
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCascade aldol condensation of an aldehyde via the aerobic oxidation of ethanol over an Au/NiO composite
Yuanyuan
Shi†
ab,
Shanli
Tian†
b,
Quanquan
Shi
*ab,
Yifei
Zhang
c,
Ammara
Waheed
bd,
Youhai
Cao
b and
Gao
Li
 *bd
*bd
aCollege of Science, Inner Mongolia Agricultural University, Hohhot 010018, China. E-mail: qqshi@dicp.ac.cn
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: gaoli@dicp.ac.cn
cInstitute of Catalysis for Energy and Environment, College of Chemistry and Chemical Engineering, Shenyang Normal University, Shenyang 110034, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 29th July 2019
Abstract
Synthesis of liquid biofuels (C11–C13) from cellulosic ethanol is regarded as a promising and versatile protocol. In this study, oxide-supported nanogold catalysts exhibit good catalytic performance in ethanol conversion with cinnamaldehyde and finally give rise to the C11–C13 hydrocarbon. High selectivity (70%) for C11–C13 hydrocarbons is achieved over Au/NiO via a one-pot cascade reaction, viz. cross-aldol condensations in the presence of oxygen and base (K2CO3) and then full hydrodeoxygenation with hydrogen gas. EtOH-TPD and TGA analyses show that the ethanol is activated to acetaldehyde (CH3CHO*) over the surface oxygen vacancies of the NiO support. The CH3CHO* then reacts with cinnamaldehyde at the interfacial perimeter of the Au/NiO composite during the cascade reactions, as evidenced by comparison of the catalytic performance with that over another oxide-supported Au NP, chemo-adsorption investigations, and in situ infrared spectroscopy investigations. This work may provide new guidelines for designing efficient catalysts to convert bioethanol into biofuels with high energy density.
Introduction
Liquid fuels are the most important source of energy in the transportation sector, to date being produced on an industrial scale from non-renewable fossil-based resources. In the last decade, the rapid emergence of the renewable plant-based industry has opened up opportunities to produce biofuels with high energy density, often with similar chemical structures to those of fossil origin.1–6 Therefore, the synthesis of sustainable biofuels from bioethanol has attracted particular interest. Bioethanol is a renewable and green chemical, commonly known as “cellulosic ethanol”, that is produced from various plant materials.7,8 The ethanol can react with an aldehyde via cross-aldol condensation, which can be promoted by base catalysts (mineral and organic bases, solid base).9–13 Cross-aldol condensation has been identified as an effective protocol for C–C bond formation to produce biofuels. Currently, the catalytic conversion pathways extend to ethanol oxidation condensation, which could provide an important breakthrough for diversification and sustainable development of hydrocarbon biofuel production.14–16Oxide-supported gold nanoparticles have emerged as a promising heterogeneous catalyst in the aerobic oxidation of alcohols.17–19 For example, Haruta et al. reported the N-formylation of amines via the aerobic oxidation of methanol over supported gold nanoparticles.20 Christensen et al. also studied the amidation of butanol with hexylamine over Au/MgAl2O4 and Au/TiO2 catalysts.21 Very recently, we studied an Au/NiO composite for a cascade reaction of furfural with bioethanol to give C7–C9 straight-chain paraffin in a one-pot synthesis.22 A very high catalytic efficiency of close to 100% furfural conversion and ca. 81% selectivity for C7–C9 paraffin production was achieved. It was found that the presence of active basic sites on the surface of the Au/NiO can potentially be employed for efficient cascade conversion. However, the detailed reaction pathway of the cascade reactions, especially the ethanol activation, has not been investigated in detail.
Herein, we explore a new one-pot cascade reaction of ethanol oxidation with cinnamaldehyde to produce C11–C13 oxygenated chemicals with furyl groups (Scheme 1). EtOH-TPD and thermogravimetric analysis (TGA) analyses showed that the activation of ethanol to acetaldehyde (CH3CHO*) occurs on the surface oxygen vacancies of the NiO support, which was further confirmed by in situ infrared spectroscopy. The formed CH3CHO* species then reacts with cinnamaldehyde at the interfacial perimeter of the Au/NiO composite during the cascade reactions. Finally, the furyl groups are chemo-selectively transferred to C11 and C13 hydrocarbons (main alkylbenzene derivatives) via a one-pot hydrodeoxygenation reaction.
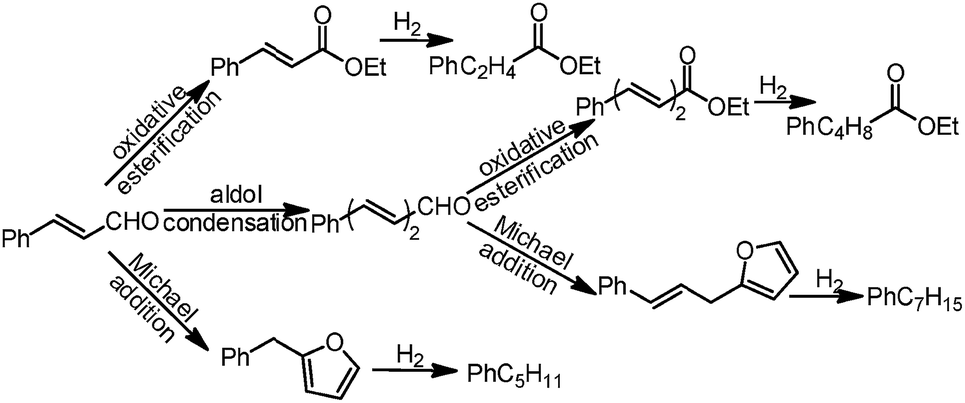 | ||
| Scheme 1 The conversion pathway of cinnamaldehyde to oxygenates and C11 and C13 hydrocarbons via the one-pot cascade reactions of oxidative esterification, aldol condensation and Michael addition over an Au/NiO catalyst in the presence of O2 and K2CO3 and ethanol, followed by full hydrodeoxygenation using H2 as the reducing agent. | ||
Experimental
Preparation of Au/oxide catalysts
The Au/oxide catalysts (where the oxide is NiO, SiO2, TiO2 or CeO2) are simply prepared via a wet-synthetic process using polyvinyl alcohol-capped Au colloids and oxide powders.23 These oxides were annealed at 300 °C in air for 3 h before the immobilization process. For example, 1 g of the NiO support was added into the colloidal gold solution under vortex-stirring for 3 h. After the impregnation process, the solid samples were washed with water and then dried at 80 °C. The final catalysts were calcined at 300 °C for 3 h in the presence of a flow of air to remove all the capping surfactant, as evidenced by temperature-programmed oxidation (O2-TPO) analysis.Characterization
Scanning transmission electron microscopy (STEM) images were obtained using a JEM-ARM200F. The lattice spacing was determined by Digital Micrograph software. Ethanol-temperature-programmed desorption (EtOH-TPD) measurements were performed on a micromeritics Autochem II chemosorption analyzer. 60 mg of the sample was loaded into a quartz reactor and heated in a flow of He gas (40 mL min−1) at 300 °C for 1 h to remove the adsorbed pollutants. Then the reactor was cooled to room temperature and the sample was saturated with ethanol vapor until no significant amount of ethanol could be detected. The temperature was ramped to 600 °C at a heating rate of 10 °C min−1, which was monitored online by gas chromatography-mass spectrometry (GC-MS) at m/z = 46 for detection of ethanol species. The amount of cinnamaldehyde adsorbed on the catalysts was determined by TGA using a STARe System (Mettler Toledo) under a N2 atmosphere and using NiO as the reference. Typically, 26 μL of cinnamaldehyde and 10 mg of support or catalyst was added in a 5 mL ethanol solution. After vortex-mixing for 2 h, the solids were collected and dried at 80 °C for 1 h, and further analyzed by TGA. We chose the boiling point of cinnamaldehyde (253 °C for 1 hour) to remove the physisorbed cinnamaldehyde from the catalysts. Next, the temperature was increased to measure the amount of chemisorbed cinnamaldehyde. X-ray photoelectron spectroscopy (XPS) spectra were recorded using a Theta Probe system (Thermo Scientific) with 400 μm diameter monochromatic Al Kα excitation at 1486.6 eV with a Hemispherical Analyzer operated in constant pass energy mode at 50 eV. The concentration of surface oxygen vacancies ([OV]%) and chemisorbed oxygen ([OC]%) was estimated based on the area under the peaks of O 1s according to the equation: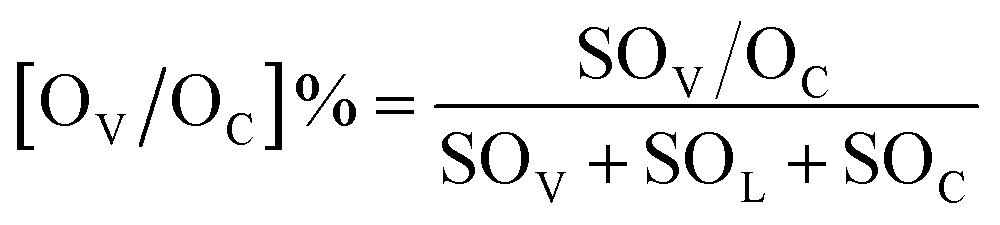
One-pot cascade tests
The one-pot cascade reactions were investigated at 130 °C with 1 MPa air in a 10 mL reactor for 4 h. The reactor was firstly loaded with 3 mL of ethanol, 40 mg of cinnamaldehyde and 10 mg of catalyst. During the hydrodeoxygenation process, the atmosphere of the reactor was changed to 1 MPa H2 gas. The supernatant was analyzed by gas chromatography-mass spectrometer (Agilent Technologies 7890B) equipped with an MS-5977 and a HP-5MS capillary column. The conversion and product selectivity were determined by GC-MS.In situ infrared spectroscopy investigation
A thin film of the catalyst was prepared for in situ IR measurements. 75 mg of Au/NiO in 10 mL of ethanol was stirred overnight to achieve a uniform suspension. 1.6 mL of the slurry was placed onto a ZnSe internal reflection element, and a thin film was formed after the evaporation of the ethanol. In situ ATR-IR spectra were obtained using a Vertex 70v spectrometer (Bruker) equipped with a liquid nitrogen-cooled mercury-cadmium-telluride (MCT) detector (ID316, ZnSe window) and an optical filter (F321). Spectra were recorded at a spectral resolution of 4 cm−1 and a scanning velocity of 60 kHz. Modulation-excitation spectroscopy was carried out by periodically changing between two different solutions: EtOH/air ↔ EtOH/He. Five cycles were tested and averaged into one cycle to enhance the S/N ratio and time resolution. Phase-sensitive detection was used to remove the noise and to obtain kinetic information on the responding surface species. The acquired time-domain spectra were mathematically treated by PSD to obtain phase-domain spectra according to the following equation:where T is the length of a cycle, ω is the demodulation frequency, φk is the demodulation phase angle, k is the demodulation index (k = 1 in this study), and A(t,
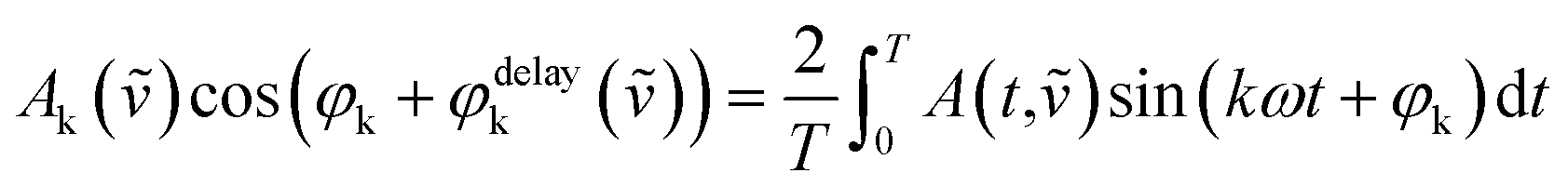
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) ) and Ak() are the active species' responses in the time and phase domains, respectively.
) and Ak() are the active species' responses in the time and phase domains, respectively.
Results and discussion
TEM analysis of the Au/NiO composite
The Au/NiO samples were prepared via the immobilization of gold colloids onto the surface of the NiO followed by annealing at 300 °C in air to remove all the organic chemicals. The STEM images show that the Au nanoparticles of 3.4 ± 0.5 nm are uniformly dispersed on the nickel oxide surface (Fig. 1a) and the gold nanoparticles are deposited on the NiO(200) plane (Fig. 1b). The prepared Au/NiO composite was subsequently investigated in the cascade reactions of cinnamaldehyde with bioethanol to generate new C–C bonds.24–26 | ||
| Fig. 1 STEM image of the Au/NiO composite. The lattice spacings of 0.23 and 0.209 nm in (b) are assigned to be the Au(111) and NiO(200) planes. The scale bars in (a) and (b) are 50 and 2 nm, respectively. | ||
Cascade catalytic reaction
The results for the cascade organic transformations of cinnamaldehyde in the presence of ethanol, O2 and K2CO3 over the gold nanoparticle catalysts are compared in Table 1. The Au/NiO catalyst exhibited a promising performance, with an 80% conversion (based on cinnamaldehyde hereafter) and 47% selectivity toward ethyl cinnamate (EC), 36% to benzylfuran (BF), 6% to ethylcinnamylideneacetate (ECDA), and 11% to cinnamylfuran (CF), Table 1, entry 1. The ester (EC and ECDA) and furan derivatives (BF and CF) are yielded through oxidative esterification and aldol condensation and Michael addition of cinnamaldehyde and 5-phenyl-2,4-pentadienal, respectively (Scheme 1). It is noteworthy that the Michael addition to generate the furan derivatives contains two steps of Robinson annulation and rearrangement.22 Interestingly, the bare NiO in the absence of gold species gave >99% selectivity toward BF at a relatively low conversion of 39% (Table 1, entry 2).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Conversionb (%) | Selectivityb (%) | ||||
| DEPB | EC | BF | ECDA | CF | ||||
| a Reaction conditions: 10 mg of 1 wt% Au catalysts, 26 μL of cinnamaldehyde, 10 mg of base, 3 mL of ethanol, 4 h, and 1 MPa air. b The cinnamaldehyde conversion and product selectivity were determined by GC-MS analysis. | ||||||||
| 1 | Au/NiO | K2CO3 | 80 | — | 47 | 36 | 6 | 11 |
| 2 | NiO | K2CO3 | 39 | — | — | >99 | — | — |
| 3 | Au/NiO | — | 68 | 100 | 8 | — | — | — |
| 4 | NiO | — | 8 | 92 | — | — | — | — |
| 5 | Au/SiO2 | K2CO3 | 83 | — | 74 | 20 | 6 | — |
| 6 | SiO2 | K2CO3 | 9 | 18 | — | 82 | — | — |
| 7 | Au/CeO2 | K2CO3 | 78 | — | 60 | 31 | 9 | — |
| 8 | CeO2 | K2CO3 | 19 | 39 | — | 61 | — | — |
| 9 | Au/TiO2 | K2CO3 | 89 | — | 89 | 8 | 3 | — |
| 10 | TiO2 | K2CO3 | 14 | 24 | — | 76 | — | — |

Furthermore, some aspects are noteworthy. First, in the absence of a base (K2CO3), the Au/NiO catalyst presents 92% selectivity for acetal production (i.e. 3,3-diethoxy-1-propen-1-ylbenzene (DEPB)) via an acetalization reaction, and the NiO showed a very low conversion of 8% and close to 100% acetal selectivity (Table 1, entries 3 and 4). These results demonstrate that the aldol condensation and Michael addition occur in basic environments (e.g., K2CO3) and the oxidative esterification is associated with gold nanoparticles. The concentration of acetaldehyde (CH3CHO) and ethyl acetate (EtOAc) products formed in the reactor system was ca. 0.11 and 0.23 wt%, respectively, which is much lower than the initial concentration of cinnamaldehyde (1.1 wt%). Of note, the acetic acid product was not detected during the GC-MS analysis.
For completion, we also evaluated the catalytic performances of the other Au/oxides (where the oxide is SiO2, CeO2 or TiO2) under the identical reaction conditions. The Au/NiO composite outperformed the Au/SiO2, Au/CeO2 and Au/TiO2 catalysts, mainly on the product selectivity (Table 1). The major product is EC (60–89% selectivity) over the Au/SiO2, Au/CeO2 and Au/TiO2 catalysts. Of note, these plain oxides of SiO2, CeO2 and TiO2 only gave the production of DEPB and BF under the identical reaction conditions (Table 1, entries 6, 8 and 10). It is noteworthy that the C13 of cinnamylfuran is only generated over the Au/NiO composite, which may be mainly due to the novel properties of the NiO support. Thus, we utilized TGA and TPD technologies (including O2-TPD, CO2-TPD and EtOH-TPD) to study the nature of the NiO and Au/NiO catalyst.
Chemo-adsorption properties of Au/NiO composite
The activity of catalysts often scales with their chemo-adsorption affinity for the reactants (O2, ethanol and cinnamaldehyde in this work), which is often deemed as the first step during catalytic reactions.27 The O2-TPD profile of the plain NiO support and the Au/NiO composite shows similar peaks at ca. 469 °C (Fig. 2a), which can be assigned to the adsorbed [O] species on the nickel oxide's surface. Of note, the lattice oxygen of NiO appears at >700 °C.22 The concentration of the [O] on the Au/NiO composite is about 1.3 times that for bare NiO, demonstrating that the presence of gold nanoparticles promotes the dissociation of oxygen molecules on the NiO. Next, it is very interesting that both the NiO and Au/NiO exhibit the same EtOH-TPD curves, demonstrating that the concentrations of chemisorbed ethanol on the NiO and Au/NiO are almost the same. Three peaks for the ethanol desorption are found in the range of 50–600 °C (Fig. 2b), which are assigned to adsorbed ethanol species at the surface of the Au/NiO. This implies that the ethanol should be selectively adsorbed on the NiO support, and then it is activated over the NiO with the active oxygen species.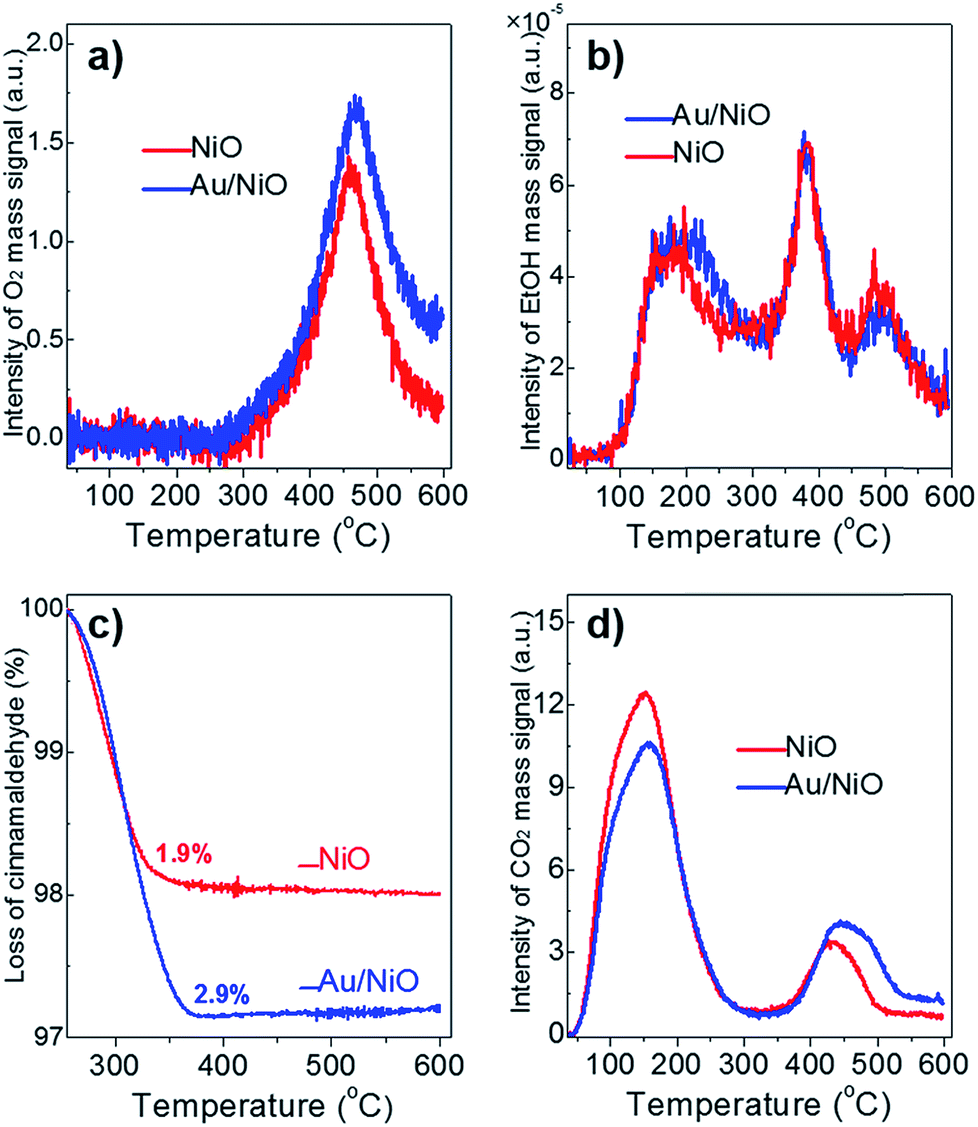 | ||
| Fig. 2 (a) O2-TPD, (b) EtOH-TPD, and (c) TGA analysis of the chemisorbed cinnamaldehyde, and (d) CO2-TPD over the free NiO (red lines) and the Au/NiO composite (blue lines). | ||
Next, TGA was exploited to measure the amount of cinnamaldehyde that was adsorbed onto the catalyst surface. Fig. 2c clearly indicates that the Au/NiO adsorbs more cinnamaldehyde in comparison to the bare NiO support (2.9 wt% for Au/NiO vs. 1.9 wt% for NiO), implying that the introduction of Au particles indeed largely improves the adsorption capacity of cinnamaldehyde, which is related to the cinnamaldehyde conversion rate discussed above. Further, the CO2-TPD results suggest that the concentration of weak base sites (associated with the peak at ∼155 °C) decreases and the concentration of the medium basic site improved (peaks at 430–455 °C) (Fig. 2d), implying that the gold NPs should be immobilized onto the weak basic sites. Taking these results of the chemo-adsorption into consideration, we deduced the aldehydes prefer to convert over the gold particles and ethanol should be reacted on the NiO support close to the gold species.
XPS analysis
Next, XPS technology was applied to study the chemical state of the surface oxygen of NiO. Only the Ni(II) species is observed in the Ni 2p XPS spectrum as the two peaks located at BE of 873.1 and 871.5 eV, corresponding to the Ni 2p1/2 of the Ni(OH)2 and the NiO, respectively (Fig. 3a). Of note, the peak at 879.7 eV is assigned to the satellites. Upon deconvolution of the O 1s peak, three O species are identified at the BE of 531.6 eV, 531.0 eV and 529.4 eV, which are attributed to the chemisorbed oxygen (OC), surface oxygen vacancies (OV), and lattice oxygen (OL), respectively (Fig. 3b). The Au/NiO composite exhibited high concentrations of surface OV (28.6%) and OC (18.2%), which can promote the activation of oxygen molecules and then the cascade aldol condensation reactions.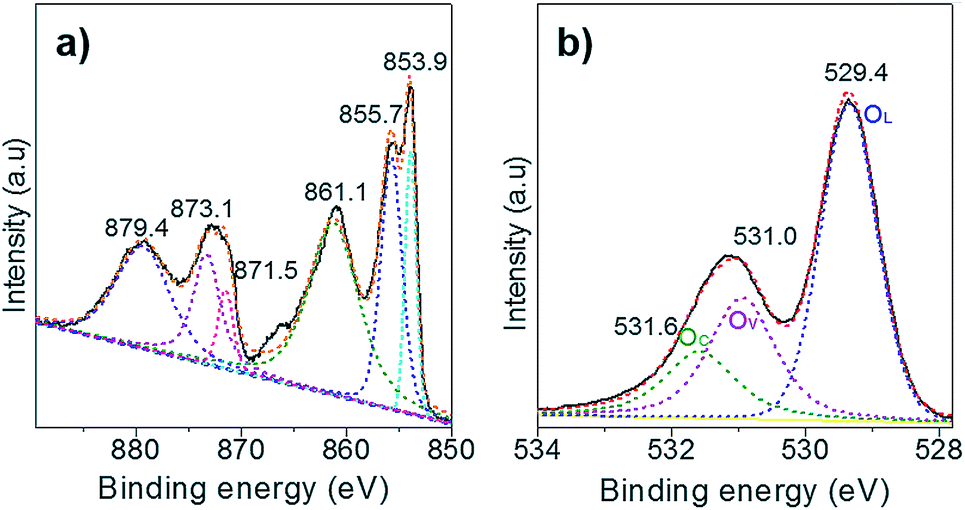 | ||
| Fig. 3 (a) Ni 2p and (b) O 1s XPS spectra of NiO support. | ||
In situ ATR-IR investigation
To support the speculation, an in situ infrared spectroscopy investigation was carried out. Fig. 4 presents the IR spectra obtained by modulation-excitation spectroscopy over the Au/NiO composite and neat NiO. Ethanol is adsorbed on the NiO surface to form ethoxy species via cleavage of the O–H bond of ethanol, evidenced by the observation of the C–O bond of ethoxy at 1067, 1019 and 1006 cm−1.28 Further, the formed ethoxy species were dissociated in the presence of oxygen, supported by the negative bands that appeared at 1097 cm−1 (attributed to ethoxy C–C bond cleavage) and 2973, 2924, 2873 cm−1 of the C–H stretching region.29 The acetaldehyde species were yielded, evidenced by a new band appearing at 1261 cm−1. Then the final over-oxidized acetate species were confirmed by the appearance of IR bands at 1376, 1338 and 1275 cm−1, presenting the stretch of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O of acetate species.30 Acetic and formic acids are not detected during the in situ infrared spectroscopic analyses. It is noteworthy that the neat NiO and Au/NiO catalyst exhibit very similar signals. Moreover, the ethoxy formation over the Au/NiO composite is more significant than that over the bare NiO, as evidenced by the peak area and intensity of the ethoxy C–O bond. Furthermore, the characteristic acetaldehyde band at 1261 cm−1 is stronger in the case of Au/NiO as compared to NiO, ensuring more dissociation of the ethoxy intermediate and the conversion to CH3CHO* species. These results give the below mechanistic framework for ethanol conversion on the supported catalyst and specifically show the competing pathways involving two key intermediates (ethoxy and acetate).
C–O of acetate species.30 Acetic and formic acids are not detected during the in situ infrared spectroscopic analyses. It is noteworthy that the neat NiO and Au/NiO catalyst exhibit very similar signals. Moreover, the ethoxy formation over the Au/NiO composite is more significant than that over the bare NiO, as evidenced by the peak area and intensity of the ethoxy C–O bond. Furthermore, the characteristic acetaldehyde band at 1261 cm−1 is stronger in the case of Au/NiO as compared to NiO, ensuring more dissociation of the ethoxy intermediate and the conversion to CH3CHO* species. These results give the below mechanistic framework for ethanol conversion on the supported catalyst and specifically show the competing pathways involving two key intermediates (ethoxy and acetate).
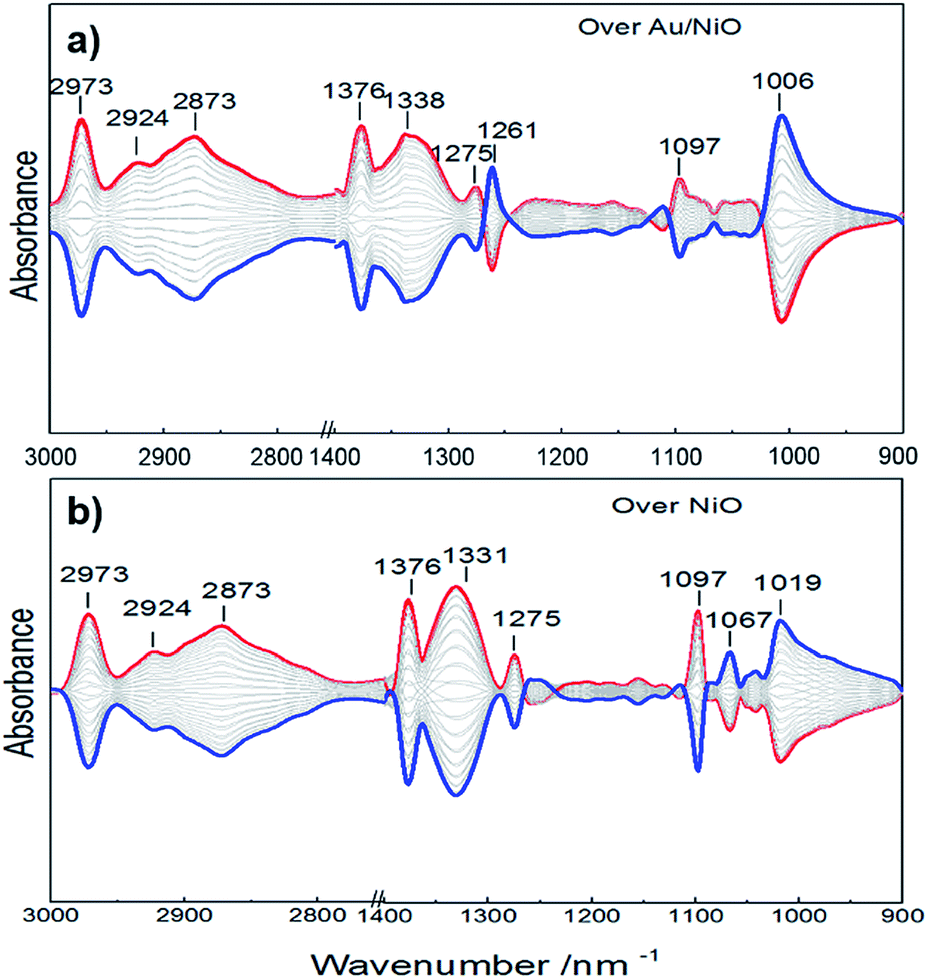 | ||
| Fig. 4 In situ ATR-IR spectra of (a) Au/NiO composite and (b) bare NiO in the presence of ethanol and O2 at 75 °C. The formed and consumed species emerge as positive and negative IR bands, respectively. | ||
Catalytic mechanism
Based on the catalytic results and the in situ infrared spectroscopy investigation, the whole reaction pathway of ethanol and cinnamaldehyde over the Au/oxides is surmised in Scheme 2. The O2 adsorption and activation occur on the surface oxygen vacancies (OV) on the oxide support.31 Then the adsorbed ethanol reacts with the activated [O] species to form the acetaldehyde (CH3CHO*) on the surface of the NiO itself, supported by the EtOH-TPD tests. The generated CH3CHO* species can react with cinnamaldehyde on the NiO surface to give benzylfuran and also further migrates to the interfacial perimeter site of gold nanoparticles and the oxides for the cascade aldol condensation. Finally, a series of products are yielded through the cascade aldol condensation and Michael addition processes; the cinnamylfuran product especially can only be generated over the NiO-supported gold nanoparticle catalyst (Table 1). | ||
| Scheme 2 The catalytic pathway of the cascade aldol condensation and Michael addition of ethanol and cinnamaldehyde in the presence of oxygen. Au, orange; O, light yellow; Ni, cyan. | ||
Hydrodeoxygenation
The gold nanoparticles have been well exploited as a novel catalyst in the hydrogenation reactions.32–35 Finally, we tried to reduce the produced oxygenates (e.g., EC, 2-benzylfuran, ethyl-5-phenyl-2,4-pentadienoate, and 2-cinnamylfuran) via a one-pot hydrodeoxygenation. The oxygenates were fully (i.e., 100%) converted to C7H16 and C9H20 hydrocarbons and ethyl ester when the atmosphere was changed to H2 (1 MPa) at 120 °C. The unsaturated esters of EC and ethyl-5-phenyl-2,4-pentadienoate are hydrogenated to saturated esters (i.e., PhC2H4COOC2H5 and PhC4H8COOC2H5, Scheme 1) after the completion of the reaction, implying that the unsaturated esters cannot be fully hydrogenated over the Au/NiO catalyst to give alkylbenzene hydrocarbon products. It is interesting that only the furan groups of 2-benzylfuran and 2-cinnamylfuran were reduced to phenylpentane and heptenylbenzene during the hydrogenolysis process when using Au/TiO2 as the catalyst for comparison under identical reaction conditions. These catalytic results demonstrate that the high performance of the hydrogenolysis process over the Au/NiO catalyst may be caused by the synergistic effect of the (AuNi)0 species at the interfacial perimeter of the gold particles and NiO support, as the reduction of Ni(II) to Ni(0) can be largely promoted by gold nanoparticles at relatively low temperatures of 100–150 °C.36,37Conclusions
In conclusion, the Au/NiO composite catalyst shows promising activity in the aerobic ethanol oxidation with cinnamaldehyde and finally gives rise to the production of alkylbenzene derivatives of pentylbenzene and heptylbenzene. 70% alkylbenzene selectivity was obtained via a one-pot cascade reaction, viz. the cross-aldol condensations in the presence of a base and then the hydrodeoxygenation with H2 gas. In situ ATR-IR analyses demonstrated that a key intermediate of acetaldehyde (CH3CHO*) is formed by the activation of ethanol on the surface oxygen vacancies of the NiO support, and then it reacts with cinnamaldehyde at the interfacial perimeter of the Au/NiO composite during the cascade reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Liaoning Revitalization Talents Program (XLYC1807121), the Postdoctoral Science Foundation of China (223232), the Natural Science Foundation of Inner Mongolia Autonomous Region (2018BS02004), BL14B and BL17B beamline of National Facility for Protein Science (NFPS), Shanghai Synchrotron Radiation Facility (SSRF) Shanghai, China for providing the beam time.Notes and references
- H. B. Goyal, D. Seal and R. C. Saxena, Renewable Sustainable Energy Rev., 2008, 12, 504–517 CrossRef CAS.
- M. I. Jahirul, M. G. Rasul, A. A. Chowdhury and N. Ashwath, Energies, 2012, 5, 4952–5001 CrossRef CAS.
- A. V. Bridgwater, Chem. Eng. J., 2003, 91, 87–102 CrossRef CAS.
- C. Zhao, T. Brueck and J. A. Lercher, Green Chem., 2013, 15, 1720–1739 RSC.
- A. N. Oumer, M. M. Hasan, A. T. Baheta, R. Mamat and A. A. Abdullah, Renewable Sustainable Energy Rev., 2018, 88, 82–98 CrossRef CAS.
- A. D. Martin, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- M. Balat and H. Balat, Appl. Energy, 2009, 86, 2273–2282 CrossRef CAS.
- S. P. S. Badwal, S. Giddey and A. Kulkarni, Appl. Energy, 2015, 145, 80–103 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- A. V. Subrahmanyam, S. Thayumanavan and G. W. Huber, ChemSusChem, 2010, 3, 1158–1161 CrossRef CAS PubMed.
- S. Guo, Q. Fang, Z. Li, J. Zhang, J. Zhang and G. Li, Nanoscale, 2019, 11, 1326–1334 RSC.
- W. Xu, X. Liu, J. Ren, H. Liu, Y. Ma, Y. Wang and G. Lu, Microporous Mesoporous Mater., 2011, 142, 251–257 CrossRef CAS.
- P. A. Zapata, J. Faria, M. P. Ruiz and D. E. Resasco, Top. Catal., 2012, 55, 38–52 CrossRef CAS.
- G. Li, W. Jiao, Z. Sun, Y. Zhao, Z. Shi, Y. Yan, L. Feng, Y. Zhang and Y. Tang, ACS Sustainable Chem. Eng., 2018, 6, 4316–4320 CrossRef CAS.
- Z. Liu, X. Tong, J. Liu and S. Xue, Catal. Sci. Technol., 2016, 6, 1214–1221 RSC.
- L. Ning, S. Liao, H. Cui, L. Yu and X. Tong, ACS Sustainable Chem. Eng., 2018, 6, 135–142 CrossRef CAS.
- G. Zhang, R. Wang and G. Li, Chin. Chem. Lett., 2018, 29, 687–693 CrossRef CAS.
- V. C. Prabhakaran, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2011, 86, 161–171 CrossRef.
- C. Zhang, Y. Chen, H. Wang, Z. Li, K. Zheng, S. Li and G. Li, Nano Res., 2018, 11, 2139–2148 CrossRef CAS.
- T. Ishida and M. Haruta, ChemSusChem, 2009, 2, 538–541 CrossRef CAS PubMed.
- S. K. Klitgaard, K. Egelblad, U. V. Mentzel, A. G. Popov, T. Jensen, E. Taarning, I. S. Nielsen and C. H. Christensen, Green Chem., 2008, 10, 419–423 RSC.
- Q. Fang, Z. Qin, Y. Shi, F. Liu, S. Barkaoui, H. Abroshan and G. Li, ACS Appl. Energy Mater., 2019, 2, 2654–2661 CrossRef CAS.
- H. Chen, Z. Li, Z. Qin, H. Kim, H. Abroshan and G. Li, ACS Appl. Nano Mater., 2019, 2, 2999–3006 CrossRef CAS.
- Y. Zhou and G. Li, Acta Phys.-Chim. Sin., 2017, 33, 1297–1309 CAS.
- G. Li and R. Jin, Nanotechnol. Rev., 2013, 5, 529–545 Search PubMed.
- J. Lin, H. Abroshan, C. Liu, M. Zhu, G. Li and M. Haruta, J. Catal., 2015, 330, 354–361 CrossRef CAS.
- E. J. Evans, H. Li, W. Y. Yu, G. M. Mullen, G. Henkelman and C. B. Mullins, Phys. Chem. Chem. Phys., 2017, 19, 30578–30589 RSC.
- A. Gazsi, A. Koós, T. Bánsági and F. Solymosi, Catal. Today, 2011, 160, 70–78 CrossRef CAS.
- T. Kratochwil, M. Wittmann and J. Küppers, J. Electron Spectrosc. Relat. Phenom., 1993, 64, 609–617 CrossRef.
- X. Liu, B. Xu, J. Haubrich, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2009, 131, 5757–5759 CrossRef CAS PubMed.
- D. Widmann and R. J. Behm, Acc. Chem. Res., 2014, 47, 740–749 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- Y. Zhang, X. Yang, Y. Zhou, G. Li, Z. Li, C. Liu, M. Bao and W. Shen, Nanoscale, 2016, 8, 18626–18629 RSC.
- C. Liu, H. Abroshan, C. Yan, G. Li and M. Haruta, ACS Catal., 2016, 6, 92–99 CrossRef CAS.
- H. Nishikawa, D. Kawamoto, Y. Yamamoto, T. Ishida, H. Ohashi, T. Akita, T. Honma, H. Oji, Y. Kobayashi, A. Hamasaki, T. Yokoyama and M. Tokunaga, J. Catal., 2013, 307, 254–264 CrossRef CAS.
- X. Xu, Q. Fu, X. Guo and X. Bao, ACS Catal., 2013, 3, 1810–1818 CrossRef CAS.
Footnote |
| † Y. S. and S. T. contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |