
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of dipeptide Boc-diphenylalanine nanotubes inside electrospun polymeric fibers with strong piezoelectric response†
Rosa M. F.
Baptista
 *a,
Etelvina
de Matos Gomes
a,
M. Manuela M.
Raposo
b,
Susana P. G.
Costa
b,
Paulo E.
Lopes
c,
Bernardo
Almeida
a and
Michael S.
Belsley
a
*a,
Etelvina
de Matos Gomes
a,
M. Manuela M.
Raposo
b,
Susana P. G.
Costa
b,
Paulo E.
Lopes
c,
Bernardo
Almeida
a and
Michael S.
Belsley
a
aCentre of Physics, Univ Minho, Campus Gualtar, 4710-057 Braga, Portugal. E-mail: batistarmf@gmail.com
bCentre of Chemistry, Univ Minho, Campus Gualtar, 4710-057 Braga, Portugal
cInst Polymers & Composites IPC, Univ Minho, Campus Azurém, 4804-533 Guimarães, Portugal
First published on 16th September 2019
Abstract
Dipeptide biomaterials are strong piezoelectric materials that can convert applied mechanical forces into electricity. We have developed large-scale hybrid electrospun arrays containing N-tert-butoxycarbonyl (Boc) diphenylalanine in the form of nanotubes embedded in biocompatible polymers. These nanofibers exhibit strong piezoelectric properties when a periodic mechanical force is applied. The nanostructured hybrid materials were produced by the electrospinning technique. Optical absorption measurements show four bands in the spectral region 240–280 nm indicating quantum confinement due to nanotube formation of Boc-diphenylalanine in dichloromethane solutions. A strong blue photoluminescence emission was observed from nanotubes crystallized inside the fiber arrays during the electrospinning process. These two dimensional hybrid biomaterial structures are able to generate voltage, current and density power of up to 30 V, 300 nA and 2.3 μW cm−2, respectively, when a periodical force of 1.5 N is applied. The dipeptide-polymer electrospun arrays can power several liquid-crystal display panels and may be used for biomedical applications and as bio-energy sources.
Introduction
Aromatic dipeptide nanostructures spontaneously form through the process of self-organization, based on efficient molecular recognition modules and constitute unique bio-inspired materials, forming a class of versatile nanostructures with unique biological and electronic properties.1–4 Among these, unprotected diphenylalanine NH2-L-phenylalanine-L-phenylalanine-OH, hereafter Phe-Phe, and protected N-tert-butoxycarbonyl diphenylalanine (Boc-L-phenylalanine-L-phenylalanine-OH), hereafter Boc-Phe-Phe, are short peptides consisting of two naturally occurring L-phenylalanine residues. They have attracted considerable and extensive research interest due to their potential applications in biomedicine and piezoelectric devices.5,6Phe-Phe, the core recognition motif of the Alzheimer's β-amyloid polypeptide, efficiently self-assembles into well-ordered nanotubular structures7,8 crystallizing in a hexagonal crystal system (space group P61) as reported by Gorbitz.9 Depending on the experimental conditions such as pH and temperature Phe-Phe may also self-assemble into nanowires,10 nanovesicles11 and nanorods.12 Boc-Phe-Phe is a member of the structural family of the aromatic dipeptide Phe-Phe, which also shows an ability to form highly ordered tubular nanostructures and nanospheres (NS) by the self-assembly of the aromatic dipeptide under different dissolution conditions.13,14 The self-association of component molecules results from noncovalent interactions leading to the formation of patterns or hierarchical structures.15 All these structures display very interesting and superior physical properties: mechanically Phe-Phe nanotubes (NTs) are very stiff with a high average point stiffness and Young's modulus of 160 N m−1 and 19–27 GPa respectively, placing them among the stiffest biological materials known.4 This remarkable rigidity is attributed to the “zipper-like” aromatic interlocks interpenetrating inside the NT backbones.16
Electromechanical coupling in biological systems is a known phenomenon exhibited in bones,17–19 amino acids,20–22 wood23 and in biological systems such as proteins, biopolymers, polysaccharides, organelles, and hair.24,25 Ferroelectricity has also been reported in porcine aortic walls26 and biomaterials.27,28
Phe-Phe NTs have been integrated in devices for electronic and biosensing applications, for nanotemplate fabrication and formation of nanoparticle composites.1,29–32 Photoluminescent dipeptide NTs were incorporated with lanthanide complexes and used for selective detection of the paraoxon neurotoxin by rapid photoluminescence quenching. The sensing mechanism occurred through the inhibition of cascaded energy transfer from NTs to lanthanide ions.33 Also, in the field of generation of environmentally clean new optical materials for short wavelength photonic devices it has been reported that quantum confinement (QC) observed in Boc-Phe-Phe nanostructures generates remarkable changes in the optical properties of semiconductors.34
Piezoelectricity in Phe-Phe nanotubes was firstly reported by Kholkin35 through PFM measurements, and a shear component of the piezoelectric tensor coefficient d15 as large as 60 pm V−1 was obtained for nanotubes with 200 nm diameter and was explained considering the dipeptide nanotubes as a crystalline nanoceramic material.
Energy harvesters based on piezoelectric Phe-Phe nanotubes were fabricated and reported to generate an output voltage and power up to 2.8 V and 8.2 nW, respectively, when a force of 42 N was periodically applied.36 On Phe-Phe vertical microrod arrays grown under an external applied electric field, an effective piezoelectric constant equal to d33 = 17.9 pm V−1 was measured. Furthermore, these microrods were able to produce up to 3.3 nW cm−2 of power at 50 MΩ, which is around 4 times higher than that measured for zinc oxide nanowires. The effect of the electric field resulted in a stretching of the peptide backbone with an increase of the dipole moment magnitude.5
The electrospinning technique is a powerful and widely used technique to fabricate low dimensional organic nanostructures and polymer fibers allowing the control of morphology and molecular orientation of embedded molecules and polymer chains along the electrospun fibers. These fibers with micro or nanometer diameters and a high ratio of surface area/volume may have in principle extremely high lengths (up to several meters) and functional properties when doped. The electrospinning process is based on the extrusion and elongation of a jet, ejected from a charged polymer solution when an intense electric field (on the order of 105–106 V m−1) is applied between the tip of a syringe pump and the collector electrodes.37–39
Electrospinning of a Phe-Phe aqueous solution was reported and nanotubes with diameters between 400 and 700 nm were obtained. However, although they were continuous in length, they were easily damaged even when nanonewton forces were applied to them.40 Electrospun membranes of poly-ε-caprolactone (PCL) with Phe-Phe self-assembled nanotubes were investigated for drug delivery.41 These hybrid composite materials were found to have enhanced mechanical strength and Young's moduli for 40% and 50% filling of PCL with the peptide, resulting in an overall enhancement of the elastic properties of the composite.41
Here we report the fabrication of electrospun hybrid structures by embedding Boc-Phe-Phe inside a polymer matrix using the high electric fields associated with the electrospinning technique. Easily handled two-dimensional polymer arrays containing dipeptide NT assemblies running along the longitudinal electrospun fiber axis were obtained. When deformed or pressed with a 1.5 N force, they were able to output a piezoelectric voltage of 30 V and power density of 2.3 μW cm−2, respectively one and three orders of magnitude higher than those reported in the literature. Three different polymers, polymethylmethacrylate (PMMA), poly-L-lactic acid (PLLA) and PCL were chosen to form, separately, the precursor solutions used in the electrospinning process. These polymers and the dipeptide are all soluble in common solvents. They are biodegradable, biocompatible42 and are drawing increasing attention due to their medical use in tissue engineering scaffolds and as materials for potential shape memory applications in the biomedical field due to their low toxicity.43,44 Furthermore, PLLA also displays piezoelectricity and a longitudinal piezoelectric coefficient value of 10 pm V−1 has been measured in nanofiber membranes.45–47 Some studies have investigated the self-assembly of cyclic peptides into poly-DL-lactide (PDLLA) solutions and found that the formation of peptide microcrystals increased the stiffness and Young's modulus of the hybrid composites.48,49
Combining the piezoelectric properties of dipeptide nanostructures and polymers, we demonstrate that it is possible to produce functional biocompatible hybrid materials with improved piezoelectric responses.
Experimental
Materials and synthesis
L-Phenylalanine (Phe), 1-hydroxybenzotriazole (HOBt), N,N-dicyclohexylcarbodiimide (DCC), thionyl chloride and di-tert-butylpyrocarbonate (Boc2O) were purchased from Sigma-Aldrich or Alfa Aesar and used as received. Polymethylmethacrylate (PMMA, Mw 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 purchased from Alpha Cimit), poly(L-lactic acid) (PLLA, Mw 217–225000 purchased from Polysciences) and poly-ε-caprolactone (PCL, Mw 80000, purchased from Aldrich). All solvents were purchased from Sigma-Aldrich and used as received.
000 purchased from Alpha Cimit), poly(L-lactic acid) (PLLA, Mw 217–225000 purchased from Polysciences) and poly-ε-caprolactone (PCL, Mw 80000, purchased from Aldrich). All solvents were purchased from Sigma-Aldrich and used as received.
Boc-Phe-Phe (Fig. 1) was synthesized by a conventional solution-phase coupling reaction between Boc-Phe-OH and H-Phe-OMe mediated by DCC/HOBt. The Boc group was used for amino acid N-terminal protection and the C-terminal was protected as a methyl ester, by standard protecting group chemistry. Removal of the methyl ester group was performed using aqueous NaOH (2 mol L−1) and the saponification progress was monitored by thin layer chromatography.50 All the intermediates were characterized by NMR spectroscopy on a Bruker Avance III 400 at an operating frequency of 400 MHz for 1H.
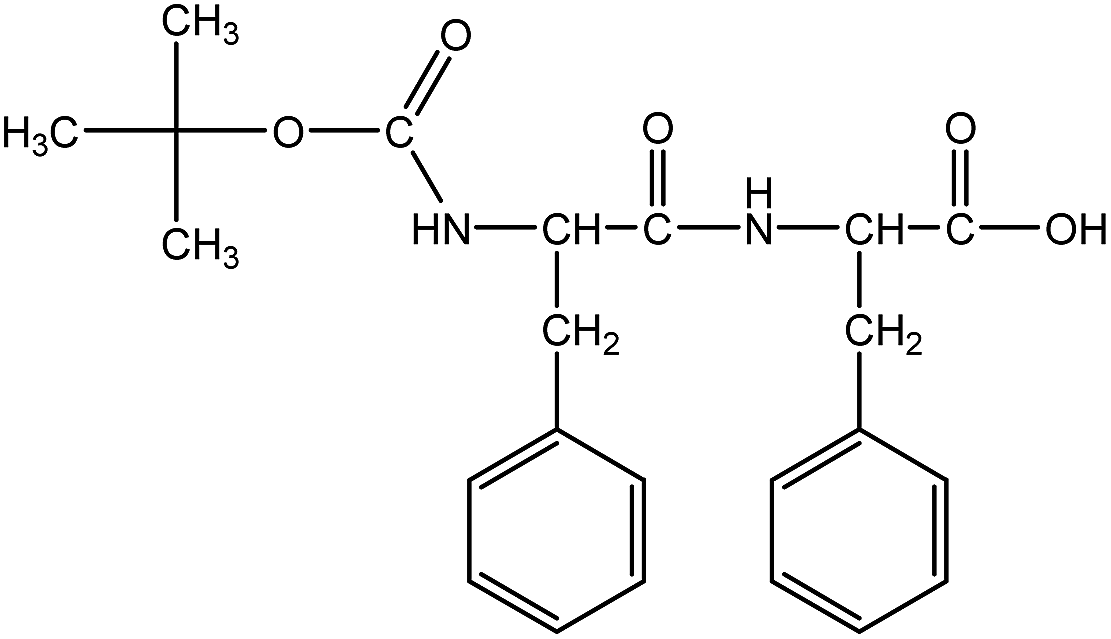 | ||
| Fig. 1 Molecular structure of Boc-Phe-Phe dipeptide. | ||
Precursor electrospinning solutions
The three precursor solutions were prepared by dissolving 0.5 g of PLLA and PMMA polymers, each separately, in 4 mL of dichloromethane (DCM) with vigorous stirring (700 rpm) at 40 °C for 1 h. After complete dissolution, 0.5 g of Boc-Phe-Phe previously dissolved in 1 mL of dimethylformamide (DMF) was added. For PCL polymer, the solution was made by dissolving 0.5 g in a mixture of chloroform/methanol (3:2 v/v) at room temperature and 0.5 g of Boc-Phe-Phe was incorporated in small amounts. The resulting clear and homogeneous solutions were stirred for several hours under ambient conditions prior to the electrospinning process.
Electrospinning of nanofibers
Boc-Phe-Phe nanofibers were electrospun by a conventional electrospinning technique described before.39 The obtained solutions were loaded into a syringe (0.5 mm diameter) with its needle connected to the anode of a high voltage power supply (Spellmann CZE2000). Electrospinning was performed at room temperature and various parameters were changed to obtain bead free fibers and stable spinning conditions, namely, the solution feeding flow rate, the electric potential difference and the needle–collector distance. An electric potential difference between 17 and 20 kV, depending on the polymer and solvent ratio, was used. The needle–collector distance was 12 cm and flow rate 0.15–0.30 mL h−1. The fibers have been collected as a random mesh on a flat plate coated with high purity aluminium foil.Scanning electron microscopy (SEM)
The morphology, distribution of diameters and thickness of the deposited polymer Boc-Phe-Phe fiber layers and crystalline Boc-Phe-Phe alone were assessed through a scanning electron microscope Nova NanoSEM operated at an accelerating voltage of 10 kV.Raman spectroscopy
Raman spectroscopy was carried out on a LabRAM HR Evolution Raman spectrometer (Horiba Scientific, France) coupled with a Horiba Scientific's Labspec 6 spectroscopy set which provides not only complete instrument control but also data processing. The Raman spectra were acquired with a 532 nm laser excitation wavelength (0.1% laser intensity), with an acquisition time and accumulation of 30 s in the range of 40–3500 cm−1.Optical absorption and photoluminescence
The measurements were made on a Shimadzu UV/2501PC spectrophotometer for optical absorption. Photoluminescence spectra were collected using a FluoroMax-4 spectrofluorometer. For optical absorption measurements, 2.5 mg mL−1 solutions of dipeptide Boc-Phe-Phe were prepared in ethanol and dichloromethane (DCM). A Phe-Phe solution was prepared in ethanol with equal concentration. Samples were measured in quartz cuvettes with 1 cm path length.Photoluminescence spectra of Boc-Phe-Phe monomers were measured in a diluted 1.0 × 10−5 mol L−1 ethanol solution using 257 nm as the excitation wavelength. The input and the output slits were fixed at 5 nm.
Boc-Phe-Phe nanotubes were deposited on quartz slides for detection of photoluminescence up to 280 nm. Electrospun fibers of Boc-Phe-Phe were also deposited on quartz slides to measure the photoluminescence at two excitation wavelengths 257 and 360 nm. The input and the output slits were fixed at 10 nm.
The photoluminescence excitation spectrum of the nanotubes was measured at the emission wavelengths of 300 and 435 nm.
Piezoelectric measurements
The piezoelectric output voltage through a load resistance of 100 MΩ was collected using a Digital Storage Oscilloscope (Agilent Technologies DS0-X-3012A) after passing a low pass filter through a low noise pre-amplifier (Research Systems SR560). The nanofibers were submitted to periodic mechanical forces imposed using a vibration generator (Frederiksen SF2185) with a frequency of 3 Hz imposed with a signal generator (Hewlett Packard 33120A). The force applied was measured using a calibrated FSR 402 Force sensing resistor (Interlink Electronics Sensor Technology). The fibers were directly deposited on high purity aluminium foil, which served as an electrode and arrays with 4 × 10−4 m2 area and 0.60 mm thickness were used.Results and discussion
Optical absorption and photoluminescence
Electron-optical QC of self-assembled Phe-Phe nanospheres (NS) and NTs, due to exciton formation have been observed for the dipeptide dissolved in 1,1,1,3,3,3-hexafluoro-2-isopropanol (HFIP)/methanol and HFIP/water solutions, respectively. The corresponding optical absorption (OA) spectra showed a peaklike optical signature in the region of 240–280 nm, with the first peak at around 248 nm (5.0 eV) followed by other peaks located at 253 nm (4.9 eV), 259 nm (4.79 eV) and 265 nm (4.68 eV), unlike the dipeptide monomer which has a narrow absorption peak at 257 nm. Similarly, Boc-Phe-Phe self-assembles into tubular or spherical nanostructures, under different dissolution conditions, using respectively HFIP/water or HFIP/ethanol as solvents.13,51Here we investigated Boc-Phe-Phe self-assembly in the organic solvents ethanol and DCM and compared it with Phe-Phe self-assembly in ethanol. DCM was the solvent used in the preparation of the electrospun precursor polymer solutions. In Fig. 2a and b, the OA spectrum of Boc-Phe-Phe, with a concentration of 2.5 mg mL−1, shows a step-like form with the characteristic four peaks located at 263, 257, 252 and 247 nm with a wavelength difference of 5–7 nm between adjacent peaks, which are evidence for nanostructure QC during the self-assembling process of Boc-Phe-Phe in DCM solutions. A small and broader peak was found in the range of 320–400 nm, ascribed to Boc-Phe-Phe nanotube formation. In Fig. 2b the spectra of Boc-Phe-Phe dissolved in ethanol and DCM, and Phe-Phe dissolved in ethanol at equal concentrations of 2.5 mg mL−1 are compared. The expansion of the band with the characteristic peaks is made.34,52–54
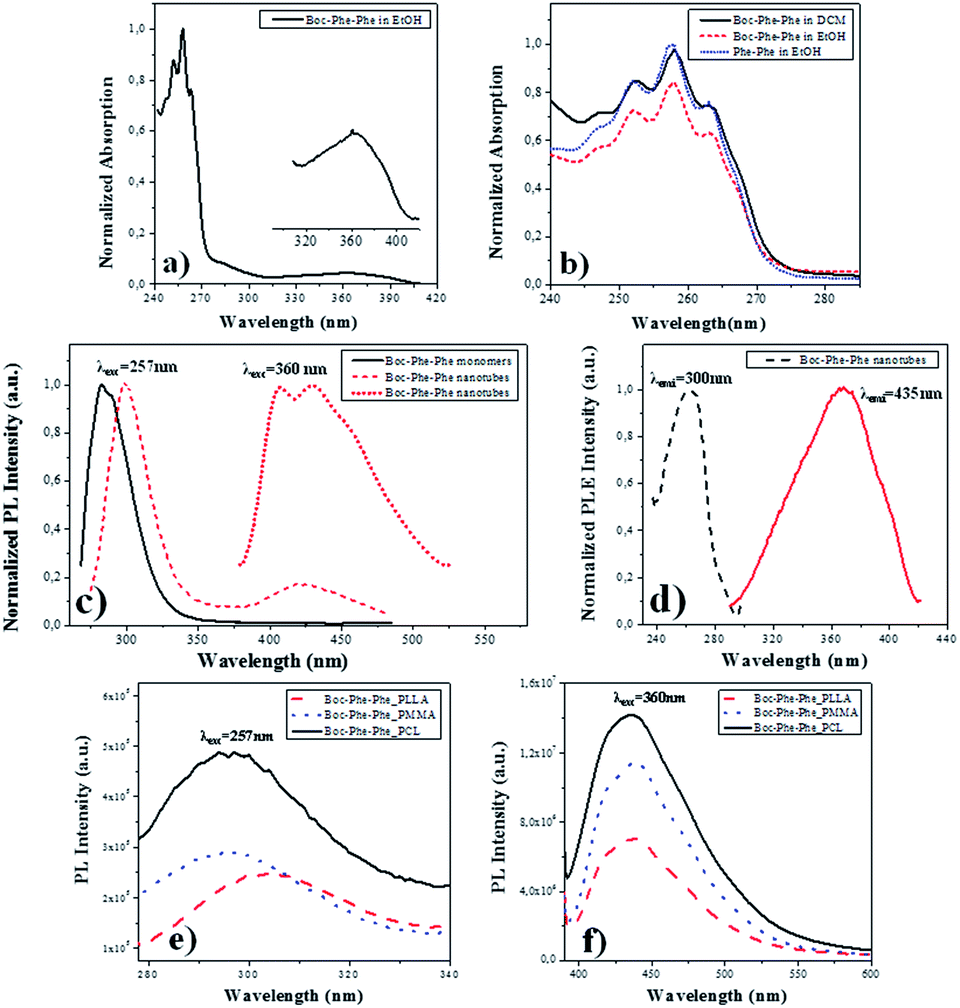 | ||
| Fig. 2 (a) Normalized optical absorption spectrum of Boc-Phe-Phe in ethanol with a concentration of 2.5 mg mL; (b) normalized optical absorption spectra of Phe-Phe in ethanol and Boc-Phe-Phe in ethanol and DCM solutions, revealing (for all the solvents) four peaks corresponding to nanostructure self-assembly; (c) normalized photoluminescence emission spectra of Boc-Phe-Phe monomers, Boc-Phe-Phe in DCM solution and Boc-Phe-Phe nanotubes showing respectively the characteristic 283 nm, 300 nm for 257 nm wavelength excitation, and 435 nm wavelength maximum for excitation at 360 nm; (d) normalized PLE spectrum of Boc-Phe-Phe while detecting the emission at two wavelengths, 300 nm (dashed line) and at 435 nm (solid line); photoluminescence emission spectra of Boc-Phe-Phe nanotubes embedded into electrospun polymer fibers at an excitation wavelength of (e) 257 nm and (f) 370 nm. Here a strong blue emission was observed. | ||
For Phe-Phe NTs, the photoluminescence (PL) spectrum was reported to be very different from the Phe-Phe monomer PL spectrum as a result of exciton-related effects.34Fig. 2c shows the PL spectra at room temperature and near-UV and visible spectral ranges, excited at 257 nm for Boc-Phe-Phe monomers and the dipeptide NTs crystallized from a DCM/ethanol solution on a glass substrate. For the monomer the PL band is centered around 283 nm while for the NTs the maximum now occurs around 305 nm. A second and much less intense and broader peak is also observed in the 400–500 nm blue spectral range. For the first peak there is a red shift of 22 nm, which may be due to a Stokes effect. The red shift in the PL spectrum is ascribed to NT formation during the crystallization process. This effect has also been observed in the PL spectrum of Phe-Phe NTs where a red shift of 11 nm has been reported. In the aggregation processes of aromatic-based molecules, a red shift in PL spectra has also been mentioned.55 The PL peak at 435 nm, under excitation at 370 nm, has an intensity which is 10 times higher than the intensity of the 305 nm peak, under excitation at 257 nm.
For an understanding of the PL nature of Boc-Phe-Phe NTs, the photoluminescence excitation (PLE) spectrum of the structures at the two emission wavelengths of 257 nm and 435 nm was measured and is shown in Fig. 2d. It indicates, as expected, that the origin of the peak at 435 nm is located at 360 nm, while that at 305 nm is located at 257 nm.
For Boc-Phe-Phe_PLLA, Boc-Phe-Phe_PMMA and Boc-Phe-Phe_PCL electrospun fiber arrays with NTs embedded, the PL spectra were measured for two excitation wavelengths of 257 nm and 360 nm and are presented in Fig. 2e and f, respectively. A near-UV band centered at around 290–305 nm was observed for excitation at 257 nm as shown in Fig. 2e. When exciting the fiber arrays at 360 nm, a strong blue PL emission with a maximum around 435 nm is observed. The peak intensity at 370 nm excitation is 15 times higher than the peak intensity at 257 nm.
We conclude from PL measurements that Boc-Phe-Phe NT spectra may be ascribed to radiative decay of excitons at 305 nm located in the UV spectral range and at 435 nm located in the blue range of the visible spectrum. These results agree with those reported for PL emission spectra of Phe-Phe NTs, which also revealed two peaks located at around 305 nm and 450 nm and ascribed to radiative decay of excitons.34 For the dipeptide electrospun fiber array, the PL bands are broader and with lower intensity than those measured for Boc-Phe-Phe NTs, which results from the embedding of dipeptide NTs inside the polymer matrix. This reduction might also be due to the influence of structural defects and electron–phonon interaction during the dipeptide NT formation process. The defects may lead to the overlapping of optical transitions accompanied by a widening of the PL spectral bands.
Fiber morphology
SEM images shown in Fig. 2a and b indicate that Boc-Phe-Phe crystallized from a DCM/ethanol (1:2) solution forms NTs with lengths of several micrometers and diameters of several hundreds of nanometers (within the range of 80–300 nm), similar to Phe-Phe which forms highly ordered NT crystalline structures with a P61 crystallographic space group.9,56,57 These results are consistent with the OA and PL spectra of Boc-Phe-Phe.
SEM analysis of Boc-Phe-Phe_PCL, Boc-Phe-Phe_PLLA, and Boc-Phe-Phe_PMMA fibers produced by the electrospinning technique, Fig. 3a, b, and c respectively, indicate that the fibers are smooth, homogeneous, and bead-free, and with no dipeptide crystals grown on their external surface. The dipeptide crystallization occurs during the electrospun fiber formation and deposition process resulting in dipeptide NT crystals embedded into the polymer matrix. The fiber diameters are within the range of nano or micrometers depending on the polymer, as indicated in Table 1.
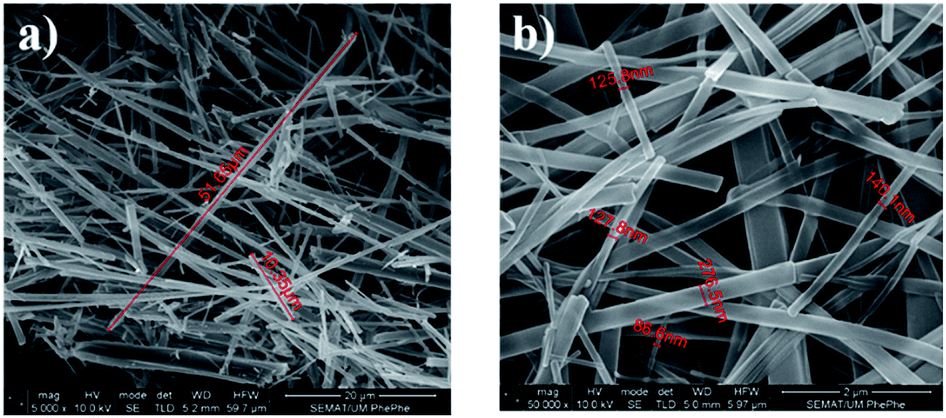 | ||
| Fig. 3 SEM images at a high magnification level (50000×) of Boc-Phe-Phe nanotubes crystallized from a DCM/ethanol (1:2) solution. The nanotubes have (a) lengths between 10 μm and 52 μm and (b) diameters ranging from 86 μm to 276 nm. | ||
| Fiber | Thickness |
|---|---|
| Boc-Phe-Phe_PMMA | 2.0 to 3.5 μm |
| Boc-Phe-Phe_PLLA | 1.3 to 2.7 μm |
| Boc-Phe-Phe_PCL | 590 to 700 nm |
Fibers formed from PCL solutions have diameters in the nanometer scale and within a 100 nm range, while for the other polymers the thickness is within micrometers, as shown in Fig. 4a, b and c for Boc-Phe-Phe_PCL, Boc-Phe-Phe_PLLA and Boc-Phe-Phe_PMMA electrospun fibers, respectively.
 | ||
| Fig. 4 SEM images at a high magnification level (50000×) of (a) Boc-Phe-Phe_PCl, (b) Boc-Phe-Phe_PLLA, and (c) Boc-Phe-Phe_PMMA electrospun fibers. | ||
For the Phe-Phe dipeptide vertically aligned tubes forming dense nanoforest arrays have been reported.58 By using the electrospinning technique, a similar alignment for the Boc-Phe-Phe dipeptide inside the polymer matrix forming hybrid nanocomposites was achieved. An example of an electrospun array of several cm2 area formed by dipeptide Boc-Phe-Phe NTs embedded in polymer fibers is shown in Fig. 5a. From SEM images, Fig. 5b, we concluded that the dipeptide NTs are embedded in the polymer and run along the fiber axis, crystallizing with their 6-fold crystallographic axis along the fiber longitudinal main axis. The nanotube section diameters are between 25 and 35 nm, well within the dimensions reported for Phe-Phe NTs which may have diameters ranging from 10 nm to more than 0.5 μm.28 We have fabricated hybrid electrospun composite structures formed by long polymer fibers containing inside Boc-Phe-Phe NTs with their hexagonal axis along the fibers' longitudinal axis. This NT alignment, we believe, is due to the very high electric field (of the order of 106 V m−1) applied during the electrospinning process, which tends to align the highest symmetry 6-fold dipeptide crystallographic axis with the main fiber axis during the process of Taylor cone formation and fiber pulling and stretching by the strong electric field applied.39,59
 | ||
| Fig. 5 a) Electrospun fiber array deposited on high purity aluminium paper. (b) SEM images at the magnification level (100000×) of Boc-Phe-Phe_PLLA fiber section cut. The inset shows the nanotube diameter sections represented by white “dots”. | ||
Raman spectra
Boc-Phe-Phe_PMMA fibers (as an example) and Boc-Phe-Phe NT crystalline powder spectra, Fig. 6, confirm the dipeptide crystallization inside the fibers in the form of NTs, as the main bands of crystalline Boc-Phe-Phe NTs are also present in the electrospun fibers. The bands at 200 cm−1 are assigned to lattice vibration modes and are the most intense bands in both Boc-Phe-Phe NTs and Boc-Phe-Phe NT doped polymer fibers. After these the more prominent bands are: 816 cm−1 torsion τ (HC9C4C3); 621 cm−1 and 1001 cm−1 ∂ring (CC); 1038 cm−1 ∂ring (CH); 1209 cm−1 ∂ring (CH); 1329 cm−1 stretching ϑring (CC) and wagging ωag (C14H2); 1586 cm−1 and 1605 cm−1 stretching ϑring (CC); stretching ϑring (CC); 2936 cm−1 stretching ϑ (C3H); 2939 cm−1 symmetric stretching ϑs (C14H2); 2979 cm−1ν(C2H); 3004 cm−1, 3036 cm−1, 3054 cm−1 and 3070 cm−1ϑring (CH).60 The band at 813 cm−1 is from PMMA polymer. The measured Raman spectra of Boc-Phe-Phe_PLLA and Boc-Phe-Phe_PCL electrospun fibers are similar to those of Boc-Phe-Phe_PMMA fiber arrays and are a further indication of dipeptide crystallization in the form of NTs inside the fibers, independent of the polymer matrix where the dipeptide is embedded.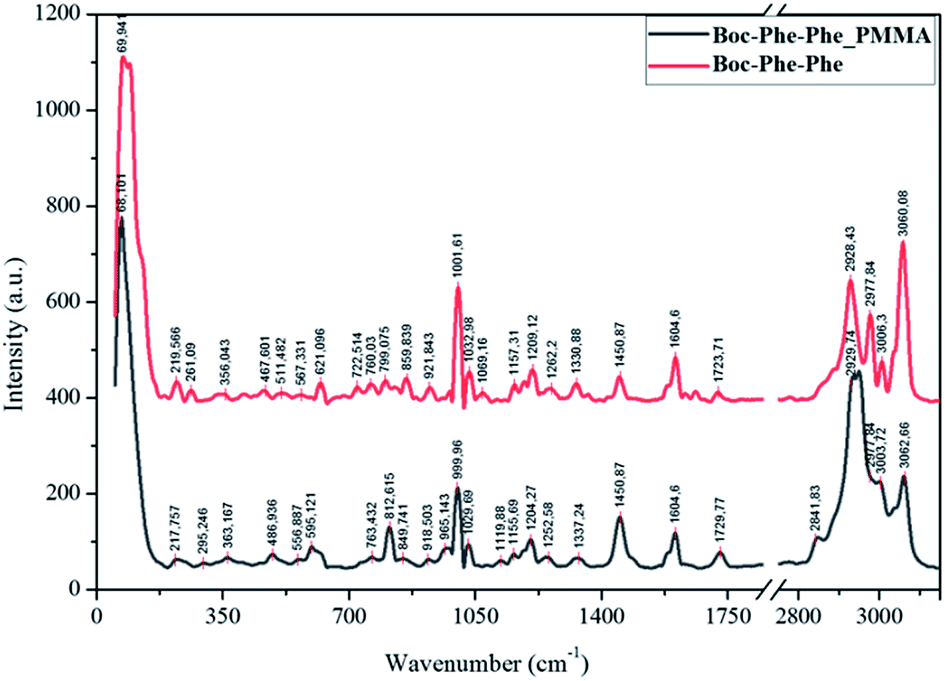 | ||
| Fig. 6 Assigned main Raman bands of crystalline Boc-Phe-Phe nanotubes (red, upper spectrum) and Boc-Phe-Phe_PMMA electrospun fibers (black). | ||
We conclude that Boc-Phe-Phe self-assembly in the form of NTs is dependent on the electrospinning process and the solvent used for the preparation of electrospun solution and does not depend on the polymer chosen, as the solvent used forms clear and homogeneous solutions containing both the dipeptide and the polymer.
The piezoelectric response of fibers
Piezoelectricity is a linear dielectric phenomenon61 and in organic materials is the process of reorienting the molecular dipoles within the crystalline material. The piezoelectric effect results from inter-conversion between mechanical and electrical stimulus inducing a charge redistribution and separation when a force is applied to the material.62 An example is the amino acid glycine in which β- and γ- polymorphs have acentric structures and thus generate shear piezoelectricity.63,64For Phe-Phe NTs crystallizing in point group 6, the piezoelectric tensor has four independent coefficients, two of them are shear components (d15 and d14) which describe deformations along two perpendicular axes relative to the NT axis. The highest and most significant piezoelectric coefficient is d15 ∼ 80 pm V−1, followed by d33 ∼ 18 pm V−1 while the other two coefficients are d14 ∼ 10 pm V−1 and d13 ∼ 4 pm V−1.65 The shear components are significant and make a high contribution to the dipeptide piezoelectric response.
The output voltage of Boc-Phe-Phe electrospun fibers was measured by application of a periodical stress perpendicular to the fiber array plane, as schematically shown in Fig. 7a. In this configuration we are taking full advantage of the high values of the shear coefficients, as the dipeptide NTs inside the fibers have their crystallographic NT axis parallel to the fiber main axis and within the fiber arrays, as discussed before. As in the case of in-plane alignment there is randomness in fiber orientation, we are applying a force perpendicular to the fiber arrays and measuring a piezoelectric voltage with main contributions from d15 and d14. Fig. 7b shows that it is possible to use Boc-Phe-Phe fiber arrays as a power source for operating an LED circuit: when pressing the electrospun fiber array the letters “nt” are turned on.
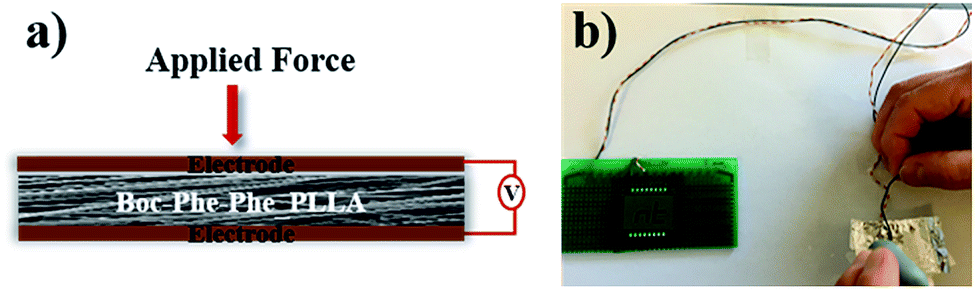 | ||
| Fig. 7 a) Schematic piezoelectric setup with Boc-Phe-Phe_PLLA fiber array and (b) LED circuit powered by the fiber array. | ||
The piezoelectric response of undoped and dipeptide doped polymer fiber arrays for a periodic applied force of 1.5 N is shown in Fig. 8a–d. When compressed and released, Boc-Phe-Phe polymer fibers converted mechanical energy into electricity. Periodic compressive forces of 0.5 N and 1.5 N were applied and released with a frequency of 3 Hz on the nanofiber arrays.
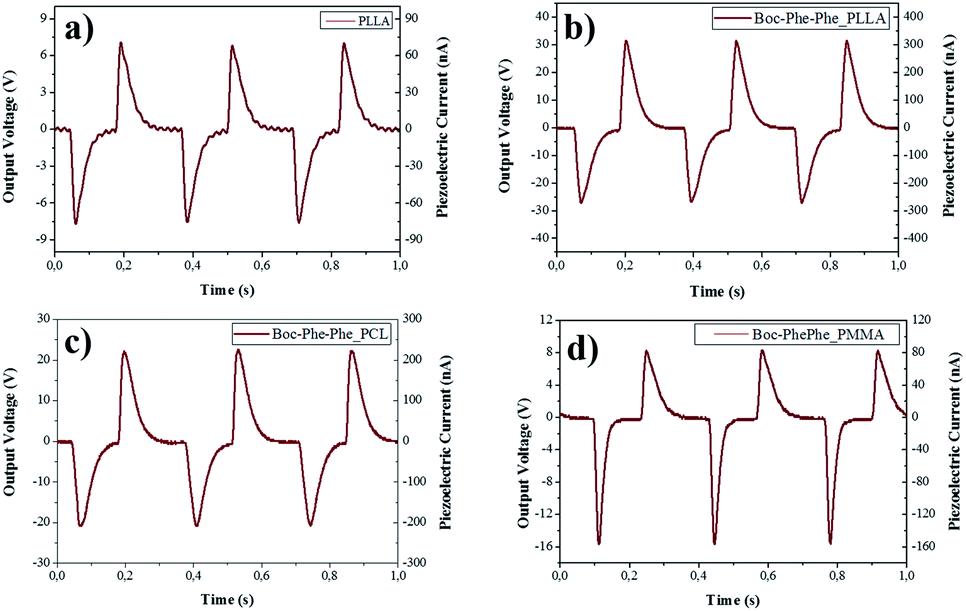 | ||
| Fig. 8 Output voltage (V) and current (nA) measured through a load resistance of 100 MΩ on electrospun fiber arrays of (a) PLLA not doped and Boc-Phe-Phe nanotubes embedded in (b) PLLA, (c) PCL, and (d) PMMA. | ||
The maximum open-circuit voltage (Vout) and current (I), measured through a load resistance of 100 MΩ reached 30 V and 300 nA, respectively, for Boc-Phe-Phe_PLLA. In order to separate the contribution from dipeptide NTs and polymer to the piezoelectric responses, the output voltage of electrospun fiber arrays of PLLA, PMMA and PCL not doped with the dipeptide was measured under similar conditions. For PLLA as expected, the response is much smaller than for dipeptide doped fibers, while for PCL and PMMA fiber arrays the response was negligible (Table 2).
| Applied force (N) | Output voltage (V) | ||
|---|---|---|---|
| PLLA | Boc-Phe-Phe_PLLA | Boc-Phe-Phe_PCL | |
| 0.5 | 6 | 24 | 15 |
| 1.5 | 8 | 30 | 22 |
For Boc-Phe-Phe_PLLA fibers a calculated instantaneous power of 2.3 μW cm−2 for a fiber array of 4.0 cm2 area was achieved. This value is very significant as it is three orders of magnitude higher than that measured in free Phe-Phe microrod arrays5 upon application of a force one order of magnitude smaller. The strong piezoelectric response from dipeptide polymer functional composite fibers to a periodic applied force may be explained as the result of a combined response from the dipeptide nanotube organization inside the polymer fibers such as the shear piezoelectric coefficients are those who make the main contribution to the piezoelectric output voltage. PLLA has been reported to form films and nanowires exhibiting shear piezoelectricity.47,66–68
For a better insight on the remarkable piezoelectric response of Boc-Phe-Phe electrospun nanofiber arrays, Table 3 presents the piezoelectric output measurements performed for some electrospun nanofiber arrays reported in the literature. Our work clearly shows that Boc-Phe-Phe NT nanofibers are able to produce an output voltage one order magnitude higher than that of P(VDF_TrFe) nanofibers which is an engineered ferroelectric polymer.
Conclusions
The optical absorption spectra of Boc-Phe-Phe self-assembly in dichloromethane and ethanol solvents showed the existence of quantum confinement as the four characteristic peaks located at 263 nm, 257 nm, 252 nm and 247 nm were observed for Boc-Phe-Phe nanostructures in solution.The photoluminescence spectrum of Boc_Phe-Phe NTs crystallized both in silica substrates and inside the electrospun polymer fibers showed a red shift of 22 nm which was due to NT formation during the crystallization process. The PL peak at 435 nm, under excitation at 370 nm has an intensity which is 10 times higher than the intensity of the 305 nm peak, under excitation at 257 nm.
Finally, our results demonstrate that embedding the dipeptide inside polymer fibers using the electrospinning technique one is able to produce macroscopy arrays of NTs and maximize their strong piezoelectric responses. Also, these electrospun arrays may be easily integrated in appropriate devices for mechanical to electric energy conversion. Using the electrospinning technique, arrays of several tens cm2 of area can be inexpensively fabricated for energy conversion. Due to its PL properties, electrospun dipeptide nanostructure arrays may also find use as optical biomarkers and biosensors.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.Acknowledgements
We are grateful to the FUNDAÇÃO PARA A CIÊNCIA E TECNOLOGIA (FCT) for a post-doctoral grant to R.M.F. Batista (SFRH/BPD/79333/2011), and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support through the Physics Centers of the Universities of Minho and Porto (Ref. UID/FIS/04650/2013) and the Chemistry Research Centre of the University of Minho (Ref. UID/QUI/00686/2013 and UID/QUI/0686/2016). The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased within the framework of the National Program for Scientific Re-equipment, contract REDE/1517/RMN/2005 with funds from POCI 2010 (FEDER) and FCT.References
- M. Yemini, M. Reches, J. Rishpon and E. Gazit, Nano Lett., 2005, 5, 183–186 CrossRef CAS PubMed.
- N. Ashkenasy, W. S. Horne and M. R. Ghadiri, Small, 2006, 2, 99–102 CrossRef CAS PubMed.
- H. A. M. Ardoña and J. D. Tovar, Bioconjugate Chem., 2015, 26, 2290–2302 CrossRef PubMed.
- N. Kol, L. Adler-Abramovich, D. Barlam, R. Z. Shneck, E. Gazit and I. Rousso, Nano Lett., 2005, 5, 1343–1346 CrossRef CAS PubMed.
- V. Nguyen, R. Zhu, K. Jenkins and R. Yang, Nat. Commun., 2016, 7, 13566 CrossRef CAS PubMed.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- C. H. Gorbitz, Chem. Commun., 2006, 2332–2334 RSC.
- J. Kim, T. H. Han, Y.-I. Kim, J. S. Park, J. Choi, D. G. Churchill, S. O. Kim and H. Ihee, Adv. Mater., 2010, 22, 583–587 CrossRef CAS PubMed.
- C. Guo, Y. Luo, R. Zhou and G. Wei, ACS Nano, 2012, 6, 3907–3918 CrossRef CAS PubMed.
- Q. Li, Y. Jia, L. Dai, Y. Yang and J. Li, ACS Nano, 2015, 9, 2689–2695 CrossRef CAS PubMed.
- L. Adler-Abramovich and E. Gazit, J. Pept. Sci., 2008, 14, 217–223 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Isr. J. Chem., 2005, 45, 363–371 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- I. Azuri, L. Adler-Abramovich, E. Gazit, O. Hod and L. Kronik, J. Am. Chem. Soc., 2014, 136, 963–969 CrossRef CAS PubMed.
- E. Fukada and I. Yasuda, J. Phys. Soc. Jpn., 1957, 12, 1158–1162 CrossRef.
- M. H. Shamos, L. S. Lavine and M. I. Shamos, Nature, 1963, 197, 81 CrossRef CAS PubMed.
- R. M. Zilberstein, Nature, 1972, 235, 174–175 CrossRef.
- E. de Matos Gomes, T. Viseu, M. Belsley, B. Almeida, M. M. R. Costa, V. H. Rodrigues and D. Isakov, Mater. Res. Express, 2018, 5, 045049 CrossRef.
- V. V. Lemanov, S. N. Popov and G. A. Pankova, Phys. Solid State, 2002, 44, 1929–1935 CrossRef CAS.
- V. V. Lemanov, S. N. Popov and G. A. Pankova, Ferroelectrics, 2003, 285, 207–216 CrossRef CAS.
- E. Fukada, J. Phys. Soc. Jpn., 1955, 10, 149–154 CrossRef.
- A. J. P. Martin, Proc. Phys. Soc., 1941, 53, 186–189 CrossRef.
- Electromechanical Behavior in Biological Systems at the Nanoscale, ed. A. Gruverman, B. J. Rodriguez and S. V. Kalinin, Springer, New York, NY, 2007 Search PubMed.
- Y. Liu, Y. Zhang, M.-J. Chow, Q. N. Chen and J. Li, Phys. Rev. Lett., 2012, 108, 078103 CrossRef.
- S. B. Lang, IEEE Trans. Dielectr. Electr. Insul., 2000, 7, 466–473 CrossRef CAS.
- N. Amdursky, P. Beker, J. Schklovsky, E. Gazit and G. Rosenman, Ferroelectrics, 2010, 399, 107–117 CrossRef.
- O. Carny, D. E. Shalev and E. Gazit, Nano Lett., 2006, 6, 1594–1597 CrossRef CAS PubMed.
- Y. Song, S. R. Challa, C. J. Medforth, Y. Qiu, R. K. Watt, D. Peña, J. E. Miller, F. v. Swol and J. A. Shelnutt, Chem. Commun., 2004, 1044–1045 RSC.
- M. Yemini, M. Reches, E. Gazit and J. Rishpon, Anal. Chem., 2005, 77, 5155–5159 CrossRef CAS PubMed.
- N. B. Sopher, Z. R. Abrams, M. Reches, E. Gazit and Y. Hanein, J. Micromech. Microeng., 2007, 17, 2360–2365 CrossRef CAS.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- N. Amdursky, M. Molotskii, D. Aronov, L. Adler-Abramovich, E. Gazit and G. Rosenman, Nano Lett., 2009, 9, 3111–3115 CrossRef CAS PubMed.
- A. Kholkin, N. Amdursky, I. Bdikin, E. Gazit and G. Rosenman, ACS Nano, 2010, 4, 610–614 CrossRef CAS PubMed.
- J.-H. Lee, K. Heo, K. Schulz-Schönhagen, J. H. Lee, M. S. Desai, H.-E. Jin and S.-W. Lee, ACS Nano, 2018, 12, 8138–8144 CrossRef CAS.
- D. H. Reneker and I. Chun, Nanotechnology, 1996, 7, 216–223 CrossRef CAS.
- A. Frenot and I. S. Chronakis, Curr. Opin. Colloid Interface Sci., 2003, 8, 64–75 CrossRef CAS.
- D. V. Isakov, E. D. Gomes, L. G. Vieira, T. Dekola, M. S. Belsley and B. G. Almeida, ACS Nano, 2011, 5, 73–78 CrossRef CAS PubMed.
- G. Singh, A. M. Bittner, S. Loscher, N. Malinowski and K. Kern, Adv. Mater., 2008, 20, 2332–2336 CrossRef CAS.
- M. S. Liberato, S. Kogikoski, E. R. da Silva, D. R. de Araujo, S. Guha and W. A. Alves, J. Mater. Chem. B, 2016, 4, 1405–1413 RSC.
- J. Kobayashi, T. Asahi, M. Ichiki, A. Oikawa, H. Suzuki, T. Watanabe, E. Fukada and Y. Shikinami, J. Appl. Phys., 1995, 77, 2957–2973 CrossRef CAS.
- S. Pandini, S. Passera, M. Messori, K. Paderni, M. Toselli, A. Gianoncelli, E. Bontempi and T. Riccò, Polymer, 2012, 53, 1915–1924 CrossRef CAS.
- L. Peponi, I. Navarro-Baena, A. Sonseca, E. Gimenez, A. Marcos-Fernandez and J. M. Kenny, Eur. Polym. J., 2013, 49, 893–903 CrossRef CAS.
- E. Fukada, IEEE Trans. Sonics Ultrason., 2000, 47, 1277–1290 CAS.
- E. Fukada, Biorheology, 1995, 32, 593–609 CAS.
- T. Ochiai and E. Fukada, Jpn. J. Appl. Phys., 1998, 37, 3374–3376 CrossRef CAS.
- D. J. Rubin, H. T. Nia, T. Desire, P. Q. Nguyen, M. Gevelber, C. Ortiz and N. S. Joshi, Biomacromolecules, 2013, 14, 3370–3375 CrossRef CAS PubMed.
- N. Even, L. Adler-Abramovich, L. Buzhansky, H. Dodiuk and E. Gazit, Small, 2011, 7, 1007–1011 CrossRef CAS PubMed.
- P. Jana, S. Maity, S. K. Maity, P. K. Ghorai and D. Haldar, Soft Matter, 2012, 8, 5621–5628 RSC.
- L. Adler-Abramovich, N. Kol, I. Yanai, D. Barlam, R. Z. Shneck, E. Gazit and I. Rousso, Angew. Chem., Int. Ed., 2010, 49, 9939–9942 CrossRef CAS PubMed.
- N. Amdursky, M. Molotskii, E. Gazit and G. Rosenman, Appl. Phys. Lett., 2009, 94, 261907-3 CrossRef.
- N. Amdursky, M. Molotskii, E. Gazit and G. Rosenman, J. Am. Chem. Soc., 2010, 132, 15632–15636 CrossRef CAS PubMed.
- A. Handelman, P. Beker, N. Amdursky and G. Rosenman, Phys. Chem. Chem. Phys., 2012, 14, 6391–6408 RSC.
- M. Pope and C. E. Swenberg, Annu. Rev. Phys. Chem., 1984, 35, 613–655 CrossRef CAS.
- C. H. Gorbitz, Chem.–Eur. J., 2001, 7, 5153–5159 CrossRef CAS.
- S. Maity, S. Nir and M. Reches, J. Mater. Chem. B, 2014, 2, 2583–2591 RSC.
- M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195 CrossRef CAS PubMed.
- S. Ramakrishna, K. Fujihara, W.-E. Teo, T.-C. Lim and Z. Ma, An Introduction to Electrospinning and Nanofibers, WORLD SCIENTIFIC, 2005 Search PubMed.
- J. G. da Silva Filho, F. E. A. Melo, J. A. Lima, G. S. Pinheiro and P. T. C. Freire, Vib. Spectrosc., 2018, 97, 75–84 CrossRef CAS.
- J. F. Nye, Physical properties of crystals: their representation by tensors and matrices, Oxford : Clarendon press, Repr. paperback ed. edn, 2004 Search PubMed.
- W. G. Cady, Piezoelectricity : an introduction to the theory and applications of electromechanical phenomena in crystals, McGraw-Hill, New York (N.Y.), 1st edn, 1946 Search PubMed.
- D. Isakov, E. D. Gomes, I. Bdikin, B. Almeida, M. Belsley, M. Costa, V. Rodrigues and A. Heredia, Cryst. Growth Des., 2011, 11, 4288–4291 CrossRef CAS.
- S. Guerin, A. Stapleton, D. Chovan, R. Mouras, M. Gleeson, C. McKeown, M. R. Noor, C. Silien, F. M. F. Rhen, A. L. Kholkin, N. Liu, T. Soulimane, S. A. M. Tofail and D. Thompson, Nat. Mater., 2018, 17, 180–186 CrossRef CAS PubMed.
- S. Vasilev, P. Zelenovskiy, D. Vasileva, A. Nuraeva, V. Y. Shur and A. L. Kholkin, J. Phys. Chem. Solids, 2016, 93, 68–72 CrossRef CAS.
- T. Yoshida, K. Imoto, K. Tahara, K. Naka, Y. Uehara, S. Kataoka, M. Date, E. Fukada and Y. Tajitsu, Jpn. J. Appl. Phys., 2010, 49, 09MC11-6 Search PubMed.
- M. Smith, Y. Calahorra, Q. S. Jing and S. Kar-Narayan, APL Mater., 2017, 5, 074105-8 Search PubMed.
- S. J. Lee, A. P. Arun and K. J. Kim, Mater. Lett., 2015, 148, 58–62 CrossRef CAS.
- L. Persano, C. Dagdeviren, Y. Su, Y. Zhang, S. Girardo, D. Pisignano, Y. Huang and J. A. Rogers, Nat. Commun., 2013, 4, 1633 CrossRef PubMed.
- D. Isakov, E. de Matos Gomes, B. Almeida, A. L. Kholkin, P. Zelenovskiy, M. Neradovskiy and V. Y. Shur, Appl. Phys. Lett., 2014, 104, 032907 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00464e |
| This journal is © The Royal Society of Chemistry 2019 |