
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoactive organic material discovery with combinatorial supramolecular assembly
Andrew M.
Levine
 abc,
Sankarsan
Biswas
abc and
Adam B.
Braunschweig
*abc
abc,
Sankarsan
Biswas
abc and
Adam B.
Braunschweig
*abc
aAdvanced Science Research Center, Graduate Center, City University of New York, 85 St. Nicholas Terrace, New York, NY 10031, USA. E-mail: adam.braunschweig@gc.cuny.edu
bDepartment of Chemistry, Hunter College, 695 Park Avenue, New York, NY 10065, USA
cGraduate Center, City University of New York, 365 5th Avenue, New York, NY 10016, USA
First published on 5th September 2019
Abstract
Organic semiconductors have received substantial attention as active components in optoelectronic devices because of their processability and customizable properties. Tailoring the organic active layer in these devices to exhibit the desired optoelectronic properties requires understanding the complex and often subtle structure–property relationships governing their photophysical response to light. Both structural organization and molecular orbitals play pivotal roles, and their interactions with each other are difficult to anticipate based upon the structure of the components alone, especially in systems comprised of multiple components. In pursuit of design rules, there is a need to explore multicomponent systems combinatorially to access larger data sets, and supramolecularly to use error correcting, noncovalent assembly to achieve long-range order. This review will focus on the use of supramolecular chemistry to study combinatorial, hierarchical organic systems with emergent optoelectronic properties. Specifically, we will describe systems that undergo excited state deactivation by charge transfer (CT), singlet fission (SF), and Förster resonance energy transfer (FRET). Adopting combinatorial, supramolecular assembly to study emergent photophysics promises to rapidly accelerate progress in this research field.
 Andrew M. Levine | Andrew M. Levine received his B.S. in Chemistry from Rensselaer Polytechnic Institute and M.S. in Science Teaching from Boston College. He is pursuing a PhD in Chemistry at the CUNY Graduate Center under the guidance of Prof. Adam B. Braunschweig at the Advanced Science Research Center (ASRC) and is an NSF CREST IDEALS graduate student fellow. Andrew's research focuses on supramolecular organic semiconductors and their fabrication into photovoltaic devices. Outside of lab he enjoys cooking, playing soccer, gardening, and astrophotography. |
 Sankarsan Biswas | Sankarsan Biswas obtained his B.Sc. in Chemistry from University of Calcutta, India and M.Sc. in Chemistry from Indian Institute of Technology Madras (IIT Madras). After his M.Sc., he joined the PhD program in Chemistry at the CUNY Graduate Center. He is currently working under the guidance of Professor Adam B. Braunschweig and Professor Rein V. Ulijn at the Advanced Science Research Center (ASRC). Sankarsan's research focuses on self-assembly of supramolecular organic semiconductors, covering fundamental studies to materials design for various applications. |
 Adam B. Braunschweig | Prof. Adam B. Braunschweig joined the Nanoscience Initiative at the Advanced Science Research Center at the City University of New York and the Department of Chemistry at Hunter College in 2016. His group investigates the photophysics of supramolecular systems, nanolithography of soft matter, and synthetic carbohydrate receptors. |
1. Introduction
Organic materials are increasingly investigated in solar energy harvesting devices,1–3 sensors,4–6 field effect transistors,7–9 and catalysts.10–13 The active components that drive these applications are organic semiconductors—small molecules or polymers that are conductive following charge injection or upon photoexcitation. Organic semiconductors offer several potential advantages over their inorganic counterparts. First, they can be cheaper to prepare because the expensive, high-energy annealing processes needed for inorganic semiconductors are not required for organics.14,15 Second, their structures can be altered to vary their frontier molecular orbital (FMO) energies, shapes, absorptions, and solid-state packing, thereby providing a degree of tailorability not available with inorganics.16 This is a major potential benefit of organic semiconductor systems because systematically altering their substituents could ideally produce relationships between their molecular structures and their responses to light and charge in solution and in films. In reality, however, deriving these relationships is far more complex than this description suggests. The optoelectronic response of organic semiconductors, and particularly multicomponent materials that are often composed of mixtures of n-type and p-type molecules, remains difficult to predict if one were to consider only the molecular structures of the components.17,18 The reason is that many important properties, such as the photophysical deactivation pathway—the way in which photoexcited electrons relax through various excited states and intermediates back to the ground state—excited state lifetimes, film conductance, and light absorption, are ensemble properties that are dependent upon the interactions between two or more molecules. As such, the properties of organic semiconducting films are sensitively dependent on the relative orientations and spacing of the components. So, predicting and optimizing the optoelectronic responses of organic semiconductors so they can realize their promise as active elements for various devices and advanced applications remains a particularly unwieldy problem. This is primarily because of the sensitive dependence of their ensemble properties on Ångstrom-scale perturbations and the necessity of reproducing these orientations across micrometer or even millimeter length scales. Meeting the structural and electronic requirements of organic optoelectronic devices becomes even more challenging in multicomponent active layers where different constituent molecules must be brought together in a specific geometry to achieve a desired function.The magnitude of the challenges involved in optimizing the photoresponse of a multicomponent organic system can be understood by considering the active layer of bulk heterojunction (BHJ) organic photovoltaics.19,20 The BHJ is an OPV device geometry where organic electron donor molecules and electron acceptor molecules are blended together into a film that is sandwiched between two electrodes (Fig. 1B). In this layer, electrons in the donor or acceptor chromophores are photoexcited to create coupled electron–hole pairs (excitons). These excitons migrate to a donor–acceptor interface where they charge separate—electrons are thermodynamically driven into the acceptor phase and holes into the donor phase.21 Following this charge separation event, charge carriers must migrate to the opposing electrodes so that they can be harnessed as electricity (Fig. 1A). When the films are cast from solution, the donor and acceptor components tend to phase-segregate,3,19,22 reducing heterojunction (i.e. donor–acceptor contact) area. As the amount of heterojunction interface decreases, the number of charge separation events decrease and/or pathways to electrodes do not form so the charges cannot be collected. Alternatively, too much mixing increases unproductive recombination events because the length of the percolation pathways to the electrodes can exceed charge carrier diffusion lengths. Ideally, the active layer could adopt a geometry (Fig. 1C) that maximizes the donor–acceptor interface, while providing contiguous and short pathways for charge migration to the electrodes.
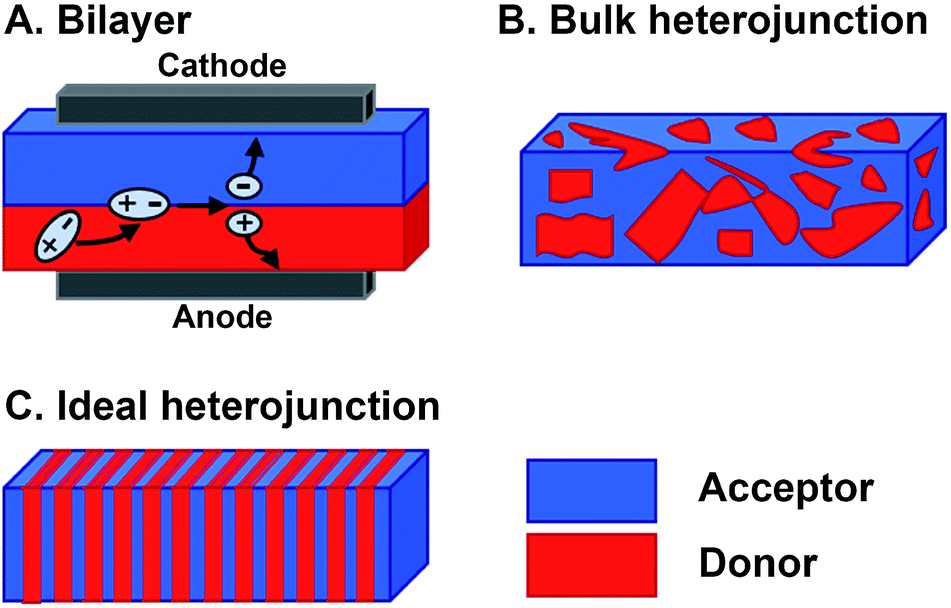 | ||
| Fig. 1 Donor–acceptor heterojunction morphologies. (A) Bilayer structure showing process of charge collection in OPV devices. (B) Bulk heterojunction (BHJ) and (C) heterojunction optimized to increase junction area, while providing contiguous pathways for charges to migrate to the electrodes. | ||
These operational criteria for organic photovoltaics dictate that the FMO energy levels and electronic coupling, which govern the dynamics of charge transfer, and film morphology are treated as equally important considerations in the design of the components of the BHJ layer, but this is not commonly reflected in practice. Rather, the donor and acceptor components are typically designed such that upon photoexcitation, the relative energies of the FMOs favor electron transfer from donor to acceptor, while morphology is an afterthought that is optimized via trial and error.20,23,24 Typically, two component systems are spin-coated together to create blended active layers, and the most common strategies for improving the mixing of donors and acceptors during this process include installing solubilizing side chains on the components,19,25 solvent annealing,26 adding insulating polymer fillers,27 adding peptide side chains,28,29 or, alternatively, covalently linking the two together to create architectures such as diads,30 triads,31 donor–acceptor alternating polymers,32 and double-cable polymers.33 Though these covalent strategies may improve heterojunction interface area, drawbacks include their cumbersome and time consuming syntheses, which limit the ability to establish structure–activity relationships, and their inability to predictably control molecular orientation on the nanoscale and film structure on the micrometer length scale, which are both necessary to create contiguous and uniform percolation pathways from the site of charge separation to the electrodes. An ideal approach towards exploring and optimizing both charge-transfer efficiency and film geometry would involve the synthesis of a library of simple components and a method to control their orientation across the molecular-to-micrometer continuum. Such an approach would inform how subtle structural modifications affect system performance, and, in turn, rapidly produce quantitative and predictive relationships between them.
Programmed supramolecular assembly of multicomponent systems is a promising approach for overcoming the phase segregation conundrum, while providing a mechanism for rapidly deriving structure–activity relationships. Supramolecular assemblies use noncovalent bonding—e.g. H-bonding, C–H⋯π interactions, π⋯π stacking, van der Waals forces, and metal–ligand coordination—to bring molecules together in a prescribed orientation (Fig. 2A).34–36 Co-assembly—where distinct components noncovalently bond with each other—can be programmed into photoresponsive organic semiconductors by incorporating complementary noncovalent bonding motifs (Fig. 2B).37 Multicomponent systems programmed to co-assemble can be combinatorial in that the photoactive cores of similar molecules can be altered with the same noncovalent bonding group so they share the same assembly with a set of complimentary components. The benefits of using supramolecular assembly to achieve order in donor–acceptor films are that (1) the cumbersome syntheses required to covalently link donor and acceptor components can be circumvented, (2) from a few donor and acceptor components with conserved noncovalent bonding groups, many combinations are attained by simply mixing different components together so structure–activity relationships can be derived, and (3) self-correcting, supramolecular assembly can also achieve hierarchical superstructures with long-range order (Fig. 2C). Because supramolecular systems are naturally combinatorial, the larger data sets that are attainable when using them to study photoactive films can provide better predictive insight into how subtle changes in molecular structure or FMO energies affect active layer performance.
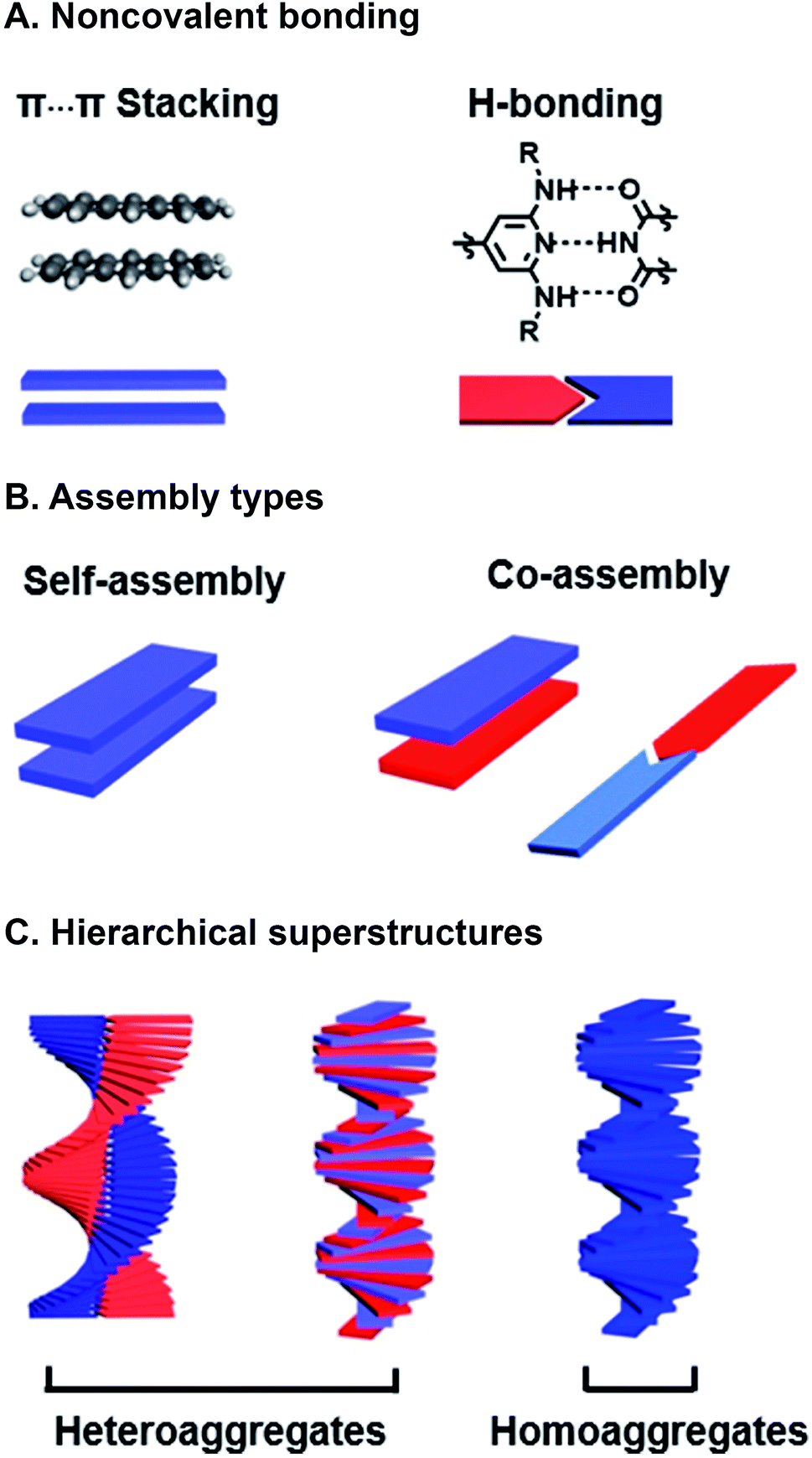 | ||
| Fig. 2 Supramolecular assembly. (A) Examples of noncovalent binding. (B) Self-assembly between like compounds or co-assembly between complementary components. (C) Superstructures with hierarchical order. | ||
Combinatorial approaches are used extensively to address some of the most vexing challenges in drug discovery,38 material science,39 and nanotechnology.40 Combinatorial science involves synthesizing libraries of components that differ in some key structural aspect, and screening them for a particular property.38 This strategy results in large data sets that are used to unearth trends and outliers without devoting exorbitant resources to time-consuming design, and, as a result, naturally lends itself to solving complex scientific challenges. Combinatorial approaches have been increasingly adopted for understanding subtle structure–activity relationships in photoresponsive organic materials, where the ability to rationally design ensemble properties continues to elude researchers.41–43 Deactivation pathways such as charge transfer (CT),44,45 singlet fission (SF),46,47 and Förster resonance energy transfer (FRET)48,49 are all in competition following the photoexcitation of organic donor–acceptor mixtures, and the factors that determine whether one occurs preferentially over another is the result of an interplay between kinetic and thermodynamic driving forces as well as the orientation of the components and the long-range film order. Here, we focus on the use of supramolecular ordering with combinatorial approaches to achieve and optimize CT, SF, and FRET in hierarchical, photoactive, organic semiconductor materials.
2. Combinatorial supramolecular photoactive assemblies
This review will present several recent examples of combinatorial, supramolecular photoactive systems that undergo CT, SF, or FRET following assembly. This section describes how chromophores were chosen to promote certain deactivation pathways and the role of combinatorial, supramolecular assembly in controlling and understanding their deactivation. In doing so we hope to show the value of adopting this approaches to study and optimize photoactive organic materials as active elements in various emerging optoelectronic applications.2.1 Charge transfer
CT involves the transfer of an electron from an electron donor to an electron acceptor (or a hole from an acceptor to a donor) upon photoexcitation, and is the most common strategy for harvesting energy from light in OPVs (Fig. 3C). Ground state CT is also possible if the electron donor and the electron acceptor sufficiently mix orbitals to produce a partial CT state.50–52 The charges generated from CT must then have contiguous pathways to diffuse to electrodes before being lost to geminate (exciton electron–hole pair) and non-geminate recombination if they are to be collected.44 This phenomena is governed by factors described in the Marcus equation,53 whose major considerations are donor–acceptor distance, electronic coupling, and the thermodynamic driving force of charge transfer. As such, designing FMOs to favor charge separation and creating ordered pathways for charge diffusion are critical considerations of donor and acceptor design.54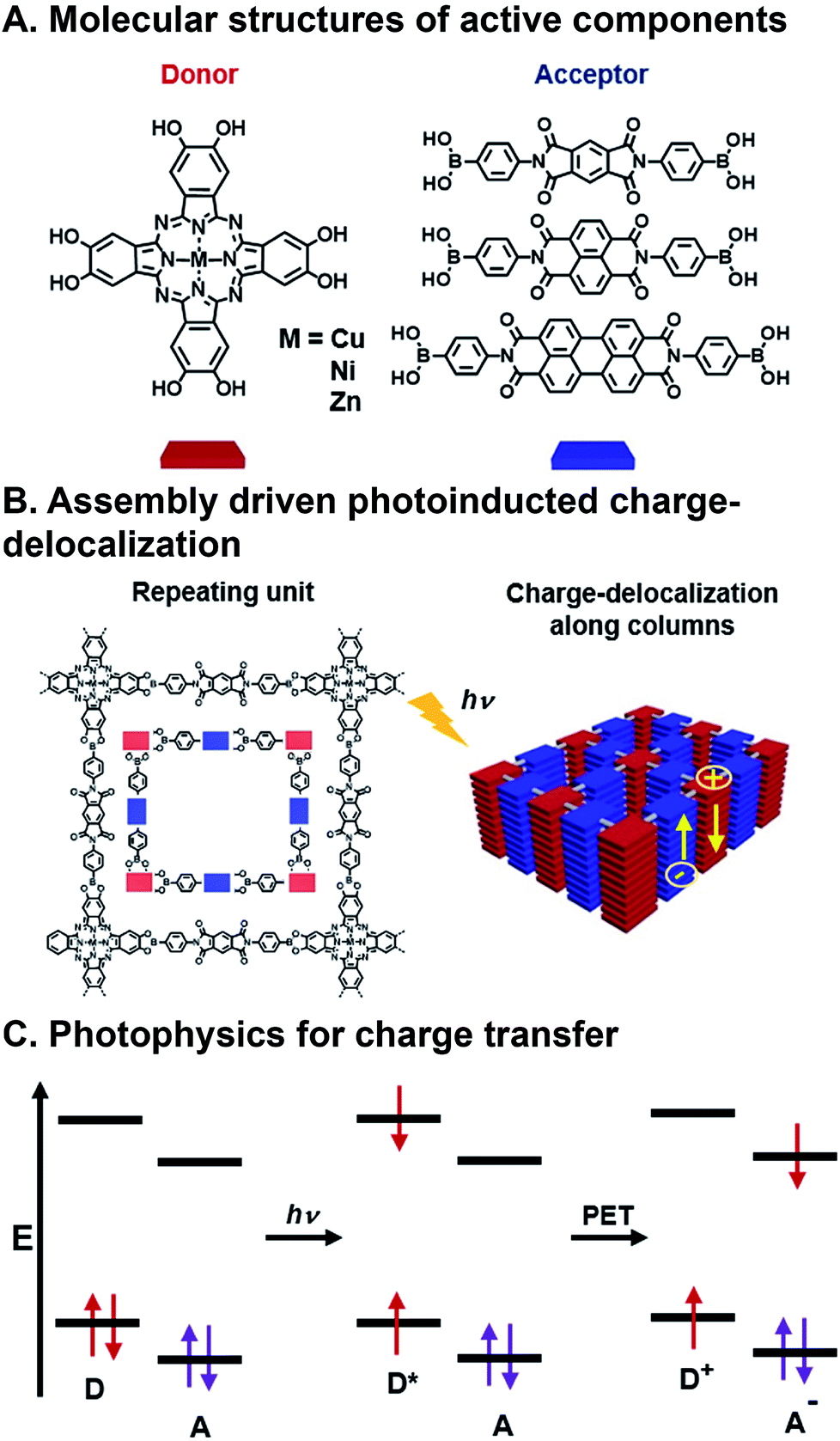 | ||
| Fig. 3 Charge transfer (CT) in a covalent organic framework (COF). (A) Metallophthalocyanine donor and diimide acceptor components. (B) Assembly into columnar arrays with emergent CT behavior. (C) Photoinduced CT mechanism via excitation and photoinduced electron transfer (PET).63 | ||
FMOs can be tuned by functionalizing the organic semiconductors with electron donating or withdrawing substituents,55,56 however, changes to molecular structure en route to tailoring FMOs will inevitably affect packing geometry, and, consequently, the electronic coupling between donors and acceptors. There are many examples of organic donor–acceptor systems that undergo photoinduced CT, and the ways in which they bring together the components involve noncovalent assembly,57 polymer blending,58,59 covalently linking,60 and inorganic bonding to form covalent organic frameworks (COFs).61,62 An example of a combinatorial system in which forming contiguous conduction pathways is also considered in conjunction with charge transfer to create active layers for OPVs as critical design criteria is the work of Jin et al.63 In this study, they describe a system in which metallophthalocyanine donors and diimide acceptors (Fig. 3A) are joined via the formation of boronate esters to form a COF with emergent photoinduced CT following assembly. This system is also significant because there has also been a push for more cost-effective, air-stable, and FMO-tunable electron acceptors because fullerenes, which are typically used as acceptors in OPVs, degrade relatively quickly in ambient conditions, absorb visible light weakly, and their isotropic structure makes using them for creating hierarchical order challenging.16,64 The donors and acceptors in these COFs assemble into columnar arrays via π⋯π stacking. These stacks promote charge migration and increase the lifetime of charges produced following photoinduced CT by providing contiguous pathways for migration (Fig. 3B). This system is combinatorial in that Cu, Ni, and Zn are explored as different metal centers in the phthalocyanine (Pc), and the three diimides have different extents of π-conjugation, but they all organize as a result of the same conserved boronate ester formation. As such, the six components produce nine donor–acceptor systems that can be formed and screened for their ability to separate charges upon photoexcitation (though only six were actually investigated). Crystal structures resolved by XRD confirm slipped stacked geometries that result in periodically aligned donor–acceptor configuration with 1D nanopores. Calculated, optimized unit cell geometries of the donor–acceptor superstructures show the most stable structure is a slip-stack geometry, which is largely dependent on the π-extension of the acceptors, and to a lesser extent, the metal centers in the donors. COFs with naphthalene diimide (NDI) and perylene diimide (PDI), allow for interlayer H-bonding, which further improves stacking stability. Transient absorption (TA) spectroscopy and time-resolved electron spin resonance (TR-EPR) were used to interrogate the lifetimes of the charge separated states. It was found that Cu metal centers enhanced CT lifetimes compared to Ni or Zn, while lattice size from changing acceptor units played a less critical role. Importantly, in this system, similar hierarchical structures are achieved for all metal/acceptor combinations, so a correlation could be drawn as to how structural and, in turn, electronic changes to individual components contribute to deactivation properties.
Another example of achieving CT with a combinatorial, supramolecular library of donor and acceptor components was reported by Mallia et al. who studied a series of nonparallel stacked dyad chromophores.65 A Suzuki–Miyaura cross-coupling reaction was used to bring together covalently naphthalimide (NI) acceptors with either naphthalene (N) or phenyl (Ph) donors (Fig. 4A). Superstructure assembly was promoted by C–H⋯π, π⋯π, and C–H⋯O interactions, while donors and acceptors tilted with respect to one another; N and Ph at 68° and 63° with respect to the NI plane, respectively. Additionally, these interactions and resulting assembled structures whose geometries were found through simulation, suggest both donor and acceptor π⋯π stack in NIN whereas only the acceptor (NI) is self-stacking in NIPh (Fig. 4B). This system is combinatorial in that the same chemistry to connect readily available donor building blocks to the NI acceptor can be used, so a large number of molecules that subsequently assembly are easily generated. As a result the same NI acceptor has also been used with other donors in supramolecular, energy harvesting studies in the Hariharan group that include triphenylamine for CT66 and perylenimide for FRET.67 The self-stacked donors and acceptors in NIN delocalize photoinduced charge carriers and decrease geminate charge recombination. In contrast, NIPh lacks contiguous pathways along Ph donors for transport of the positive charge carriers produced from CT. These expectations are substantiated by (1) the bathochromic shifting in emission spectra of NIN when varying solvent polarity, whose ground state CT character was higher in NIN (39%) than NIPh (21%), (2) the population of radical ion state found in NIN, when probed with femtosecond transient absorption (fsTA), which is not present in NIPh, and (3) more positive electrochemical reduction potential of NIN in increasing concentrations of NIN, as a result of radical ion delocalization along D–A stacks, while NIPh reduction potential was largely unaffected by concentration. As a consequence, nsTA reveals radical ion pair intermediates surviving 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times longer in their aggregated state than as monomers, demonstrating how photophysical properties were rapidly understood and optimized by adopting combinatorial supramolecular assembly.
000 times longer in their aggregated state than as monomers, demonstrating how photophysical properties were rapidly understood and optimized by adopting combinatorial supramolecular assembly.
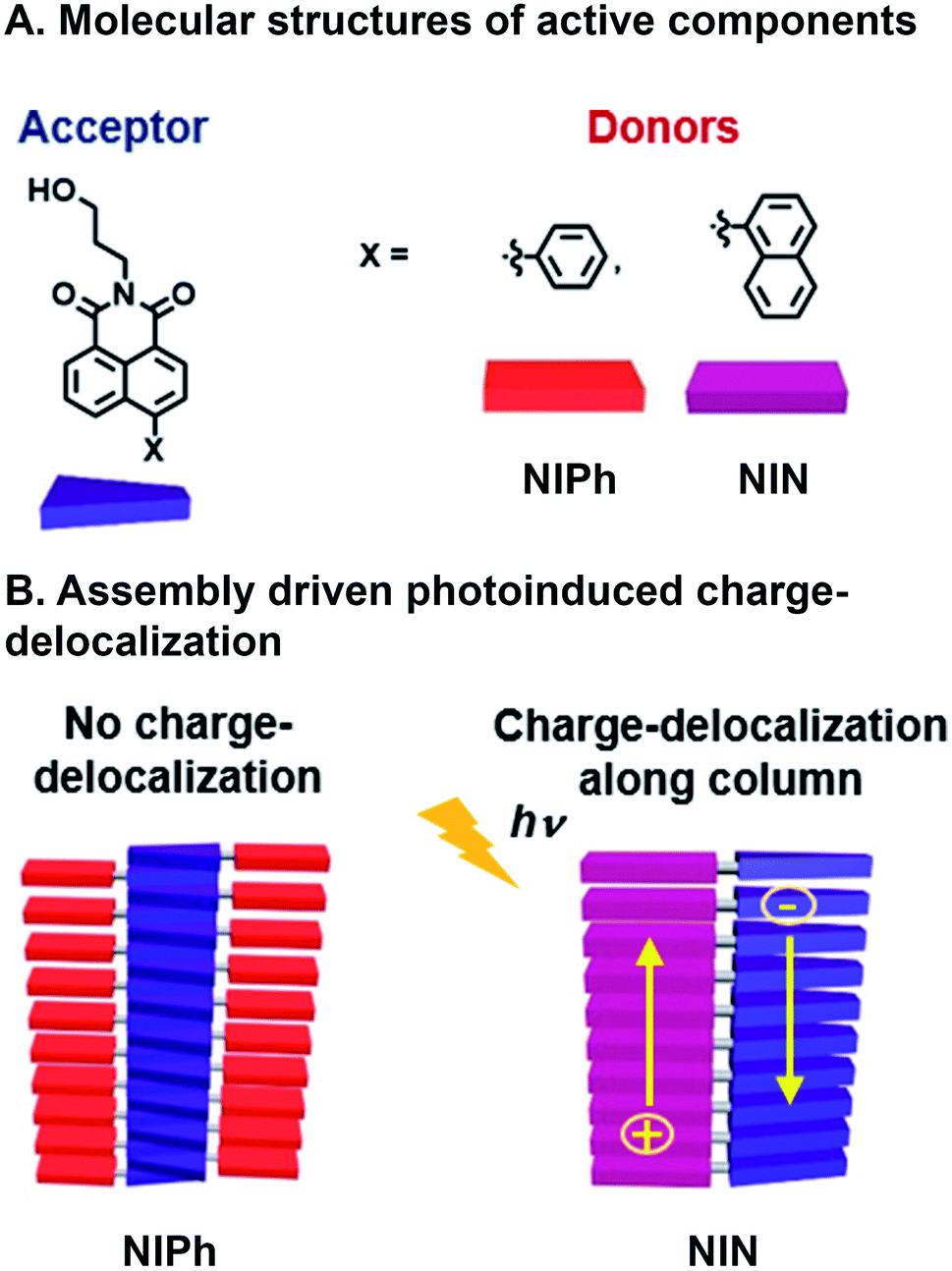 | ||
| Fig. 4 Charge transfer (CT) in a supramolecular dyad system. (A) Naphthalimide (NI) acceptor with either naphthalene (N) or phenyl (Ph) appended donors. (B) Emergent photoinduced charge separation occurs in NIN superstructures but not NIPh.65 | ||
2.2 Singlet fission
SF is the process by which two electronically coupled chromophores in the ground state (S0S0) are photoexcited by a single photon to a singlet state (S1S0) and share energy to form a spin-allowed state comprised of two coupled triplets (T1T1), each roughly half the energy of the initial singlet state (Fig. 5C).46,47 One of the big advantages to SF is that one photon is used to produce two excited electrons, but there are caveats including that the resulting triplet state energy must be roughly half the energy of the initial singlet state, and bound triplet pairs must decouple for the SF process to be complete and the triplets to be harnessed.68 SF is an attractive deactivation process because it has the potential to increase the theoretical efficiency limit of solar energy materials to 44% (ref. 46) compared to traditional single band-gap material, which is 33%.69 SF has been studied extensively since the 1960's in organic materials, primarily in acenes68,70 and more recently in rylenes71,72 and diketopyrrolopyrroles (DPPs).73,74 Others have shown SF dynamics in various acene derivatives, with75 and without76,77 linkers to affect coupling, to be highly sensitive to packing, spacing, and orientation. “Ideal” SF chromophore organization could provide a 200% triplet yield. Importantly, optimized geometries would maximize coupled triplet formation, but also promote their decoupling—a necessary event for harvesting these excited states without losing them to recombination. For harvesting decoupled triplets, fullerenes have been used with pentacene, essentially promoting CT after the SF event, to make solar cells.78 Control over the heterojunction organization and overall active layer morphology, and stability in ambient conditions, however, are still ongoing challenges whose resolution could lead to higher SF yields as well as greater collection of decoupled triplets.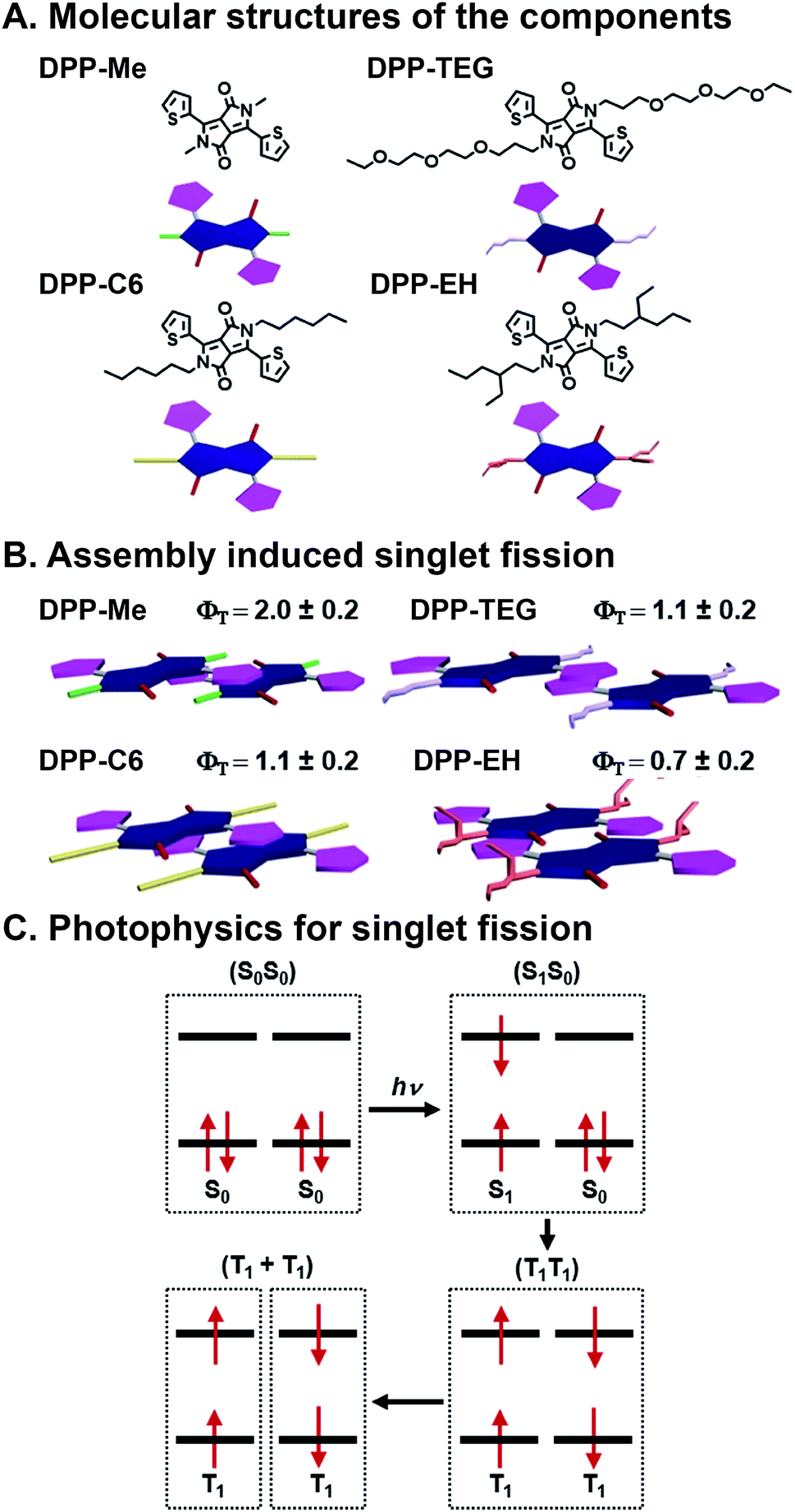 | ||
| Fig. 5 Singlet fission (SF) in self-assembled diketopyrrolopyrrole (DPP). (A) Methyl (Me), n-hexyl (C6), triethylene glycol (TEG), and 2-ethylhexyl (EH) substituents appended to the DPP core nitrogen. (B) Varying slip-stack geometries caused by sterics, which affect intermolecular orbital coupling and ultimately SF yields (ΦT) and lifetimes. (C) SF mechanism, where one photon generates two excitons.73 | ||
Mauck et al. used a combinatorial approach to study SF kinetics and triplet yields in DPP by installing different side chains off the DPP core nitrogen (Fig. 5A).73 This is a combinatorial, supramolecular system in that in their work, DPP derivatives with methyl (Me), n-hexyl (C6), triethylene glycol (TEG), and 2-ethylhexyl (EH) substituents on the nitrogen of the DPP core are prepared using the same straightforward chemistry. These DPPs assemble into stacks, and crystal structures of these materials show that the side chains alter the DPP stacking torsion angle offset along with transverse and longitudinal displacement with the thiophene substituent of one molecule over the DPP core of its neighbor. This slip-stack geometry is known as J-aggregation and is preferable for SF over H-aggregation, where molecules are stacked parallel, because of the antisymmetric orbital overlap between coupled neighbors.73 This π⋯π stacking geometry is altered by the sterics imposed by side chains off the core, and these relatively subtle offsets in packing geometry play critical roles in orbital overlap, and thus, SF yields and lifetimes (Fig. 5B). Increased sterics of the side chains separates the DPP cores and leads to slower formation (τ1) of an intermediate state that precedes the formation of (T1T1) via SF (τ2). The Me, C6, and TEG systems demonstrate SF yields over 100%, although τ2 increases with increasing side chain size of 22.1 ± 0.9, 336 ± 7, and 195 ± 8 ps, respectively. The bulkier EH system, however, presents a τ2 of 1600 ± 500 ps and is the only system with <100% triplet yield. This longer deactivation lifetime is assigned to fluorescence, and based on the 70% triplet yield, τ2 is calculated to be 1.2 ns. This work demonstrates precisely the importance of control over molecular packing geometry in promoting SF, and because of DPP's structural customizability, this study showed how supramolecular assembly with combinatorial components leads to insight into how structure affects the complex photophysics of SF.
We have also recently developed a supramolecular system to explore how SF yields and lifetimes are affected by subtle changes in stacking geometry.79 The active component that undergoes SF is based upon a DPP core that is substituted with diamidopyridine (DAP) H-bonding moieties (Fig. 6A). In addition, orthogonal π⋯π stacking between the cores of the aromatic DPP dyes drives organization into hierarchical structures. The appended DAP groups form H-bonds with rylene diimides, which serve as scaffolds for altering relative DPP orientations in the stack. Both DPP and rylene structures are easily modified, providing a lever for altering superstructure geometry in the solid state (Fig. 6B). In our system molecular and long-range geometry were affected in two ways: (1) DPP was monodentate (mDPP) or bidentate (dDPP) depending on the number of DAP groups appended to the DPP core, or (2) core and bay substitution of rylenes were varied to affect their packing geometry, and, as a result, the way they scaffold DPP in the superstructures. From two DPPs and three rylenes, six superstructures with fiber, sheet, and scroll morphologies (Fig. 6C) in the solid state were obtained, and these were all interrogated with fs- and nsTA spectroscopy. Compared to samples composed solely of DPP, some heterosuperstructures show increased decoupled triplet yields and lifetimes. In the case of monodentate mDPP, most decoupled triplet lifetimes from SF were longer (as long as 9.2 μs) in combinatorial, supramolecular thin films compared to films composed solely of mDPP (1.9 μs). And in the case of dDPP, most decoupled triplet yields were higher in mixed films (as high as 28%) compared to films of dDPP (23%). To date, the most common approaches to explore and optimize SF focus solely on dye structure, and orientation of dyes in films is an afterthought. As such, very few groups use secondary components to control their geometry, despite the importance of packing on SF yield. Our work showed that subtle changes in DPP geometry that result from controlled noncovalent binding manifest as changes in SF yields and lifetimes.
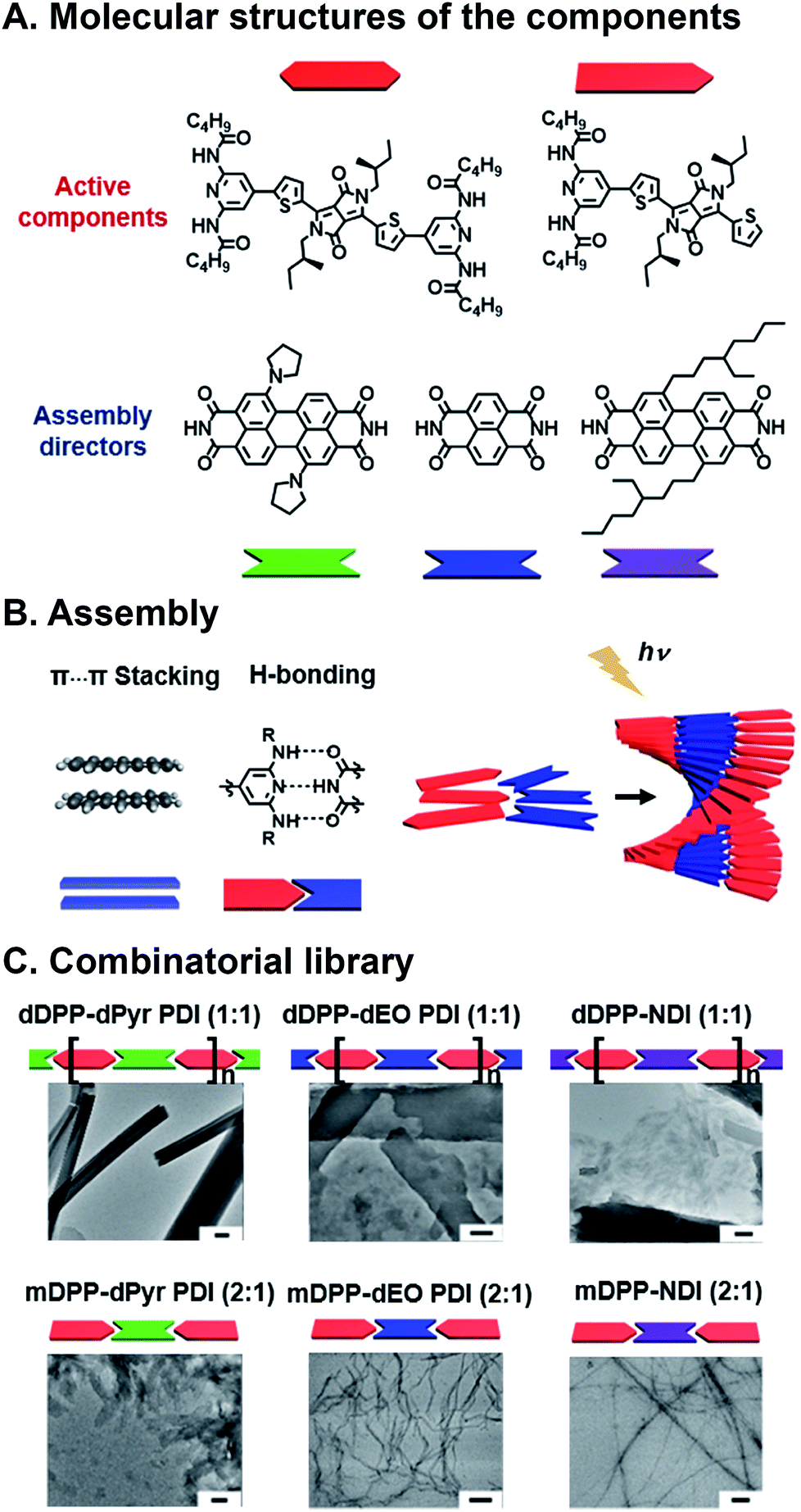 | ||
| Fig. 6 SF in combinatorial, supramolecular DPP–rylene superstructures. (A) DPP SF components and rylenes used for scaffolding. (B) Noncovalent interactions lead to cooperative assembly for forming hierarchical superstructure. (C) Different superstructure morphologies including fibers, sheets, and scrolls. Scale bars are 200 nm.79 | ||
2.3 Förster resonance energy transfer
FRET occurs when the energy released nonradiatively by an excited chromophore is absorbed by a neighboring molecule (Fig. 7C). This happens when donor and acceptor are in close proximity, ∼10–100 Å, and the absorbance of the acceptor overlaps with the emission of the donor.80 The nonradiative transfer of energy manifests as a decrease in the donor's fluorescence and an increase in the acceptor's. FRET is not as sensitive to electronic coupling as CT or SF and is less affected by orientation changes, but is highly sensitive to chromophore distance and spectral overlap. Therefore a wider range of modifications can be done to donors and acceptors to better control FMOs without as much concern for how the changes in structure affect neighbor orientation with respect to the FRET partner. FRET has many applications including sensitizing triplet oxygen into its excited singlet state, which can be used for photodynamic cancer therapy,81 as a photocatalyst for synthesis,82 and for solar energy harvesting.83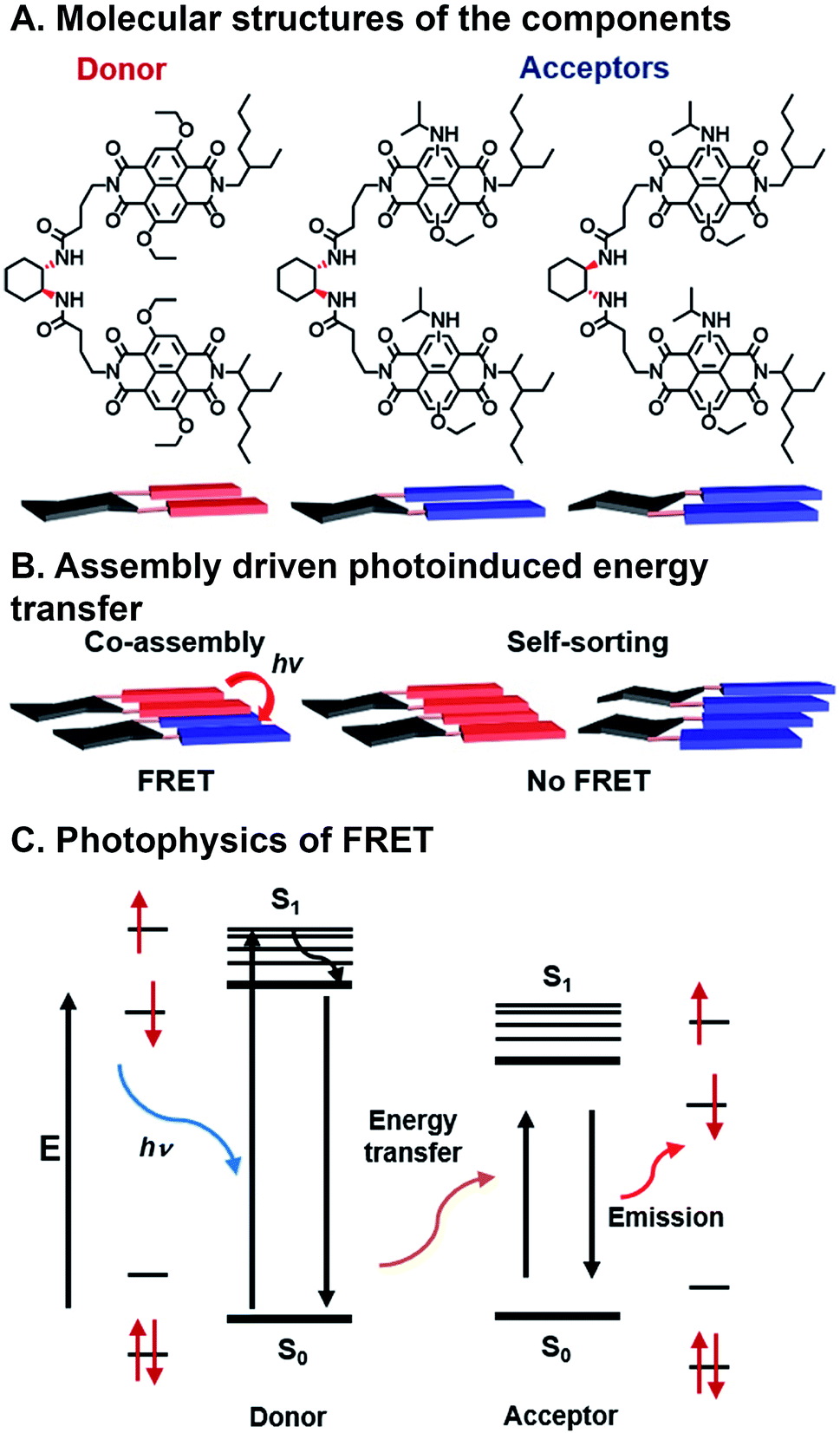 | ||
| Fig. 7 Förster resonance energy transfer (FRET) in assembling naphthalene dimers. (A) Donors and acceptors synthesized by varying core substitution. (B) Chiral linkers promote either co-assembly or self-sorting structures. (C) FRET mechanism showing energy transfer from donor to acceptor.84 | ||
Sarkar et al. have used combinatorial, supramolecular assembly to explore a chiral system that undergoes FRET that uses stacked NDI dimers (Fig. 7A).84 In this system, NDI donors possessed symmetrical ethoxy substituents in the NDI core positions, while NDI acceptors had asymmetric ethoxy and N-isopropyl amine substituents. Fibrous superstructures formed in nonpolar solvents as a result of π⋯π stacking between NDI components. The chiral 1,2-diaminocyclohexane linkers dimerizing the NDI donors or acceptors were used to facilitate self-assembly or co-assembly. When linkers had the same stereochemistry, co-assembly is possible, whereas different stereochemistry precluded co-assembly. Self-assembled, or “self-stacked”, dimers did not result in FRET while co-assembled, or “co-stacked”, systems exhibited FRET because donors and acceptors were brought into close proximity (Fig. 7B). This system is combinatorial in nature because FMOs can be modulated by varying chromophore substituents, effectively tailoring FRET absorption and emission, and programmed assembly can be used to bring donor and acceptor in close enough range for energy transfer.
Ji et al. have recently reported chiral light-harvesting nanotube antennas and studied cooperative assembly and energy transfer amongst its water soluble donor and acceptor components.85 In their work, cyanostilbene-appended glutamate (CG) self-assembled into helical nanotubes, as seen in SEM and TEM, whose chirality was controlled by switching between L-CG and D-CG (Fig. 8A). This CG bilayer structure, which was substantiated with XRD, is the basic building block that formed lamellar structures, which then roll into chiral nanohelices and nanotubes (Fig. 8B). When achiral acceptor components thioflavin T (ThT) and/or acridine orange (AO) acceptors were inserted into the L-CG or D-CG to form superstructures, FRET occurs where the CG is the FRET donor. This system is combinatorial because of the variety, and multitude, of acceptors that can be incorporated into the superstructure resulting in 14 possible combinations from five components. Co-assembly was confirmed by emergent signals in circular dichroism (CD) measurements. CG emission overlaps with ThT absorption, making them a suitable FRET pair, and ThT emission overlaps with AO absorption, but CG emission does not overlap with AO absorption. As such, FRET can be seen in CG/ThT dyads but not CG/AO dyads. In CG/ThT/AO triads, ThT absorbs energy from CG, which is then transferred to AO, essentially acting as a bridge, resulting in two sequential FRET (S-FRET) events. In addition to transferring chirality from donor to acceptor, circularly polarized luminescence (CPL)—the circularly polarized emission of light—is also amplified through CG/ThT and triads. All three components in the CG/ThT/AO triad were independently excited and CPL was measured for chirality transfer (ChT) and compared to solutions of the individual components. In the co-assembled superstructures, a CPL signal was seen when only exciting AO, which was attributed to the chirality imparted by the superstructure. AO CPL further increased when directly exciting ThT, and even more so when directly exciting CG. As such, CPL was enhanced for each additional FRET event in the triad system. More generally, this is an elegant example of how biopolymer assembly, like the peptide assembly that's used to create nanotube scaffolding in this study, or DNA assembly86 used in other photoactive systems, can be combinatorial given the diverse library of components, their ease of fabrication into oligomers, and their chirality can be used for investigating emergent deactivation pathways in combinatorial, supramolecular systems.
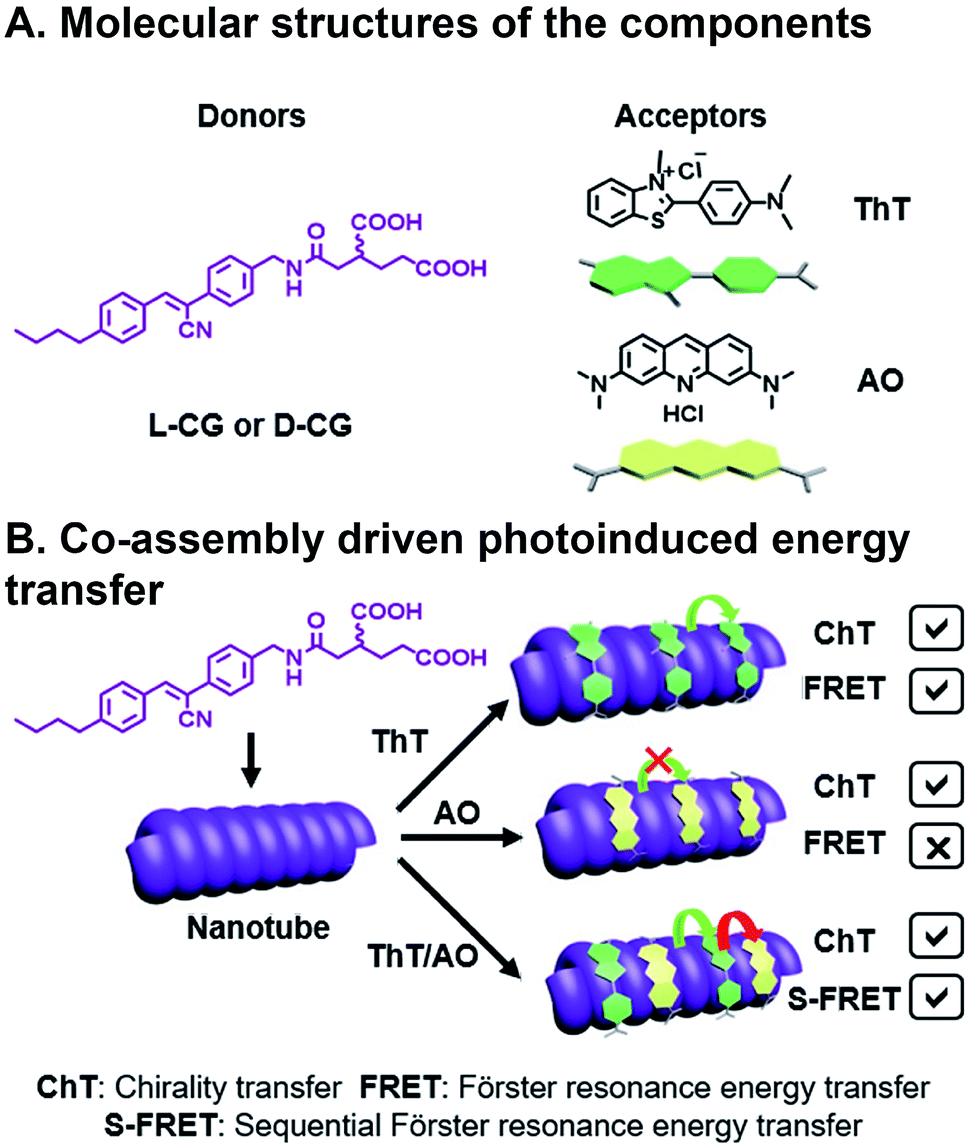 | ||
| Fig. 8 Three component FRET system. (A) Cyanostilbene-appended glutamate (CG) donor, thioflavin T (ThT), and acridine orange (AO) acceptors. (B) Supramolecular assembly with a bridging acceptor facilitates two sequential FRET (S-FRET) events due to absorption and emission overlap. Chirality of emissive photons is also transferred (ChT).85 | ||
3 Conclusion and outlook
Optimizing the photophysical deactivation pathways of organic semiconductors requires understanding their structure–property relationships. The packing geometry of the molecular components can be tailored using programmed supramolecular assembly to form self-assembled and co-assembled materials to achieve emergent photophysics that are absent in individual components. Understanding the subtle and complex relationships that govern how hierarchical structure affects overall device performance can be accelerated by adopting a combinatorial approach where libraries of components assemble by error-correcting, noncovalent assembly that can be screened quickly and in parallel to reveal trends that would otherwise be difficult to derive. Here we show how CT, SF, and FRET were studied and the insights revealed by adopting this strategy. Although we have only selected a few examples, this approach is being increasingly adopted to understand complex photoactive organic systems and promises to rapidly accelerate progress in this important research area. Combinatorial, supramolecular libraries can have orders of magnitude more components, and it will be then that we have truly tapped into its potential for material discovery. To do so, however, will require concomitant advances in how we study and understand complex photophysics in hierarchical organic systems.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Science Foundation CREST Center for Interface Design and Engineered Assembly of Low Dimensional systems (IDEALS), NSF grant number HRD-1547830.References
- Y. Li, G. Xu, C. Cui and Y. Li, Flexible and Semitransparent Organic Solar Cells, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Organic solar cells based on non-fullerene acceptors, Nat. Mater., 2018, 17, 119 CrossRef CAS.
- P. Cheng and X. Zhan, Stability of organic solar cells: challenges and strategies, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC.
- C. Zhang, P. Chen and W. Hu, Organic field-effect transistor-based gas sensors, Chem. Soc. Rev., 2015, 44, 2087–2107 RSC.
- Z. Chen, Q. Wang, X. Wu, Z. Li and Y.-B. Jiang, Optical chirality sensing using macrocycles, synthetic and supramolecular oligomers/polymers, and nanoparticle based sensors, Chem. Soc. Rev., 2015, 44, 4249–4263 RSC.
- T. Sekitani, T. Yokota, U. Zschieschang, H. Klauk, S. Bauer, K. Takeuchi, M. Takamiya, T. Sakurai and T. Someya, Organic Nonvolatile Memory Transistors for Flexible Sensor Arrays, Science, 2009, 326, 1516–1519 CrossRef CAS.
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Organic field-effect transistor sensors: a tutorial review, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC.
- Y.-H. Chou, H.-C. Chang, C.-L. Liu and W.-C. Chen, Polymeric charge storage electrets for non-volatile organic field effect transistor memory devices, Polym. Chem., 2015, 6, 341–352 RSC.
- J. Zhang, J. Jin, H. Xu, Q. Zhang and W. Huang, Recent progress on organic donor–acceptor complexes as active elements in organic field-effect transistors, J. Mater. Chem. C, 2018, 6, 3485–3498 RSC.
- T. Bach, More Chemistry with Light! More Light in Chemistry!, Angew. Chem., Int. Ed., 2015, 54, 11294–11295 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- T. Zhang and W. Lin, Metal–organic frameworks for artificial photosynthesis and photocatalysis, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Catalysis and photocatalysis by metal organic frameworks, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Reducing the efficiency–stability–cost gap of organic photovoltaics with highly efficient and stable small molecule acceptor ternary solar cells, Nat. Mater., 2016, 16, 363 CrossRef PubMed.
- R. García-Valverde, J. A. Cherni and A. Urbina, Life cycle analysis of organic photovoltaic technologies, Prog. Photovolt. Res. Appl., 2010, 18, 535–558 CrossRef.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Next-generation organic photovoltaics based on non-fullerene acceptors, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- A. Mishra and P. Bäuerle, Small Molecule Organic Semiconductors on the Move: Promises for Future Solar Energy Technology, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS.
- F. Zhao, C. Wang and X. Zhan, Morphology Control in Organic Solar Cells, Adv. Energy Mater., 2018, 8, 1703147 CrossRef.
- X. Guo, N. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Polymer solar cells with enhanced fill factors, Nat. Photonics, 2013, 7, 825 CrossRef CAS.
- A. Facchetti, Polymer donor–polymer acceptor (all-polymer) solar cells, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Material insights and challenges for non-fullerene organic solar cells based on small molecular acceptors, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- J. Huang, J. H. Carpenter, C.-Z. Li, J.-S. Yu, H. Ade and A. K.-Y. Jen, Highly Efficient Organic Solar Cells with Improved Vertical Donor–Acceptor Compositional Gradient Via an Inverted Off-Center Spinning Method, Adv. Mater., 2016, 28, 967–974 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Over 16% efficiency organic photovoltaic cells enabled by a chlorinated acceptor with increased open-circuit voltages, Nat. Commun., 2019, 10, 2515 CrossRef.
- J. Lee, S.-J. Ko, H. Lee, J. Huang, Z. Zhu, M. Seifrid, J. Vollbrecht, V. V. Brus, A. Karki, H. Wang, K. Cho, T.-Q. Nguyen and G. C. Bazan, Side-Chain Engineering of Nonfullerene Acceptors for Near-Infrared Organic Photodetectors and Photovoltaics, ACS Energy Lett., 2019, 4, 1401–1409 CrossRef CAS.
- S. Miller, G. Fanchini, Y.-Y. Lin, C. Li, C.-W. Chen, W.-F. Su and M. Chhowalla, Investigation of nanoscale morphological changes in organic photovoltaics during solvent vapor annealing, J. Mater. Chem., 2008, 18, 306–312 RSC.
- B. Kang, F. Ge, L. Qiu and K. Cho, Effective Use of Electrically Insulating Units in Organic Semiconductor Thin Films for High-Performance Organic Transistors, Adv. Electron. Mater., 2017, 3, 1600240 CrossRef.
- C. Berdugo, S. K. M. Nalluri, N. Javid, B. Escuder, J. F. Miravet and R. V. Ulijn, Dynamic Peptide Library for the Discovery of Charge Transfer Hydrogels, ACS Appl. Mater. Interfaces, 2015, 7, 25946–25954 CrossRef CAS.
- S. K. M. Nalluri, C. Berdugo, N. Javid, P. W. J. M. Frederix and R. V. Ulijn, Biocatalytic Self-Assembly of Supramolecular Charge-Transfer Nanostructures Based on n-Type Semiconductor-Appended Peptides, Angew. Chem., Int. Ed., 2014, 53, 5882–5887 CrossRef CAS.
- T. L. Nguyen, T. H. Lee, B. Gautam, S. Y. Park, K. Gundogdu, J. Y. Kim and H. Y. Woo, Single Component Organic Solar Cells Based on Oligothiophene–Fullerene Conjugate, Adv. Funct. Mater., 2017, 27, 1702474 CrossRef.
- P. E. Hartnett, S. M. Dyar, E. A. Margulies, L. E. Shoer, A. W. Cook, S. W. Eaton, T. J. Marks and M. R. Wasielewski, Long-lived charge carrier generation in ordered films of a covalent perylenediimide–diketopyrrolopyrrole–perylenediimide molecule, Chem. Sci., 2015, 6, 402–411 RSC.
- D. Dang, D. Yu and E. Wang, Conjugated Donor–Acceptor Terpolymers Toward High-Efficiency Polymer Solar Cells, Adv. Mater., 2019, 31, 1807019 CrossRef.
- G. Feng, J. Li, F. J. M. Colberts, M. Li, J. Zhang, F. Yang, Y. Jin, F. Zhang, R. A. J. Janssen, C. Li and W. Li, “Double-Cable” Conjugated Polymers with Linear Backbone toward High Quantum Efficiencies in Single-Component Polymer Solar Cells, J. Am. Chem. Soc., 2017, 139, 18647–18656 CrossRef CAS.
- E. Mattia and S. Otto, Supramolecular systems chemistry, Nat. Nanotechnol., 2015, 10, 111 CrossRef CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS.
- W. Wang, Y.-X. Wang and H.-B. Yang, Supramolecular transformations within discrete coordination-driven supramolecular architectures, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- S. R. Peurifoy, C. X. Guzman and A. B. Braunschweig, Topology, assembly, and electronics: three pillars for designing supramolecular polymers with emergent optoelectronic behavior, Polym. Chem., 2015, 6, 5529–5539 RSC.
- R. Liu, X. Li and K. S. Lam, Combinatorial chemistry in drug discovery, Curr. Opin. Chem. Biol., 2017, 38, 117–126 CrossRef CAS PubMed.
- D. A. Keller, A. Ginsburg, H.-N. Barad, K. Shimanovich, Y. Bouhadana, E. Rosh-Hodesh, I. Takeuchi, H. Aviv, Y. R. Tischler, A. Y. Anderson and A. Zaban, Utilizing Pulsed Laser Deposition Lateral Inhomogeneity as a Tool in Combinatorial Material Science, ACS Comb. Sci., 2015, 17, 209–216 CrossRef CAS PubMed.
- M. Sarikaya, C. Tamerler, A. K. Y. Jen, K. Schulten and F. Baneyx, Molecular biomimetics: nanotechnology through biology, Nat. Mater., 2003, 2, 577–585 CrossRef CAS.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach, Nat. Mater., 2016, 15, 1120 CrossRef.
- P. Heinrichová, J. Pospíšil, S. Stříteský, M. Vala, M. Weiter, P. Toman, D. Rais, J. Pfleger, M. Vondráček, D. Šimek, L. Fekete, P. Horáková, L. Dokládalová, L. Kubáč and I. Kratochvílová, Diketopyrrolopyrrole-Based Organic Solar Cells Functionality: The Role of Orbital Energy and Crystallinity, J. Phys. Chem. C, 2019, 123, 11447–11463 CrossRef.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Self-Organization in Coordination-Driven Self-Assembly, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, Light Harvesting for Organic Photovoltaics, Chem. Rev., 2017, 117, 796–837 CrossRef CAS.
- O. Ostroverkhova, Organic Optoelectronic Materials: Mechanisms and Applications, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Singlet Fission, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Recent Advances in Singlet Fission, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS.
- S. Kundu and A. Patra, Nanoscale Strategies for Light Harvesting, Chem. Rev., 2017, 117, 712–757 CrossRef CAS.
- X. Liu and J. Qiu, Recent advances in energy transfer in bulk and nanoscale luminescent materials: from spectroscopy to applications, Chem. Soc. Rev., 2015, 44, 8714–8746 RSC.
- K. Deepthi, R. R. B. Amal, V. R. Rajeev, K. N. N. Unni and E. B. Gowd, Directed Assembly of Hierarchical Supramolecular Block Copolymers: A Strategy To Create Donor–Acceptor Charge-Transfer Stacks, Macromolecules, 2019, 52, 2889–2899 CrossRef CAS.
- Z. Gao, Z. Li, Z. Gao and F. Wang, Supramolecular alternate donor–acceptor copolymers mediated by Pt⋯Pt metal–metal interactions and their photocatalytic applications, Nanoscale, 2018, 10, 14005–14011 RSC.
- M. A. Kobaisi, R. S. Bhosale, M. E. El-Khouly, D. D. La, S. D. Padghan, S. V. Bhosale, L. A. Jones, F. Antolasic, S. Fukuzumi and S. V. Bhosale, The sensitivity of donor–acceptor charge transfer to molecular geometry in DAN – NDI based supramolecular flower-like self-assemblies, Sci. Rep., 2017, 7, 16501 CrossRef.
- P. F. Barbara, T. J. Meyer and M. A. Ratner, Contemporary Issues in Electron Transfer Research, J. Phys. Chem., 1996, 100, 13148–13168 CrossRef CAS.
- S. Chandrabose, K. Chen, A. J. Barker, J. J. Sutton, S. K. K. Prasad, J. Zhu, J. Zhou, K. C. Gordon, Z. Xie, X. Zhan and J. M. Hodgkiss, High Exciton Diffusion Coefficients in Fused Ring Electron Acceptor Films, J. Am. Chem. Soc., 2019, 141, 6922–6929 CrossRef CAS.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Rylene and Related Diimides for Organic Electronics, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- L. Zöphel, V. Enkelmann and K. Müllen, Tuning the HOMO–LUMO Gap of Pyrene Effectively via Donor–Acceptor Substitution: Positions 4, 5 Versus 9, 10, Org. Lett., 2013, 15, 804–807 CrossRef PubMed.
- Y. Liu, C. e. Zhang, D. Hao, Z. Zhang, L. Wu, M. Li, S. Feng, X. Xu, F. Liu, X. Chen and Z. Bo, Enhancing the Performance of Organic Solar Cells by Hierarchically Supramolecular Self-Assembly of Fused-Ring Electron Acceptors, Chem. Mater., 2018, 30, 4307–4312 CrossRef CAS.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Single-Junction Binary-Blend Nonfullerene Polymer Solar Cells with 12.1% Efficiency, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- Y. Guo, Y. Li, O. Awartani, H. Han, J. Zhao, H. Ade, H. Yan and D. Zhao, Improved Performance of All-Polymer Solar Cells Enabled by Naphthodiperylenetetraimide-Based Polymer Acceptor, Adv. Mater., 2017, 29, 1700309 CrossRef PubMed.
- A. R. Mallia, A. M. Philip, V. Bhat and M. Hariharan, Persistent Charge-Separated States in Self-Assembled Twisted Nonsymmetric Donor–Acceptor Triads, J. Phys. Chem. C, 2017, 121, 4765–4777 CrossRef CAS.
- S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Charge Dynamics in A Donor–Acceptor Covalent Organic Framework with Periodically Ordered Bicontinuous Heterojunctions, Angew. Chem., Int. Ed., 2013, 52, 2017–2021 CrossRef CAS PubMed.
- N. Keller, D. Bessinger, S. Reuter, M. Calik, L. Ascherl, F. C. Hanusch, F. Auras and T. Bein, Oligothiophene-Bridged Conjugated Covalent Organic Frameworks, J. Am. Chem. Soc., 2017, 139, 8194–8199 CrossRef CAS PubMed.
- S. Jin, M. Supur, M. Addicoat, K. Furukawa, L. Chen, T. Nakamura, S. Fukuzumi, S. Irle and D. Jiang, Creation of Superheterojunction Polymers via Direct Polycondensation: Segregated and Bicontinuous Donor–Acceptor π-Columnar Arrays in Covalent Organic Frameworks for Long-Lived Charge Separation, J. Am. Chem. Soc., 2015, 137, 7817–7827 CrossRef CAS PubMed.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Nonfullerene Acceptor Molecules for Bulk Heterojunction Organic Solar Cells, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- A. R. Mallia, P. S. Salini and M. Hariharan, Nonparallel Stacks of Donor and Acceptor Chromophores Evade Geminate Charge Recombination, J. Am. Chem. Soc., 2015, 137, 15604–15607 CrossRef CAS PubMed.
- A. R. Mallia and M. Hariharan, Self-Assembled Donor–Acceptor Trefoils: Long-Lived Charge Separated State through Aggregation, J. Phys. Chem. C, 2017, 121, 4778–4788 CrossRef CAS.
- R. T. Cheriya, K. Nagarajan and M. Hariharan, Single-Component Organic Light-Harvesting Red Luminescent Crystal, J. Phys. Chem. C, 2013, 117, 3240–3248 CrossRef CAS.
- S. N. Sanders, A. B. Pun, K. R. Parenti, E. Kumarasamy, L. M. Yablon, M. Y. Sfeir and L. M. Campos, Understanding the Bound Triplet-Pair State in Singlet Fission, Chem, 2019, 5, 1988–2005 CAS.
- W. Shockley and H. J. Queisser, Detailed Balance Limit of Efficiency of p–n Junction Solar Cells, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- C. Hetzer, D. M. Guldi and R. R. Tykwinski, Pentacene Dimers as a Critical Tool for the Investigation of Intramolecular Singlet Fission, Chem.–Eur. J., 2018, 24, 8245–8257 CrossRef CAS.
- E. A. Margulies, J. L. Logsdon, C. E. Miller, L. Ma, E. Simonoff, R. M. Young, G. C. Schatz and M. R. Wasielewski, Direct Observation of a Charge-Transfer State Preceding High-Yield Singlet Fission in Terrylenediimide Thin Films, J. Am. Chem. Soc., 2017, 139, 663–671 CrossRef CAS.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Enabling singlet fission by controlling intramolecular charge transfer in π-stacked covalent terrylenediimide dimers, Nat. Chem., 2016, 8, 1120 CrossRef CAS.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, Singlet Fission via an Excimer-Like Intermediate in 3,6-Bis(thiophen-2-yl)diketopyrrolopyrrole Derivatives, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS.
- C. M. Mauck, P. E. Hartnett, Y.-L. Wu, C. E. Miller, T. J. Marks and M. R. Wasielewski, Singlet Fission within Diketopyrrolopyrrole Nanoparticles in Water, Chem. Mater., 2017, 29, 6810–6817 CrossRef CAS.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, B. T. Phelan, M. D. Krzyaniak, S. R. Reddy, P. B. Coto, N. E. Horwitz, R. M. Young, F. J. White, F. Hampel, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Unified model for singlet fission within a non-conjugated covalent pentacene dimer, Nat. Commun., 2017, 8, 15171 CrossRef CAS.
- K. Miyata, Y. Kurashige, K. Watanabe, T. Sugimoto, S. Takahashi, S. Tanaka, J. Takeya, T. Yanai and Y. Matsumoto, Coherent singlet fission activated by symmetry breaking, Nat. Chem., 2017, 9, 983 CrossRef CAS.
- N. R. Monahan, D. Sun, H. Tamura, K. W. Williams, B. Xu, Y. Zhong, B. Kumar, C. Nuckolls, A. R. Harutyunyan, G. Chen, H.-L. Dai, D. Beljonne, Y. Rao and X. Y. Zhu, Dynamics of the triplet-pair state reveals the likely coexistence of coherent and incoherent singlet fission in crystalline hexacene, Nat. Chem., 2016, 9, 341 CrossRef.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, External Quantum Efficiency Above 100% in a Singlet-Exciton-Fission-Based Organic Photovoltaic Cell, Science, 2013, 340, 334–337 CrossRef CAS.
- A. M. Levine, C. Schierl, B. S. Basel, M. Ahmed, B. A. Camargo, D. M. Guldi and A. B. Braunschweig, Singlet Fission in Combinatorial Diketopyrrolopyrrole–Rylene Supramolecular Films, J. Phys. Chem. C, 2019, 123, 1587–1595 CrossRef CAS.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, FRET-Based Small-Molecule Fluorescent Probes: Rational Design and Bioimaging Applications, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, The triplet excited state of Bodipy: formation, modulation and application, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- Y. Lei, Y. Xue, Y. Li, X. Liu, F. Wang and S. Min, High-performance Förster resonance energy transfer-based dye-sensitized photocatalytic H2 evolution with graphene quantum dots as the homogeneous energy donor, Photochem. Photobiol. Sci., 2018, 17, 1147–1152 RSC.
- T. Goh, J.-S. Huang, E. A. Bielinski, B. A. Thompson, S. Tomasulo, M. L. Lee, M. Y. Sfeir, N. Hazari and A. D. Taylor, Coevaporated Bisquaraine Inverted Solar Cells: Enhancement Due to Energy Transfer and Open Circuit Voltage Control, ACS Photonics, 2015, 2, 86–95 CrossRef CAS.
- A. Sarkar, S. Dhiman, A. Chalishazar and S. J. George, Visualization of Stereoselective Supramolecular Polymers by Chirality-Controlled Energy Transfer, Angew. Chem., Int. Ed., 2017, 56, 13767–13771 CrossRef CAS.
- L. Ji, Y. Sang, G. Ouyang, D. Yang, P. Duan, Y. Jiang and M. Liu, Cooperative Chirality and Sequential Energy Transfer in a Supramolecular Light-Harvesting Nanotube, Angew. Chem., Int. Ed., 2019, 58, 844–848 CrossRef CAS.
- P. P. Neelakandan, Z. Pan, M. Hariharan, Y. Zheng, H. Weissman, B. Rybtchinski and F. D. Lewis, Hydrophobic Self-Assembly of a Perylenediimide-Linked DNA Dumbbell into Supramolecular Polymers, J. Am. Chem. Soc., 2010, 132, 15808–15813 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |