
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of microporous silica nanoparticles to study water phase transitions by vibrational spectroscopy†
Daniel J.
Rosenberg
 *ab,
Selim
Alayoglu
a,
Robert
Kostecki
c and
Musahid
Ahmed
*a
*ab,
Selim
Alayoglu
a,
Robert
Kostecki
c and
Musahid
Ahmed
*a
aChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA-94720, USA. E-mail: mahmed@lbl.gov
bGraduate Group in Biophysics, University of California, Berkeley, California 94720, USA
cEnergy Storage & Distributed Resources Division, Lawrence Berkeley National Laboratory, Berkeley, CA-94720, USA
First published on 7th November 2019
Abstract
Silica can take many forms, and its interaction with water can change dramatically at the interface. Silica based systems offer a rich tapestry to probe the confinement of water as size and volume can be controlled by various templating strategies and synthetic procedures. To this end, microporous silica nanoparticles have been developed by a reverse microemulsion method utilizing zinc nanoclusters encapsulated in hydroxyl-terminated polyamidoamine (PAMAM-OH) dendrimers as a soft template. These nanoparticles were made tunable within the outer diameter range of 20–50 nm with a core mesopore of 2–15 nm. Synthesized nanoparticles were used to study the effects of surface area and microporous volumes on the vibrational spectroscopy of water. These spectra reveal contributions from bulk interfacial/interparticle water, ice-like surface water, liquid-like water, and hydrated silica surfaces suggesting that microporous silica nanoparticles allow a way to probe silica water interactions at the molecular scale.
1. Introduction
Water and silica are two molecular systems, whose interactions manifest on geological timescales in rock and clay in the Earth crust geosphere, chemical formulations of tablets for drug delivery, concrete, lubricants, and in thermal heat transfer systems, amongst many others.1 Silica water interactions can lead to the formation of astrochemical grains,2 where it is postulated, that the first organic compounds were processed. In addition, it appears that water confined by solid walls to spaces smaller than a few nanometers begin to exhibit effects including delocalization and quantum coherence.3 These properties are strikingly different from those of bulk water and can influence the resulting physical and chemical behavior. For instance, it has been suggested more than a decade ago that water moves through nanoscale pores, orders of magnitude faster than through larger pores.4 Confinement organized the water network to behave differently by lowering the chemical potential and local electric fields can also influence water's orientation to the pore walls. The properties of water can also be influenced dramatically at the interface when walls and pores reach molecular dimensions. When water has been confined in more regular systems such as SBA-15 and MCM-41,5–7 it appears that the water dynamics is not influenced by surface interactions, as compared to more irregular systems such as silica gels and Vycor glass. However, to date, there have been very few studies of probing confinement of water with silica systems of tunable size.8–13The use of colloidal particles as hard templates is perhaps the oldest strategy for formation of hollow nanostructures and is still widely used today. An extensive amount of research has gone into the development of colloidal particles with the specifications needed to act as good templates. Many different hard templates are used for the creation of microporous (<2 nm pores) and mesoporous (2–50 nm pores) silica nanospheres, but the most commonly used are metal nanoparticles,14–19 polystyrene nanospheres,20,21 and solid silica or carbon nanospheres.22 Hard template methods are probably the most commonly used as they are easily formed and many are commercially available. Since these pre-formed templates have a ridged molecular structure, they tend to lead to nanostructures that are more monodisperse than other methods. However, having better control over polydispersity and morphology comes at a price in the removal of the hard template, which can be challenging and typically involves either chemical etching or thermal calcination (sintering) under high temperatures and pressures requiring high energy and complicated procedures. Additionally, limited template diversity narrows the overall size range, which has motivated researchers to focus on developing other synthetic techniques in recent years.
Soft template methods have many advantages over hard templates including simplified procedures, more template diversity, and easy removal of template after synthesis. Thus, microporous and mesoporous silica nanosphere synthesis by a soft template strategy is of rapidly growing interest to researchers. There are many different soft templates, but those commonly used in the synthesis of microporous silica nanospheres involve self-assembling template in an emulsion system. These techniques involve the use of surfactant or copolymers in their formation. A wide variety of ionic or non-ionic surfactants and copolymers can be utilized in this process. The diversity of surfactants and copolymer systems has led to a wide variety of organized structures including micelles,23–29 vesicles,30–32 and ionic (lyotropic) liquid crystals33–36 to be utilized as soft templates in the synthesis of microporous and mesoporous silica nanostructures, tailor made to suit individual research needs. These methods are often simpler than those using hard templates, often accomplished by a one pot synthesis with the easy removal of the template after the reaction. The major disadvantage, however, is that the polydispersity and overall morphology of generated nanostructures cannot be as carefully controlled due to structural defects and the inherent dynamic nature of these soft templates.
Herein, we seek to develop a strategy to synthesize a series of microporous silica nanoparticles that can be tuned within the outer diameter range of 20–50 nm with a core mesopore of 2–15 nm to study the effects of surface area and microporous volumes on water dynamics using transmission Fourier-transform infrared spectroscopy (FTIR). To this end, a reverse microemulsion method has been developed utilizing zinc nanoclusters encapsulated in PAMAM-OH dendrimers as a soft template. These soft templates were selected for their standardized size range and controllable metal packing to provide careful control of the overall nanoparticle size and the size of internal mesopores. The major peaks analyzed via vibrational spectroscopy represent contributions from bulk interfacial/interparticle water, ice-like surface water, liquid-like water, and hydration of the silica surface to give a complete picture of the water dynamics within and without the individual silica nanoparticle species.
2. Experimental section
Materials, sources, and purity are detailed in the ESI.† Provided here are experimental procedures for synthesis and characterization.2.1 Preparation of zinc encapsulated x = 4–7 generation hydroxyl terminated dendrimer (Zn@Gx-OH) by varying generation
Different dendrimer generations and zinc concentrations were utilized in this study to tune the sizes of internal mesopores (ESI Fig. S1†). The general method for the preparation of Zn@Gx-OH was to add 1 mL of the appropriate concentration of ZnSO4 solution in water to a 1 mL solution of the appropriate concentration of Xth generation hydroxyl terminated dendrimer in water. Solutions were then mixed by slow shaker for 20 min to allow for impregnation of zinc into dendrimer before moving on to the silanization step. Copper species have also been prepared in the same way by substituting the same concentration of CuSO4 solution in water.2.2 Preparation of zinc encapsulated 4th generation hydroxyl terminated dendrimer (Zn@G4-OH) by varying disperse phase composition
Changes to the composition of the solvent was varied during preparation of zinc encapsulated 4th generation hydroxyl terminated dendrimer (Zn@G4-OH) which allowed for further tuning of the overall nanoparticle sizes and the sizes of internal mesopores (ESI S1†). The general method for the preparation of Zn@G4-OH was to add 1 mL of 21.6 mM ZnSO4 solution in water to a 1 mL 1.351 mM solution of 4th generation hydroxyl terminated dendrimer in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water to either methanol, ethanol, or 1-hexanol. Solutions were then mixed by slow shaker for 20 min to allow for impregnation of zinc into dendrimer before moving on to the silanization step.
:1 water to either methanol, ethanol, or 1-hexanol. Solutions were then mixed by slow shaker for 20 min to allow for impregnation of zinc into dendrimer before moving on to the silanization step.
2.3 Synthesis of Zn@Gx-OH@SiO2 (x = 4–7) by reverse microemulsion
In a typical silanization step, 4.99 g IGEPAL CO-520 (∼5 mL, 11.3 mmol) was dissolved via sonication in 85 mL cyclohexane making a 0.13 M solution. 2 mL of prepared Zn@Gx-OH solution was then slowly added to a stirring IGEPAL solution. The solution became cloudy and slowly turns transparent showing that the reverse microemulsion had formed. The mixture was stirred for 30 min and then the appropriate amount of TEOS was added for Zn@Gx-OH@SiO2 (ESI S1†). The mixture was stirred for 30 min then 400 μL of 5 M ammonia hydroxide was added. The mixture was stirred at room temperature for 36 h. 5 mL Ethanol was then added to destabilize the microemulsion. Suspension was then centrifuged at 10000 rpm for 20 min, the supernatant was decanted, and the pellet washed in 15 mL ethanol by sonication. This washing process was repeated thrice to remove surfactant. Nanoparticles were redispersed in ethanol by sonication for template and metal etching step.
2.4 Removal of zinc and dendrimer by acid catalyzed etching
To remove the zinc and the dendrimer from the core of the formed mesoporous silica nanoparticles, 19 mL of Zn@Gx-OH@SiO2 dispersed in ethanol was brought to reflux. Then 1 mL of 3.7% HCl was added and the system was allowed to reflux for 90 min. 15 mL D.I. H2O was added and the reaction mixture was cooled to ambient temperature. The solution was then centrifuged at 10000 rpm for 20 min, the supernatant was decanted, and the pellet washed in 15 mL D.I. H2O by sonication. This washing process was repeated thrice. Nanoparticles were redispersed in 1:1 v/v ethanol:IPA by sonication for storage and dropcasting.
2.5 Infrared spectroscopy data collection
All synthesized nanoparticles were analyzed by FTIR using a Thermo Fisher Nicolet 6700 FTIR Spectrometer run in transmission mode. For hydration experiments, FTIR was utilized in conjunction with a custom-built vapor cell to monitor the adsorption and desorption of water vapor, the design for which is illustrated in ESI Fig. S2.† The system used ZnSe windows providing a spectral range of 4000–650 cm−1 with a pathlength of 338 mm. For these experiments, samples were dropcast on mirror finished Ge windows which were suspended at the center of the cell attached to a Z-stage to make it possible to adjust sample position to maximize signal. Dropcast samples were created from suspensions of silica nanoparticles in a 1:1 ratio of EtOH to IPA. Both the cell and the sample chamber of the FTIR were purged with dry N2 at a flow rate of 5 SCFH and 50 SCFH respectively and the vapor cell could be reduced to 50 mTorr by using a Pfeiffer pump station when required. The cell was monitored for temperature using an Extech EasyView 15 thermometer datalogger with a thermocouple inserted directly into the sample chamber near the Ge window. The percent relative humidity was first measured using a Vaisala HMI41 humidity probe and later calculated by using the water vapor peak at 3724 cm−1 which was found to give an accurate dependence on relative humidity. Maximum percent relative humidity for all samples was found to be 50 ± 5%. To control the vapor pressure of the water entering the cell, the dried N2 was split between two identical rotameters, each with a maximum flow of 5 SCFH, one leading to a water bubbler and one which bypassed the bubbler. These gas lines were then recombined before reaching the vapor cell to provide careful control of relative humidity in the sample chamber. All spectra were recorded using Thermo Fisher OMNIC 9 software with 32 scans, 8 cm−1 resolution, and a laser frequency of 15798.25 cm−1. The background for subtraction was produced with 64 scans of the original dry silica NPs immediately before the addition of water vapor to the chamber.
2.6 Infrared spectroscopy analysis and fittings
All spectra were processed and analyzed using Origin 9.1 and MS Excel. To properly analyze the individual spectra, first the baseline for each individual spectrum was normalized to zero to account for drift over time. Next a hydration series was taken of blank Ge under the same vapor conditions as the samples. These spectra were interpolated and used to suppress the contribution of atmospheric water vapor in the chamber that was not interacting with the samples. Finally, the whole spectral series was normalized to the 3250 cm−1 peak to more easily make comparisons between individual nanoparticle species. The process of data manipulation can be seen in ESI Fig. S3.† Once the spectra were finalized, the peaks of interest were fit using CasaXPS.2.7 Transmission electron microscopy
Bright field and annular dark field TEM images, STEM/EDS maps and STEM/EELS spectra were acquired using a Jeol 2100F microscope equipped with an Oxford EDS spectrometer and a GATAN Tridem EELS spectrometer. An accelerating voltage of 200 kV was used for both the TEM, STEM/EDS, and STEM/EELS. EELS resolution was 1 eV, measured as FWHM of zero loss peak in vacuum, and collection angle was 12 mrad. High resolution SEM images were collected using a Zeiss Gemini Ultra-55 Analytical Field Emission SEM with electron accelerating voltages of 1 and 3 kV.3. Results and discussion
3.1 Synthesis and characterization of silica nanoparticles
Two discrete series of metal encapsulated dendrimer microporous silica nanoparticles were created for this study, both through a reverse microemulsion method similar to that developed by Lin et al. 2015,27 as illustrated by Fig. 1. For the purposes of this paper, these individual nanoparticle species are denoted M@Gx-OH@SiO2 where M = Zn or Cu and x = 4, 5, 6, or 7 representing the generation of dendrimer used, which correspond to approximate dendrimer diameters of 4.5, 5.4, 6.7, or 8.1 nm respectively. Variations of the dendrimer generation, metal packing within the dendrimer, and the composition of the dispersed phase of the reverse microemulsion system, allowed for careful control of both the overall particle size between 20–50 nm and more importantly the sizes of the internal mesopores between 2 nm and 15 nm. These variations in sample preparation are tabulated in ESI S1.† The final samples as well as several of their intermediate steps were analyzed by transmission electron microscopy (TEM) for morphology, STEM/EDS or STEM/EELS for chemical composition, and FTIR to study the effects of surface area and microporous volumes on the vibrational behavior of water within and without the individual nanoparticle species. | ||
| Fig. 1 Scheme for total synthesis of dendrimer encapsulated mesoporous silica NPs. Step (1) inoculation of dendrimer with metal ions, (2) formation of reverse microemulsion with disperse phase (blue) and continuous phase (orange), (3) base catalyzed silica formation, (4) acid catalyzed etching of metal encapsulated dendrimers. | ||
M@Gx-OH@SiO2 nanoparticles were synthesized starting with the encapsulation of various concentrations of zinc or copper proportional to the generation of PAMAM-OH dendrimer. The preparation of metal encapsulated dendrimers was accomplished by utilizing methods pioneered by Zhao et al. 1998 in their work on dendrimer encapsulated Cu nanoclusters.36 It was found in this seminal work that Cu2+ would coordinate with the outermost tertiary amine groups within each of the PAMAM-OH dendrimers, allowing the generation of dendrimer to effectively bind a proportional number of Cu2+ ions. Thus, the generation 4, 5, 6, and 7 PAMAM-OH dendrimers could effectively bind 16, 32, 64, and 128 Cu2+ ions respectively due to this phenomenon and demonstrated in this study (vide infra), to work similarly with Zn2+.
Copper encapsulated nanoparticles were initially useful for visualization, however it was found that the size of the internal mesopore after etching could not be controlled and was independent of metal packing whereas zinc led to the formation of reproducible, size controlled mesopores forming the final silica nanoparticle series necessary for this work. The enhanced ability of zinc to produce these nanoparticles species was possibly due to catalytic effects on either silica nucleation, amide hydrolysis, or both.37 The contrast between the use of these two metals in the context of silica nucleation is an interesting topic for a future study but is beyond the scope of this work. In both cases however, it was found that metal inoculation was integral in the formation of hollow nanoparticles, as empty dendrimers did not result in any internal mesopores. After the inoculation of the desired metal ions, these M@Gx-OH dendrimers were encapsulated within microporous silica via the reverse microemulsion technique to form M@Gx-OH@SiO2 nanoparticles (Fig. 1). Fig. 2A shows STEM/EDS maps at Si K, N K, and Cu Kα lines, indicating an encapsulation of the metal (Cu) and N based dendrimer localized to the core of the silica nanoparticle. The metal was subsequently removed by washing with dilute 100 mM HCl to ensure that it would not influence hydration experiments. For the Cu@G4-OH@SiO2 nanoparticles, the internal mesopores were about 15 nm, shown in Fig. 2B, irrespective of the dendrimer generation (x = 4–7) and was not used in further studies as a diverse enough series could not be developed for comparison. The vibrational spectra in Fig. 2C shows an attenuation of amide I and amide II bands at 1650 cm−1 and 1590 cm−1, respectively, and N–H stretch at 3300 cm−1 after the acid treatment, indicating the breakup and removal of dendrimer.
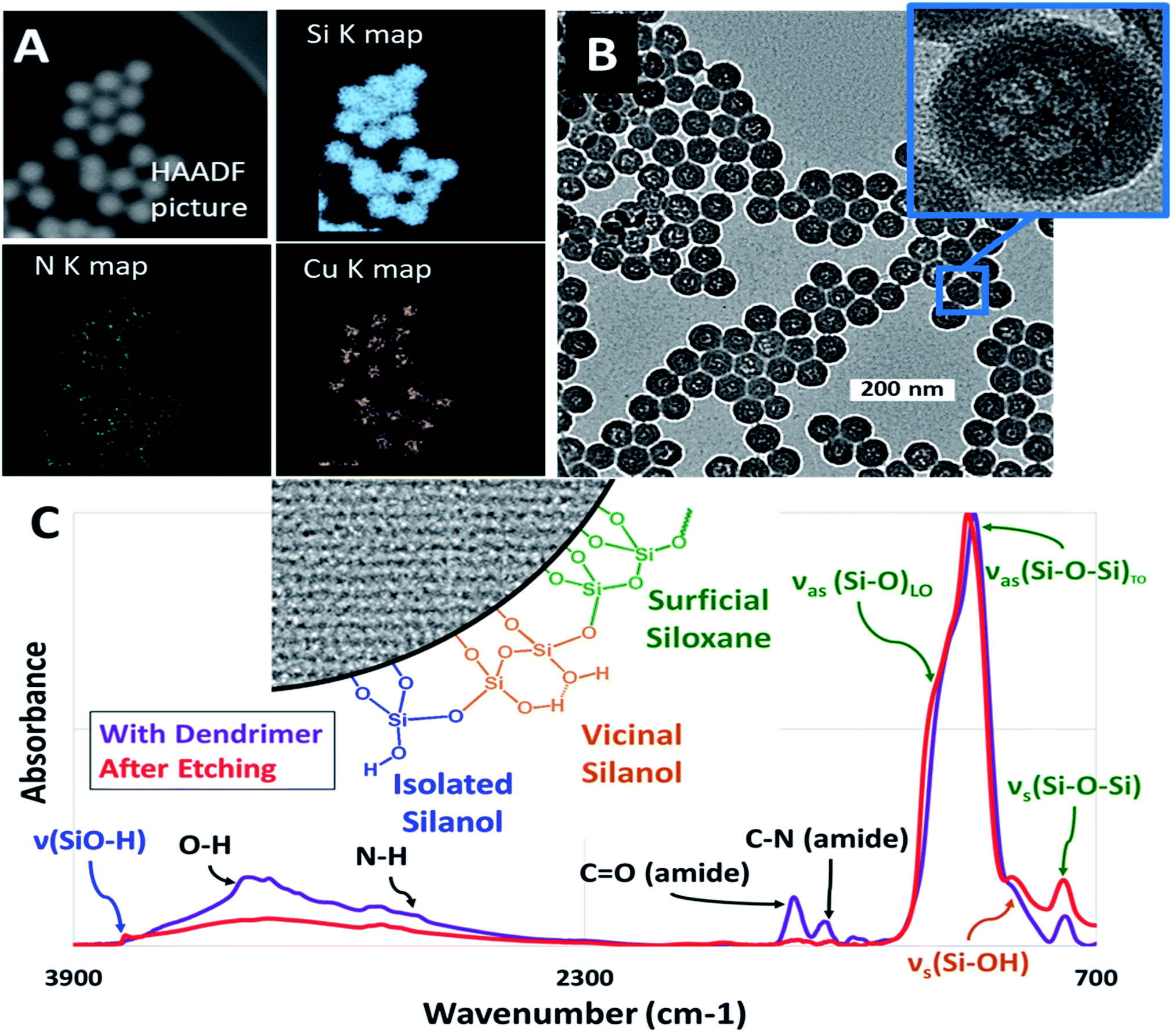 | ||
| Fig. 2 Figure shows (A) STEM/EDS phase maps for Cu encapsulated G4OH in microporous silica nanoparticles (NPs), (B) TEM picture of silica inorganic micelles after removal of the metal and dendrimer with enlarged inset of single NP, and (C) FTIR spectra of as-synthesized silica NPs before (purple) and after (red) the removal of Cu2+ and G4OH. Inset defines functional group variations at the silica surface. | ||
High-angle annular dark-field (HAADF) images and STEM/EDS spectra of a cluster of nanoparticles, taken before and after the acid treatment, indicated the removal of zinc (Fig. 3A and B). Furthermore, the removal of both zinc and dendrimer was locally confirmed on single nanoparticles by STEM/EELS spectra obtained at N K and Zn L edges. STEM/EELS at O K and Si K edges showed no discernible changes to silica microstructure (Fig. 3C). Importantly, this method produced sharply monodisperse microporous silica nanospheres with six discrete internal mesopore sizes and one species without noticeable mesopores. Measurements for overall particle and internal mesopore sizes was determined by statistical analysis of TEM images (ESI Fig. S4†). One species displayed no visible mesopores thus suggesting a continuous microporous structure of pores <1.5 nm which is below the resolution visible in both bright field and annular dark field TEM images. The average overall diameter (OD), internal mesopore diameter (MD), and the ratio between the two (OD/MD) have been tabulated in Table 1. Representative mesopore size histograms and TEM pictures are shown in Fig. 4. Mesopore sizes between 4.7 nm and 2.2 nm were controlled by increasing the dendrimer generation and corresponding metal packing for the Zn@Gx-OH@SiO2 (x = 4–6) nanoparticles and <1.5 nm in the case of Zn@G7-OH@SiO2 nanoparticles, as represented by Fig. 5A. It should also be noted that hydrodynamic size of a 4th generation dendrimer is about 4 nm (as per manufacturer specifications) which closely matches the measured mesopores size, but no such correlation could be drawn for the larger dendrimers suggesting that zinc packing contributes more to morphological changes. In addition to alterations in dendrimer generation and metal packing, a second series of samples was created utilizing modifications to the dispersed phase of the reverse microemulsion system. These changes to the dispersed phase demonstrated a reproducible effect on the size of the internal mesopore while also systematically changing the overall nanoparticle size (Fig. 5B). In this case, a 1:1 ratio of alcohol to water was substituted for pure water as the dispersed phase. Three different alcohols were used with increasing hydrocarbon chain length of 1, 2, and 6 and an inverse relationship was observed between increasing chain length and both the increase in overall nanoparticle size and the size of the internal mesopore, as shown in Fig. 5B. As the pure water system displays size order near the middle of the series, it is apparent that overall polarity of the dispersed phase plays less of a role in the size evolution than stabilization effects of the localized polarity of the alcohols themselves. Two separate phenomena seem to govern the overall nanoparticle size in this series. For the short chain alcohols (methanol and ethanol) which are miscible with the aqueous phase, the alcohols likely stabilize the micelles created by the surfactants38,39 leading to larger nanoparticles, while the longer chain alcohol (n-hexanol) acts as a co-surfactant leading to smaller water droplet sizes, producing an overall reduction in nanoparticle size as seem in previous works.27,40 Lastly, the addition of methanol and ethanol to the disperse phase makes the continuous oil phase pre-saturated with ethanol. Thus, the ethanol produced during the hydrolysis of the silica precursors must remain in the water phase, keeping the reaction in the core from going to product. These factors would make the shell less prone to etching while the less condensed core can be easily removed leading to larger internal mesopores. In the case of methanol and ethanol, it is reasonable to assume that changes to the disperse phase did not lower the packing density of the silica shell which would have led to larger micropores that would be detectable in TEM images. It is unclear however, whether the n-hexanol increases packing density producing smaller micropores.
 | ||
| Fig. 3 Figure shows (A) STEM/HAADF images for Zn encapsulated G4OH in microporous silica NPs before (purple) and after (red) etching, and (B) STEM/EDS spectra of a collection of NPs, and (C) local STEM/EELS spectra taken from the centers of single NPs. The same color code as in (A) applies to all spectra in (B and C). | ||
| Sample ID | Overall diameter (OD) (nm) | Mesopore diameter (MD) (nm) | Ratio (OD/MD) |
|---|---|---|---|
| Zn@G4-OH@SiO2 (methanol/water) | 47.9 ± 3.5 | 15.0 ± 2.2 | 3.1 |
| Zn@G4-OH@SiO2 (ethanol/water) | 44.6 ± 4.5 | 14.2 ± 3.6 | 3.2 |
| Zn@G4-OH@SiO2 (hexanol/water) | 22.0 ± 2.5 | 4.0 ± 0.9 | 5.6 |
| Zn@G4-OH@SiO2 (water) | 30.9 ± 2.4 | 4.7 ± 1.2 | 6.6 |
| Zn@G5-OH@SiO2 | 30.2 ± 2.4 | 2.7 ± 0.6 | 11.2 |
| Zn@G6-OH@SiO2 | 29.5 ± 2.3 | 2.3 ± 0.5 | 13.0 |
| Zn@G7-OH@SiO2 | 28.2 ± 2.0 | <1.5 | >18.8 |
 | ||
| Fig. 4 Figure shows mesopore size histograms of a select inorganic silica micelles. Insets show single particle TEM pictures for the corresponding micelles. | ||
 | ||
| Fig. 5 Figure demonstrates both nanoparticle series created by altering (A) dendrimer generation and metal packing and (B) dispersed phase of the reverse microemulsion system. | ||
3.2 Probing size effects on the vibrational behavior of water
Well-characterized silica nanoparticles were dropcast onto mirror polished Ge windows forming nanoparticle films. The film thickness across the window varied from 10–20 μm as shown in ESI Fig. S5† but had no noticeable effect on the overall IR spectra as shown in ESI Fig. S6.† These films were used for the evaluation of water uptake on various synthesized nanoparticle species as a function of morphological changes. Fig. 6A shows the FTIR spectral series for all microporous silica nanoparticles at a maximum relative humidity of ∼50% at 309 K. It is readily apparent that the FTIR spectra were different particularly in the 2400–3600 cm−1 region for the nanoparticles studied. The interaction of water with mesoporous silica can give rise to spectral signatures in the IR which represent both the hydrogen bonding of water and its connectivity to the surface. An early example of this was the seminal sum frequency generation work performed on the water/α-quartz interface where, depending on pH, ice like features could be discerned on the interface.41 A molecular dynamics study of water adsorbed on silica42 also identified “ice-like” and “liquid-like” vibrational spectral signatures confirming the earlier sum frequency results. Similar results showing the evolution of adsorbed water layers on silicon oxide surfaces as a function of relative humidity was obtained using attenuated total reflection (ATR) spectroscopy.8 Starting with ice like features at low humidity, at around 30%, liquid water features were discerned, and an adsorption isotherm was obtained via the changes in the OH stretching vibrational peak positions and intensities. | ||
| Fig. 6 Figure shows (A) representative FTIR spectral series of the hydration of all synthesized nanoparticle species, and (B) representative FTIR spectrum of the vibrational energies of water as individually fit. (C) Plot of the vibrational stretching regions of water, and the hydration of the surface silica, as they change as a function of the ratio of outer to inner nanoparticle diameter. (D) A visual representation of the ratio of the outer diameter to the internal mesopore diameter. | ||
To obtain a quantitative picture for the hydration of silica within and on the surface of the individual nanoparticle species, several regions of the vibrational spectrum were examined and compared. These regions included, the O–H stretching band, from about 2600–3600 cm−1, and the silanol (Si–OH) and siloxane (Si–O) stretches at 956 and 876 cm−1 respectively.9 The O–H stretching band was deconvoluted into four O–H stretch peaks at 3624, 3450, 3250, and 3018 cm−1 to show how water's interaction with silica changes its vibrational spectra and hence phase. The deconvolution of the O–H stretching band is based on the fact that water molecules can make up distinct H-bonding environments which correspond to different water conformations and phases in the bulk and at the silica surface. A representative collection of the decomposed peaks is shown in Fig. 6B and more are shown in ESI Fig. S7.† Additionally, a detailed list of peak assignments can be found in ESI Fig. S8.†
There are four major known conformations of interfacial and confined water in silica micelles which exhibit distinct IR absorption peaks: (1) bulk interfacial/interparticle water at 3018 cm−1,10 (2) ice-like surface water (first 3 water monolayers) at 3241 cm−1,8,11 (3) bulk confined water at 3459 cm−1,8,11 and (4) vicinal SiO–H bonding, at about 3624 cm−1 on the silica surface.9 For the purpose of this analysis, the spectra were normalized to the 3250 cm−1 peak as depicted in Fig. 6B. The peak fittings centered at 3018 and 3241 cm−1 were constrained to isolate the change in the peak centered at 3459 cm−1. This isolation of the 3459 cm−1 was used to rule out the effects of slight changes in the surface area and interparticle space to focus solely on the changes in the rise in liquid-like water due to changes in the percentage of overall porous volume of the individual species. Additional information was taken from the silanol siloxane stretch at 956 cm−1 which forms an isosbestic point with the siloxane stretch at 876 cm−1 due to the hydration of the bridged surficial siloxane bonds (Si–O–Si).9
The fitted peak areas for 3459 and 956 cm−1 (Fig. 6C) were plotted as a function of the ratio between the average overall/outer diameter to the internal mesopore diameter of each of the nanoparticles. This relationship between outer and inter-diameter was selected to simultaneously demonstrate the relationship of increasing porous volume in the case of the 3459 cm−1 peak and the decrease in overall surface area of the 956 cm−1 peak. It was determined that as the amount of overall porous volume increased, the contribution of liquid-like water also increased and as expected, the decrease in overall surface area demonstrated a decrease in hydration at the silica surface. The major outlier in these trends was observed in the case of Zn@G4-OH@SiO2 (OD/MD = 5.6) prepared with 1:1 n-hexanol to water as the disperse phase. This may signify that the microporous structure has in fact been altered as discussed.
The steady rise in liquid-like water at 3459 cm−1 while the contribution of ice-like (3241 cm−1) and silica–water interactions (3018 cm−1) remain consistent at maximum hydration would suggest that the initial ice-like layer is formed on the silica surface, followed by the formation of interfacial layers (as suggested by Asay and Kim8), which reach a saturation point and remains constant, leading to a liquid-like layer that can develop just from gas phase water uptake relative to available microporous volumes. These findings suggest that most of the dynamics are occurring within the microporous volume of the nanoparticles.
In other studies, Musat et al.12 used ultrafast IR pump probe spectroscopy to probe fully hydrated controlled pore glasses (CPC). They found an increase in the lifetime of OH vibration and a blue shift in the frequency spectrum as the size of the pore decreased, suggesting slower rotational dynamics with increased confinement. Huang et al.10 performed Raman spectroscopy on hydration of Vycor glass (6.9 nm average diameter), and observed a new peak at 3590 cm−1, multimeric water forms on the inner surface of mesoporous hydrophilic materials, and a small confinement effect (water pool less than 4 nm) conceals interfacial effects. Le Caër et al.13 used attenuated total reflectance on water confined on CPC and suggested that there is substantial perturbation of the hydrogen bond network even with these fairly large dimensions. Using a core–shell model, they came to the surprising conclusion that an interfacial water layer of 3, 12, 13 and 30 nm form for pores with diameters of 8, 25, 55 and 320 nm respectively. Also an analysis of the connectivity band between 30–350 cm−1 suggested that the water network becomes more organized upon confinement. Very recently, an anomalously low dielectric constant was measured for water sandwiched between two atomically flat walls separated between 1–100 nm.43 The authors invoked a layered structure for water near a solid surface, reminiscent of that observed here and by other experimental groups to explain these results.
Here we posit, that our synthetic method of generating tunable microporous silica nanoparticles with a tunable mesoporous core and performing vibrational studies with them provides a new way to probe the dynamics of interfacial and confined water on silica systems. Future work with these nanoparticles includes performing temperature and humidity dependent measurements using attenuated total reflectance and terahertz spectroscopy where nanoparticle thin films lend themselves well for such studies. To clearly distinguish between the microporous shells and interior mesopore, as well as intra-particle surface regions and inter-particle space, synchrotron based nano-infrared spectroscopy44 will be employed to study our inorganic micelles during hydration and dehydration. Another approach would be to use synchrotron based X-ray photoelectron spectroscopy of nanoparticles to probe the local electronic structure of both the silica and water confined within the pores.45,46 Functionalization of these silica nanoparticles will also allow one to tune the wetting properties of the surfaces to provide a gradient between hydrophobic and hydrophilic interactions between water and these surfaces.47 Beyond probing confinement, these nanoparticles could also be used to probe evaporation and condensation from surfaces, dynamics at the solid liquid vapor interface (SLV), a topic of much interest in thermal science.48–50 The complex chemical and structural properties of the SLV interface under non-equilibrium conditions make molecular level modeling and experimentation very challenging. At a SLV interface, there is a transition from strong ionic and covalent bonds to weaker van der Waals and hydrogen bonds, which governs thermal and mass transport at the interface. We suggest that synthesized nanoparticles coupled with vibrational spectroscopy could be a way forward to directly probe these interfaces.
4. Conclusion
Silica nanoparticles with controlled overall sizes and internal mesoporous volumes were generated by using a metal loaded dendrimer-template silica nucleation and growth strategy. Two discreet series of metal encapsulated dendrimer mesoporous silica nanoparticles have been created by utilizing (1) variations to the dendrimer generation, and metal packing, and (2) the composition of the dispersed phase of the reverse microemulsion system. These modifications allowed for careful control of both the overall particle size between 20–50 nm and the sizes of the internal mesopores between 2 nm and 15 nm. Control of the size of the internal mesopore and overall nanoparticle size has been attributed to three contributing factors, (1) the different amounts of metal (Zn) ions that can bind within the dendrimers, (2) the different internal pore volumes of various dendrimer generations, and (3) variations in the packing arrangements of various dendrimer generations and aqueous droplet (micelle) size in the microemulsion. These factors can be explained by three different observations, (1) dendrimers complexed with Cu ions produced much larger mesopores, (2) empty dendrimers did not produce any mesopores at all, and (3) distinct morphological changes were obtained through variations to the dispersed phase effecting the packing density of the dendrimers, silica nucleation, and the size of the aqueous droplet in the microemulsion. Finally, these nanoparticles were dropcast as thin films and water uptake on them were performed with FTIR. Distinct spectral features revealed signatures of liquid, interfacial and ice-like water which were correlated to mesoporous vs. microporous structures within these nanoparticles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory under U.S. Department of Energy Contract No. DE-AC02-05CH11231. MA also acknowledges support from the Condensed Phase, Interfaces and Molecular Sciences program of DOE under the same contract. TEM, STEM/EELS, and FESEM imaging and measurements were performed at the Imaging and Manipulation facility in the Molecular Foundry. Work performed at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231.References
- H. Zhang, W. Gu, M. J. Li, Z. Y. Li, Z. J. Hu and W. Q. Tao, Int. J. Heat Mass Transfer, 2014, 78, 947–959 CrossRef CAS.
- A. Rosu-Finsen, D. Marchione, T. L. Salter, J. W. Stubbing, W. A. Brown and M. R. S. McCoustra, Phys. Chem. Chem. Phys., 2016, 18, 31930–31935 RSC.
- M. D. Fayer and N. E. Levinger, Annu. Rev. Anal. Chem., 2010, 3, 89–107 CrossRef CAS PubMed.
- A. Noy, H. G. Park, F. Fornasiero, J. K. Holt, C. P. Grigoropoulos and O. Bakajin, Nano Today, 2007, 2, 22–29 CrossRef.
- S. Cerveny, F. Mallamace, J. Swenson, M. Vogel and L. Xu, Chem. Rev., 2016, 116, 7608–7625 CrossRef CAS PubMed.
- A. W. Knight, N. G. Kalugin, E. Coker and A. G. Ilgen, Sci. Rep., 2019, 9, 8246 CrossRef PubMed.
- T. Takei, K. Mukasa, M. Kofuji, M. Fuji, T. Watanabe, M. Chikazawa and T. Kanazawa, Colloid Polym. Sci., 2000, 278, 475–480 CrossRef CAS.
- D. B. Asay and S. H. Kim, J. Phys. Chem. B, 2005, 109, 16760–16763 CrossRef CAS PubMed.
- S. L. Warring, D. A. Beattie and A. J. McQuillan, Langmuir, 2016, 32, 1568–1576 CrossRef CAS PubMed.
- X. F. Huang, Q. Wang, X. X. Liu, S. H. Yang, C. X. Li, G. Sun, L. Q. Pan and K. Q. Lu, J. Phys. Chem. C, 2009, 113, 18768–18771 CrossRef CAS.
- J. Peixinho, G. Lefèvre, F.-X. Coudert and O. Hurisse, J. Colloid Interface Sci., 2013, 408, 206–211 CrossRef CAS PubMed.
- R. Musat, J. P. Renault, M. Candelaresi, D. J. Palmer, S. Le Caër, R. Righini and S. Pommeret, Angew. Chem., Int. Ed., 2008, 47, 8033–8035 CrossRef CAS PubMed.
- S. Le Caër, S. Pin, S. Esnouf, Q. Raffy, J. Ph. Renault, J.-B. Brubach, G. Creff and P. Roy, Phys. Chem. Chem. Phys., 2011, 13, 17658 RSC.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- J. Wang, K. P. Loh, Y. L. Zhong, M. Lin, J. Ding and Y. L. Foo, Chem. Mater., 2007, 19, 2566–2572 CrossRef CAS.
- Q. Yu, J. Hui, P. Wang, B. Xu, J. Zhuang and X. Wang, Nanoscale, 2012, 4, 7114 RSC.
- C.-L. Fang, K. Qian, J. Zhu, S. Wang, X. Lv and S.-H. Yu, Nanotechnology, 2008, 19, 125601 CrossRef PubMed.
- Z. Teng, X. Su, Y. Zheng, J. Sun, G. Chen, C. Tian, J. Wang, H. Li, Y. Zhao and G. Lu, Chem. Mater., 2013, 25, 98–105 CrossRef CAS.
- J. Dou and H. C. Zeng, J. Am. Chem. Soc., 2012, 134, 16235–16246 CrossRef CAS PubMed.
- X. Zhang, L. Zhang and Q. Yang, J. Mater. Chem. A, 2014, 2, 7546–7554 RSC.
- X. Mei, D. Chen, N. Li, Q. Xu, J. Ge, H. Li and J. Lu, Microporous Mesoporous Mater., 2012, 152, 16–24 CrossRef CAS.
- I. Lee, M. A. Albiter, Q. Zhang, J. Ge, Y. Yin and F. Zaera, Phys. Chem. Chem. Phys., 2011, 13, 2449–2456 RSC.
- J. Zhu, J. Tang, L. Zhao, X. Zhou, Y. Wang and C. Yu, Small, 2010, 6, 276–282 CrossRef CAS PubMed.
- Y.-S. Lin, S.-H. Wu, C.-T. Tseng, Y. Hung, C. Chang and C.-Y. Mou, Chem. Commun., 2009, 3542 RSC.
- D. K. Yi, S. T. Selvan, S. S. Lee, G. C. Papaefthymiou, D. Kundaliya and J. Y. Ying, J. Am. Chem. Soc., 2005, 127, 4990–4991 CrossRef CAS PubMed.
- J. Liu, Q. Yang, L. Zhang, H. Yang, J. Gao and C. Li, Chem. Mater., 2008, 20, 4268–4275 CAS.
- C.-H. Lin, J.-H. Chang, Y.-Q. Yeh, S.-H. Wu, Y.-H. Liu and C.-Y. Mou, Nanoscale, 2015, 7, 9614–9626 RSC.
- D. K. Yi, S. S. Lee, G. C. Papaefthymiou and J. Y. Ying, Chem. Mater., 2006, 18, 614–619 CrossRef CAS.
- Y. Ren, X. Xin, W. Tang, Y. Zhang, J. Shen and L. Wang, Colloid Polym. Sci., 2015, 293, 1695–1703 CrossRef CAS.
- J. Peng, Y. Yao, X. Zhang, C. Li and Q. Yang, Chem. Commun., 2014, 50, 10830 RSC.
- H. Djojoputro, X. F. Zhou, S. Z. Qiao, L. Z. Wang, C. Z. Yu and G. Q. Lu, J. Am. Chem. Soc., 2006, 128, 6320–6321 CrossRef CAS PubMed.
- L. Zhang, S. Z. Qiao, Y. G. Jin, Z. G. Chen, H. C. Gu and G. Q. Lu, Adv. Mater., 2008, 20, 805–809 CrossRef CAS.
- S. Z. Qiao, C. X. Lin, Y. Jin, Z. Li, Z. Yan, Z. Hao, Y. Huang and G. Q. Lu, J. Phys. Chem. C, 2009, 113, 8673–8682 CrossRef CAS.
- C. X. Lin, P. Yuan, C. Z. Yu, S. Z. Qiao and G. Q. Lu, Microporous Mesoporous Mater., 2009, 126, 253–261 CrossRef CAS.
- Y. Wan and S.-H. Yu, J. Phys. Chem. C, 2008, 112, 3641–3647 CrossRef CAS.
- M. Zhao, L. Sun and R. M. Crooks, J. Am. Chem. Soc., 1998, 120, 4877–4878 CrossRef CAS.
- S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS.
- R. Nagarajan and E. Ruckenstein, Langmuir, 2000, 16, 6400–6415 CrossRef CAS.
- F. J. Arriagada and K. Osseo-Asare, Colloids Surf., 1992, 69, 105–115 CrossRef CAS.
- L. Yang, R. Xie, L. Liu, D. Xiao and J. Zhu, J. Phys. Chem. C, 2011, 115, 19507–19512 CrossRef CAS.
- V. Ostroverkhov, G. A. Waychunas and Y. R. Shen, Chem. Phys. Lett., 2004, 386, 144–148 CrossRef CAS.
- M. Sulpizi, M.-P. Gaigeot and M. Sprik, J. Chem. Theory Comput., 2012, 8, 1037–1047 CrossRef CAS PubMed.
- L. Fumagalli, A. Esfandiar, R. Fabregas, S. Hu, P. Ares, A. Janardanan, Q. Yang, B. Radha, T. Taniguchi, K. Watanabe, G. Gomila, K. S. Novoselov and A. K. Geim, Science, 2018, 360, 1339–1342 CrossRef CAS PubMed.
- H. A. Bechtel, E. A. Muller, R. L. Olmon, M. C. Martin and M. B. Raschke, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7191–7196 CrossRef CAS PubMed.
- M. I. Jacobs, O. Kostko, M. Ahmed and K. R. Wilson, Phys. Chem. Chem. Phys., 2017, 19, 13372–13378 RSC.
- O. Kostko, B. Xu, M. I. Jacobs and M. Ahmed, J. Chem. Phys., 2017, 147, 013931 CrossRef CAS PubMed.
- R. K. Sharma, S. Sharma, S. Dutta, R. Zboril and M. B. Gawande, Green Chem., 2015, 17, 3207–3230 RSC.
- Y. Nagata, K. Usui and M. Bonn, Phys. Rev. Lett., 2015, 115, 236102 CrossRef PubMed.
- R. Wan, C. Wang, X. Lei, G. Zhou and H. Fang, Phys. Rev. Lett., 2015, 115, 195901 CrossRef PubMed.
- J. B. Boreyko and C.-H. Chen, Phys. Rev. Lett., 2009, 103, 184501 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00544g |
| This journal is © The Royal Society of Chemistry 2019 |