailless phage (the example of bacteriophage ΦX174) that acted as a template for the design of our antibacterial nanoparticles.31 TEM image analysis revealed that the small and large SiO2 cores had diameters of approximately 45 ± 8.25 nm and 130 ± 10.36 nm, respectively, and were covered with 3.5 ± 1.57 nm gold-nanospheres (Fig. 2C–E). The amount of gold nanospheres covering the phage-mimicking ANPs varied from ∼50 for the 45 nm core size to ∼105 for the larger 130 nm SiO2 core (Fig. 2F). For a more detailed comparison of the phage-mimicking ANPs with the head structure of tailed and tailless phages, Table 2 analyzes 9 phages and lists the number of protein-turrets on the viral capsid of the phages, their capsid diameter, the interspacing between the protein turrets, the surface area of the capsid, and the surface density of the protein turrets. The surface area of the capsid was calculated for the icosahedral shape. Cryo-electron microscopy imaging data from the protein database (PDB) was used to determine the dimensions of the individual capsid proteins.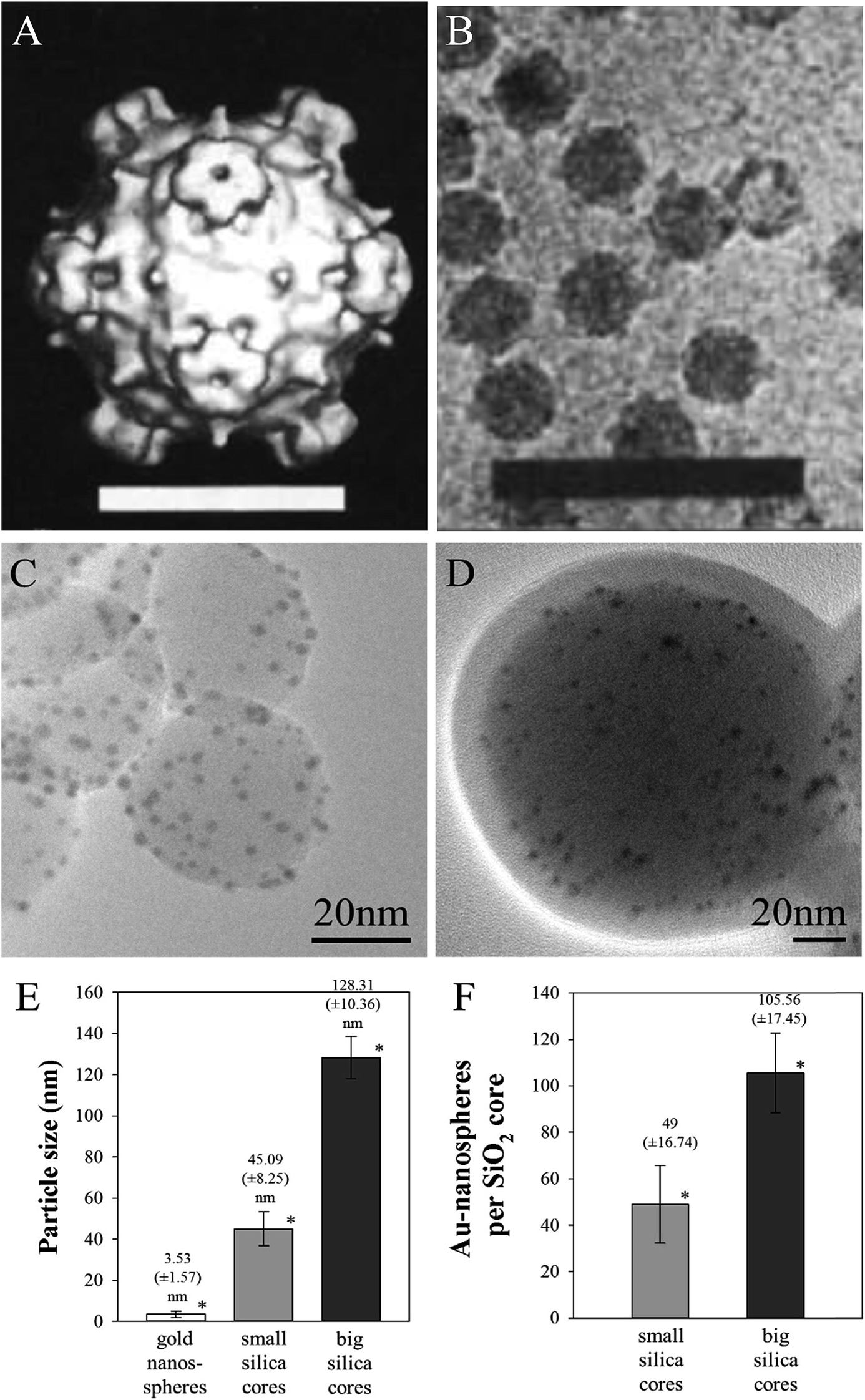 | ||
| Fig. 2 The procapsid particles of bacteriophage ΦX174 presented as (A) a surface-rendered image (scale bar: 20 nm) from (B) cryo-electron microscopy images (HRTEM) (scale bar: 100 nm) (reproduced with permission).70 SiO2@Au@Ag NPs with (C) small 45 nm and (D) large 130 nm core sizes are shown with the example of the high silver coating on the gold nanospheres. (E) The mean diameter of the gold nanospheres and SiO2 cores. (F) The number of gold nanospheres on silica cores. *P ≤ 0.01 (t- and f-test). | ||
| Tailed bacteriophage | Number of protein turrets/capsid | Capsid diameter (nm) | Interspacing between protein turrets on capsid (nm) | Surface area of capsid (nm2) | Surface density of protein turrets (nm−2) |
|---|---|---|---|---|---|
| P68 | 12 | 48 | 11.33 | 5191 | 0.0023 |
| phi812 | 12 | 90 | 29.58 | 24916 |
0.0005 |
| φKZ | 12 | 145 | 59.80 | 46407 |
0.0003 |
| Tailless bacteriophage | Number of protein turrets/capsid | Capsid diameter (nm) | Interspacing between protein turrets on capsid (nm) | Surface area of capsid (nm2) | Surface density of protein turrets (nm−2) |
|---|---|---|---|---|---|
| ΦX174 | 12 | 28 | 8.28 | 3068 | 0.0039 |
| PM2 | 12 | 59 | 28.61 | 10120 |
0.0012 |
| PRD1 | 12 | 64 | 17.32 | 11898 |
0.0010 |
| STIV | 12 | 70 | 27.25 | 14496 |
0.0008 |
| SH1 | 12 | 82 | 36.44 | 15827 |
0.0008 |
| SpV4 | 18 | 31 | 9.31 | 2635 | 0.0068 |
| Phage-mimicking ANPs | Number of Au nanospheres/silica core | Silica core diameter (nm) | Interspacing between Au nanospheres on silica core (nm) | Surface area of silica core (nm2) | Surface density of Au nanospheres (nm−2) |
|---|---|---|---|---|---|
| Small (45 nm) | 49 | 45 | 2.02 | 6362 | 0.0077 |
| Large (130 nm) | 106 | 130 | 9.87 | 53093 |
0.0020 |
In general, the size of the smaller phage-mimicking ANPs (45 nm) relates better to the capsid sizes of tailless bacteriophages than that of the larger 130 nm phage-mimicking ANPs. Both the 45 nm and 130 nm core sizes carry more gold nanospheres per unit area in comparison to the number of protein turrets on the heads of the bacteriophage's capsids (∼4× and 8× higher, respectively). Regardless, the 2.02 nm interspacing between the gold nanospheres of the small 45 nm phage-mimicking ANPs is at least 4× smaller than the geometrically consistent distance between the protein-turrets covering the surface of the phage capsids. The gold nanosphere interspacing of 9.87 nm on the larger 130 nm phage-mimicking ANPs, on the other hand, is comparable to the protein turret interspacing found on the smaller bacteriophages P68, ΦX174, and SpV4 (11.33 nm, 8.28 nm, and 9.31 nm) (Table 2).32–34
Interestingly, the bacteriophage SpV4 possesses a surface density of protein turrets (0.0068 nm−2) that is 88% similar to the surface density of gold nanospheres (0.0077 nm−2) on small 45 nm phage-mimicking ANPs.33 The SpV4 bacteriophage belongs to the viral family of Microviridae which are small (25–27 nm), tailless bacteriophages.35 One genus of this family is named Chlamydiamicrovirus and as the name suggests it infects various species of the obligate intracellular and pathogenic Chlamydia.36 Bacteriophages of the genus Chlamydiamicrovirus are known to initiate infection of the bacterial host cell by pilus-mediated adsorption, which is dependent on the external bacteriophage structure as well as the host cell receptors.37–40 Additionally, the surface density of protein turrets on the Staphylococcus aureus infecting bacteriophage P68 (0.0023 nm−2) is 85% similar to the surface density of gold nanospheres on the larger 130 nm phage-mimicking ANPs (0.0020 nm−2).32 Thus, the surface density of silver-coated gold nanospheres on the silica cores of both phage-mimicking ANP sizes is closely related to the surface density of protein turrets of pathogen-infecting bacteriophages which is indicative of the phage structure mimicking properties of our phage-mimicking ANPs. Importantly, in our manufacturing technique, we used redox plating to alloy silver41 with randomly distributed gold nanospheres already immobilized on silica cores resulting in an anisotropic silver shell on the gold nanospheres. Our synthesis protocol led to robust retention of the phage-mimicking structure under physiological conditions as opposed to techniques that employ only electrostatic immobilization of nanospheres on silica cores (Table 3).
| Single point of contact on silica cores by immobilizing pre-synthesized Au@Ag core–shell NPs (nm2) | Robust hemispherical contact on silica cores by anisotropic plating of Ag on immobilized Au nanospheres (nm2) | |
|---|---|---|
| (a) Schematic |

|

|
| (b) Surface area of the interface | 0 | ≤9.81 |
| (c) Surface area of silver | ≥39.24 | ≤19.62 |
3.2 Antibacterial activity of phage-mimicking nanoparticles against four nosocomial strains
:Ag = 70:30) (Table 4) which will result in <1 μg ml−1 silver ions leaching into solution for every 100 μg ml−1 70:30 Au:Ag nanoalloy used.45 In fact using ICP-OES we were not able to detect any Ag ions in the supernatant of phage-mimicking ANPs exposed to bacterial cell culture conditions. The negligible leaching of silver ions from our phage-mimicking ANPs will lead to less off-target toxicity. The unique architecture of our phage-mimicking nanoparticles will achieve similar (if not better) results than silver nanoparticles in actively inhibiting the growth of Staphylococcus aureus USA300.
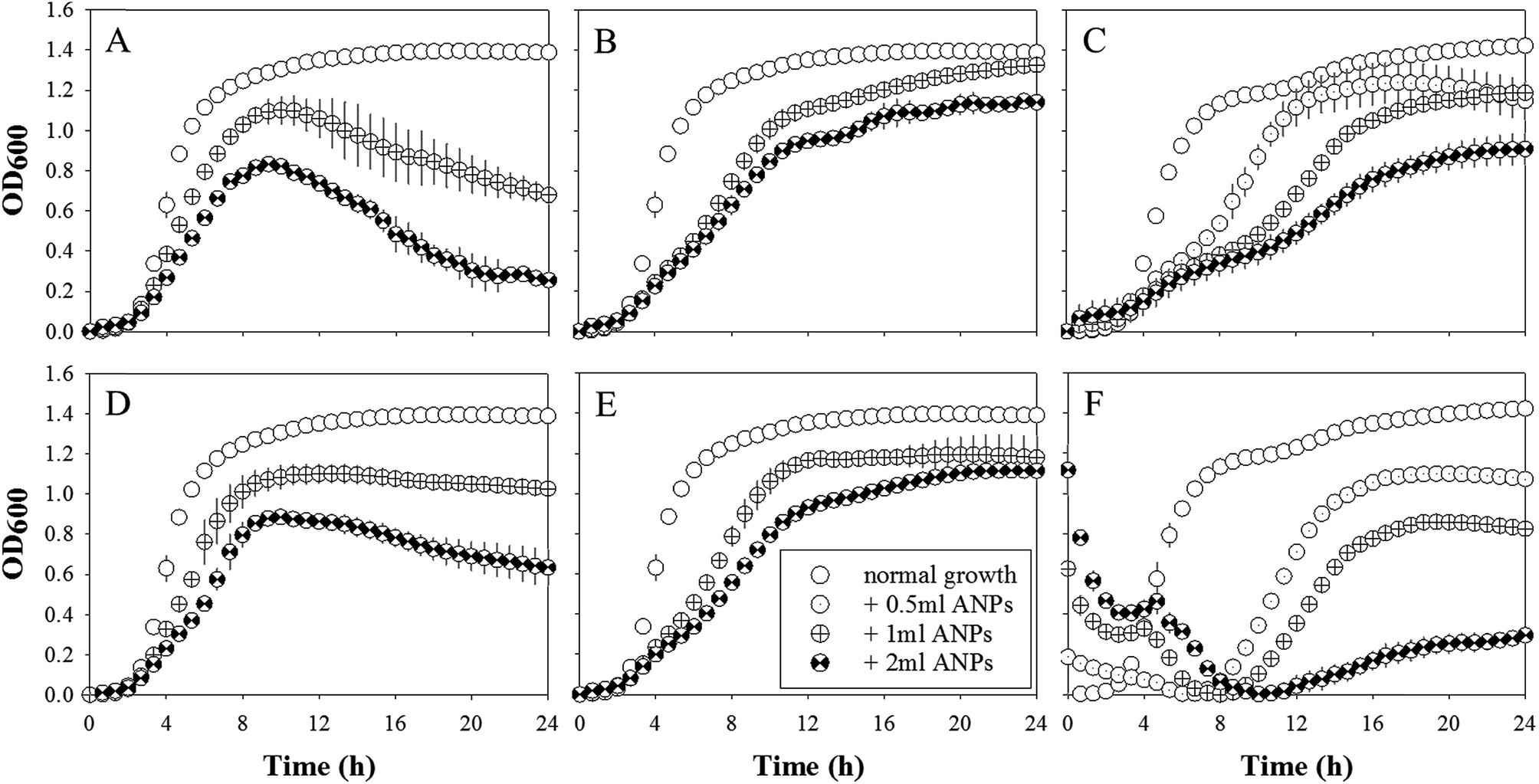 | ||
| Fig. 3 Growth of Staphylococcus aureus USA300 in the presence of (A–C) small 45 nm and (D–F) large 130 nm phage-mimicking ANPs with various silver coatings containing (A and D) low, (B and E) medium, and (C and F) high silver concentrations. | ||
| Phage-mimicking ANPs | Gold concentration, μg ml−1 (±SD) | Silver concentration, μg ml−1 (±SD) |
|---|---|---|
| 45 nm LS | 637.61 (±52.91) | 17.64 (±9.77) |
| 130 nm LS | 679.59 (±51.04) | 26.62 (±13.57) |
| 45 nm MS | 624.86 (±39.65) | 92.15 (±58.60) |
| 130 nm MS | 652.48 (±67.73) | 141.63 (±81.42) |
| 45 nm HS | 665.21 (±66.93) | 123.25 (±87.91) |
| 130 nm HS | 635.32 (±50.35) | 189.22 (±122.13) |
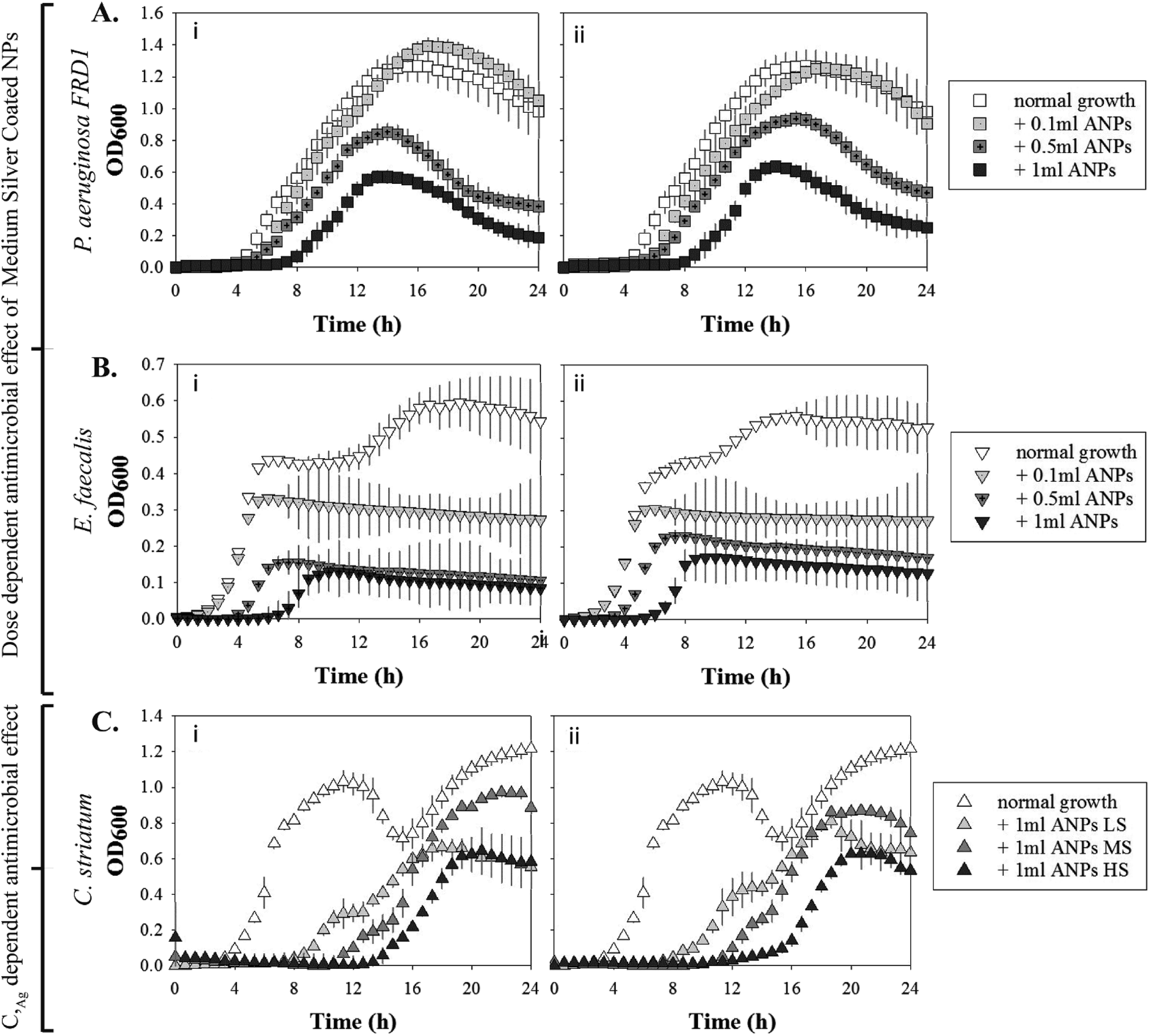 | ||
| Fig. 4 P. aeruginosa FRD1 and E. faecalis exhibited dose-dependent log phase inhibition when treated with medium silver-coated NPs. Growth of P. aeruginosa FRD1 in the presence of (A(i)) small 45 nm and (A(ii)) large 130 nm NPs with the medium silver-coating in three different dosages (0.1, 0.5, and 1 ml). Growth of E. faecalis in the presence of (B(i)) small 45 nm and (B(ii)) large 130 nm NPs with the medium silver coating in three different dosages (0.1, 0.5, and 1 ml). C. striatum exhibited a silver coverage dependent retardation of log-phase initiation for a given dose (e.g., 1 ml). Growth of C. striatum in the presence of a 1 ml dose of either (C(i)) small 45 nm or (C(ii)) large 130 nm phage-mimicking ANPs with silver coatings varied from low (LS) to medium (MS) to high (HS). | ||
| Bacteria | NP diameter (nm) | Ag coverage | Dosage (ml) | Linear slope log phase (h−1) | % Log phase retardation w.r.t control | Log phase start (h) | Duration of log phase (h) |
|---|---|---|---|---|---|---|---|
| S. aureus USA300 | 45 nm | Zero | 0 | 0.280 | 0% | 2.1 | 4.5 |
| LS | 1 | 0.190 | 32.3% | 2.6 | 5.5 | ||
| 2 | 0.118 | 57.8% | 1.4 | 7.8 | |||
| MS | 1 | 0.111 | 60.3% | 1.3 | 11.3 | ||
| 2 | 0.094 | 66.5% | 1.4 | 10.4 | |||
| HS | 1 | 0.074 | 73.6% | 2.7 | 12.6 | ||
| 2 | 0.043 | 84.7% | 2.6 | 21.4 | |||
| 130 nm | Zero | 0 | 0.280 | 0% | 2.1 | 4.5 | |
| LS | 1 | 0.177 | 36.8% | 2.6 | 4.7 | ||
| 2 | 0.129 | 53.8% | 1.9 | 7.3 | |||
| MS | 1 | 0.138 | 50.8% | 2.0 | 8.5 | ||
| 2 | 0.100 | 64.3% | 1.9 | 9.3 | |||
| HS | 1 | 0.105 | 62.5% | 8.5 | 8.8 | ||
| 2 | 0.028 | 90.1% | 10.6 | 9.5 |
S. aureus USA300 demonstrated a dosage-dependent and a silver coverage dependent sensitivity to phage-mimicking ANPs as indicated by the increased log phase inhibition with increasing dosage and increasing Ag coverage (Table 5). There were no clear trends that would indicate whether there was a size-dependent enhancement of phage-mimicking ANPs' antimicrobial efficacy. The maximum log phase inhibition achieved was 84.7% for 2 ml of 45 nm SiO2@Au@Ag HS and 90.1% for 2 ml of 130 nm SiO2@Au@Ag HS. In the presence of 2 ml of 45 nm SiO2@Au@Ag HS phage-mimicking ANPs, S. aureus USA300 reached the lag phase after ∼22 h, which was delayed 4-fold in comparison to the control. This indicates that the HS variant of the 45 nm SiO2@Au@Ag was the most effective in inhibiting the growth of S. aureus USA300.
E. faecalis exhibited the maximum sensitivity to the phage-mimicking ANPs (Table 6). The log phase inhibition of E. faecalis when exposed to 3 different dosages (0.1 ml, 0.5 ml, and 1 ml) of MS coated phage-mimicking ANPs showed the same dose-dependent inhibition as S. aureus USA300. In E. faecalis there was additionally a size dependency in the efficacy of the phage-mimicking ANPs. 1 ml of the smaller MS coated phage-mimicking ANPs (45 nm) was ∼20% more efficient in inhibiting the log growth phase in comparison to 1 ml of the larger MS coated phage-mimicking ANPs (130 nm). P. aeruginosa FRD1 in comparison was the least sensitive to MS coated phage-mimicking ANPs. Although there was a dose-dependent and size-dependent inhibition of the log growth phase, the maximum log phase inhibition was only 21.2% for P. aeruginosa FRD1 in comparison to the 60.3% for S. aureus USA300 and 72.5% for E. faecalis for the same dose. These results suggest that the mucoid strain P. aeruginosa FRD1 is more adept in preventing the interaction of the structure-based phage-mimicking ANPs with the bacterial cell membrane and more resistant to such phage-mimicking ANP designs once they form a mucinous mass.
| Bacteria | Phage-mimicking ANP core diameter (nm) | Ag coverage | Dosage (ml) | Linear slope log phase (h−1) | % Log phase retardation w.r.t control | Log phase start (h) | Duration of log phase (h) |
|---|---|---|---|---|---|---|---|
| S. aureus USA300 | 45 nm | MS | 0 | 0.2798 | 0% | 2.1 | 4.5 |
| 1 | 0.1112 | 60.3% | 1.3 | 11.3 | |||
| 2 | 0.09373 | 66.5% | 1.4 | 10.4 | |||
| 130 nm | MS | 0 | 0.2798 | 0% | 2.1 | 4.5 | |
| 1 | 0.1376 | 50.8% | 2.0 | 8.5 | |||
| 2 | 0.0998 | 64.3% | 1.9 | 9.3 | |||
| P. aeruginosa FRD1 | 45 nm | MS | 0 | 0.131 | 0% | 3.8 | 9.4 |
| 0.1 | 0.1268 | 3.2% | 6.8 | 8.6 | |||
| 0.5 | 0.1104 | 15.7% | 5.4 | 6.7 | |||
| 1 | 0.1032 | 21.2% | 7.3 | 5.5 | |||
| 130 nm | MS | 0 | 0.131 | 0% | 3.8 | 9.4 | |
| 0.1 | 0.1273 | 2.8% | 6.1 | 7.9 | |||
| 0.5 | 0.1229 | 6.2% | 6.1 | 6.6 | |||
| 1 | 0.1205 | 8.0% | 8.1 | 4.6 | |||
| E. faecalis | 45 nm | MS | 0 | 0.1394 | 0% | 2.9 | 2.4 |
| 0.1 | 0.0911 | 34.6% | 2.0 | 3.9 | |||
| 0.5 | 0.0522 | 62.6% | 3.4 | 2.7 | |||
| 1 | 0.0383 | 72.5% | 6.0 | 3.4 | |||
| 130 nm | MS | 0 | 0.1394 | 0% | 2.9 | 2.4 | |
| 0.1 | 0.1248 | 10.5% | 2.7 | 2.0 | |||
| 0.5 | 0.074 | 46.9% | 3.4 | 3.2 | |||
| 1 | 0.0634 | 54.5% | 6.0 | 2.6 | |||
| C. striatum | 45 nm | MS | 0 | 0.158 | 0% | 3.3 | 6.7 |
| 1 | 0.082 | 48.0% | 10.6 | 12.2 | |||
| 130 nm | MS | 0 | 0.158 | 0% | 3.3 | 6.7 | |
| 1 | 0.111 | 29.7% | 11.3 | 7.4 |
C. striatum exhibited a size-dependent susceptibility to the smaller diameter phage-mimicking ANPs, inducing a higher inhibition of the log phase growth (Table 7). However, there was no apparent dependence of C. striatum susceptibility to the silver coverage on the phage-mimicking ANPs. The percent log phase inhibitions in the presence of LS, MS, and HS variants of the phage-mimicking ANPs were less than 3% apart. But, with nearly 50% inhibition of the log phase growth of C. striatum in the presence of 1 ml of 45 nm phage-mimicking ANPs, it was clear that C. striatum along with S. aureus USA300 and E. faecalis were the most susceptible to the antimicrobial effect of our rationally designed phage-mimicking nanoparticles. P. aeruginosa FRD1 also exhibited a significant decrease in log phase growth and a 2-fold decrease in the duration of the log phase (Table 6).
| Bacteria | Phage-mimicking ANP core diameter (nm) | Ag coverage | Dosage (ml) | Linear slope log phase (h−1) | % Log phase retardation w.r.t control | Log phase start (h) | Duration of log phase (h) |
|---|---|---|---|---|---|---|---|
| S. aureus USA300 | 45 nm | Zero | 0 | 0.280 | 0% | 2.1 | 4.5 |
| LS | 1 | 0.190 | 32.3% | 2.6 | 5.5 | ||
| MS | 1 | 0.111 | 60.3% | 1.3 | 11.3 | ||
| HS | 1 | 0.074 | 73.6% | 2.7 | 12.6 | ||
| 130 nm | Zero | 0 | 0.280 | 0% | 2.1 | 4.5 | |
| LS | 1 | 0.177 | 36.8% | 2.6 | 4.7 | ||
| MS | 1 | 0.138 | 50.8% | 2.0 | 8.5 | ||
| HS | 1 | 0.105 | 62.5% | 8.5 | 8.8 | ||
| C. striatum | 45 nm | Zero | 0 | 0.158 | 0% | 3.3 | 6.7 |
| LS | 1 | 0.086 | 45.3% | 7.8 | 8.2 | ||
| MS | 1 | 0.082 | 48.0% | 10.6 | 12.2 | ||
| HS | 1 | 0.082 | 48.0% | 13.3 | 6.0 | ||
| 130 nm | Zero | 0 | 0.158 | 0% | 3.3 | 6.7 | |
| LS | 1 | 0.113 | 28.2% | 9.3 | 9.5 | ||
| MS | 1 | 0.111 | 29.7% | 11.3 | 7.4 | ||
| HS | 1 | 0.108 | 31.7% | 14.1 | 5.9 |
The growth curve results confirm the broad-spectrum capability of our phage mimicking ANPs. Our tests also revealed a mucus-like secretion employed as a potential defense mechanism by the mucoid bacterial strain P. aeruginosa FRD1 to overcome our structure-based phage-mimicking ANPs. In future iterations of the phage-mimicking nanoparticles, design consideration will be given to include anti-mucogenic ligands on the phage-mimicking ANPs to test if that might improve the antibacterial effect against mucoid strains of pathogenic bacteria.
3.3 Mechanism of antibacterial activity
Several studies have confirmed that the bactericidal effect of AgNPs is due to their ability to efficiently bind and reduce disulfide (S–S) bridges on the bacterial cell membrane.52,53 In the absence of S–S bridges, the electron transport across the membrane is disrupted, leading to interference with bacterial energetics and rise in reactive oxygen species (ROS) in the bacteria.52,54–56 Our phage-mimicking ANPs have been designed to be 1/10th to 1/5th the size of the bacteria. Zeta-potential (ζ-potential) measurements of the amine-functionalized silica core confirmed the positive surface charge with the 130 nm silica core having double the surface charge compared to the 45 nm silica core. The ζ-potential of the 45 nm silica core was 10.2 ± 3.4 mV, and the ζ-potential of the 130 nm silica core was 20.1 ± 7.4 mV. Upon addition of the negatively charged nanospheres to form the patchy shell on the silica-cores, the ζ-potentials of the 45 nm silica core and the 130 nm silica core dropped to −6.16 ± 3.4 mV and −4.07 ± 5.0 mV. While the net ζ-potential of the phage-mimicking ANPs was negative, the ζ-potential range was near zero mV, like that of zwitterionic molecules.57 The zwitterionic nature of the phage-mimicking ANPs implies that the negatively charged membrane of the bacteria will interact with the phage-mimicking ANPs for durations long enough to allow Au/Ag on the phage-mimicking ANPs to bind to cysteine residues on the bacterial membrane without any hindrance from charge–charge repulsion.58,59We hypothesize that the chemisorption of thiols from cysteine residues onto multiple Au/Ag nanospheres will immobilize the phage-mimicking nanoparticles on the bacteria, eventually leading to the physical disruption of the bacterial cell membrane.60 The bacteria might initially sense the phage-mimicking nanoparticles as ≤5 nm Au/Ag nanospheres and thereby are slow to initiate their defense mechanisms.50 Darkfield imaging was used to track the localized surface plasmon resonance signal (LSPRS) from Au and Ag on the phage-mimicking ANPs (Fig. 5) using established techniques.50,61–63 The LSPRS of the phage-mimicking ANPs lies in the visible spectrum (blue to red).64 Darkfield imaging confirmed that the SiO2@Au@Ag HS NPs exhibited LSPRS colors of blue, green, yellow, orange, and red (Fig. 5A and B). Additionally, darkfield imaging confirmed that untreated S. aureus USA300 appeared spherical, as expected (Fig. 5C).65 However, S. aureus USA300 treated with SiO2@Au@Ag HS phage-mimicking ANPs exhibited elliptical morphology with a dense number of NPs associated with the bacteria as confirmed through darkfield imaging (Fig. 5D), which showed that the LSPR signal of the phage-mimicking ANPs was colocalized with the bacteria.
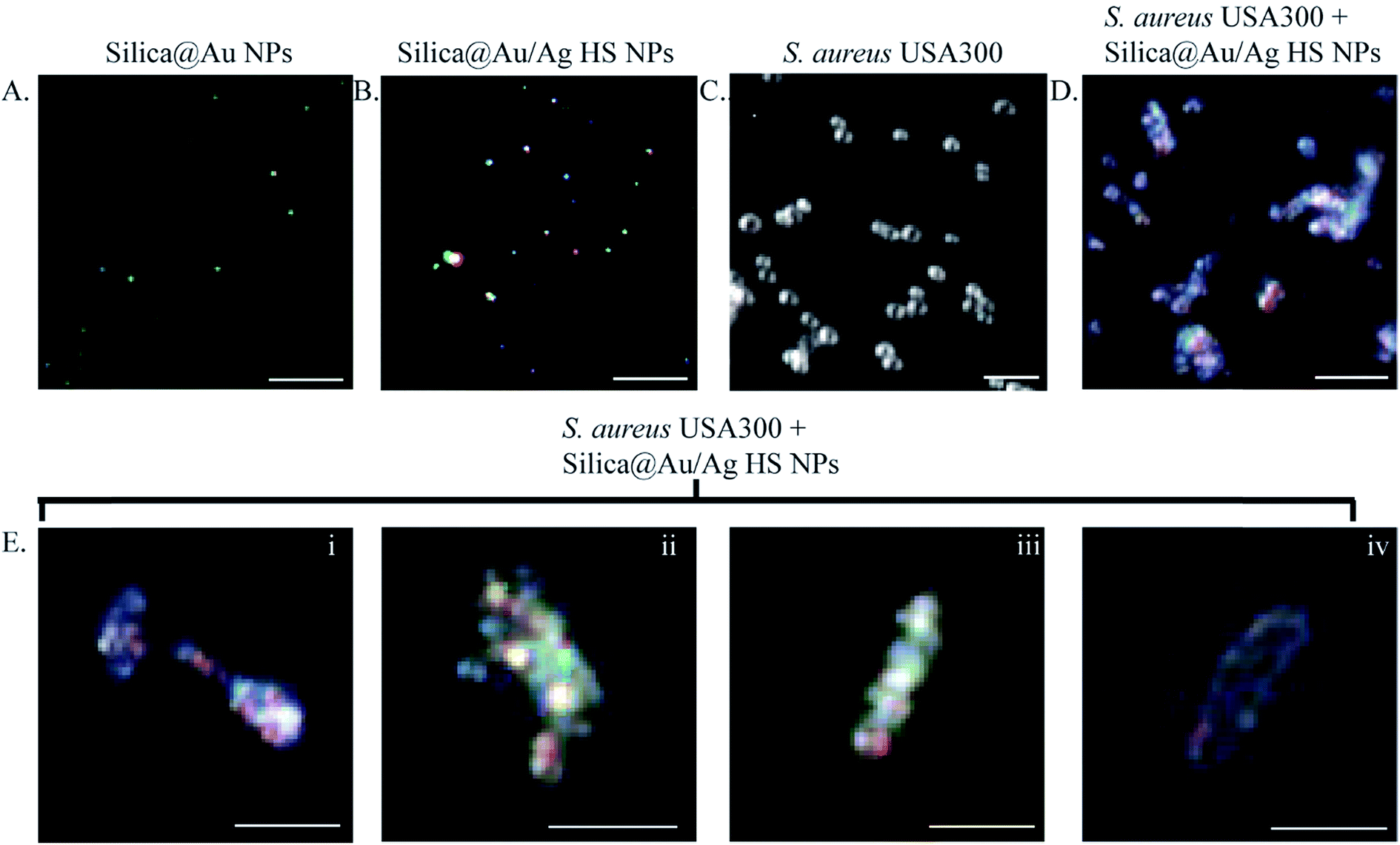 | ||
| Fig. 5 Darkfield microscopy was used to assess the effect of the phage-mimicking nanoparticle treatment on the morphology of S. aureus USA300. (A) Darkfield images of the localized surface plasmon resonance (LSPR) of Au nanoclusters on the 45 nm silica cores. The LSPRS of Au was either green or orange, which is in good accordance with the existing literature. (B) The LSPRS of high silver-coated Au nanoclusters on the 45 nm silica core was a rainbow of colors with blue, green, yellow, and red being prominent. (C) Darkfield images showed that untreated S. aureus USA300 appeared spherical. (D) However, S. aureus USA300 treated with SiO2@Au@Ag HS phage-mimicking ANPs exhibited elliptical morphology with a high density of the NPs associated with the bacteria as confirmed through dark-field imaging of the bacteria as well as the LSPR signal of NPs from the bacteria. (E) Magnified images of elliptical S. aureus USA300 with the characteristic LSPR signal of silver on SiO2@Au@Ag HS phage-mimicking ANPs. Scale bars are all 10 μm. The LSPR images of the nanoparticles are all diffraction-limited and not the actual size of the nanoparticles. | ||
Untreated control samples of S. aureus USA 300 had an average diameter of 1.39 μm and an aspect ratio of 1 in all directions. Samples of S. aureus USA 300 exposed to SiO2@Au@Ag HS phage-mimicking ANPs were elliptical and possessed a short axis and a long axis with an aspect ratio of 3.57. In bacteria exposed to phage-mimicking ANPs, the diameter along the short axis was 1.77 μm, and the diameter along the long axis was 6.30 μm. SiO2@Au@Ag HS phage-mimicking ANPs were co-localized with the bacterial membrane as seen in the representative darkfield images in Fig. 5E. This provides strong support for our hypothesis that the phage-mimicking structure of our phage-mimicking ANPs allows them to structurally and chemically prolong their interaction with the bacterial membrane, thus interrupting bacterial cell division, which will ultimately lead to physical disruption and death of the bacteria, as evidenced initially by inhibition of bacterial growth in our growth curve studies (Fig. 3 and 4).
3.4 Biocompatibility with a human skin cell line
All ten synthesized nanoparticle variations (SiO2, SiO2@Au, SiO2@Au@Ag LS, SiO2@Au@Ag MS, and SiO2@Au@Ag HS; 45 nm or 130 nm) are highly biocompatible with human skin cells (HaCaT) Fig. 6. Subsamples of the HaCaT cell line incubated with the nanoparticles for 16 h showed growth and a healthy appearance of the cells (Fig. 6B and C).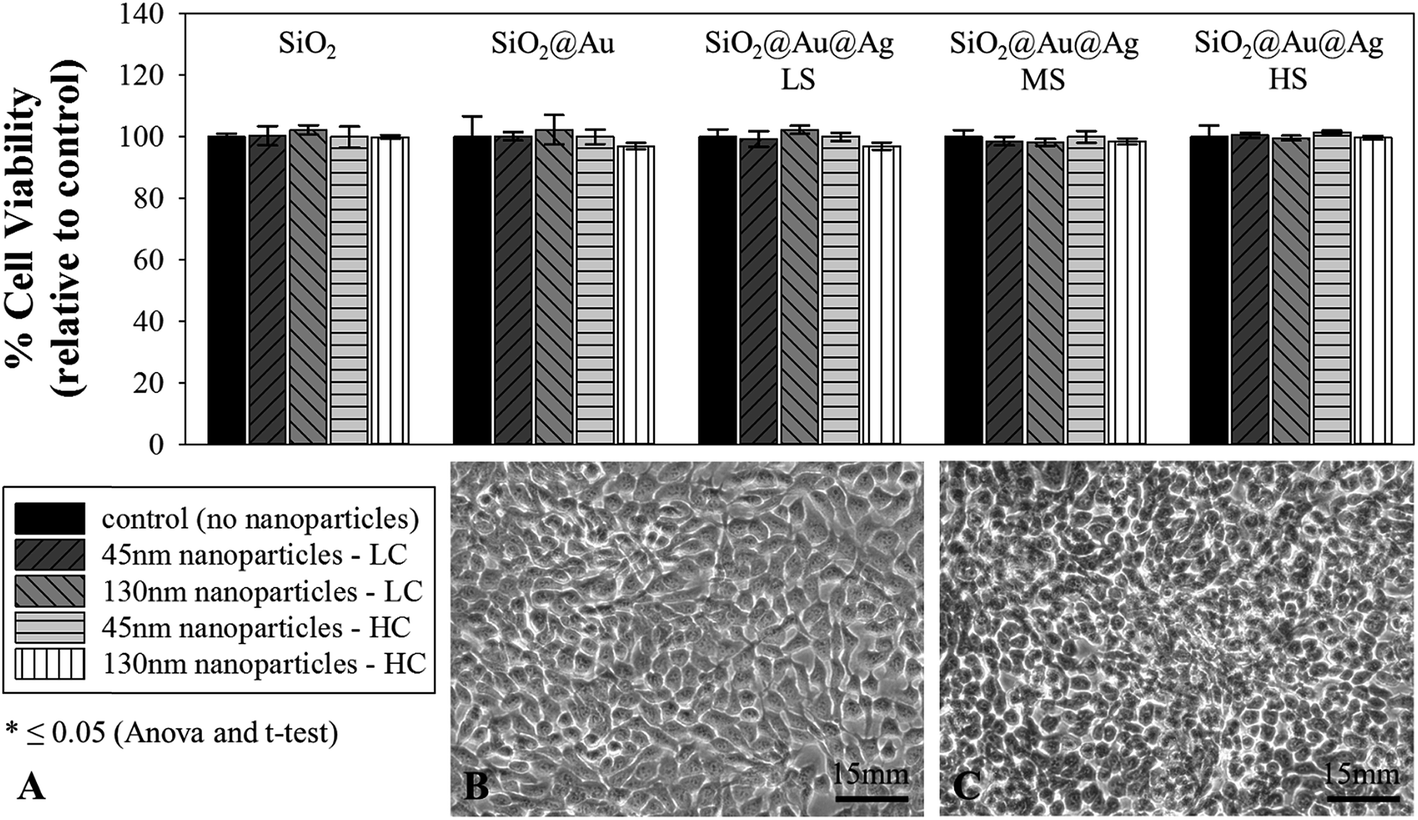 | ||
| Fig. 6 (A) Cell viability (% of control) for all ten synthesized NP variations: SiO2, SiO2@Au, SiO2@Au@Ag LS (low silver), SiO2@Au@Ag MS (medium silver), and SiO2@Au@Ag HS (high silver), with both 45 and 130 nm SiO2 cores, and at two concentrations (low concentration and high concentration). LC, low concentration = approximately 6.99 × 1011 particles per ml of 45 nm phage-mimicking ANPs and 1.09 × 1011 particles per ml of 130 nm phage-mimicking ANPs; HC, high concentration = approximately 1.40 × 1012 particles per ml of 45 nm phage-mimicking ANPs and 2.19 × 1011 particles per ml of 130 nm phage-mimicking ANPs. Optical microscope images showing (B) HaCaT cells in DMEM and (C) after treatment with 45 nm NPs with HS coating. | ||
In general, the use of silica-based materials is not considered harmful to humans.66 Depending on the size and concentration as well as the exposure/drug delivery routes in tested cell lines, silica nanoparticles cover a wide spectrum between biocompatible and slightly toxic.66 But nonporous amorphous silica NPs (>10 nm) with APTES molecules, similar to the ones employed as cores in our phage-mimicking ANPs, have proven biocompatibility with human cells (e.g., Calu-3).67 Silver nanoparticles have also been recommended as a good alternative for the control of bacterial strains, with less risk of toxicity to mammalian cells.68 But silver nanoparticle toxicity to eukaryotic cells is size-dependent. Using data from dose-dependent toxicity studies of AgNPs to eukaryotic cells, we extrapolated the size- and shape-dependent toxicity of nanomaterials.69 The LC50 of AgNPs ≤10 nm (1180 ± 140 μg ml−1) against eukaryotic fibroblast cells is lower than that of AgNPs >10 nm (1760 ± 110 μg ml−1) since the smaller AgNPs are easily taken up by cells and concentrated intracellularly, much more than the >10 nm AgNPs.51
Despite choosing Au/Ag nanospheres <5 nm for our phage-mimicking ANPs, we hypothesize that the lack of abundance of cysteine residues on eukaryotic cell membranes, the zwitterionic nature of the nanoparticles (similar to PEG-coated nanoparticles) as determined from zeta potential measurements, the ∼10-fold increase in the effective diameter of the immobilized Au/Ag nanospheres on the silica cores, and the size of the eukaryotic cells still being ∼200-fold larger than the phage-mimicking ANPs, will increase the repulsion of our phage-mimicking nanoparticles away from the surface of eukaryotic cells.51 The arrangement of the <5 nm Au/Ag nanospheres on a >45 nm silica core also ensures that the Au/Ag nanospheres do not present themselves as ≤5 nm AgNPs, thereby reducing the probability of the phage-mimicking ANPs permeabilizing the eukaryotic cell membrane. The phage-mimicking structure of our ANPs increased the biocompatibility towards eukaryotic cells as was evident from our tests on model skin cell lines (HaCaT).66 Additionally, the Environmental Protection Agency (EPA) has established a chronic oral Reference Dose (RfD) of 5 μg per kg per day for silver which broadly translates to 250 μg to 750 μg per person per day.48 Our phage-mimicking ANPs exhibit a maximum silver content of 189.22 μg ml−1 and a predicted amount of <1 μg ml−1 silver ions leaching into solution which are well below the EPA prescribed safety limits for exposure to silver.
4. Conclusions
Rational nanoparticle engineering revealed that the previously hypothetical phage-mimicking nanoparticles could be synthesized with very specific properties (bactericidal, biocompatible, etc.). The structural approach and the modular assembly of the nanoparticles designed by our lab are effective against four different nosocomial strains (Staphylococcus aureus USA300, Pseudomonas aeruginosa FRD1, Corynebacterium striatum, and Enterococcus faecalis), strongly inhibiting and suppressing their growth. All four strains grow slower (inhibition of log-phase growth) and do not reach their maximal growth potential (optical density@600 nm cut down by approximately 1/2 to 1/3 compared to nanoparticle-free growth). Growth inhibition is dependent on the silver coating concentration on the gold nanospheres, with a higher silver concentration positively correlated with higher bactericidal and increased antibacterial efficacy of the nanoparticles. However, the bactericidal properties of our phage-mimicking ANPs were not a function of Ag concentration alone. Previously existing literature that has utilized AgNPs in the size range of 8–100 nm demonstrated that bacterial defenses (P. aeruginosa and B. subtilis) were highly efficient in the efflux of individual AgNPs.50 To confirm the importance of our structure-based antibacterial design approach, phage-mimicking ANPs were tracked using the signal from the LSPR of Au/Ag nanoclusters in dark-field imaging to conclusively demonstrate that our phage-mimicking ANPs, much like their biological counterparts, remain irreversibly associated with the bacterial cell wall. The phage-mimicking ANPs bound to the bacterial membranes played a disruptive role during multiple bacterial divisions resulting in an abnormally elongated morphology being exhibited by S. aureus USA300. Phage-mimicking ANPs induced a dose-dependent 21% to 90% log phase inhibition when tested against the four different nosocomial bacterial strains. In addition to the phage-mimicking ANPs' advantageous bacteriostatic activity, the inorganic phage-mimicking nanoparticles are also biocompatible with human skin cells. Our phage-mimicking ANPs are the first to demonstrate an antibacterial activity that is dependent on the structure of the NPs, resulting in a bactericidal activity that is greater than that of the sum of the individual components that constitute the nanoparticles. Our results demonstrate an innovative and proactive approach to deal with the rising threat of antibiotic-resistant bacteria. Our robust, modularly assembled, antibacterial NPs can be applied to infectible surfaces to pre-empt an infection from even developing at the tissue/material interface. This study where we employed modular assembly to engineer our phage-mimicking ANPs hierarchically and to gain a clearer understanding of the component-to-structure-to-function relationship has laid the foundation for future structure-based, antibiotic-free antibacterial nanoparticles with higher bactericidal efficiency and bacterial specificity to stem the continued rise of antibiotic-resistant strains of bacteria.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
The Nallathamby lab would like to acknowledge the financial and material support of the Notre Dame Center for Nano Science and Technology (NDnano) and the Notre Dame Center for Advanced Diagnostics and Therapeutics (AD&T). Dr Nallathamby's research was supported by a Project Development Team grant from the Indiana Clinical and Translational Sciences Institute (CTSI-PDT: 373037-31005-FY19CTSIK), an Institutional Research Grant from the American Cancer Society (ACS IRG-17-182-04) and a seed grant from the Notre Dame Advanced Diagnostics and Therapeutics Center (372333-31025). Dr Hopf's research was supported by a grant from the Advanced Diagnostics and Therapeutics research initiative at the University of Notre Dame (372717-43310-FY17RFP). Margo Waters was funded through an NDnano Undergraduate Research Fellowship (NURF) and Veronica Kalwajtys was funded through a College of Science-Summer Undergraduate Research Fellowship (COS-SURF).References
- C. L. Ventola, Pharm. Therapeut., 2015, 40, 277–283 Search PubMed.
- S. Santajit and N. Indrawattana, BioMed Res. Int., 2016, 2016, 2475067 CrossRef PubMed.
- J. N. Pendleton, S. P. Gorman and B. F. Gilmore, Expert Rev. Anti-Infect. Ther., 2013, 11, 297–308 CrossRef CAS PubMed.
- D. Pletzer, S. C. Mansour, K. Wuerth, N. Rahanjam and R. E. Hancock, mBio, 2017, 8, e00140 CrossRef CAS PubMed.
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71–109 CrossRef CAS PubMed.
- L. O'Neill, Vet. Rec., 2016, 178, 590 Search PubMed.
- S. B. Zaman, M. A. Hussain, R. Nye, V. Mehta, K. T. Mamun and N. Hossain, Cureus, 2017, 9, e1403 Search PubMed.
- H. A. Hemeg, Int. J. Nanomed., 2017, 12, 8211–8225 CrossRef CAS PubMed.
- N. Beyth, Y. Houri-Haddad, A. Domb, W. Khan and R. Hazan, J. Evidence-Based Complementary Altern. Med., 2015, 2015, 246012 Search PubMed.
- A. Gupta, R. F. Landis and V. M. Rotello, F1000Research, 2016, 5, 1–10 Search PubMed.
- R. Y. Pelgrift and A. J. Friedman, Adv. Drug Delivery Rev., 2013, 65, 1803–1815 CrossRef CAS PubMed.
- A. J. Huh and Y. J. Kwon, J. Controlled Release, 2011, 156, 128–145 CrossRef CAS PubMed.
- R. P. Allaker and G. Ren, Trans. R. Soc. Trop. Med. Hyg., 2008, 102, 1–2 CrossRef PubMed.
- B. Ramalingam, T. Parandhaman and S. K. Das, ACS Appl. Mater. Interfaces, 2016, 8, 4963–4976 CrossRef CAS PubMed.
- P. V. Baptista, M. P. McCusker, A. Carvalho, D. A. Ferreira, N. M. Mohan, M. Martins and A. R. Fernandes, Front. Microbiol., 2018, 9, 1441 CrossRef PubMed.
- A. Sirelkhatim, S. Mahmud, A. Seeni, N. H. M. Kaus, L. C. Ann, S. K. M. Bakhori, H. Hasan and D. Mohamad, Nano-Micro Lett., 2015, 7, 219–242 CrossRef CAS PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- N. G. Bonine, A. Berger, A. Altincatal, R. Wang, T. Bhagnani, P. Gillard and T. Lodise, Am. J. Med. Sci., 2019, 357, 103–110 CrossRef PubMed.
- E. M. Windels, J. E. Michiels, M. Fauvart, T. Wenseleers, B. Van den Bergh and J. Michiels, ISME J., 2019, 13, 1239–1251 CrossRef CAS PubMed.
- H. W. Ackermann, Adv. Virus Res., 1998, 51, 135–201 CrossRef CAS PubMed.
- E. B. Gordon, C. J. Knuff and B. A. Fane, J. Virol., 2012, 86, 9911–9918 CrossRef CAS PubMed.
- F. C. Tenover and R. V. Goering, J. Antimicrob. Chemother., 2009, 64, 441–446 CrossRef CAS PubMed.
- A. Neemuchwala, D. Soares, V. Ravirajan, A. Marchand-Austin, J. V. Kus and S. N. Patel, Antimicrob. Agents Chemother., 2018, 62, e01776 CrossRef CAS PubMed.
- A. A. Weaver, N. A. Hasan, M. Klaassen, H. Karathia, R. R. Colwell and J. D. Shrout, J. Med. Microbiol., 2019, 68, 1507–1516 CrossRef PubMed.
- G. Ryan, T. Dolling and S. Barnet, Med. Educ., 2004, 38, 638–645 CrossRef PubMed.
- D. E. Ohman and A. M. Chakrabarty, Infect. Immun., 1981, 33, 142–148 CAS.
- W. Wang, P. D. Nallathamby, C. M. Foster, J. L. Morrell-Falvey, N. P. Mortensen, M. J. Doktycz, B. Gu and S. T. Retterer, Nanoscale, 2013, 5, 10369–10375 RSC.
- T. H. L. Nghiem, T. N. Le, T. H. Do, T. T. D. Vu, Q. H. Do and H. N. Tran, J. Nanopart. Res., 2013, 15, 2091 CrossRef.
- P. Manivel, A. Balamurugan, N. Ponpandian, D. Mangalaraj and C. Viswanathan, Spectrochim. Acta, Part A, 2012, 95, 305–309 CrossRef CAS PubMed.
- J. M. Laing, M. D. Gober, E. K. Golembewski, S. M. Thompson, K. A. Gyure, P. J. Yarowsky and L. Aurelian, Mol. Ther., 2006, 13, 870–881 CrossRef CAS PubMed.
- F. Fenner, Virology, 1976, 71, 371–378 CrossRef CAS PubMed.
- M. Takac, A. Witte and U. Blasi, Microbiology, 2005, 151, 2331–2342 CrossRef CAS PubMed.
- P. R. Chipman, M. Agbandje-McKenna, J. Renaudin, T. S. Baker and R. McKenna, Structure, 1998, 6, 135–145 CrossRef CAS PubMed.
- P. Willingmann, S. Krishnaswamy, R. McKenna, T. J. Smith, N. H. Olson, M. G. Rossmann, P. L. Stow and N. L. Incardona, J. Mol. Biol., 1990, 212, 345–350 CrossRef CAS PubMed.
- K. L. Brentlinger, S. Hafenstein, C. R. Novak, B. A. Fane, R. Borgon, R. McKenna and M. Agbandje-McKenna, J. Bacteriol., 2002, 184, 1089–1094 CrossRef CAS PubMed.
- S. R. Bhattarai, S. Y. Yoo, S. W. Lee and D. Dean, Biomaterials, 2012, 33, 5166–5174 CrossRef CAS PubMed.
- J. Ren, Y. Guo, L. Shao, Y. Liu and Q. Liu, Exp. Ther. Med., 2018, 16, 3286–3292 Search PubMed.
- S. Wei, Q. Liu, T. Lian and L. Shao, Virus Res., 2019, 267, 1–8 CrossRef CAS PubMed.
- J. Sliwa-Dominiak, E. Suszynska, M. Pawlikowska and W. Deptula, Arch. Microbiol., 2013, 195, 765–771 CrossRef CAS PubMed.
- T. D. Read, C. M. Fraser, R. C. Hsia and P. M. Bavoil, Microb. Comp. Genomics, 2000, 5, 223–231 CrossRef CAS PubMed.
- S. Fischer, J. K. Swabeck and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 12325–12332 CrossRef CAS PubMed.
- E. Z. Gomaa, J. Genet. Eng. Biotechnol., 2017, 15, 49–57 CrossRef PubMed.
- E. Z. Gomaa, J. Gen. Appl. Microbiol., 2017, 63, 36–43 CrossRef CAS PubMed.
- D. P. Tamboli and D. S. Lee, J. Hazard. Mater., 2013, 260, 878–884 CrossRef CAS PubMed.
- G. A. Sotiriou, G. D. Etterlin, A. Spyrogianni, F. Krumeich, J. C. Leroux and S. E. Pratsinis, Chem. Commun., 2014, 50, 13559–13562 RSC.
- M. Hentzer, G. M. Teitzel, G. J. Balzer, A. Heydorn, S. Molin, M. Givskov and M. R. Parsek, J. Bacteriol., 2001, 183, 5395–5401 CrossRef CAS PubMed.
- X. Yan, B. He, L. Liu, G. Qu, J. Shi, L. Hu and G. Jiang, Metallomics, 2018, 10, 557–564 RSC.
- R. Krishnan, V. Arumugam and S. K. Vasaviah, J. Nanomed. Nanotechnol., 2015, 6, 4 Search PubMed.
- R. P. Smith, A. L. Baltch, P. B. Michelsen, W. J. Ritz and R. Alteri, Antimicrob. Agents Chemother., 2003, 47, 1081–1087 CrossRef CAS PubMed.
- P. D. Nallathamby, K. J. Lee, T. Desai and X. H. Xu, Biochemistry, 2010, 49, 5942–5953 CrossRef CAS PubMed.
- A. Ivask, I. Kurvet, K. Kasemets, I. Blinova, V. Aruoja, S. Suppi, H. Vija, A. Kakinen, T. Titma, M. Heinlaan, M. Visnapuu, D. Koller, V. Kisand and A. Kahru, PLoS One, 2014, 9, e102108 CrossRef PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- O. Gordon, T. Vig Slenters, P. S. Brunetto, A. E. Villaruz, D. E. Sturdevant, M. Otto, R. Landmann and K. M. Fromm, Antimicrob. Agents Chemother., 2010, 54, 4208–4218 CrossRef CAS PubMed.
- M. Akter, M. T. Sikder, M. M. Rahman, A. Ullah, K. F. B. Hossain, S. Banik, T. Hosokawa, T. Saito and M. Kurasaki, J. Adv. Res., 2018, 9, 1–16 CrossRef CAS PubMed.
- H. L. Su, C. C. Chou, D. J. Hung, S. H. Lin, I. C. Pao, J. H. Lin, F. L. Huang, R. X. Dong and J. J. Lin, Biomaterials, 2009, 30, 5979–5987 CrossRef CAS PubMed.
- W. Wang, Y. Lu, Z. Yue, W. Liu and Z. Cao, Chem. Commun., 2014, 50, 15030–15033 RSC.
- M. E. Bayer and J. L. Sloyer Jr, J. Gen. Microbiol., 1990, 136, 867–874 CrossRef CAS PubMed.
- H. Ou, T. Cheng, Y. Zhang, J. Liu, Y. Ding, J. Zhen, W. Shen, Y. Xu, W. Yang, P. Niu, J. Liu, Y. An, Y. Liu and L. Shi, Acta Biomater., 2018, 65, 339–348 CrossRef CAS PubMed.
- E. Pensa, E. Cortés, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benítez, A. A. Rubert and R. C. Salvarezza, Acc. Chem. Res., 2012, 45, 1183–1192 CrossRef CAS PubMed.
- K. J. Lee, L. M. Browning, T. Huang, F. Ding, P. D. Nallathamby and X. H. Xu, Anal. Bioanal. Chem., 2010, 397, 3317–3328 CrossRef CAS PubMed.
- P. D. Nallathamby and X. H. Xu, Nanoscale, 2010, 2, 942–952 RSC.
- T. Huang, P. D. Nallathamby, D. Gillet and X. H. Xu, Anal. Chem., 2007, 79, 7708–7718 CrossRef CAS PubMed.
- P. D. Nallathamby, T. Huang and X. H. Xu, Nanoscale, 2010, 2, 1715–1722 RSC.
- B. K. Monson, J. Stringham, B. B. Jones, S. Abdel-Aziz, C. M. Cutler Peck and R. J. Olson, J. Ocul. Pharmacol. Ther., 2010, 26, 133–136 CrossRef CAS PubMed.
- H. Jaganathan and B. Godin, Adv. Drug Delivery Rev., 2012, 64, 1800–1819 CrossRef CAS PubMed.
- J. McCarthy, I. Inkielewicz-Stepniak, J. J. Corbalan and M. W. Radomski, Chem. Res. Toxicol., 2012, 25, 2227–2235 Search PubMed.
- R. Salomoni, P. Leo, A. F. Montemor, B. G. Rinaldi and M. Rodrigues, Nanotechnol., Sci. Appl., 2017, 10, 115–121 Search PubMed.
- W. Liu, Y. Wu, C. Wang, H. C. Li, T. Wang, C. Y. Liao, L. Cui, Q. F. Zhou, B. Yan and G. B. Jiang, Nanotoxicology, 2010, 4, 319–330 CrossRef CAS PubMed.
- L. L. Ilang, N. H. Olson, T. Dokland, C. L. Music, R. H. Cheng, Z. Bowen, R. McKenna, M. G. Rossmann, T. S. Baker and N. L. Incardona, Structure, 1995, 3, 353–363 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00461k |
| This journal is © The Royal Society of Chemistry 2019 |