
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrolyte selection for supercapacitive devices: a critical review
Bhupender
Pal
a,
Shengyuan
Yang
 b,
Subramaniam
Ramesh
c,
Venkataraman
Thangadurai
d and
Rajan
Jose
*a
b,
Subramaniam
Ramesh
c,
Venkataraman
Thangadurai
d and
Rajan
Jose
*a
aNanostructured Renewable Energy Materials Laboratory, Faculty of Industrial Sciences and Technology, Universiti Malaysia Pahang, 26300 Gambang Kuantan, Malaysia. E-mail: bhupender@ump.edu.my; rjose@ump.edu.my.
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, International Joint Laboratory for Advanced Fiber and Low-dimension Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China
cCentre for Ionics University of Malaya, Department of Physics, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia
dDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, AB, Canada
First published on 27th August 2019
Abstract
Electrolytes are one of the vital constituents of electrochemical energy storage devices and their physical and chemical properties play an important role in these devices' performance, including capacity, power density, rate performance, cyclability and safety. This article reviews the current state of understanding of the electrode–electrolyte interaction in supercapacitors and battery–supercapacitor hybrid devices. The article discusses factors that affect the overall performance of the devices such as the ionic conductivity, mobility, diffusion coefficient, radius of bare and hydrated spheres, ion solvation, viscosity, dielectric constant, electrochemical stability, thermal stability and dispersion interaction. The requirements needed to design better electrolytes and the challenges that still need to be addressed for building better supercapacitive devices for the competitive energy storage market have also been highlighted.
1. Introduction
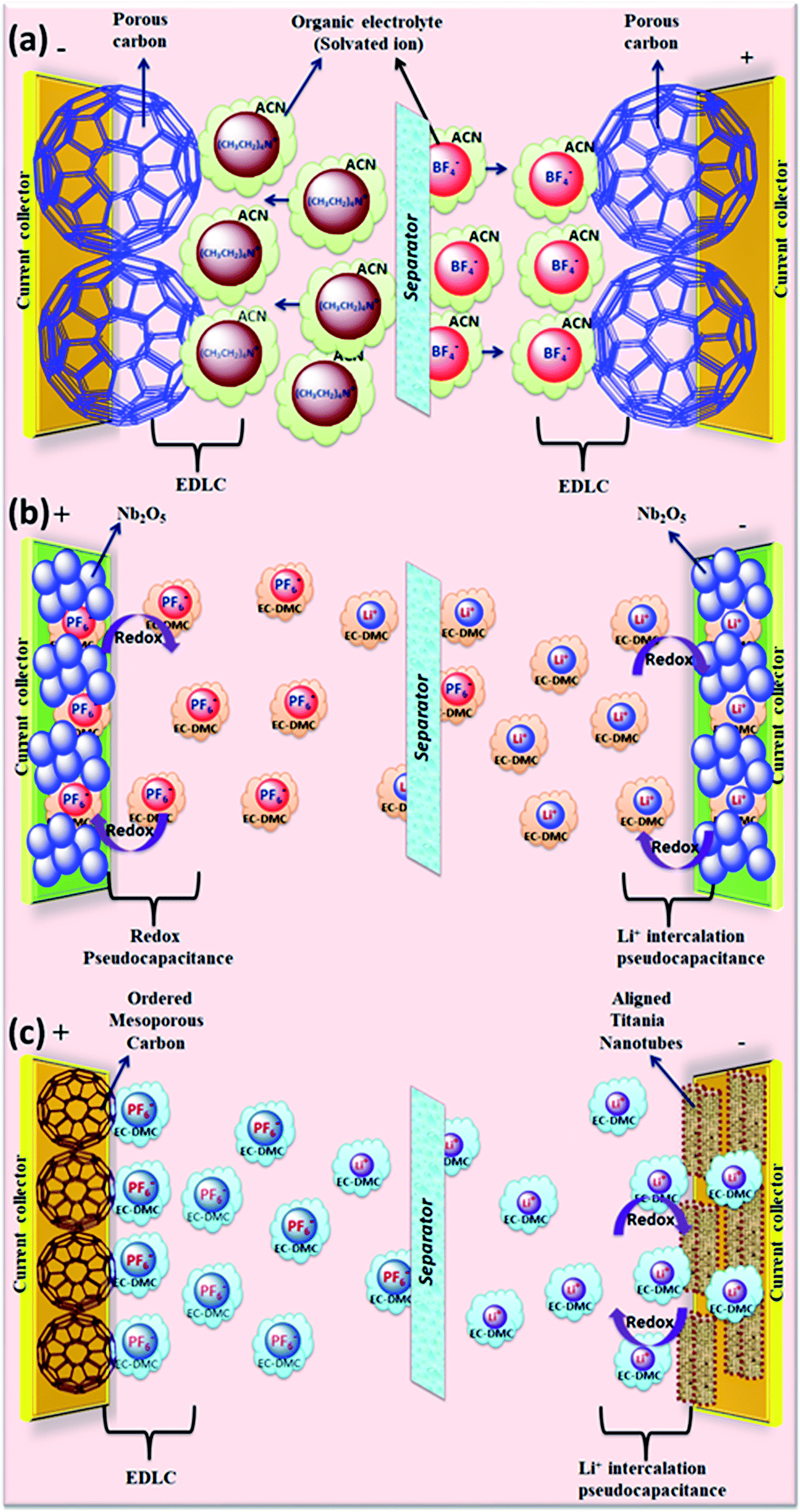
The rapid growth of the global economy, urbanization, and significant increase in the quality of living standards induce some major issues worldwide, such as accelerated depletion of fossil fuel reserves and life-threatening environmental hazards (e.g. increased greenhouse gas emission and water and air pollution). As a result, globally, zero-emission and renewable energy technologies are given paramount importance. High performance electrochemical energy storage (EES) materials1 and devices are intensively researched. In this regard, EES devices such as batteries2–4 and supercapacitors5 are the “first choice” due to their high efficiency, stability, and environmental friendliness.6–8 Electrochemical capacitors (synonymously, supercapacitors or ultracapacitors) are a preferred choice for applications demanding high power (such as electric vehicles, satellites, robots etc.) due to their high specific power (PS > 10 kW kg−1), fast charge/discharge capability (within seconds) and long cycling stability (>105 cycles) as compared to batteries.9–11Supercapacitor devices (SCDs) are mainly classified into three categories based on their charge storage mechanism: (i) electric double-layer capacitors (EDLCs), (ii) pseudocapacitors, and (iii) hybrid electrodes5,12 (Fig. 1).13,14 The mechanisms of capacitive energy storage in these devices are shown in Fig. 2. EDLCs store physically charges (physisorption) via the formation of an electric double layer (EDL) at the interface between the electrode and electrolyte (Fig. 2(a and b)).15–17 Traditionally, various allotropes of carbon with high porosity and surface area are the preferred choice as the EDLC electrode owing to their high electrical conductivity. The double layer here is due to adsorption of negative ions from the electrolyte on the positively charged electrode and vice versa. The fast diffusion of ions in the electrolyte to the electrode surface thereby achieved is responsible for their high power density (PS). However, EDLCs have relatively low specific energy (ES) compared with batteries. By contrast, pseudocapacitors can store charge not only via the formation of an EDL but also via reversible oxidation–reduction (redox) reactions (Fig. 2(c)) with fast insertion of the electrolytes on the surface of the electrode.18–20 Pseudocapacitive mechanisms include redox pseudocapacitance (Fig. 2(c)), as occurring in metal oxides, and intercalation pseudocapacitance, where Li+ ions are inserted into the host material as shown in Fig. 2(d).21 While the charge intercalation is surface limited in pseudocapacitive electrodes, the intercalation is deep and diffusion limited in battery-type electrodes and consequently, battery-type electrodes show that highest ES among the three types of electrodes.22,23
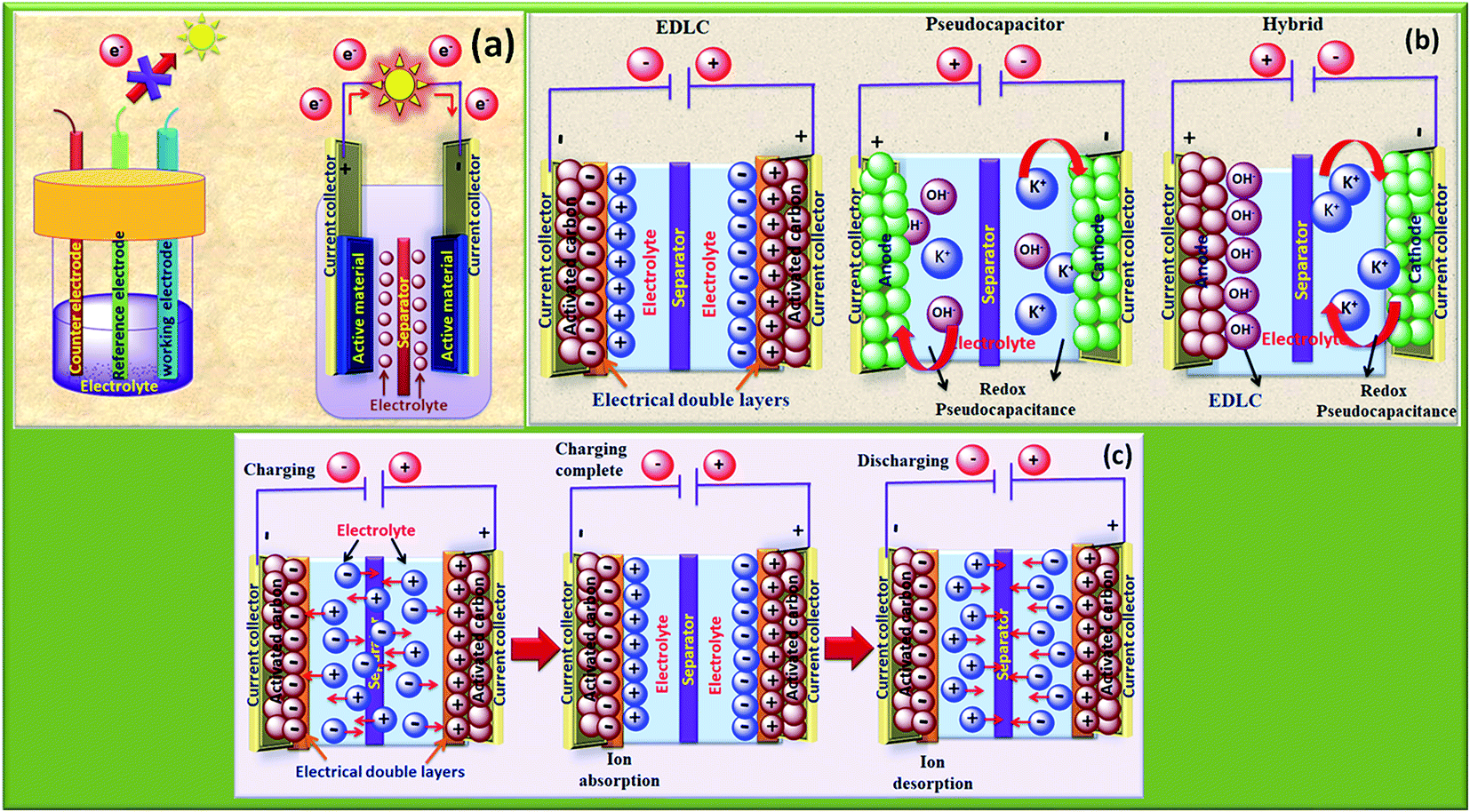 | ||
| Fig. 1 (a) Schematics of the three-electrode configuration and two-electrode configuration, (b) type of supercapacitors: EDLC, pseudocapacitor and hybrid, (c) charge storage mechanism in EDLCs, ion absorption and desorption on the surface. | ||
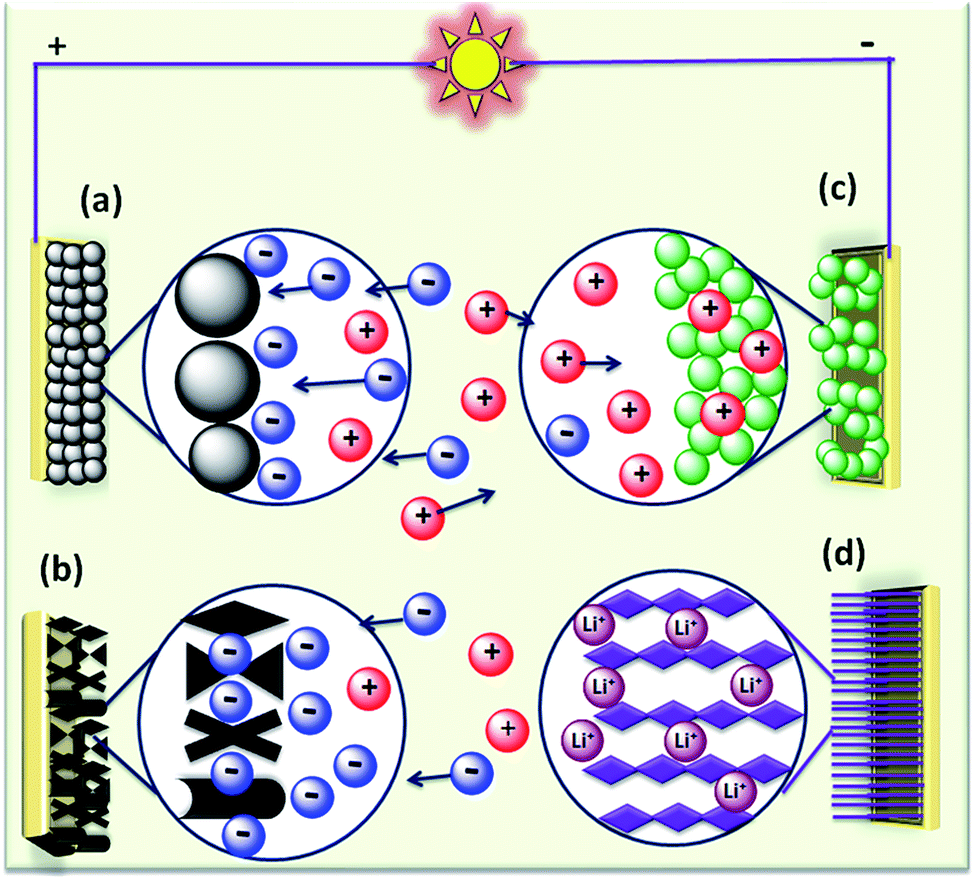 | ||
| Fig. 2 (a–d) Mechanisms of capacitive energy storage by double-layer capacitance developed at electrodes comprising (a) carbon particles and (b) porous carbon. Pseudocapacitive mechanisms include (c) redox pseudocapacitance, as in hydrous metal oxides, and (d) intercalation pseudocapacitance, where Li+ ions are inserted into the host material. | ||
Among SCDs, EDLCs are matured technologically and being used in commercial applications. However, these supercapacitors have an order of magnitude lower specific energy (ES ≤ 10 W h kg−1) than batteries (ES ∼ 200–250 W h kg−1).24,25 Research to increase the performance of supercapacitive devices to similar values to those of batteries showed impressive progress, and an ES as high as ∼100 W h kg−1 has been reported for them.26ES is expressed as 1/2CV2, where C is the capacitance and V is the voltage of the devices; ES could be increased by increasing the C (which is determined by the amount of charge stored by the electrode) and voltage (V) (defined as the difference in electrochemical potentials between the electrodes).27–29 Consequently, intensive research has been undertaken worldwide for the development of highly capacitive electrode materials (for high C) and electrolytes of lower electrochemical potential (for high V) or both.30,31Fig. 3 shows the statistics of publications on electrodes and electrolytes as well as their regional distribution. The majority of the studies were concentrated on developing EDLC32 electrodes; the research on electrolytes was the next major activity. Research on battery–supercapacitor devices,5 in which a battery-type electrode or pseudocapacitive electrode is used as the positive electrode and an EDLC electrode is used as the negative electrode, and pseudocapacitor electrodes were the next major activities. Fiber-shaped materials for supercapacitors33,34 have attracted a huge amount of attention and were utilized as effective energy storage devices because of their extended cycling life, high power density, rapid charge/discharge capability and excellent flexibility.35–38
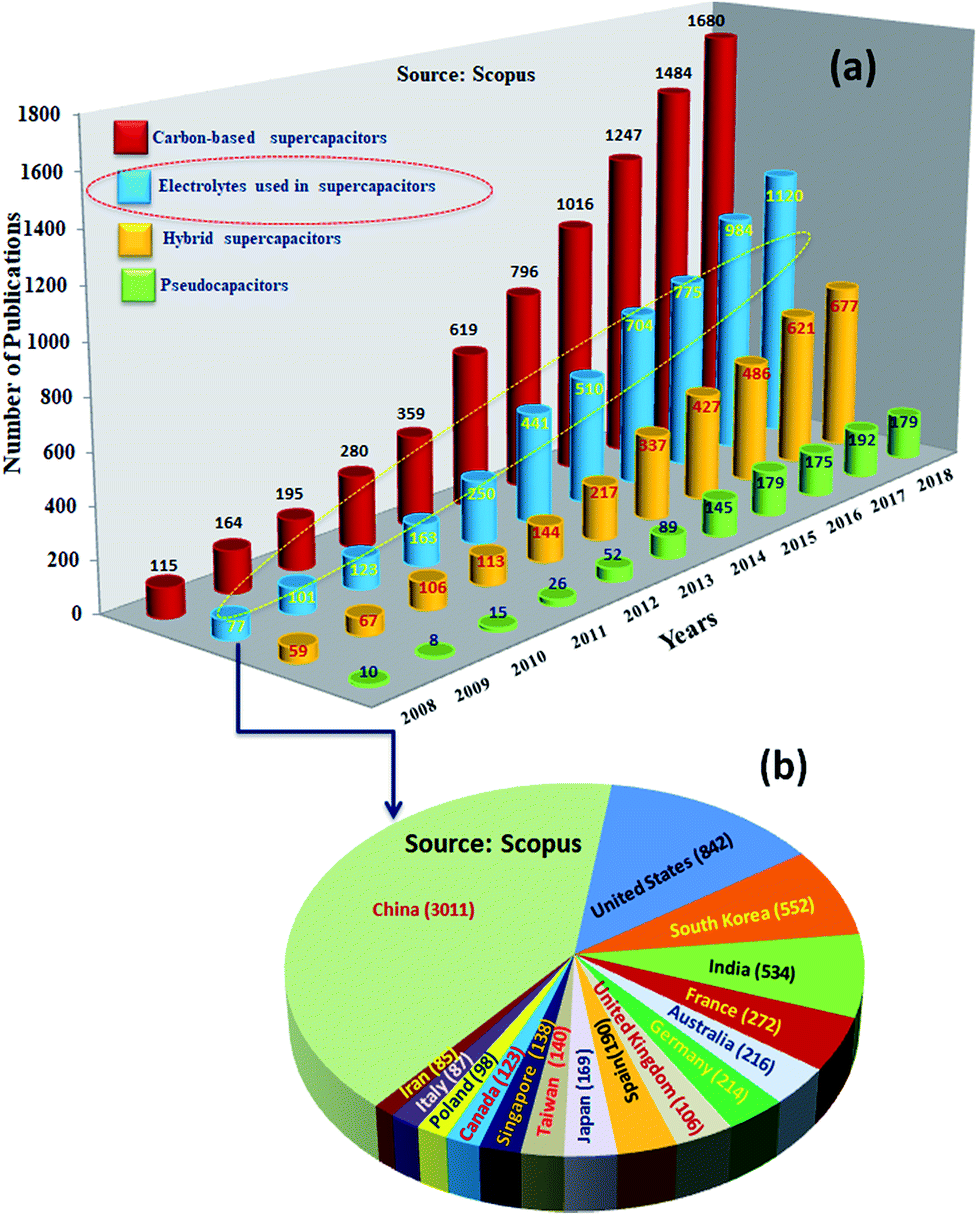 | ||
| Fig. 3 (a) Total number of reported literature studies related to carbon-based supercapacitors, electrolytes for supercapacitors, hybrid supercapacitors and pseudocapacitors from 2008 to 2018 and (b) countrywise distribution of publications related to electrolytes for supercapacitors (shown within brackets). Search formulation: (“carbon-based supercapacitors*, electrolytes for supercapacitors*, hybrid supercapacitors* and pseudocapacitors*”); search from Scopus; search date: 17, May, 2019. | ||
The electrolyte is an essential and significant constituent in supercapacitors and plays a very crucial role in transferring and balancing charges between the two electrodes.39 The electrolytes for electrochemical supercapacitors are classified into various categories (Fig. 4). The interaction between the electrolyte and electrodes in all electrochemical processes significantly influences the electrode–electrolyte interface state and internal structure of active materials. The choice of electrolyte is the key to safe and high performance supercapacitive devices. To date, there has been no perfect electrolyte meeting all the requirements for electrochemical devices. For example, supercapacitive devices using aqueous electrolytes possess both high conductivity and capacitance,40 but a low energy density, cycling stability and leakage problem.41 Although organic electrolytes and ionic liquids (ILs) can operate at higher voltages, and these normally suffer from much lower ionic conductivity.42 Solid-state electrolytes may avoid the potential leakage problem of liquid electrolytes, but also suffer from low ionic conductivity.43 Non-flammable, wide electrochemical stability windows and moisture tolerance of highly concentrated “water in salt”44 electrolytes are making them promising electrolytes for energy storage devices (Fig. 4). However, their high viscosity, low conductivity and salt precipitation at low temperature affect the performance of energy storage devices. Dou et al.45 introduced acetonitrile as a co-solvent in “water in salt” electrolytes to formulate a hybrid electrolyte that provides significantly reduced viscosity, improved conductivity and expanded applicable temperature range. Therefore, the rapid development and application of electrode materials depends on finding compatible electrolytes. Hence, the reaction mechanism, design, and development of electrolytes occupy a crucial position for developing safe and efficient supercapacitive devices. Few reviews have been published on electrolytes for electrochemical supercapacitors.46–49 However, progress in designing electrolytes considering their electrochemical interaction with the electrode material in determining a supercapacitive device's performance indicators such as achievable capacitance (amount of charge stored), voltage window, charge storage time constants, characteristic resistances, rate performance, cyclability, and safety has not been reviewed.
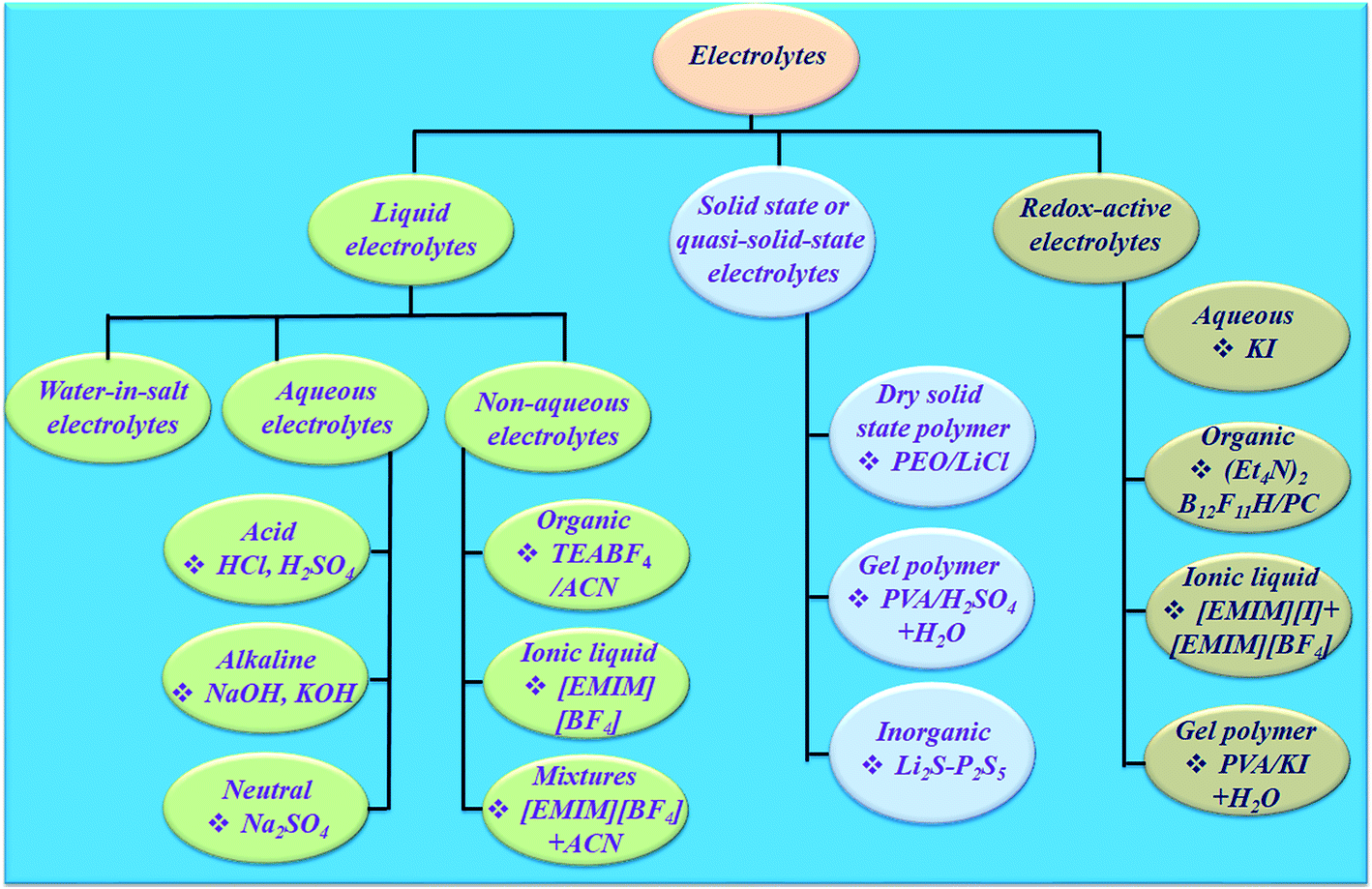 | ||
| Fig. 4 Classification of electrolytes for electrochemical supercapacitors. | ||
A comprehensive understanding of the electrode–electrolyte interaction and the factors affecting the overall performance of SCDs such as ionic conductivity and mobility, diffusion coefficient, radius of hydrated spheres, solvation, dielectric constant, electrochemical stability, thermal stability, and dispersion interaction is the primary objective of this review. Consequently, this article is divided into six main sections (including this section of introduction). In the next section (section #2), requirements for an ideal electrolyte for SCDs are defined; section #3 explains conductivity of state-of-the-art electrolytes for SCDs; a comprehensive review of the role of cations and anions of various electrolytes (aqueous, organic, ionic liquids, solid and redox electrolytes) in charge storage for SCDs is detailed in section #4; section #5 summarizes challenges of electrochemical supercapacitor electrolytes, and section #6 concludes the article by providing a brief summary and outlook for future endeavors.
2. Requirements for designing electrolytes for SCDs
2.1 Electrolyte conductivity
Electrolytes are the key for supercapacitive devices to have a high power and energy density, long cycling life, and safety. Ionic mobility and conductivity are the important factors to evaluate the performance of electrolytes. The conductivity of aqueous electrolytes is generally high as compared to that of non-aqueous and solid electrolytes due to the low dynamic viscosity of aqueous electrolytes. The conductivity (σ) of species (i) is basically related to the ionic mobility (μi), concentration of charge carriers (ni), elementary charge (e) and magnitude of valence of the mobile ion charges (zi), i.e.,39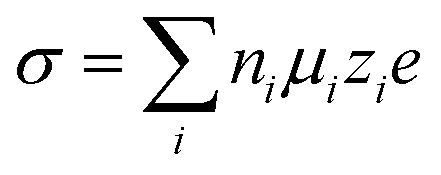 | (1) |
The variables in the above equation depend on the solvation effect, migration of the solvated ion, and lattice energy of the salt. Thus, all the constituents including solvents, additives, and salts will affect the conductivity of the liquid/gel electrolytes.
2.2 Salt effect
The conductivity can be different for different salts in the same solvent due to the interaction between the anion and cation and the volume difference of various anions.50–52 The conductivity also varied with the salt concentration in the same system. The number of free ions is dominant at a low concentration of the salt, so the optimum concentration of the salt can also increase the ionic conductivity.53 When there is balance between viscosity and free ions, the conductivity will also be very high. If the salt concentration is very high in the solvent, the anions and cations in the solvent will associate strongly with neutral ions, which reduces the number of free ions, thus decreasing the electrolyte conductivity. However, the conductivity usually decreases monotonically with an increase of the solution concentration in a high-viscosity ionic liquid system.54–572.3 Solvent effect
The solvent constituents greatly affect the conductivity of liquid/gel electrolytes. The two main properties of solvents which directly affect the conductivity of the electrolyte are the dielectric constant and viscosity because dielectric constant decides the dissociation of the salts and viscosity decides the ionic mobility of the salt.58 The literature data showed that the solvent with high viscosity also has a high dielectric constant.59 Therefore, the common method to find a suitable solvent system for supercapacitive devices is to mix a solvent with low viscosity and a high dielectric constant. Various research groups have done a systematic study of electrolyte conductivity with different solvents for supercapacitive devices;60–64 these studies show that single-component solvent-based electrolytes had much lower conductivity than binary solvent-based electrolytes. Therefore, the mixing of a high dielectric constant solvent such as ethylene carbonate, which dominates the dissociation of salts, with a low viscosity solvent (for e.g. dimethyl carbonate) will enhance the ionic conductivity. A computational-experimental approach to investigate the kinetic and thermodynamic factors for a single-component solvent and a binary solvent was conducted by Kamath et al.58 and they identified that the mixtures of ethylene carbonate–dimethyl carbonate and ethylene carbonate–ethyl methyl carbonate (which possess kinetic advantages) were the best formulations for high-power sodium-ion batteries.2.4 Electrochemical stability
Electrochemical stability is strongly related to safety and cycling life of SCDs. The upper and lower limits of the redox reactions of the electrolytes are the expression of the electrochemical stability window. The electrochemical stability not only depends on the constituents of the electrolytes but also depends on the compatibility of the electrolyte with the electrode. Linear sweep voltammetry and cyclic voltammetry are the most frequently used measurement techniques, despite some changes of the testing results when using many test systems.39 A systematic study on the electrochemical stability of electrolytes at various temperatures is important. The specific decomposition potentials of some significant electrolytes are summarized in the literature;65–70 these studies have shown that solvent systems, type of salt, and working electrodes all affect the electrochemical stability window.2.5 Thermal stability
Some electrolytes are prone to decomposition during the charging–discharging process, which is connected with the rise in operating temperature and heat release, and this rise in temperature brings about safety problems for the SCDs. Hence, the thermal stability of the supercapacitor electrolyte is a crucial parameter to study. The thermal stability of SCDs depends upon two aspects: the interaction of the electrolyte and electrode with each other and the thermal stability of the electrolyte itself. The thermal stability of an electrolyte is mainly determined by its composition: salt, solvent, and any additive. It is often analyzed by TGA/DSC studies.71–783. Conductivity of state-of-the art electrolytes for electrochemical supercapacitors
The concentration of free charge carriers (cations and anions) and ionic mobilities are the principal factors which affect the conductance of electrolytes. These two factors further depend upon the solubility of the salt in the solvent and its degree of dissociation “α” or pairing of the ions of the dissolved salt. There are three stages of ion pairing as shown in Fig. 5(a–c). Other factors are viscosity of the solvent and long-range electrostatic interactions between free or dissociated ions, which are determined by the dielectric constant of the solvent.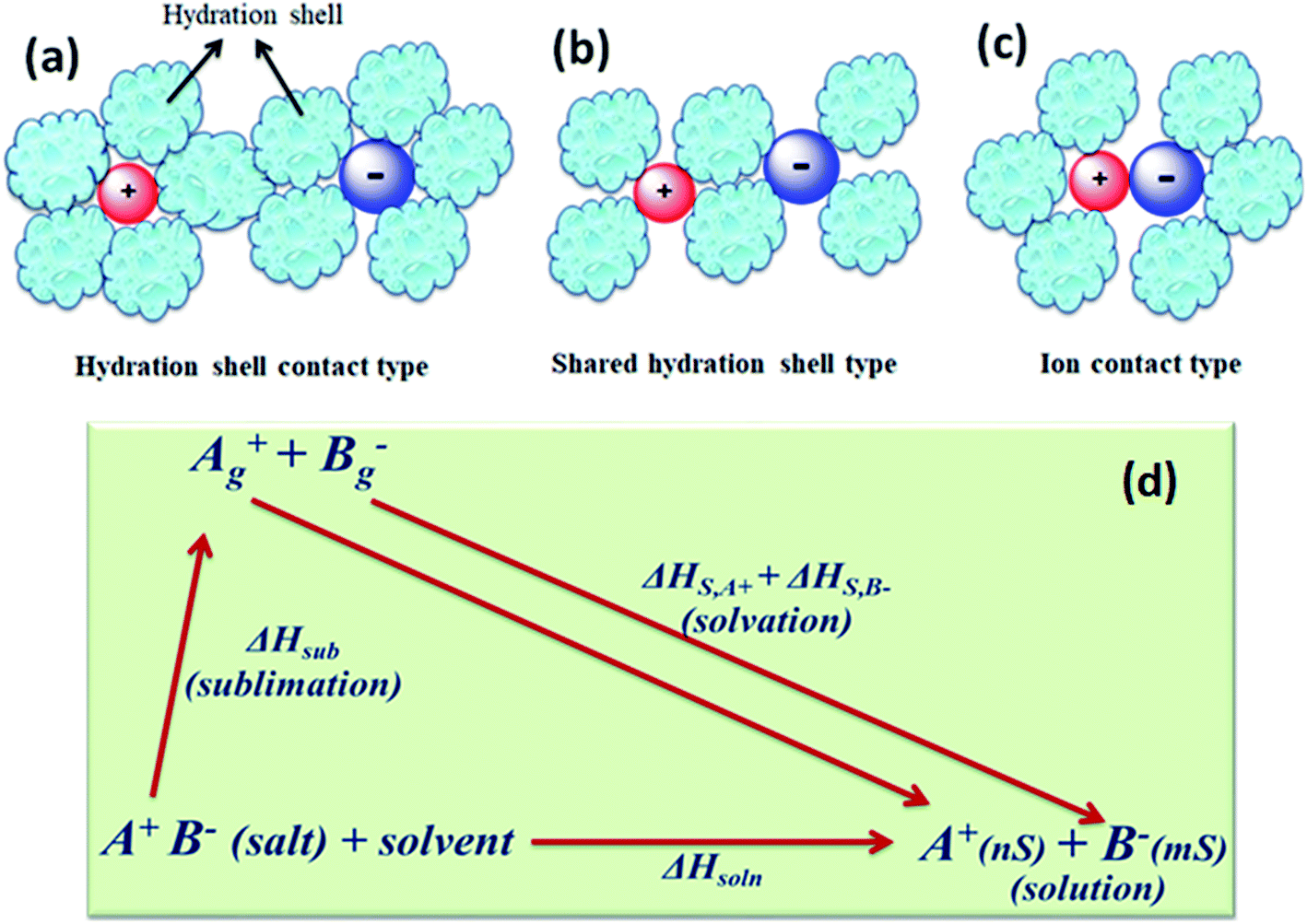 | ||
| Fig. 5 Types of ion pairs in an electrolyte solution: (a) solvated-ion pairs, (b) solvent shared ion pairs, and (c) contact ion pairs. (d) Thermodynamic cycle involving the dissolution of the salt. | ||
3.1 Electrolyte conductance and dissociation
The specific conductance (k) of a solution depends on the ion mobility and the number of conducting ions per cubic centimeter; therefore k is related to the ionic concentration. An electrochemically well-suited electrolyte salt (strongly soluble in the solvent, minimum ion pairing in the dissolved state and maximum free dissociated ion mobility) can achieve the best performance for supercapacitors by minimizing the internal resistance. The dissociation of any salt AB with concentration c into free ions can be determined by the equilibrium: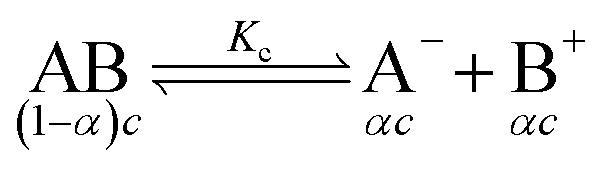 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt):
:1 salt):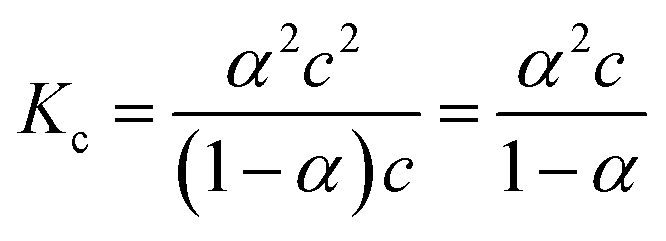 | (3) |
3.2 Mobility of the free ions
The free ion movement under an electric field E in a solvent is treated as a classical hydrodynamic relation (Stokes' law).| v/E = ze/6πηri(s) | (4) |
It involves the solvent viscosity, η (force on ion = zeE = hydrodynamic resistance (6πηri(s))v, where ri(s) is the solvated ion radius and v is the ion velocity under field E, v/E is the ion mobility per unit field). Eqn (4) predicts that (v/E) × η should be a constant for ion mobility in various solvents (having different viscosities). Eqn (4) shows that a low-viscosity solvent offers better conductance of an electrolyte than a more viscous solvent, though this is not always the case, because the viscosity is also associated with the dipole moment and molecular interaction of the solvent. These two factors determine the dielectric constant (ε) of the solvent: a higher dielectric constant reduces ion pairing and improves the conductivity of a given salt. Hence, although a lower viscosity improves the actual mobility of the free ions, the lower dielectric constant that is usually associated with low viscosity solvents (because of weak intermolecular interactions) tends to diminish the conductivity. Therefore, the use of mixed solvents often achieves optimization of conductance.
3.3 Role of the dielectric constant (ε) of the solvent in ion pairing and dissociation
The interaction energy, U, between ions at some distance is determined by the dielectric constant of the solvent.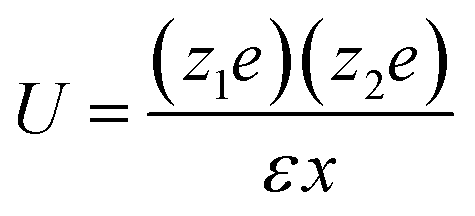 | (5) |
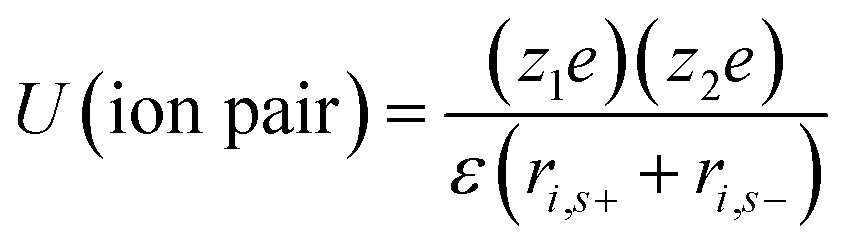 | (6) |
When the energy U in the above equation is ≫kT, the scope of ion pairing is large (α ≪ 1). Likewise, dissociation (α) usually increases with increase in temperature and leads to improved conductance. At lower temperature, the internal resistance increases and the power delivery capability decreases in the case of supercapacitors and batteries. In some cases, when solvated ions are paired, their solvation shells become shared (Fig. 5(b)) and come into contact (Fig. 5(c)) ,which increases positive and negative electrostatic repulsion that is counterbalanced by a considerable loss of solvation energy of the ions. The ion pairing in various solvents can be quantitatively characterized by infrared, Raman, and NMR spectroscopic studies.79
3.4 Ion solvation
The interaction of solvent molecules with ions determines the solubility of salts, dissociation of salts, ion pairing, and mobility of free ions. These are the important factors which directly influence the conductivity of electrolytes. The difference between the ion's energy in the solvent and in the gas phase is defined as the hydration (ion solvation) energy. In the solvent, the ion experiences a high interaction with the electric dipoles (dielectric) of solvent molecules. Experimentally, the heats of solvation (ΔHs) of the ions of a salt are derived from the thermodynamic cycle involving the dissolution of the salt (Fig. 5(d)). The sublimation energy (ΔHsub) of the salt is a large negative (exothermic) quantity (−320 to −500 kJ mol−1 for monovalent ions). The sum of the solvation energies of the two ions of the salt is equal to ΔHsoln − ΔHsub. The solvation energy provides a large driving force for the dissolution of the salt. The standard Gibbs energy (ΔG°) of a solution determines the equilibrium solubility of a salt. The conductance of a salt also depends on the dielectric (polarity) properties of the solvent. The solubility product Ksp (equilibrium constant) is related to ΔG° (ΔG° = −RTlnKsp) and large differences of Ksp (10−13 to 101 for a variety of salts and from one solvent to another). Therefore, large differences of conductance can arise, so for supercapacitor or battery electrolytes, either well-dissociated salts or largely soluble electrolytes must be used.
4. Role of cations and anions of various electrolytes in charge storage
4.1 Aqueous electrolytes (alkaline, neutral, and acidic)
Aqueous electrolytes have been used extensively in research and development due to their easy handing in the laboratory as compared to organic electrolytes and ILs, which require purification procedures. Aqueous electrolytes81 exhibit high conductivity as compared to organic and ionic electrolytes which is beneficial for lowering the equivalent series resistance and leads to better power delivery of SCDs.82 Materials whose working potential is located between the O2 evolution potential and H2 evolution potential are suitable for aqueous alkali-metal ion batteries. In addition, the electrode materials should be carefully selected with consideration of the pH, which is strongly associated with the O2 and H2 evolution potentials of an aqueous system.83 Aqueous-based electrochemical supercapacitors and their performance are listed in Table 1. The electrochemical performance of aqueous-based asymmetric supercapacitors has been studied by Jabeen et al.84 and Xiong et al.40 A 2.6 V aqueous asymmetric supercapacitor is demonstrated by using Na0.5MnO2 nanowall arrays as the cathode and carbon-coated Fe3O4 nanorod arrays as the anode.84 Aqueous electrolytes are divided into three categories: (1) alkaline, (2) neutral, and (3) acidic solutions. The most frequently used aqueous electrolytes are KOH,85 NaOH, LiOH, Na2SO4, H2SO4, (NH4)2SO4, K2SO4, Li2SO4, MgSO4, CaSO4, BaSO4, KCl, NaCl, LiCl, HCl, CsCl, CaCl2, KNO3, LiNO3, Na(CH3COO), Li(CH3COO), Mg(CH3COO)2, Na2HPO4, NaHCO3, Na2B4O7, Li-SiW, Na-SiW, and K-SiW. Graphical representation of the reaction mechanism between an alkaline electrolyte (KOH) and carbon–MnO2 electrodes for EDLCs, pseudocapacitors and hybrid supercapacitors is shown in Fig. 6.| Electrode materials | Electrolyte | Cell voltage (V) | Power density (W kg−1) | Energy density (W h kg−1) | Specific capacitance (F g−1) | Temp. (°C) | Ref. |
|---|---|---|---|---|---|---|---|
| CNFs with radially grown graphene sheets | 1 M Na2SO4 | 1.8 | 450 | 29.1 | — | — | 86 |
| Hydrous RuO2 | 1 M Na2SO4 | 1.6 | 500 | 18.77 | 52.66 at 0.625 A g−1 | — | 87 |
| MnO2@carbon nanofiber composites | 0.5 M Na2SO4 | 0.85 | — | — | 551 at 2 mV s−1 (75 °C) | 0–75 | 88 |
| Well-ordered mesoporous carbon/Fe2O3 nanoparticle composites | 1 M Na2SO3 | 1 | — | 39.4 | 235 at 0.5 A g−1 | RT | 89 |
| Mesoporous MnO2 | 0.65 M K2SO4 | 1 | ∼70 | ∼24.1 | 224.88 at 1 mV s−1 | RT | 90 |
| Mesoporous MnO2 | 1 M Li2SO4 | 1 | ∼70 | ∼28.8 | 284.24 at 1 mV s−1 | RT | 90 |
| Mesoporous MnO2 | 1 M Na2SO4 | 1 | ∼70 | ∼28.4 | 278.8 at 1 mV s−1 | RT | 90 |
| MnCl2-doped PANI/SWCNT nanocomposites | 1 M KCl | 1.6 | ∼550 | 194.13 | 546 at 0.5 A g−1 | RT | 91 |
| AC | 0.5 M Na2SO4 | 1.6 | — | ∼10 | 135 at 0.2 A g−1 | — | 92 |
| Seaweed carbons | 0.5 M Na2SO4 | 1.6 | — | 10.8 | 123 at 0.2 A g−1 | — | 93 |
| AC | 4 M NaNO3-EG | 2 | ∼500 | 14–16 | 22.3 at 2 mV s−1 | 0–60 | 94 |
| Microporous carbon | 0.5 M Na2SO4 | 1.8 | ∼40 | ∼7 | ∼60 at 0.2 A g−1 | — | 95 |
| AC | 1 M NaNO3 | 1.6 | — | — | 116 at 2 mV s−1 | RT | 96 |
| 3D FHPC | 1 M Na2SO4 | 1.8 | 317.5 | 15.9 | — | — | 97 |
| MMPGC | 2 M H2SO4 | 0.8 | 20 | 4 | 105 at 4 mV s−1 | RT | 98 |
| AC fibers | 1 M H2SO4 | 0.9 | — | — | 280 at 0.5 A g−1 | RT | 99 |
| GQD/3DG composite | 1 M H2SO4 | 0.8 | — | — | 268 at 1.25 A g−1 | RT | 100 |
| 3D heteroatom-doped carbon nanofiber networks | 2 M H2SO4 | 1 | ∼100 | 7.76 | 204.9 at 1 A g−1 | RT | 101 |
| Phosphorus-enriched carbons | 1 M H2SO4 | 1.3 | 33 | 16.3 | 220 at 1 A g−1 | — | 102 |
| ANS–rGO | 1 M H2SO4 | 2.0 | 1328 | 213 | 375 at 1.3 A g−1 | RT | 103 |
| RuO2–graphene | 0.5 M H2SO4 | 1.2 | 600 | 20.28 | 479 at 0.25 A g−1 | — | 104 |
| PANI-grafted rGO | 1 M H2SO4 | 0.8 | 60000 |
8.3 | 1045.51 at 0.2 A g−1 | — | 105 |
| PPy thin films | 0.5 M H2SO4 | 1 | 758 | 133 | 510 at 0.25 mA cm−2 | — | 106 |
| Graphene/mPANI | 1 M H2SO4 | 0.7 | 106.7 | 11.3 | 749 at 0.5 A g−1 | — | 107 |
| Nitrogen and oxygen-containing hierarchical porous carbon frameworks | 1 M H2SO4 | 0.8 | 197 | 37.4 | 428.1 at 0.5 A g−1 | — | 108 |
| 3D FHPC | 6 M KOH | 1 | — | — | 294 at 2 mV s−1 | — | 97 |
| Highly porous graphene planes | 6 M KOH | 1 | ∼50 | 6.5 | 303 at 0.5 A g−1 | — | 109 |
| p-CNTn/CGBs | 6 M KOH | 0.9 | 150 | 4.9 | 202 at 0.325 A g−1 | RT | 110 |
| Sub-3 nm Co3O4 nanofilms | 2 M KOH | 0.47 | — | — | 1400 at 1 A g−1 | RT | 111 |
| Porous NiCo2O4 nanotubes | 2 M KOH | 0.41 | 205 | 38.5 | 1647.6 at 1 A g−1 | — | 112 |
| MnO2 nanoflowers | 1 M LiOH | 0.6 | — | — | 363 at 2 mV s−1 | — | 113 |
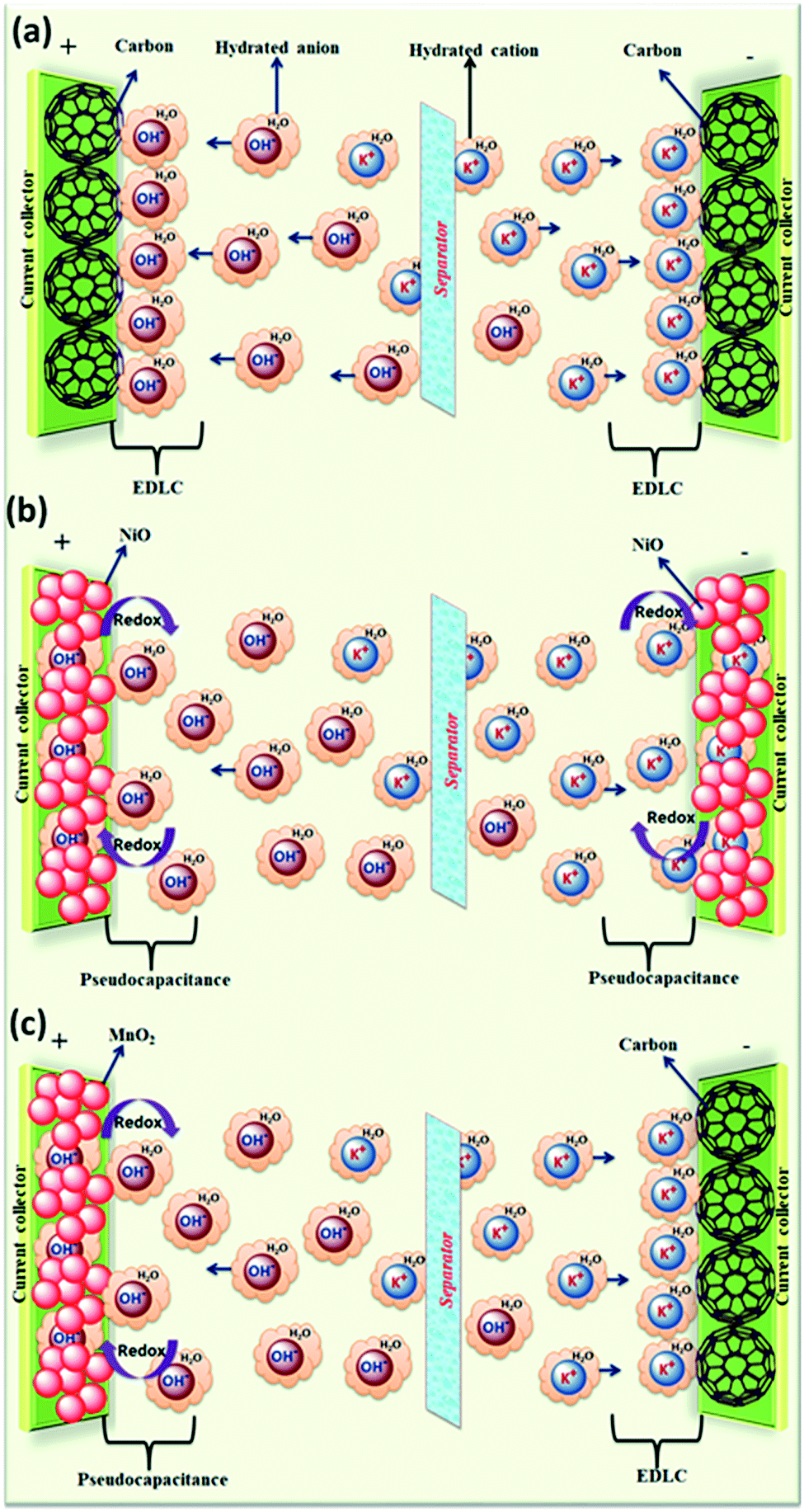 | ||
| Fig. 6 Graphical representation of reaction mechanism between alkaline electrolyte (KOH) and carbon–MnO2 electrodes for (a) EDLC (b) pseudocapacitor (c) hybrid supercapacitor. | ||
The effect of various electrolyte cations (H+, Li+, Na+, and K+) on the electrochemical performance of graphene/polypyrrole based supercapacitors has been studied by Zhu et al.114 The results showed a significant difference in the specific capacitance (280.3, 255.4, 210.4, and 197.9 F g−1 at 5 mV s−1 for HCl, KCl, NaCl, and LiCl as shown in Fig. 7(B)) only by changing the cationic species in various electrolytes. The significant difference in the specific capacitance was due to the cationic mobility, hydrated cationic radius, and conductivity, and their effect on charge/ion exchange and diffusion as shown in Fig. 7(A). The hydrated cationic radius was in the order H+-H2Oδ− < K+-H2Oδ− < Na+-H2Oδ− < Li+-H2Oδ−. The Li+ ions had the largest hydration sphere radius because of the strong Li+-H2Oδ− interactions caused by the large surface charge density and their ionic mobility decreased accordingly from K+ to Li+. The large ionic mobility and small hydration sphere radius of H+ were due to the jumping transference mode between water molecules by hydrogen bonds. The H+ ion also had the highest molar ionic conductivity as compared to K+, Na+, and Li+. The higher conductivity and ionic mobility help in fast charge transfer, and the smaller hydration sphere radius also offers more ion adsorption at the electrolyte/electrode interface to further facilitate the Faraday reaction. Therefore, due to the smallest hydrated ions and highest conductivity and ionic mobility, the supercapacitor with HCl as the electrolyte shows the largest specific capacitance of up to 280.3 F g−1. The long-term cycling stability was also strongly dependent on the cationic species. After 10000 cycles, the specific capacitance of HCl, LiCl, NaCl, and KCl electrolytes was 92.0%, 73.9%, 58.7%, and 37.9%, respectively, due to the significant composition damage caused by the insertion/de-insertion of large size cations (H+ < Li+ < Na+ < K+) during the charging–discharging cycles as shown in Fig. 7(C). Wang et al.115 have studied the effect of various electrolyte anions (OH−, SO42−, Cl−, and NO3−) on the electrochemical performance of calcium carbide-derived carbon (CCDC) as the electrode; a significant difference in the specific capacitance only by changing the anionic species in various electrolytes (KOH, KCl, KNO3 and K2SO4) due to higher conductivity and ionic mobility was observed. The higher conductivity leads to the higher current response during CV in KOH than those during CV in KCl, KNO3 and K2SO4 electrolytes as shown in Fig. 8(a). The OH− ion showed higher conductivity (198 cm2 Ω mol−1) and ionic mobility (2.06 × 10−6 cm2 s−1 v−1) than Cl−, NO3− and SO42− ions. Hence, the higher conductivity and ionic mobility of OH− ions can cause much better capacitive behavior. The relationship between pores and hydration sphere ions is shown in Fig. 8(c). The radius of the hydration sphere of OH− was slightly smaller than the radii of the hydration sphere of Cl− and NO3− ions and the most probable apertures are equal to about 2.1× the size of the radii of hydration spheres. The radius of the hydration sphere of SO42− ions was bigger (the most probable aperture is just equal to about 1.8 × the size of the radius of the hydration sphere as shown in Fig. 4(b)), and leads to a decrease in the quantity of ions entering into the pores, and thus a lower electric double layer is formed. Furthermore, the conductivity and ionic mobility of SO42− ions were also low as compared to other anions. Therefore, the activated CCDC supercapacitor in a 0.5 M K2SO4 aqueous electrolyte displays a poor capacitive behavior.
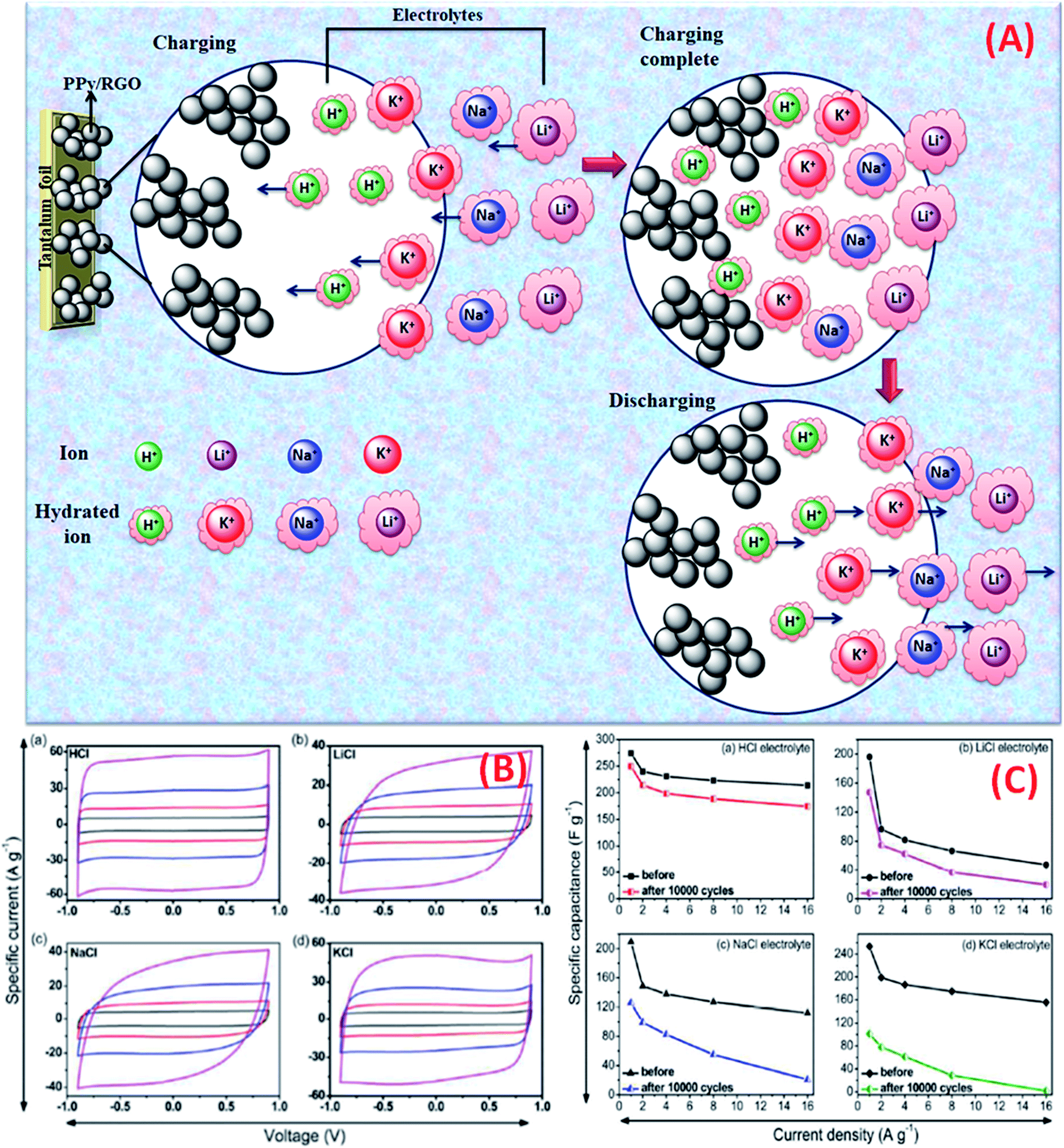 | ||
| Fig. 7 (A). Schematic representation of mobility/diffusion of hydrated H+, K+, Na+ and Li+ electrolyte ions in a PPy/RGo electrode according to their hydrated radius during the charging and discharging process; (B) CV curves of the supercapacitors in various electrolytes at different scan rates: 20, 50, 100, and 200 mV s−1: (a) HCl, (b) LiCl, (c) NaCl, and (d) KCl; (C) capacitance retention after 10000 cycles of SCs in various electrolytes: (a) HCl (b) LiCl (c) NaCl and (d) KCl. (B) and (C) have been adapted from ref. 114 with permission from the Royal Society of Chemistry. | ||
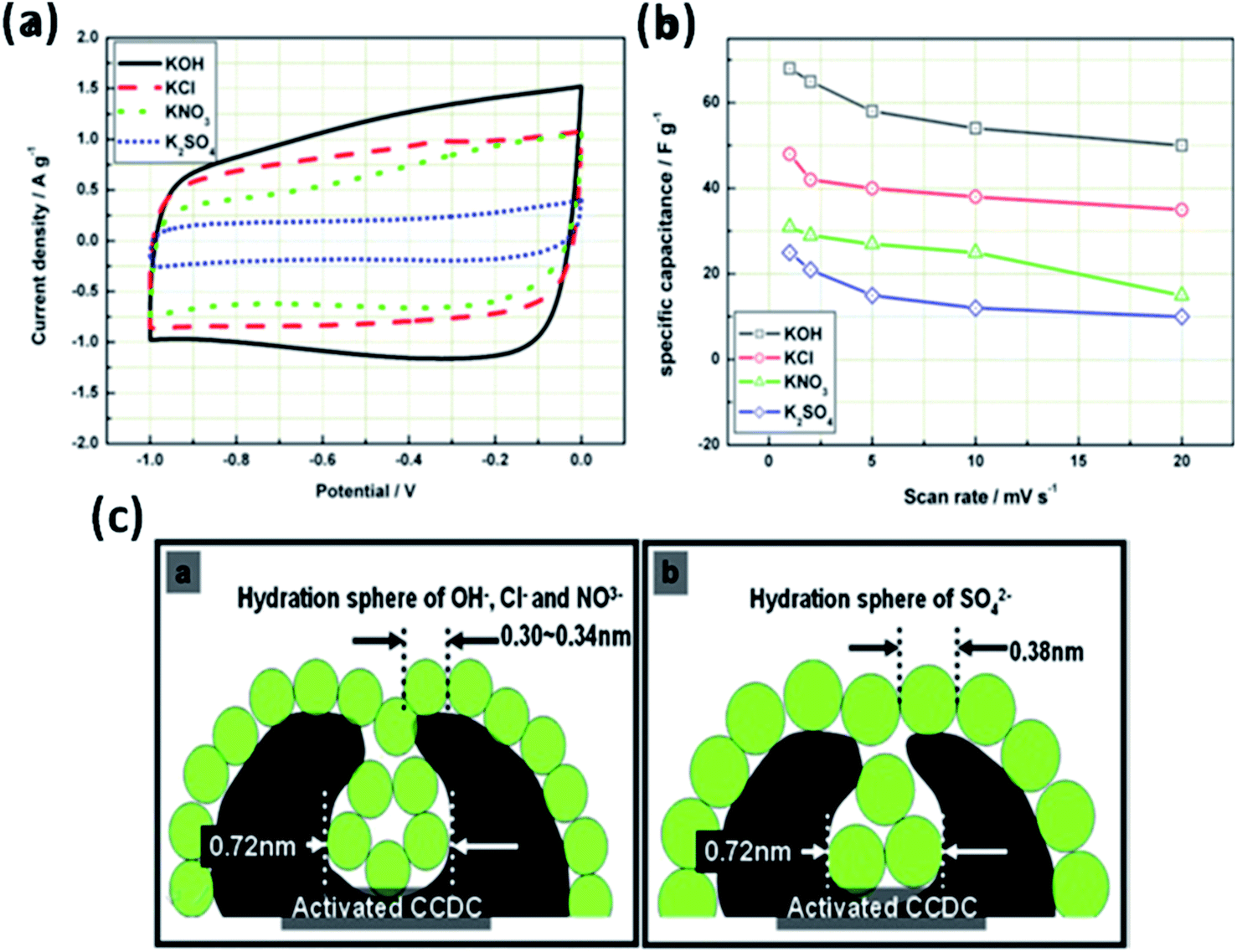 | ||
| Fig. 8 (a) Cyclic voltammograms of activated CCDC supercapacitors in the aqueous electrolytes 0.5 M K2SO4, 2 M KCl, 1 M KNO3 and 6 M KOH (20 mV s−1) and (b) the specific capacitance of the supercapacitor at different scan rates. (c) Schematic diagram of the relationship of hydration sphere ions and pores. This figure has been adapted from ref. 115 with permission from Elsevier. | ||
Nithya et al.116 have studied the electrochemical performance of Bi2WO6 nanoparticles in various aqueous electrolytes such as 1 M NaOH, 1 M LiOH, 1 M Na2SO4, 1 M KOH and 6 M KOH solutions. The Bi2WO6 based electrodes exhibited an enhanced electrochemical performance in the KOH electrolyte due to their high ionic mobility, smaller hydration sphere radius and lower equivalent series resistance. The Bi2WO6 based electrodes exhibited an excellent coulombic efficiency and a specific capacitance of ∼304 F g−1 at 3 mA cm−2 (potential range from −0.9 to 0.1 V vs. Hg/HgO) in 1 M KOH electrolyte. CV curves for all the electrolytes are shown in Fig. 9(a–e). The current response of Bi2WO6 in various electrolytes decreases in the order 6 M KOH > 1 M KOH > 1 M NaOH > 1 M Na2SO4. This enhanced current response of KOH compared to other electrolytes is due to the difference in the hydration sphere radius of K+ ions (3.31 Å), Na+ ions (3.58 Å) and Li+ ions (3.82 Å), respectively. It was noted that the reported conductivity of K+ (73 cm2 Ω−1 mol−1) ions is greater than that of Na+ ions (50 cm2 Ω−1 mol−1) and Li+ ions (38 cm2 Ω−1 mol−1) at 25 °C and hence the mobility of ions would be higher for K+ compared with Na+ and Li+. The smaller hydration sphere radius of K+ ions favours enhanced ionic mobility and interaction with the electrode material, thereby resulting in enhanced electrochemical performance. The K+ ions also acquire a small charge density (weak solvation interactions with water molecules which favour easier polarization during the de-solvation processes) which further helps K+ ions go easily into the electrode during the redox reactions, concluding that the cations had a major role in the electrochemical reaction processes. This study also compared the CV results of Na2SO4 and NaOH electrolytes to understand the role of anions in the electrochemical performance. Results show that the redox peak was dominant in NaOH compared with the Na2SO4 electrolyte, which was due to the increased anionic size of sulphate ions (1.49 Å) as compared to hydroxyl ions (1.10 Å).
 | ||
| Fig. 9 CV curves at various scan rates of (a) 1 M LiOH (b) 1 M NaOH (c) 1 M Na2SO4 (d) 1 M KOH and (e) 6 M KOH and (f) capacitance vs. scan rate for different electrolytes. This figure has been adapted from ref. 116 with permission from Elsevier. | ||
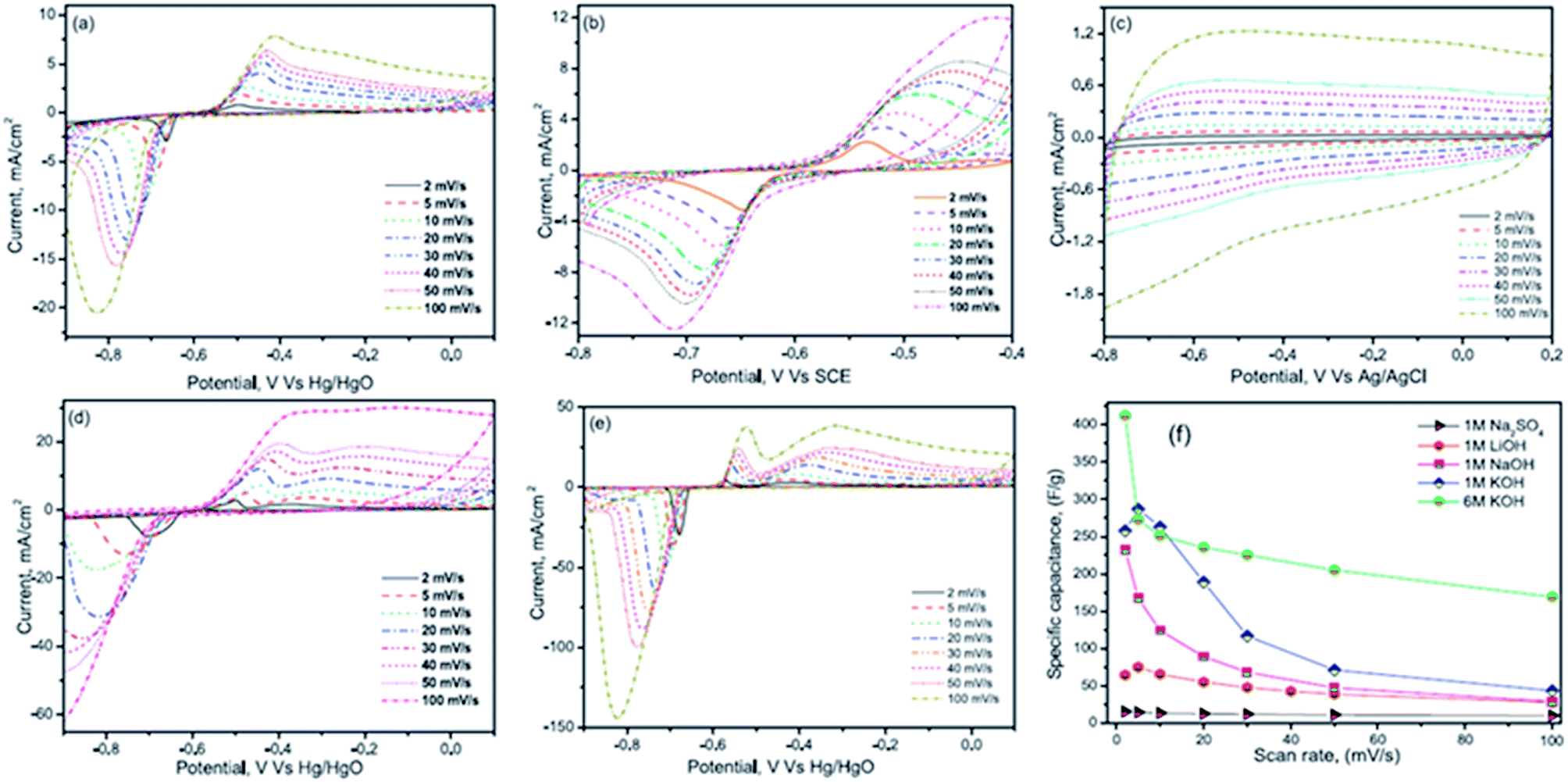
Wang et al.117 reported the electrochemical performances of symmetric supercapacitors assembled with MnFe2O4 colloidal nanocrystal clusters (CNCs) in aqueous electrolytes (KOH, NaOH, LiOH and Na2SO4). The effect of the hydrated ionic radius and the electrolyte concentration was investigated based on electro-chemical characterization of the supercapacitors; the specific capacitances of CNC electrodes were found to increase with the decrease in hydrated ionic radii in alkaline electrolytes under the same conditions as shown in Fig. 10(a and b). The hydrated ionic radius in the electrolytes and their concentrations were suggested to play important roles in determining the electrochemical performances of ferrite-based supercapacitors. Compared with the capacitances of the electrode materials using the electrolytes containing KOH, NaOH, LiOH, LiNO3 and Na2SO4 with the same concentration, the maximum specific capacitance is obtained when using the KOH electrolyte, followed by NaOH, LiOH and LiNO3 and the use of Na2SO4 shows minimal specific capacitance. Generally, for ions with the same charge, the smaller the radius is, the stronger the polarization is in a polar solvent, leading to a stronger ionic solvation. Thereafter, the more the ions will attract the surrounding solvent molecules, the greater the radius of hydrated ions as shown in Fig. 10(c). The electrolyte ion transport rate thus decreased with the ion radius, reducing the capacity of the electrode material. K+, Na+ and Li+ have the same electric charge; however, the existence of SO42− ions should further impact the energy storage at the interface due to the large size of the ions, leading to the smallest capacitance of CNC-based electrodes in the Na2SO4 electrolyte. Thus the capacitances of the electrode material displayed variation with the electrolytes at the same concentration. Misnon et al.113 reported a relationship between the solvated ion size of alkaline electrolytes (LiOH, NaOH and KOH) and the pore size in the electrode material favoring a high specific capacitance and faster electrode kinetics. This study reported that LiOH showed a higher specific capacitance (LiOH > NaOH > KOH) than NaOH and KOH due to the differences in the ion diffusion coefficients of Li+, Na+ and K+. The ion interaction becomes stronger on the surfaces of δ-MnO2 as the ion diffusion coefficient increases. The unsolvated Li+, Na+ and K+ ions have diameters of ∼0.6 Å, ∼0.95 Å, and 1.33 Å, respectively; therefore, intercalation–deintercalation reactions easily occurred in the LiOH (smaller ionic radius of Li+) electrolyte compared to other electrolytes.
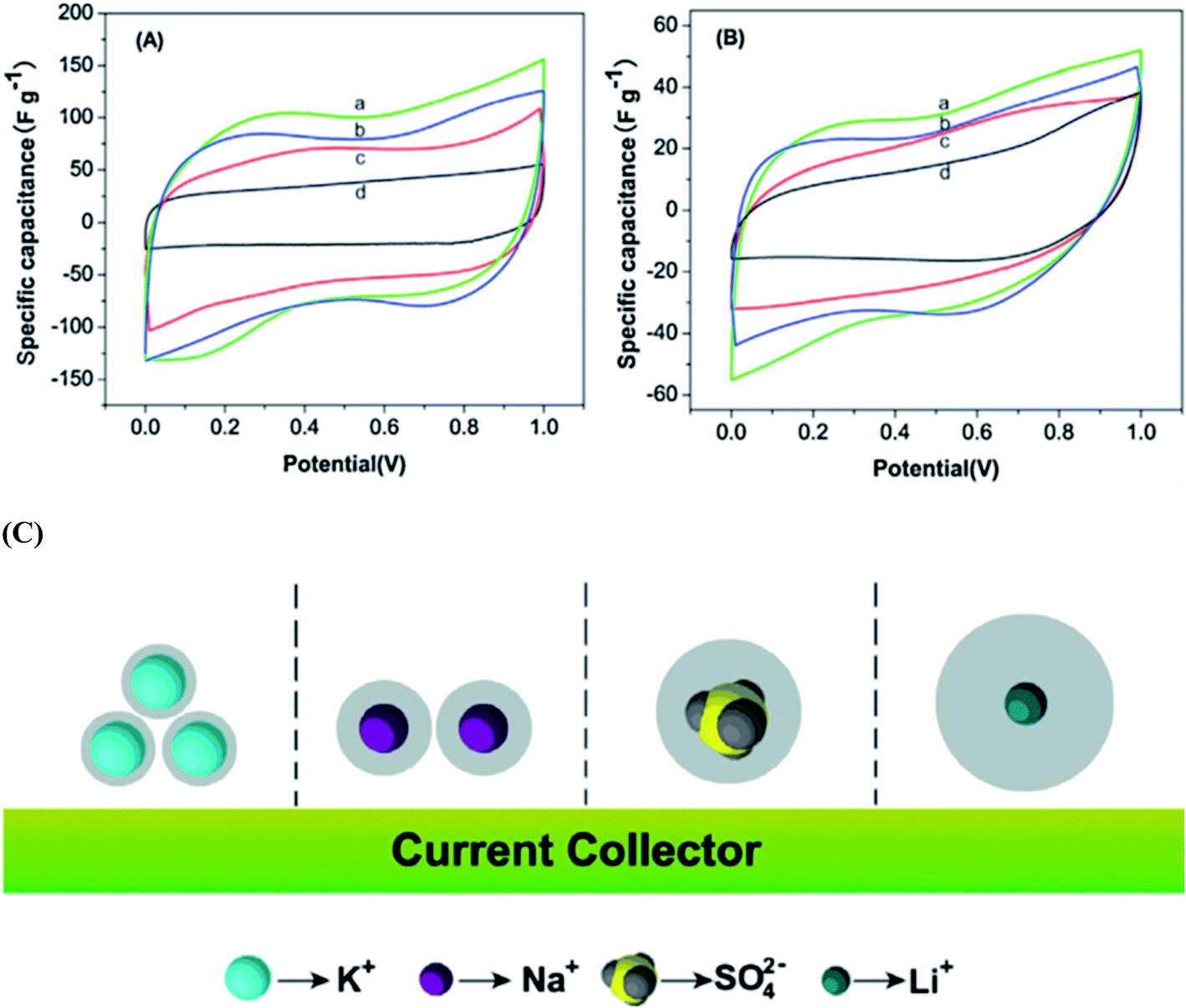 | ||
| Fig. 10 CV curves of the supercapacitors with the electrolytes (2 M) (a) KOH, (b) NaOH, (c) LiOH and (d) Na2SO4 at scan rates of (A) 5 mV s−1 and (B) 100 mV s−1. (C) The ion of hydrated ionic radius on the MnFe2O4-based supercapacitors. This figure has been adapted from ref. 117 with permission from Elsevier. | ||
Fic et al.118 investigated three alkali metal sulfate solutions such as Li2SO4, Na2SO4, and K2SO4 for carbon based electrodes. The best capacitance and charge propagation values were achieved for Li2SO4 solution as shown in Fig. 11(a), and this was due to the mobility of alkali metal ions, increasing in the following order: Li+ < Na+ < K+. The charging and discharging of the EDL in EDLCs do not need very high ion mobility like pseudocapacitive systems where fast transfer of ions is needed for quick redox processes at the electrode/electrolyte interface. This study also found that the ions already located at the electrolyte/electrode interface are attracted and pushed off from the electrode to a small distance when the porous structure of the electrode is well-saturated by the electrolyte; therefore, high mobility might enhance fast and efficient charge propagation. The specific capacitance value was also higher for Li2SO4 (170 F g−1) as compared to Na2SO4 (105 F g−1) and K2SO4 (78 F g−1) at both slow and fast scan rates as shown in Fig. 11(a) and it is explained on the basis of ion dimensions. The highest capacitance values obtained for the largest and most solvated ions, i.e. Li+ being surrounded by even 27 molecules of water, might be explained with their poor mobility and low diffusion coefficient. As discussed earlier, rather than remaining near the electrode/electrolyte interface during discharging and then quickly appearing at this interface during charging, both Na+ and K+ cations are characterized by higher mobilities and diffusion coefficients, and therefore the ions can migrate into the electrolyte bulk during discharging and their return to the electrode/electrolyte interface during charging might not be so fast. Additionally, when considering the ion dimensions and solvation energy, it might also be assumed that solvated Li+ cations rather avoid desolvation. Na+ and K+ cations are characterized by a smaller ion–solvent complex diameter and solvation/desolvation energy; therefore, these ions can be sorbed differently in the micropores as shown in Fig. 11(c). Sorption in the pores is a diffusion-limited process; hence, lower capacitance values and poor charge propagation can be observed for both of them in cyclic voltammograms and capacitance vs. current load dependencies for capacitors operating in appropriate alkali metal solutions, especially at higher scan rates and for higher discharge current densities. It is also worth noting that the sulfate anion, SO42−, is also one of the biggest (even 40 molecules of water in the considered solvated structure) and strongest solvated inorganic anions, with a desolvation energy of about 108 kJ mol−1 per one SO42−–H2O bond. Chae et al.119 found that for electrolytes with the same cations and concentration, changing anions from SO42− ions to Cl− ions could increase the specific capacitance due to the smaller size of Cl− ions compared to SO42− ions. Most recently, some new types of electrolytes, such as lithium, sodium, and potassium salts of silicotungstic acid (Li-SiW, Na-SiW and K-SiW), were explored as aqueous neutral electrolytes for EDLCs by Gao et al.120 These electrolytes exhibited much higher ionic conductivities when compared to their counterparts with Cl−, SO42− or NO3− anions. This is because of the larger amount of dissociated cations and the much greater anion mobility of Keggin anions. A cell voltage of 1.5 V was achieved for carbon-based EDLCs with these neutral electrolytes.
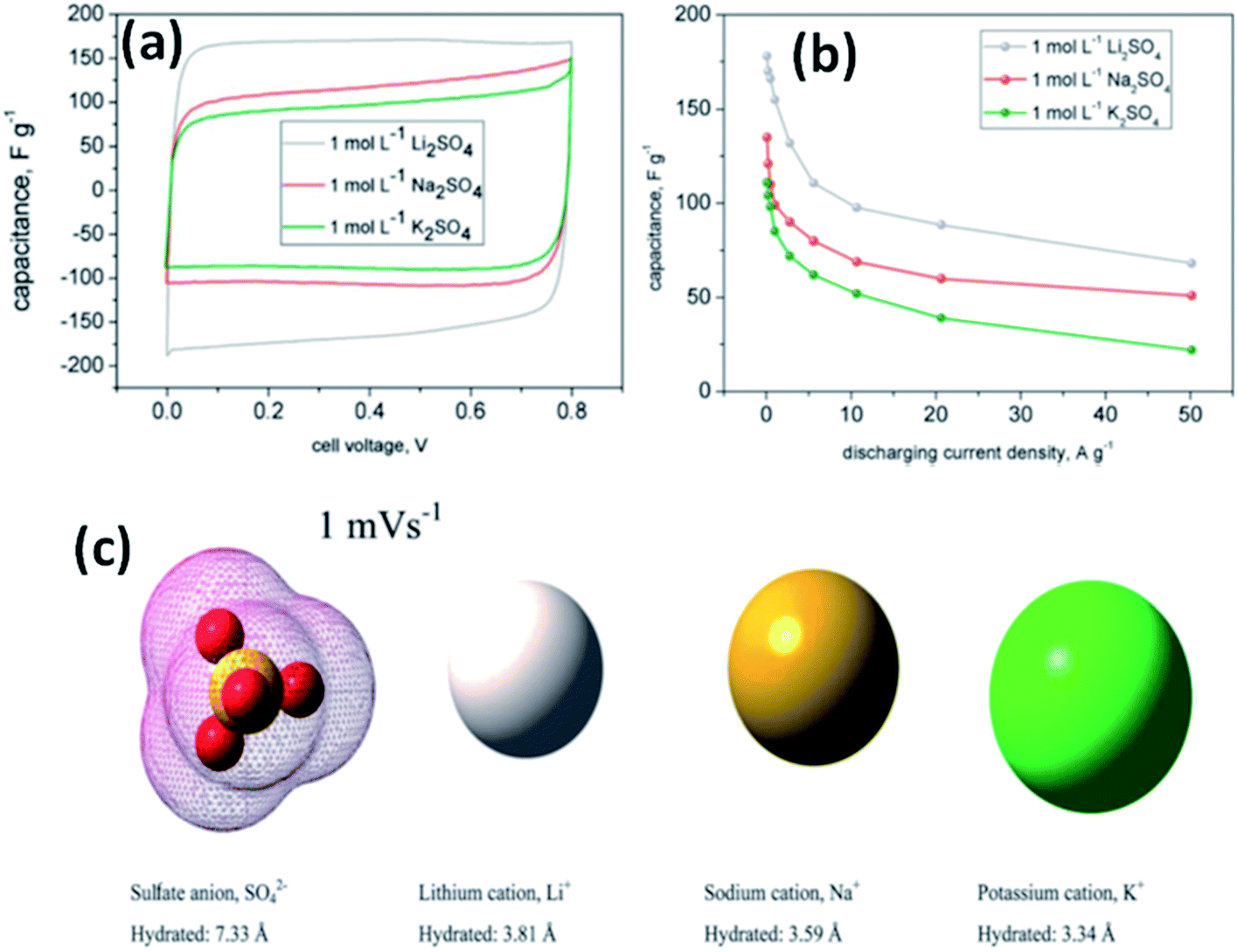 | ||
| Fig. 11 (a) CV curves of a carbon based supercapacitor in 1 mol L−1 alkali metal sulfate solutions (Li, Na, K) at 1 mV s−1, (b) capacitance vs. current load for different alkali metal sulfates solutions, and (c) ion shape and dimensions in aqueous solutions. This figure has been adapted from ref. 118 with permission from the Royal Society of Chemistry. | ||
To conclude, various factors of aqueous electrolytes, such as types of cationic and anionic species, salt concentrations, hydrated ion size, conductivity, and ion mobility, have been found to have a great influence on the electrochemical supercapacitor performance. Regarding the cationic species, various alkaline metal or alkaline-earth metal cations have different ionic sizes and hydrated ion sizes, and thus have different diffusion coefficients and ionic conductivities, which are expected to have strong influences on both the specific capacitance and ESR of the electrochemical supercapacitors. However, more improvements in the electrochemical supercapacitor performance with aqueous electrolytes are still needed to increase the energy density and cycle-life further.
4.2 Organic electrolytes
Organic electrolytes for SCDs usually consist of organic solvents and conducting salts dissolved in them. The organic electrolyte-based SCDs and their performances are listed in Table 2. Because of their high voltage window (2.6 to 2.9 V), devices based on organic electrolytes are currently dominating the commercial market. The disadvantage of organic electrolytes is their higher resistivity than aqueous electrolytes due to their large size molecules, which require a large pore size in the electrodes. Similar to aqueous electrolyte-based SCDs, the nature of solvents and salts, such as the ion size, ion–solvent interaction, conductivity, and viscosity, has profound influences on the performance of organic electrolyte-based SCDs. Graphical representation of the reaction mechanism between organic electrolytes and various electrodes for EDLCs, pseudocapacitors and hybrid supercapacitors is shown in Fig. 12.| Electrode materials | Electrolyte | Cell voltage (V) | Power density (W kg−1) | Energy density (W h kg−1) | Specific capacitance (F g−1) | Temp. (°C) | Ref. |
|---|---|---|---|---|---|---|---|
| MnO2 nanorods-rGO//V2O5 | 1 M LiTFSI/ACN | 2 | 436.5 | 15.4 | 36.9 | RT | 124 |
| Porous graphitic carbon//Li4Ti5O12 | 1 M LiPF6/EC–DMC (1:1) |
3 | ∼110 | ∼55 | — | — | 125 |
| Fe3O4–graphene//3D graphene | 1 M LiPF6/EC–DEC–DMC (1:1:1) |
3 | 150 | 147 | — | RT | 126 |
| AC (MSP-20)//mesoporous Nb2O5-carbon | 1 M LiPF6/EC–DMC (1:1) |
3.5 | ∼100 | 74 | — | — | 127 |
| Non-porous activated mesophase carbon microbeads//graphitized carbon | 1.5 M TEMABF4/PC | 4 | ∼30 | ∼60 | — | RT | 128 |
| Non-porous activated mesophase carbon microbeads//AC | 1 M SBPBF4/PC | 3.5 | ∼100 | ∼47 | — | — | 129 |
| Highly porous interconnected carbon nanosheets | 1 M TEABF4/ACN | 2.7 | 2500–2700 | 25 | 120–150 at 1 mV s−1 | — | 130 |
| Graphene–CNT composites | 1 M TEABF4/PC | 3 | 400 | 34.3 | 110 at 1 A g−1 | — | 131 |
| AC | 1 M TEABF4/HFIP | — | — | — | 110 at 1 A g−1 | — | 132 |
| AC | 0.7 M TEABF4/ADN | 3.75 | — | ∼28 | 25 at 20 mV s−1 | RT | 133 |
| AC | 1.6 M TEAODFB/PC | 2.5 | ∼1000 | ∼28 | 21.4 at 1 A g−1 | −40–60 | 134 |
| Microporous TiC–CDC | 1 M TEMABF4/(PC–PS 95:5) |
2.7 | ∼1000 | ∼25–27 | 100 at 10 mV s−1 | −45–60 | 135 |
| Carbon (provided by Batscap) | 1 M SBPBF4/ACN | 2.3 | — | — | 109 | −30–60 | 136 |
| AC | 1.5 M SBPBF4/PC | 3.5 | — | 52 | 122 at 0.1 A g−1 | RT | 137 |
| Heteroatom doped porous carbon flakes | 1 M LiPF6/(EC–DEC 1:1) |
3 | 2243 | 29 | 126 at 1 A g−1 | RT | 138 |
| Microporous carbide derived carbon | 1 M NaPF6/(EC–DMC–PC–EA 1:1:1:0.5) |
3.4 | ∼90 | ∼40 | 120 at 1 mV s−1 | −40–60 | 139 |
| Nanoporous Co3O4–graphene composites | 1 M LiPF6/(EC–DEC 1:1) |
— | — | — | 424.2 at 1 A g−1 | RT | 140 |
| MoO3 nanosheets | 1 M LiClO4/PC | — | — | — | 540 at 0.1 mV s−1 | — | 141 |
| Heterostructured poly(3,6-dithien-2-yl-9H-carbazol-9-yl acetic acid)/TiO2 nanocomposite | 0.5 M Bu4NBF4/ACN | 1.2 | — | 89.98 | 462.88 at 2.5 mA cm−2 | RT | 142 |
| PANI/graphite | 0.5 M LiClO4/PC | 1 | — | — | ∼420 at 50 mV s−1 | RT | 143 |
 | ||
| Fig. 12 Graphical representation of reaction mechanism between organic electrolytes and various electrodes for (a) EDLC (b) pseudocapacitor (c) hybrid supercapacitor. | ||
The effect of the ionic size of small quaternary ammonium BF4 salts on electrochemical double layer capacitors has been reported by Koh et al.;121 trimethylethylammonium BF4 and trimethylpropylammonium BF4 showed 10% higher capacitance than tetraethylammonium BF4 using microporous activated carbon electrodes (Fig. 13). The study also found that the capacitance depends on the size of the cations rather than the BF4 anion due to the smaller size of the anion than quaternary ammonium ions. The capacitance was found to be proportional to the reciprocal radii of the bare cations. The ions in narrow pores were found to lose their solvation shells as they enter into deeper and narrower spaces, and then these small desolvated ions moved easily within narrow pores and enhanced the capacitance. A large capacitance was obtained for 1 M TBA-BF4 in acetonitrile compared to 1 M TEA-BF4 using carbon as the working electrode by McDonough et al.122 This study found that larger alkyl chains of the TBA+ cation result in better separation of ions and counter-ions at the charged carbon surface, which allows for better charge screening. The influence of solvent properties on the characteristics of EDLCs was also studied by Arulepp et al.123
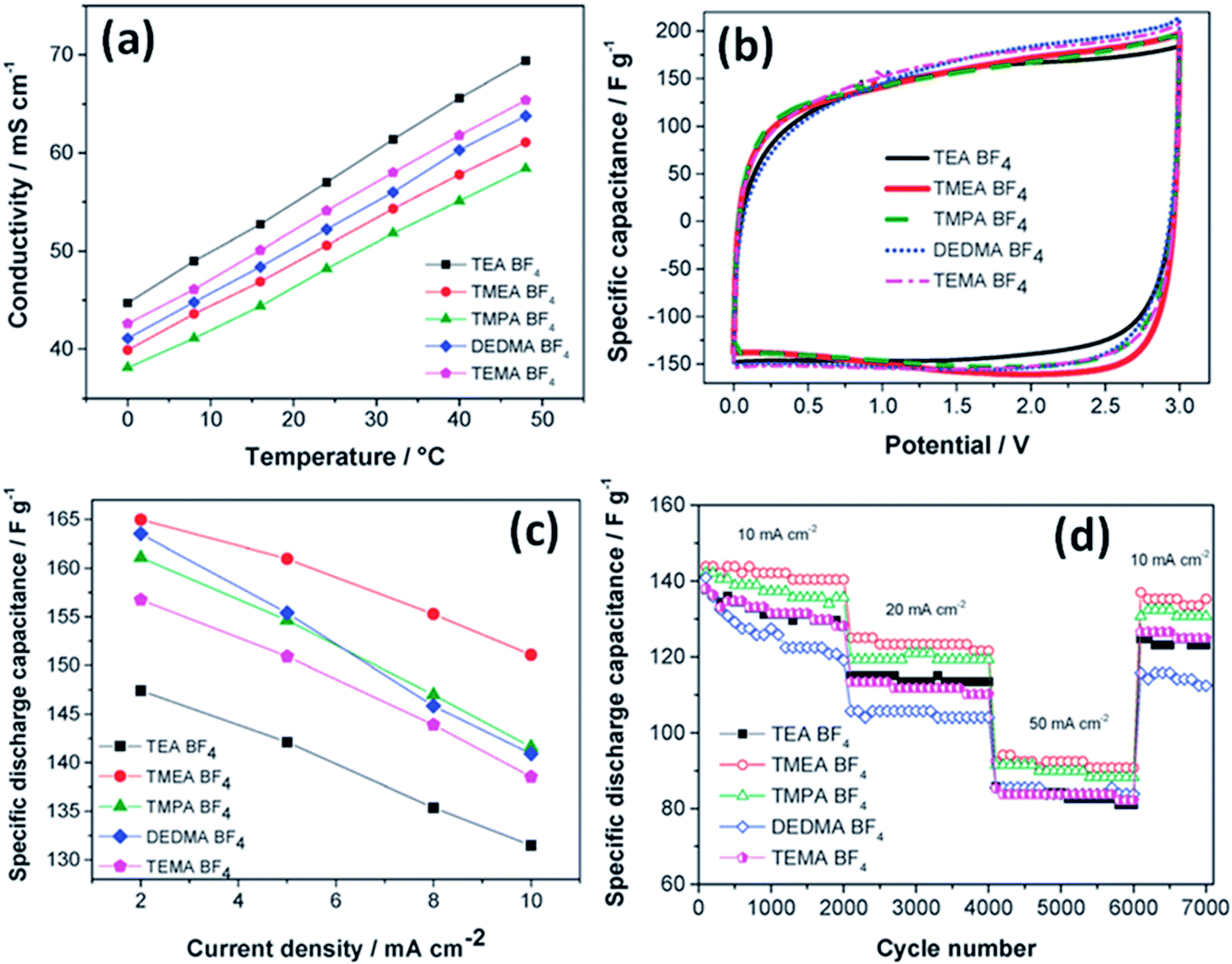 | ||
| Fig. 13 (a) Conductivity values vs. temperature, (b) CV in various electrolytes at 2 mV s−1, (c) capacitance values at low currents, and (d) cycle life tests. This figure has been adapted from ref. 121 with permission from the Royal Society of Chemistry. | ||
Different electrolyte solutions 1 M triethylmethylammonium tetrafluoroborate in acetonitrile (AN), acetone, γ-butyrolactone (GBL), and propylene carbonate (PC) with nanoporous carbon electrodes were used in the above report. It was observed that most of the significant parameters such as series resistance, series capacitance, and phase angle remarkably depend on the electrolyte composition and cell potential applied. It was found that the internal distribution of the electrode resistance increases in the following order of solvent: AN < GBL < PC.
Organic electrolytes have also been used for pseudocapacitors with pseudocapacitive materials144–147 such as metal oxides,148 composite materials, and conductive polymers.141–143 Most of the organic electrolytes used for pseudocapacitors contain Li ions due to their small bare ion size and easy ion intercalation/de-intercalation. The most used salts for organic electrolytes are LiPF6 and LiClO4 as reported in the literature.60,64 The common solvents used in organic electrolytes were acetonitrile, propylene carbonate, and a mixture of various solvents such as ethylene carbonate–ethyl methyl carbonate, ethylene carbonate–diethyl carbonate, ethylene carbonate–dimethyl carbonate,149 ethylene carbonate–dimethyl carbonate–ethyl methyl carbonate, and ethylene carbonate–dimethyl carbonate–diethyl carbonate.140,150 Organic electrolytes with asymmetric electrochemical supercapacitors have also attracted significant attention due to further enhancement in the energy density. A number of organic electrolytes with asymmetric electrochemical supercapacitors such as carbon//TiO2 (1 M LiPF6/EC–DMC), graphite//AC (electrolyte: 1.5 M TEMABF4/PC), carbon//V2O5 (1 M LiTFSI/ACN), carbon//ECP (1 M TEABF4/PC), and carbon//Li4Ti5O12 (1 M LiPF6/EC–EMC) have been reported by Amatucci et al.151 These organic electrolytes with asymmetric electrochemical supercapacitors can deliver a higher energy density due to having a much wider voltage window (3–4 V) than aqueous-based asymmetric supercapacitors. The electrochemical operational voltage windows of seven organic electrolytes for EDLCs using ab initio molecular orbital theory were studied by Maeshima et al.152 as shown in Fig. 14(a–d). Four types of modes were used to investigate the effect of intermolecular interactions: (i) a single-ion-in-solvent model, (ii) a single-ion in vacuo model, (iii) an ion-pair-in-solvent model, and (iv) an ion-pair in vacuo model as shown in Fig. 14(a–d). The ion pairs with optimized structures are shown in Fig. 15 with the shortest distance between the F atom in the anionic species and H atom in the cationic species. These distances vary from 2.30 to 2.46 Å and are shorter than the van der Waals radii of F and H (2.7 Å) which show that hydrogen bonds exist between F and H atoms in the solute species. This study also has verified that intermolecular interactions play very a crucial role in determining the operational voltage windows of the electrolytes and cannot be neglected in any quantitative approach.
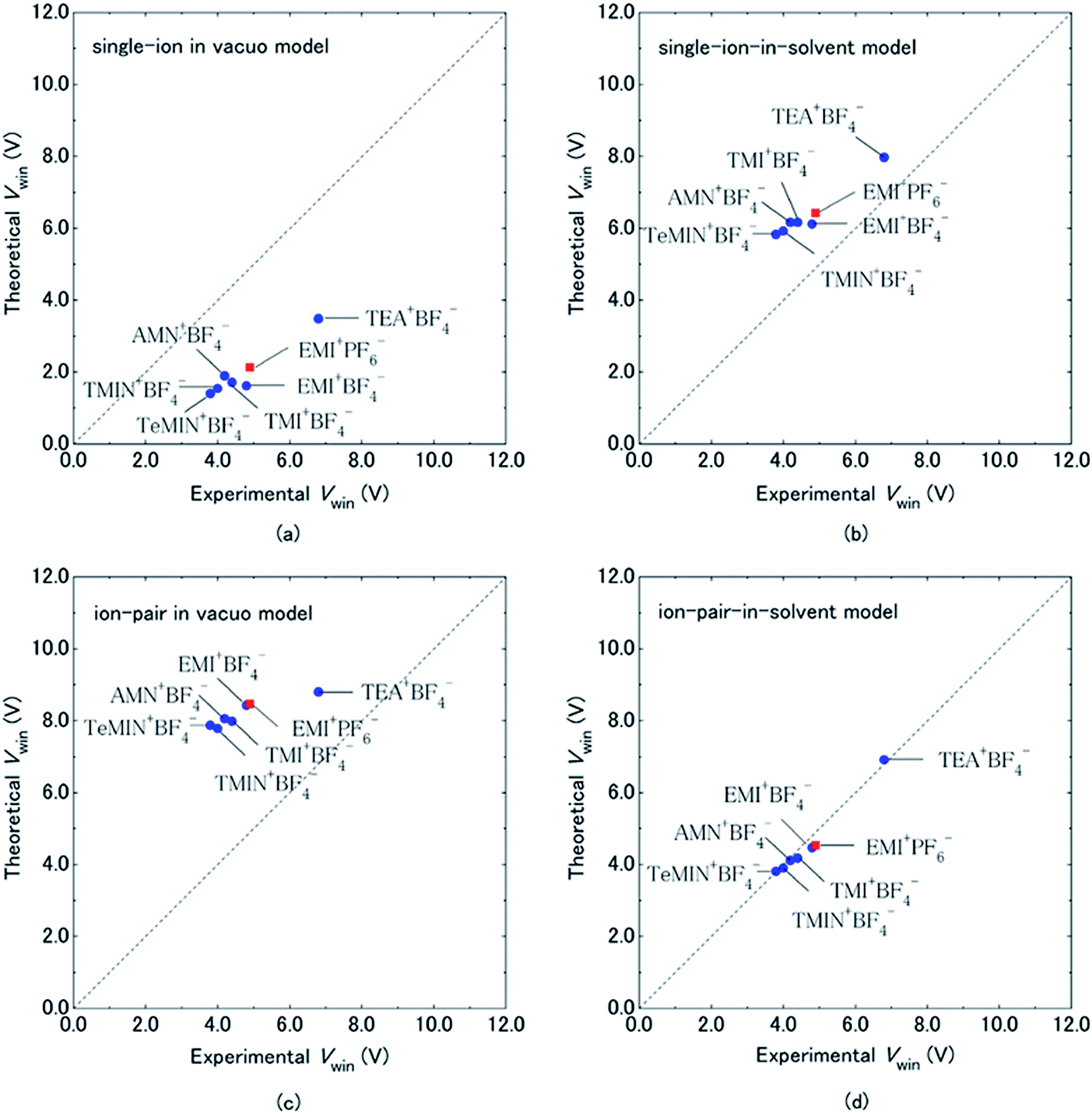 | ||
| Fig. 14 Comparison of electrochemical potential windows of seven electrolytes evaluated by CV measurement (experimental) and HF/6-31 + G(d,p) calculations using four types of models (theoretical): (a) single-ion in vacuo model, (b) single-ion-in-solvent model, (c) ion-pair in vacuo model, and (d) ion-pair-in-solvent model. Cation–anion interactions were treated by forming a cation–anion pair in (c) and (d), and solvent effects were treated using IPCM (with a dielectric constant of 69.0 for the PC solvent) in (b) and (d). Circles are for BF4−-based electrolytes and squares are for the PF6−-based electrolyte. This figure has been adapted from ref. 152 with permission from The Electrochemical Society. | ||
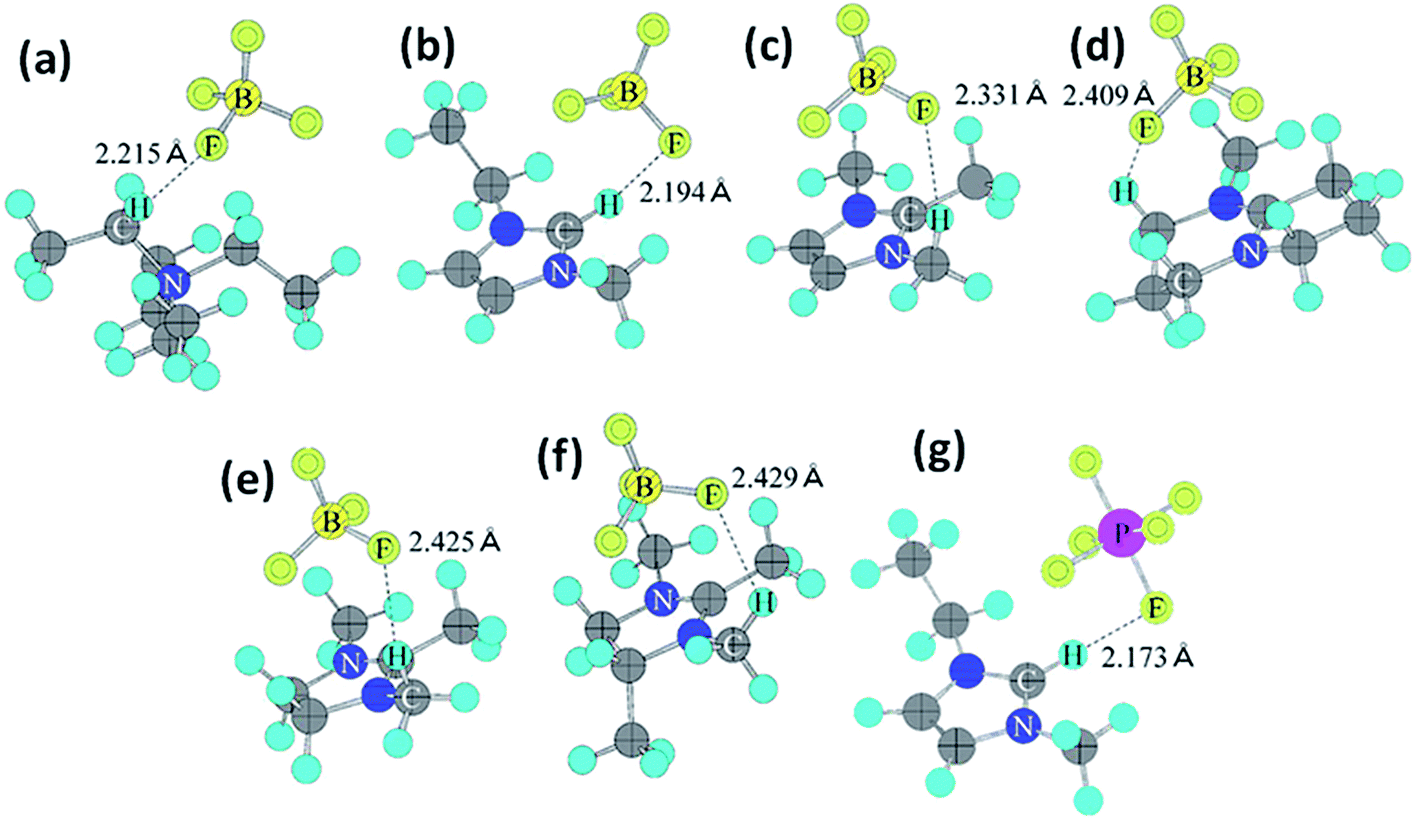 | ||
| Fig. 15 Ion-pair structures in vacuo optimized using HF/6-31 + G(d,p), (a) TEA+BF4−, (b) EMI+BF4−, (c) TMI+BF4−, (d) AMN+BF4−, (e) TMIN+BF4−, (f) TeMIN+BF4−, and (g) EMI+PF6−. This figure has been adapted from ref. 152 with permission from The Electrochemical Society. | ||
It is concluded that the specific capacitance of an EDLC depends not only on the specific surface area but also on the pore size distribution of the carbon materials. The properties of organic electrolytes such as sizes of anionic and cationic species and ion–solvent interactions are closely related to the pores of working electrodes. The small size pores in carbon materials may increase the specific surface area but they can limit the accessibility of electrolyte ions to go in and out of the electrodes, and individually the larger organic ions cannot easily access the small pores and decrease the specific capacitance. Hence, it is essential to match the size of electrolyte ions with the pore size of carbon materials to maximize the specific capacitance. Additionally, the sizes of both bare and solvated ions should also be considered. It was observed that the pores with a size close to the size of desolvated ions could increase the specific capacitance of EDLCs. Furthermore, other properties such as the power density, charging/discharging rate, and ESR should also be considered when designing the electrolytes and optimizing the pore structures of electrodes.
4.3 Ionic liquid electrolytes
The salts composed of ions (organic cations and organic/inorganic anions) with melting points below 100 °C (ref. 153) are called ionic liquids (ILs). Due to their unique structures and properties, ionic liquids have received significant interest as alternative electrolytes.154 The physical and chemical properties of ILs can be tuned easily due to their variety of combinations of cations and anions.42 The supercapacitor performance can be improved (tuning the voltage window and working temperature range) by the tenability properties of ILs.155–157 Ionic liquids have various potential benefits such as non-flammability, higher thermal and electrochemical stability, high voltage window (>3 V), and insignificant volatility as compared to organic electrolytes. Ionic liquids can be classified as protic, aprotic and zwitter ionic types based on their composition.158 Ionic liquids used for supercapacitors are based on ammonium, sulfonium, imidazolium, pyrrolidinium, and phosphonium cations and hexafluorophosphate (PF6), tetrafluoroborate (BF4), bis(trifluoromethanesulfonyl)imide (TFSI), bis(fluorosulfonyl)imide (FSI), and dicyanamide (DCA) anions. Graphical representation of the reaction mechanism between IL electrolytes and various electrodes for EDLCs, pseudocapacitors and hybrid supercapacitors is shown in Fig. 16. | ||
| Fig. 16 Graphical representation of reaction mechanism between ionic-liquid electrolytes and various electrodes for (a) EDLC (b) pseudocapacitor (c) hybrid supercapacitor. | ||
Protic ionic liquids have received limited interest as electrolytes for EDLCs due to their much lower operative cell voltage (1.2–2.5) as compared to aprotic ILs.159–161 The protic ILs are triethylammonium bis(trifluoromethylsufonyl)imide ([Et3NH][TFSI]), pyrrolidinium nitrate (PyNO3),159 diisopropyl-ethyl-ammonium bis(trifluoromethanesulfonyl)-imide ([DIPEA][TFSI]) and pyrrolidinium bis(trifluoromethanesulfonyl)imide ([Pyrr][TFSI]).160 Demarconnay et al.159 have reported the optimization of the performance of supercapacitors based on protic ILs as electrolytes and carbon as electrodes. The electrochemical performance of carbon electrodes in basic media (pH 11, 208 F g−1) was better than that in neutral media (pH 7 i.e. 121 F g−1), respectively. The voltage window with the IL used here (triethylammonium bis(trifluoromethylsufonyl)imide-NEt3H TFSI) was only 1.2 V due to the presence of a small amount of water in it. Therefore, this protic ionic liquid was dried to expand the potential voltage window and it achieved maximum voltage (2.5 V). Timperman et al.160 reported a comparative study of two protic ionic liquids as an electrolyte for EDLCs. This study describes the preparation, characterization, and application of two protic ionic liquids (pyrrolidinium bis(trifluoromethanesulfonyl)imide ([Pyrr][TFSI]) and diisopropyl-ethyl-ammonium bis(trifluoromethanesulfonyl)imide ([DIPEA][TFSI]) as the electrolyte for supercapacitors and showed that the cationic nature of protic ionic liquids was a key parameter for the electrochemical behavior of the supercapacitor.
Kurig et al.162 thoroughly studied various IL electrolytes with [EMIM]+ cations and different anions for EDLCs as shown in Fig. 17(a–d). The study showed that both electrochemical stable potential window and operative cell voltage were profoundly influenced by the structure and chemical composition of the IL anion. [EMIM][B(CN)4] (1-ethyl-3-methylimidazolium tetracyanoborate) and [EMIM][BF4] (1-ethyl-3-methylimidazolium tetrafluoroborate) ILs have shown the best performance among other ILs. A series of ILs composed of [EMIM]+ cations and various anions including NTF2, BF4, EtSO4, DCA, and OAc with graphene based EDLCs were studied by Shi et al.155 It was found that the EDLC with the [EMIM][DCA] IL electrolyte showed higher rate capability, specific capacitance, and smallest resistance due to the ion size, lower viscosity, and molecular weight compared to other ILs, but it has a narrower potential window (2.3 V) as compared to the [EMIM][BF4] IL-based EDLC (∼4 V). As a result, the [EMIM][BF4] IL-based EDLC delivered a higher specific energy density (67 W h kg−1 at 1 A g−1) than the [EMIM][DCA] IL-based ELDC (20 W h kg−1). The [BMIM][NTf2]-based supercapacitor showed a higher specific capacitance than those based on [C12MIM][NTf2], [EMIM][NTf2] and 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl) imide ([BPyr][NTf2])) as reported by Bettini et al.163 The high specific capacitance was due to the shortest alkyl chain in the case of the [BMIM][NTf2] electrolyte. The two azepanium-based ILs, (N-methyl, N-hexylazepanium bis(trifluoromethanesulfonyl)imide ([AZP16][TFSI])) and (N-methyl, N-butyl-azepanium bis(trifluoromethanesulfonyl) imide ([AZP14][TFSI]) were compared with the [PYR14][TFSI] IL by Pohlmann et al.164 for EDLCs. The results showed the all three ILs have the same operative working voltage (3.5 V); however, the [PYR14][TFSI] IL as the electrolyte for EDLCs showed a higher energy density than both the azepanium-based ILs, which was attributed to the larger cation sizes of these two ILs and thus lower accessibilities to the electrode surfaces. The ammonium-based ILs such as N,N-diethyl-N-methyl-N-(2-methoxyethyl)ammonium tetrafluoroborate ([DEME][BF4]) and [DEME][TFSI] were compared with 1 M TEABF4/PC organic electrolytes and the [EMIM][BF4] IL by Sato et al.165 for the EDLC based electrodes and found that the [DEME][BF4] IL showed a wider operational voltage window (6 V) than [EMIM][BF4] (4.5 V) ILs. The [DEME][TFSI] and [DEME][BF4] ILs showed a wider voltage window than conventional [EMIM][BF4] IL and TEABF4/PC (tetraethylammonium tetrafluoroborate in propylene carbonate) organic electrolytes as reported by Kim et al.166 Five fluorohydrogenate ILs (FHILs) namely 1-methoxymethyl-1-methylpyrrolidinium (MOMMPyr), 1-ethyl-3-methylimidazolium (EMIM), 1,3-dimethylimidazolium (DMIM), 1-butyl-3-methylimidazolium (BMIM), and 1-ethyl-1-methylpyrrolidinium (EMPyr), respectively, were studied by Senda et al.167 and they reported higher specific capacitances for these fluorohydrogenate ILs as compared to conventional [EMIM][BF4] IL and 1 M TEABF4/PC organic electrolytes. The ILs with different types of cations such as 1-butyl-3-methylpyrrolidinium tris(pentafluoroethyl) trifluorophosphate ([BMPy][FAP]), 1,2-dimethyl-3-propylimidazolium bis(trifluoromethylsulfonyl) imide ([DMPIM][TFSI]), [EMIM][BF4], and 1-ethyl-3-methylimidazolium dicyanamide ([EMIM][N(CN)2]) were studied by Sillars et al.168 and they compared them with the [BMPy][FAP] IL and 1 M TEABF4/PC organic electrolyte using EDLC electrodes. The capacitive performances of EDLCs using these ILs decreased in the following order: [EMIM][BF4] > [DMPIM][TFSI] > [BMPy][FAP] > [EMIM][N(CN)2], which was attributed to the ion size and electrolyte viscosity as shown in Fig. 18(a and b). Various types of cations in ILs were investigated by Rennie et al.169 and it was found that the introduction of an ether bond into the cation alkyl side chain could effectively increase the specific capacitance and reduce the equivalent series resistance of EDLCs.
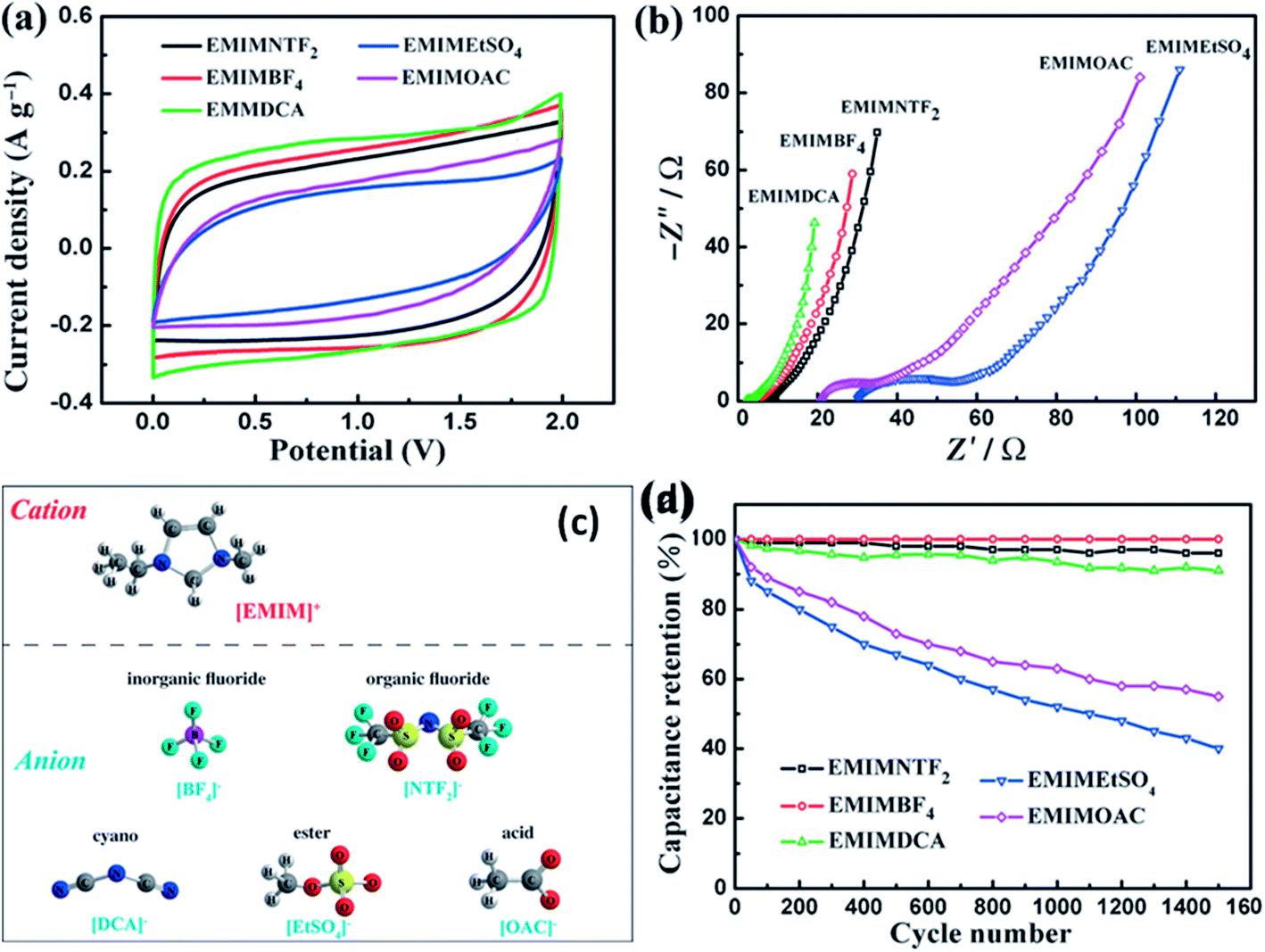 | ||
| Fig. 17 (a) CV curves of the GS electrodes in [EMIM][NTF2], [EMIM][BF4], [EMIM][DCA], [EMIM][OAc], and [EMIM]-[EtSO4] scanning with an operation voltage region of 0–2 V at 10 mV s−1 and (b) EIS plots of the GS electrodes measured in the corresponding electrolytes. (c) Molecular structures and abbreviations of the same cation and different anions of ILs used in this study. (d) The cycling stability of the GS electrodes in five IL electrolytes with the same potential window of 2 V at a current density of 1 A g−1. This figure has been adapted from ref. 155 with permission from Wiley. | ||
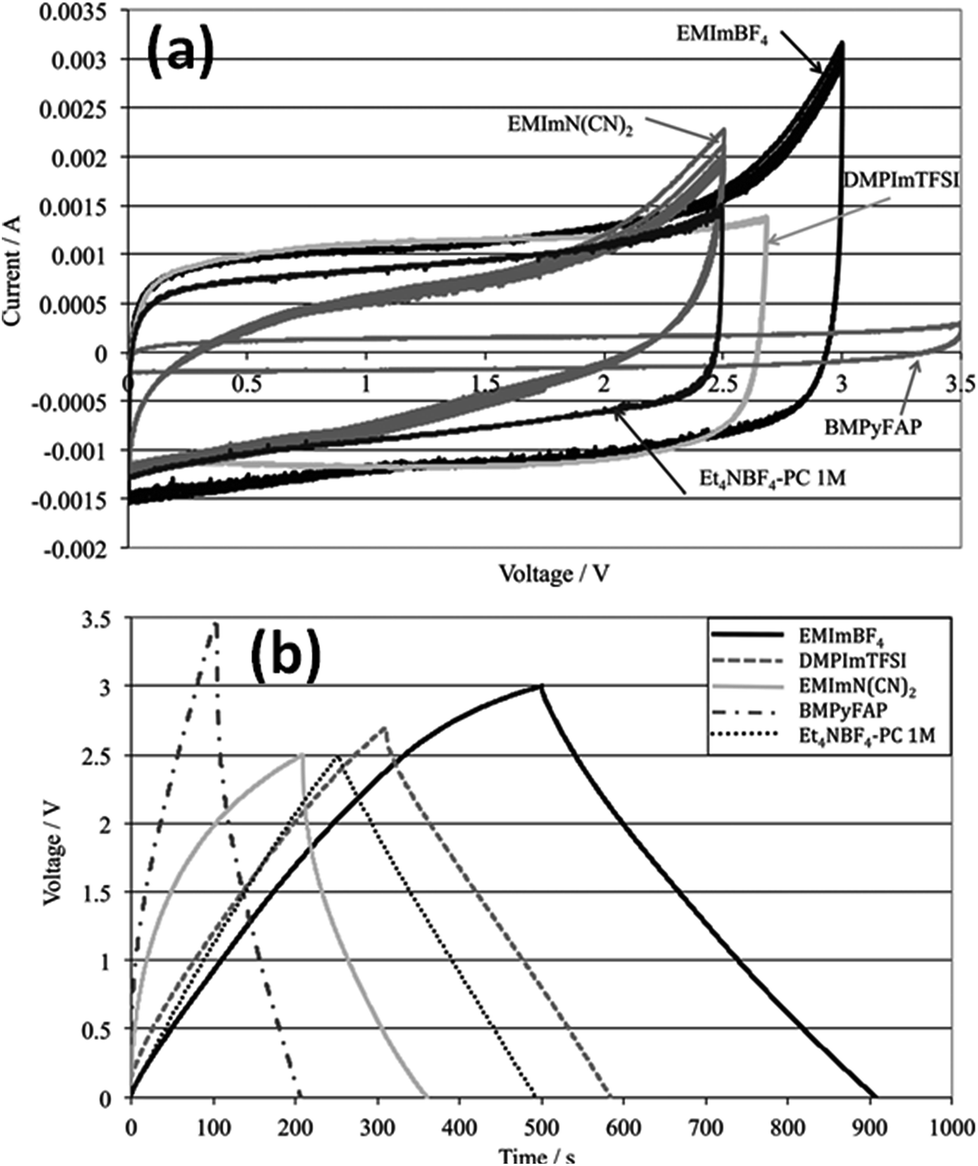 | ||
| Fig. 18 (a) CV of various electrolytes at 5 mV s−1 and (b) galvanostatic charge–discharge at 2 mA cm−2. This figure has been adapted from ref. 168 with permission from the Royal Society of Chemistry. | ||
In order to increase the conductivity and reduce the viscosity of ILs, solution mixtures containing organic solvents and ILs have been explored as electrolytes for supercapacitive devices. Imidazolium-based ILs have been studied extensively due to their relatively high conductivity compared to other used ILs. In this aspect, a series of alkyl functionalized ILs mixed with organic solvents was investigated by Orita et al.170 for EDLCs. The imidazolium cations with allyl groups (diallylimidazolium (DAIM)) and TFSA anions and imidazolium cations with saturated alkyl groups (EMIM) and TFSA anions were used as IL electrolytes for EDLCs, and it was found that the stability of the EDLC using 1.4 M[DAIM][BF4]/PC was lower than that of the one using [EMIM][BF4]/PC. The molecular sizes of [EMIM]+ and [TFSI] are similar but they have different solvated ion sizes in the ACN solvent: [TFSI]− in ACN > [EMI]+ in ACN > [EMI]+ = [TFSI], as reported by Lin et al.171 Therefore, the optimization of the size distribution and pore structure of porous carbon with respect to the ionic liquid size in an organic solvent should consider the solvated ion size, which might be different from the size of the bare IL electrolyte. Furthermore, organic solvents mixed with imidazolium-based ILs have also been explored as electrolytes for pseudocapacitors and hybrid supercapacitive devices. The introduction of DMF into the [BMIM][PF6] IL increased the capacitance and decreased the internal resistance of asymmetric AC//MnO2 supercapacitive devices as reported by Zhang et al.172 The study showed that the improvement was attributed to the improved ion mobility and electrolyte penetration as compared to the pure IL. The addition of organic solvents to the pyrrolidinium-based ionic liquids significantly decreased the viscosity and increased the conductivity of the electrolytes due to the solvation effect provided by the organic solvents.
4.4 Solid and quasi-solid-state electrolytes
Solid-state electrolyte based SCDs have attracted great interest in recent years due to the rapidly growing demand of power for wearable electronics, portable electronics, printable electronics, microelectronics, and especially flexible electronic devices.173–177 The solid-state electrolytes have lot of advantages over other electrolytes such as good ionic conducting media, simple packaging, and liquid-leakage free etc.178 The solid-state electrolytes developed for SCDs have been based on polymer electrolytes. The polymer-based solid electrolytes are divided into three types: solid polymer electrolytes (SPEs), gel polymer electrolytes (GPEs), and polyelectrolytes. The gel polymer electrolytes are also called quasi-solid state electrolytes due to the presence of a liquid phase in them.179 As shown in Fig. 19, the gel polymer electrolyte is composed of a polymer host (e.g., PEO, PEG, PVA etc.) and an aqueous electrolyte (e.g., KOH, H2SO4, K2SO4etc.) or a conducting salt dissolved in a solvent.180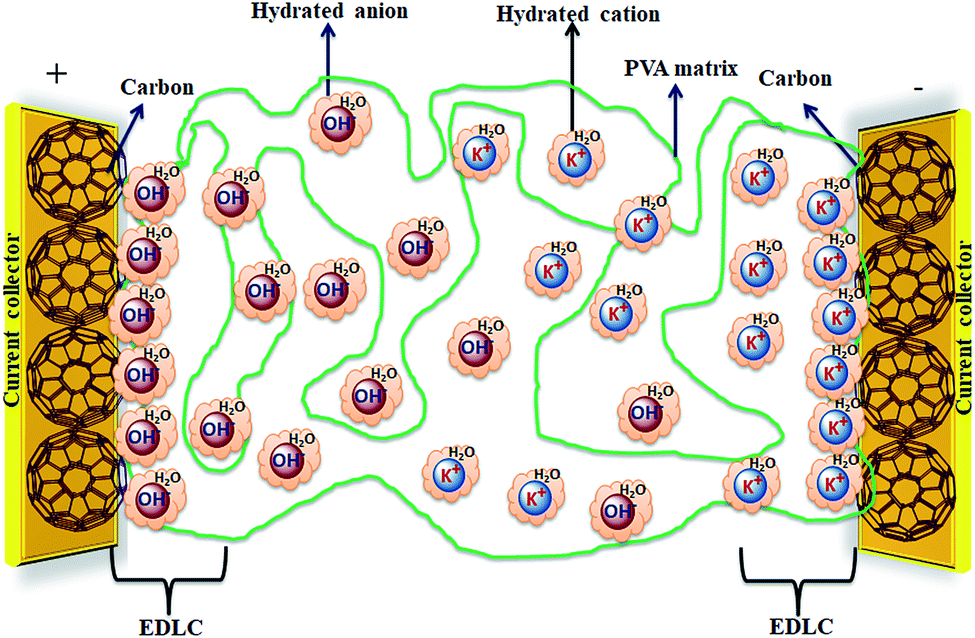 | ||
| Fig. 19 Graphical representation of a hydrogel polymer electrolyte (PVA/KOH/H2O) and a carbon electrode for EDLCs. | ||
Generally, gel polymer electrolytes have the highest ionic conductivity among these three types of solid-state electrolytes due to the presence of a liquid phase. Due to their high ionic conductivity, gel polymer electrolytes are currently dominating solid electrolyte-based SCDs, and the usage of solid polymer electrolytes (SPEs) is very limited.181 However, gel polymer electrolytes may also suffer from narrow operative temperature due to the presence of water and relatively poor mechanical strength. A number of polymer hosts have been explored for preparing gel polymer electrolytes such as poly(acrylic acid) (PAA), poly(vinyl alcohol) (PVA), poly(ethyl oxide) (PEO), potassium polyacrylate (PAAK), poly(ether ether ketone) (PEEK), poly-(methylmethacrylate) (PMMA), poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP), and poly(acrylonitrile)-block-poly(ethylene glycol)-block-poly(acrylonitrile) (PAN-b-PEG-b-PAN). The gel polymer electrolyte is also called a hydrogel polymer electrolyte due to the usage of water as a plasticizer. Some organic solvents such as EC, DMF, PC, and their mixtures (PC–EC and PC–EC–DMC) have also been used as plasticizers in gel polymer electrolytes.182–185 The gel polymer electrolytes have many significant advantages like tunable shapes and bendable structures for various applications. Various PVA-based hydrogel SCDs have been developed including stretchable SCDs, flexible supercapacitors, printable micro-SCDs, flexible micro-SCDs, on chip micro-SCDs, 3D micro-SCDs, transparent SCDs, wire or fiber-shaped SCDs, ultrathin SCDs, paper-like SCDs, and integrated SCDs with other devices.186–197 Poly(vinyl alcohol) has been extensively studied as a polymer matrix with various aqueous solutions such as alkaline (KOH), neutral, and strong acid (H2SO4 and H3PO3) electrolytes to prepare hydrogels.198 The thickness of the electrode layer for PVA/H3PO3 and H2SO4-based supercapacitors has played an important role in achieving a high capacitance as reported by Kaempgen et al.199 Ion penetration can be varied with the thickness of the electrode layer. Electrodes with various material structures such as 3D graphene networks, porous graphene ribbons, CNTs grown on carbon cloth, graphene/porous carbon aerogels, and activated carbon cloth have been studied using PVA-based hydrogel electrolytes.200–203 It was observed that these advanced carbon structures can simplify the penetration of a hydrogel electrolyte into the porous electrode and improve the performance of SCDs. Careful selection of the electrolyte solution for PVA also plays a very important role in the performance of devices. Six different PVA-based hydrogels using various electrolytes such as KOH, H2SO4, H3PO4, NaOH, NaCl, and KCl for graphene-based supercapacitors have been studied by Chen et al.204 as shown in Fig. 20(a–e). The results showed that the PVA–H3PO4 hydrogel electrolyte displayed the best capacitive performance among other PVA-based hydrogels based on electrochemical measurements. The reason for the higher capacitance in the case of the H3PO4–PVA electrolyte was the smaller ionic radius of H+ as compared to other ions (Na+, K+, OH−, and Cl−) in the gel electrolyte. Due to the smaller ionic radius, H+ ions can diffuse quickly and easily between the graphene layers. H3PO4 can also produce more free ions than NaCl or NaOH at the same molar concentration. Therefore, H3PO4–PVA gel electrolyte based supercapacitors showed a higher specific capacitance than other salt electrolytes.
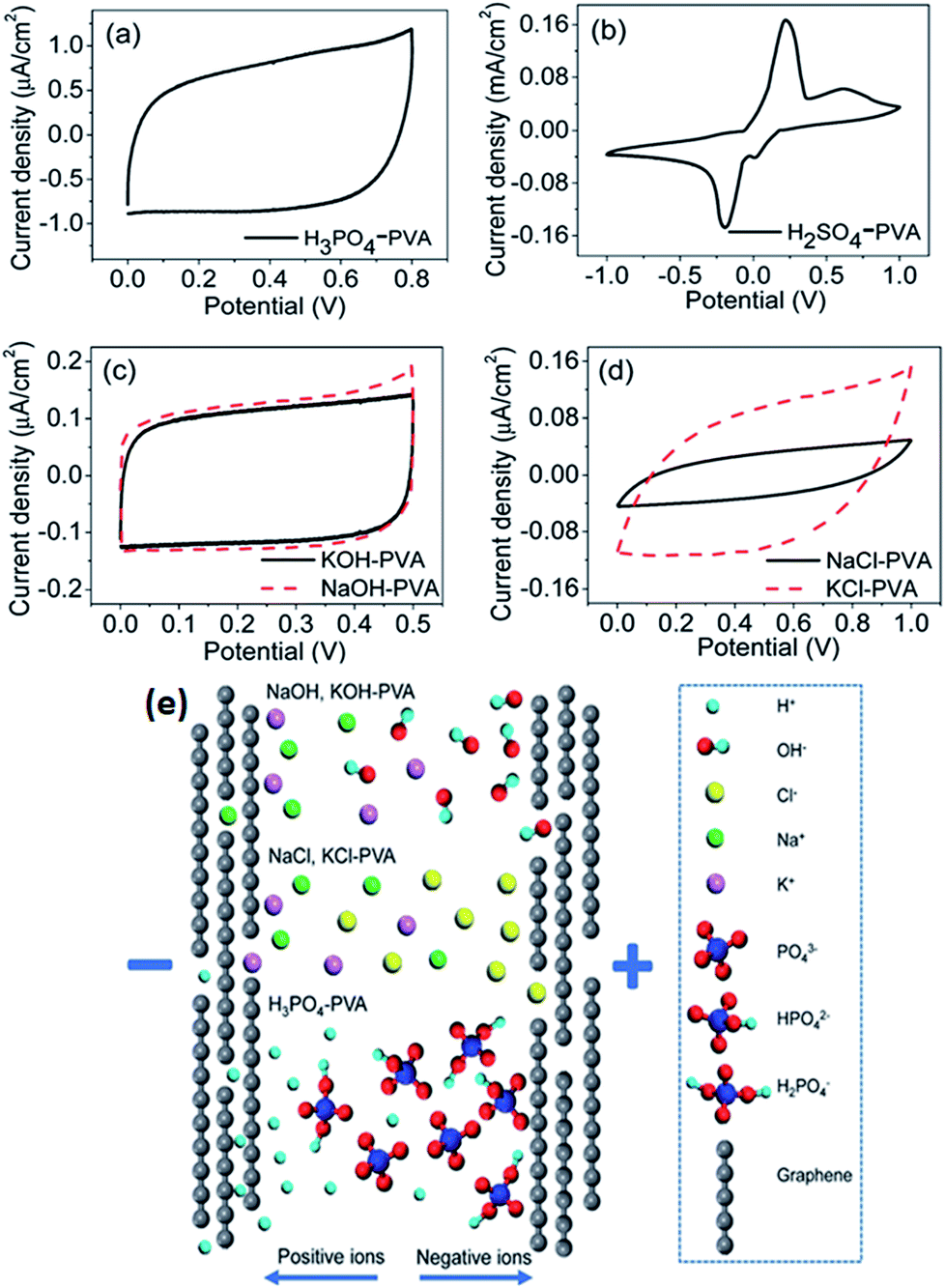 | ||
| Fig. 20 CV curves of the as-prepared solid-state supercapacitors with different electrolytes at 100 mV s−1 scan rate. (a) H3PO4–PVA and (b) H2SO4–PVA electrolyte. (c) Base gel electrolytes: KOH (black) and NaOH (red dashed) and (d) neutral gel electrolytes: NaCl (black) and KCl (red dashed). (e) Schematic illustration of ion diffusion in different electrolytes. This figure has been adapted from ref. 204 with permission from the Royal Society of Chemistry. | ||
Other polymers such as poly(acrylic acid) and poly(acrylate)-based hydrogel electrolytes have also been used for SCDs. Since the protons in their side chains can be easily withdrawn in an aqueous medium, these polymers can promote proton conduction.205–209 In this regard, Kim et al.205 have studied the performance of RuO2-based supercapacitors using various acrylic hydrogel electrolytes, such as potassium polyacrylate (PAAK), poly(acrylic acid) (PAA), and poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMPS). The specific capacitance of these electrolytes was found to decrease in the following order: PAMPS/H2O > 1 M H2SO4 (aqueous electrolyte) > PAA/H2SO4 > PAAK/H2SO4 > PAMPS/H2SO4. This highest capacitance is attributed to the most favorable proton accommodation in the side chain groups of PAMPS. The effects of acrylic gel polymer electrolytes in the polyaniline supercapacitor have been studied by Ko et al.206 as shown in Fig. 21(a–d). This study used potassium polyacrylate (PAAK), poly(acrylic acid) (PAA), and poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMPS) as gel polymer electrolytes for polyaniline based electrodes. The interaction between the acrylic polymer and H2SO4 solvent molecule was shown to be more advantageous than the H2O-based electrolyte due to the higher specific capacitance (400–500 F g−1) even at high scan rates as shown in Fig. 21(a–d). The possible interaction mechanism between solvent molecules and acrylate components is shown in Fig. 22. Organic solvents have also been used in gel polymer based electrolytes for supercapacitors to increase the working cell voltage of the device. These organic solvent based gel polymer electrolytes are also known as organogel electrolytes. Various polymer hosts such as PMMA, polyvinylpyrrolidone (PVP), PEO, PEEK, and copolymers have been reported in the literature for organogel electrolytes.210–216 Copolymers with various constituent units such as poly(acrylonitrile)-block-poly(ethylene glycol)-block-poly(acrylonitrile) (PAN-b-PEG-b-PAN) have shown to be most promising polymeric hosts in organogel electrolytes-based supercapacitors. Hsueh et al.213 stated a synergistic effect between poly(ethylene glycol) and poly(acrylonitrile) when used as the gel polymer electrolyte host. The carbon-based EDLCs using these copolymer based organogel electrolytes could deliver higher energy and power densities (20 W h kg−1 at 10 kW kg−1) than EDLCs using organic liquid electrolytes. The typically studied organic solvents include DMC, PC, EC, CAN, DMSO, DMF, and their mixtures; they increased the operational voltage window up to 2.5–3 V,217 which was much higher than hydrogel electrolyte-based supercapacitors. The methacrylate-based organogel electrolyte improved the operative voltage window up to 4 V as reported by Schroeder et al.217 and further increased the energy density. Considerable research efforts have been put into learning about the ionic liquid-based gel polymer (ionogel electrolytes) electrolytes for the improvement of thermal stability and the electrochemical stable potential window. Presently, the variety of host polymers studied for ionogel electrolytes include PEO, PMMA, PVA, poly(ethylene glycol)diacrylate, and PVDF-HFP.218–222 The highly transparent and stretchable PMMA/[BMIM][TFSI] ionogel was prepared by Tamilarasan et al.219 which showed four-fold stretchability compared to conventional gel polymer electrolytes. Yamagata et al.223 also reported a positive effect of a chitosan host on the performance of ionogel-based EDLCs. The incorporation of nanoscale inorganic fillers into the polymer matrix of solid polymer electrolytes to form a composite solid polymer electrolyte is one of the most effective approaches to improve the ionic conductivity. The studies224 on composite solid polymer electrolytes have demonstrated that the improvement of ionic conductivity in composite solid polymer electrolytes originates from the interactions at the interface between the polymer matrix and ceramic fillers.
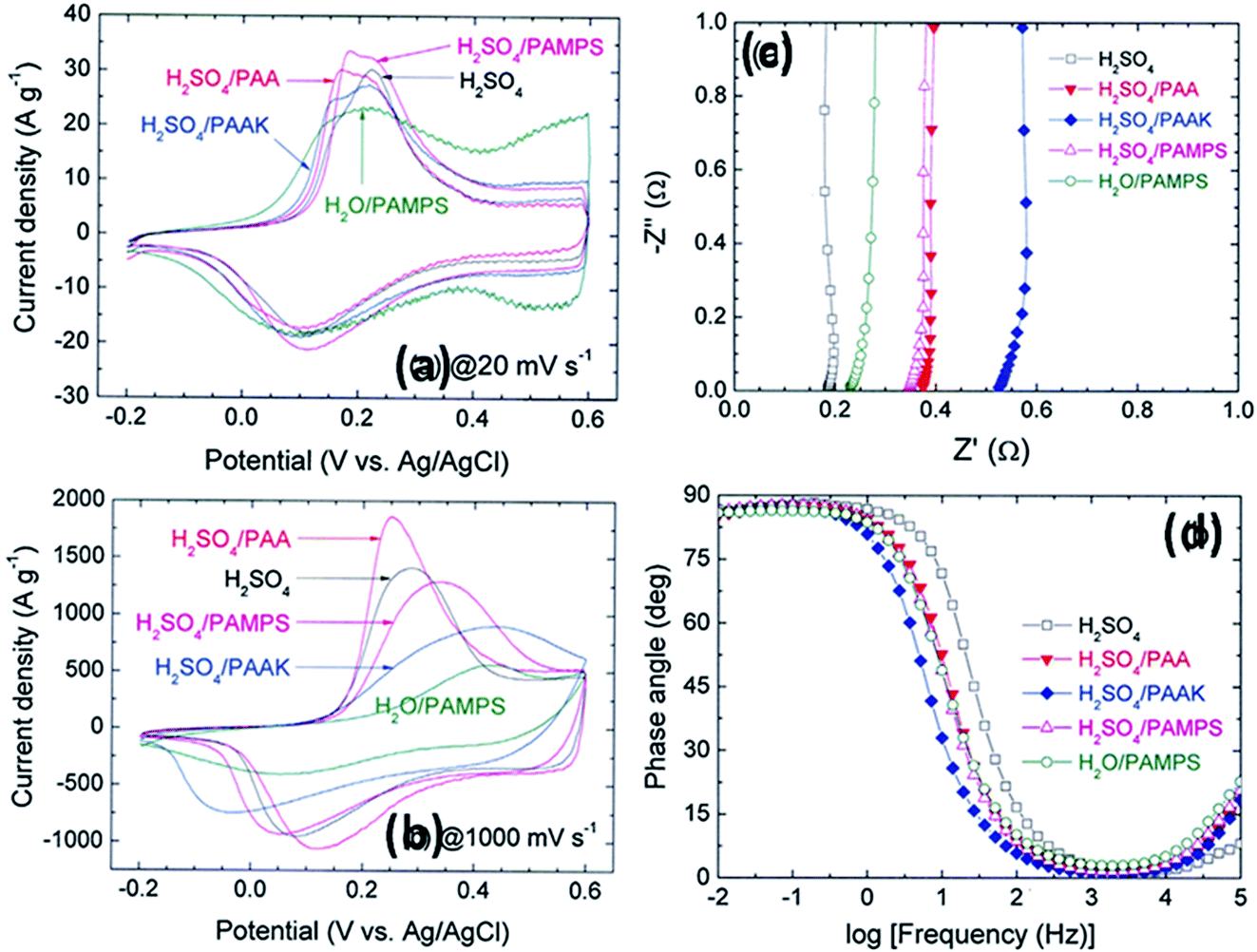 | ||
| Fig. 21 CV of the ePAn electrode for the supercapacitor adopting H2SO4, H2O/PAMPS, H2SO4/PAMPS, H2SO4/PAA, and H2SO4/PAAK as the electrolyte system, at scan rates of (a) 20 and (b) 1000 mV s−1. (c) Nyquist and (d) Bode plots for the ePAn electrode for the supercapacitor adopting various acrylic gel polymer electrolytes. This figure has been adapted from ref. 206 with permission from Elsevier. | ||
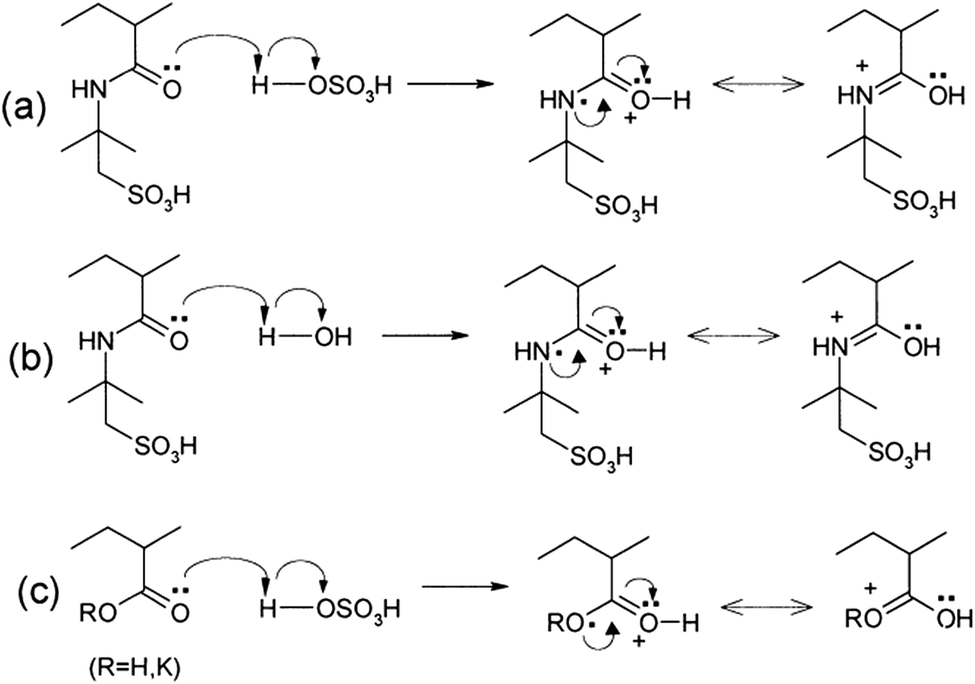 | ||
| Fig. 22 Possible interaction mechanisms between the acrylate components and solvent molecules in the acrylic gel polymer electrolytes for the H2SO4/PAMPS electrolyte. This figure has been adapted from ref. 206 with permission from Elsevier. | ||
4.5 Redox-active electrolytes
The capacitance of supercapacitors can be increased by inducing pseudocapacitive contribution from the redox-active electrolytes. The pseudocapacitance is not only contributed by the pseudocapacitive electrode materials but also can be contributed by the electrolytes (redox mediator in the electrolyte).225,226 For example, an iodide/iodine redox pair was used by Lota et al.227 as a redox-active aqueous electrolyte for carbon-based supercapacitor. Due to the pseudocapacitive capacitance contribution from the electrolyte, a high capacitance (1840 F g−1) has been reported. However, the high capacitance value was observed only at the positive electrode (with a narrow potential range). With the decrease in the van der Waals radius of alkali metals, the capacitance of the positive electrode decreased in the following order: RbI (2272 F g−1) > KI (1078 F g−1) > NaI (492 F g−1) > LiI (300 F g−1).228 To increase the total capacitance, Frackowiak et al.229 used 1 M VOSO4 for the negative electrode and 1 M KI aqueous electrolyte for the positive electrode. A high pseudocapacitance contribution was made at the negative electrode by the VOSO4 electrolyte due to faradaic reactions. Silicotungstic acid and phosphotungstic acid (heteropoly acids) were explored as the redox mediators for redox-active electrolytes. These acids provide multiple rapid electron transfer redox processes and high proton conductivities.230–232 The phosphotungstic acid-based electrolyte not only provides pseudocapacitance for carbon based supercapacitors but also increases the voltage window up to 1.6 V as reported by Suárez-Guevara et al.232 Cu2+ reduction by the surface oxygen group on the carbon electrode enhances the specific capacitance as reported by Mai et al.233 In addition, organic redox mediators as pseudocapacitive sources have been studied including methylene blue, hydroquinone, p-phenylenediamine, indigo carmine, m-phenylenediamine, lignosulfonates, humic acid, and sulfonated polyaniline.234–242 Chen et al.235 reported that the fast self-discharge in two electrodes was due to the migration of a redox-active electrolyte between them. Organic and IL-based electrolytes have been shown in the literature to achieve a high cell voltage and high energy density.243–246 The introduction of redox-active polyfluorododecaborate cluster ions into an organic PC–DMC mixture solvent could effectively contribute additional pseudocapacitance to the total capacitance of the carbon-based supercapacitors as reported by Ionica-Bousquet et al.245 A tetraethylammonium undecafluorododecaborate/PC–DMC electrolyte was able to provide overcharge protection for supercapacitors as compared to the TEABF4/PC–DMC organic electrolyte, due to the presence of redox-active anions in the former electrolyte. The addition of 5 wt% 1-ethyl-3-methylimidazolium iodide ([EMIM][I]) to the [EMIM][BF4] IL could result in a nearly 50% increase in the specific capacitance of the supercapacitor when compared to a bare [EMIM][BF4] IL electrolyte as reported by Tooming et al.243 An N-ethyl-N-methylpyrrolidinium fluorohydrogenate IL electrolyte could significantly contribute extra specific capacitance to the supercapacitor through the redox reaction of the electrolyte as reported by Taniki et al.246The charge storage mechanisms in the porous carbon electrode of a supercapacitor with a redox electrolyte are shown in Fig. 23.48 In Fig. 23, R and O are the reduced and oxidized states of the redox species in the electrolyte. The redox species in the bulk solution (R bulk or O bulk) need to first enter the pores of the electrode to participate in the charge storage process, accompanied by the process (1) or (1′). These processes represent the equilibria of de-solvation and solvation of the species when entering or exiting the pores. The redox species in the pore (Rpore and Opore) must reach the transition state (R* or O*) via (2′) or (2) as in an electrode reaction before electron transfer reactions. The conversion from the transition states to adsorbed states (Rads and Oads) is shown by processes (3′) and (3) which are a necessary process for the application in supercapacitors. The conversion between the adsorbed redox species is caused by electron transfer via (4) on the internal and external surfaces of the electrode, which is responsible for the improved charge storage capacity. The redox species (R* and O*) may also undergo an electron transfer reaction via (5′) or (5) without involving adsorption once in the transition state. At the time of electron transfer, the adsorbed species (Rads or Oads) is transformed via (5′) or (5) to a soluble product (Rpore or Opore) that can diffuse through the pores and enter the bulk electrolyte. The electron transfer reaction occurs not only inside the pores, but also on the outer surface of the electrode via (6′) or (6). Field-emission scanning electron microscopy images of various types of supercapacitive electrodes are shown in Fig. 24.
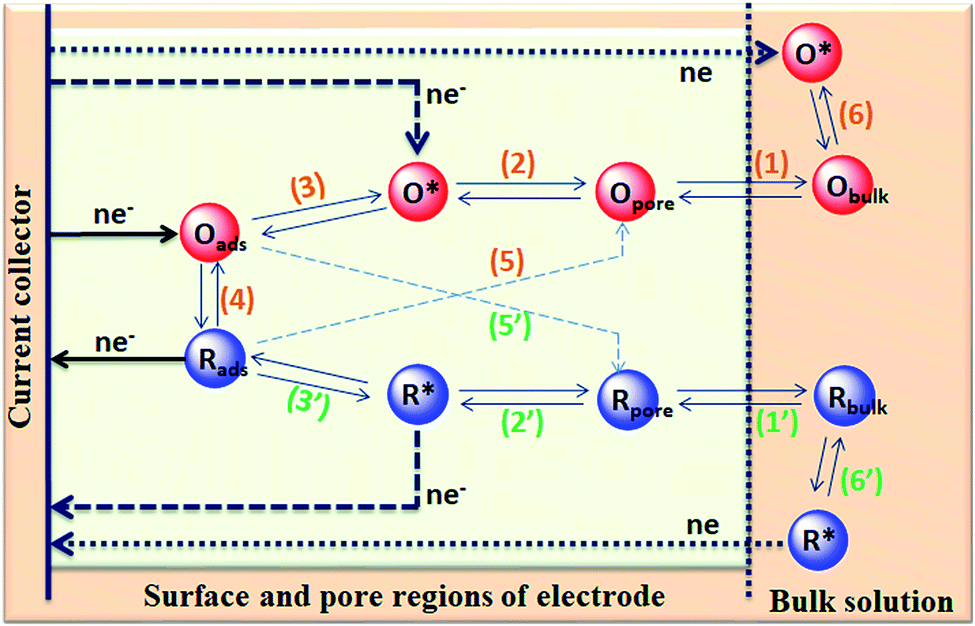 | ||
| Fig. 23 Illustration of charge storage mechanisms in the porous carbon electrode of a supercapacitor with a redox electrolyte. | ||
 | ||
| Fig. 24 FE-SEM images of various electrode materials: (a and b) porous carbon, (c and d) SnO2/TiO2 composite flowers and (e and f) Cu2O nanowires. (a and b) have been adapted from ref. 81 with permission from Elsevier. (c and d) have been adapted from ref. 180 and (e and f) have been adapted from ref. 29 with permission from the American Chemical Society. | ||
5. Challenges of electrochemical supercapacitive electrolytes
Electrolytes have been recognized as one of the very crucial components in the electrochemical supercapacitor performance as discussed earlier. The major challenges in electrochemical supercapacitor development are to increase the cycle life and energy and power densities and safety issues. The main factors which affect both performance and practical applications of electrochemical supercapacitors are ionic conductivity, ion mobility, operating voltage window, operating temperature range, and electrochemical and thermal stabilities of the electrolytes. Although good progress has been made in electrochemical supercapacitor electrolytes, there are various challenges, which obstruct the commercial application maturity of the technology. Some challenges are discussed below:(1) The charging–discharging mechanisms of the newly developed electrode and electrolyte systems (such as solid-state, ionic liquid and redox-active electrolytes) have not been clearly addressed in majority of the reports. The compatibility or optimization of the interaction between these newly developed electrolytes and the electrode material is essential for better performance of electrochemical supercapacitors. However, some in situ experimental advancements and significant theoretical studies have been achieved, but still some controversies exist about the capacitance at the ionic liquid electrolyte–electrode interface, effects of organic solvent addition on the EDL structure and effects of temperature. There is also a lack of complete understanding of the dynamics of electrolyte ions in more complex electrode structures (e.g., hybrid nanostructures) during the charging–discharging process. Furthermore, compared to the fundamental studies on the EDL charge storage mechanisms, fundamental understanding such as theoretical modelling for pseudocapacitors and hybrid EE devices with certain electrolytes is rather limited.247
(2) The energy and power densities of the electrochemical supercapacitors are directly affected by the operating cell voltage of the electrolytes. The electrolytes with higher electrochemically stable potential window values can increase the cell voltage of the supercapacitors which further can improve the energy density. However, this higher cell voltage often degrades other properties of electrolytes. For example, ionic liquids and organic electrolytes have much higher cell voltages (up to 4 V) compared to aqueous electrolytes but simultaneously these have very low ionic conductivity. Furthermore, these non-aqueous electrolytes (ionic liquids and organic) also showed a smaller specific capacitance value compared to aqueous electrolytes. Therefore, it is a big challenge to develop electrochemical supercapacitors with improved energy densities without deteriorating other properties.
(3) Ionic liquids and organic electrolytes are suitable candidates for increasing the energy density of electrochemical supercapacitors but due to their lower ionic conductivity, the equivalent series resistance is also higher than that of aqueous electrolytes. As a result, the application of electrolytes with a higher electrochemically stable potential window of electrochemical supercapacitors often leads to increased ESR values and decreases the performance of the device. The flexible and all solid-state supercapacitor also suffers from poor mechanical properties and low electrochemical performances.
(4) The absence of appropriate and standardized characterization methods to evaluate the performance of electrolytes for electrochemical supercapacitors: there is an urgent need to establish standardized methods suitable to characterize the performance of many new electrolytes (such as ionic liquids, solid-state and redox-active electrolytes). It is also not easy to identify suitable electrolytes from different literature studies because the performance metrics are usually obtained under different conditions. Additionally, the electrochemical supercapacitor energy and power densities and gravimetric capacitance are generally reported based on the mass of active electrode materials. However, the electrolytes also contribute a lot to the total mass which should not be neglected.
(5) Compatibility between the electrolyte–electrode materials and their effect on overall electrochemical supercapacitor performance: for achieving a high performance for electrochemical supercapacitors, proper compatibility between the electrolyte–electrode materials is very crucial. For example, Largeot et al.248 and Chmiola et al.13 reported that the specific capacitance can be improved significantly when the average pore size of the carbon electrode was close to the electrolyte ion size (TEABF4/CAN and ionic liquids). However, Centeno et al.249 reported that the specific capacitance was relatively constant when the pore size of porous carbons and TEABF4/ACN electrolytes was almost the same. Additionally, with the development of various new electrolytes (such as ionic liquid mixtures, ionic liquid-organic solvent mixtures and solid-state electrolytes), the optimization of matching between these electrolytes and new electrode materials is important.
6. Summary and outlook
This paper provides a complete overview of the reaction mechanism, electrolyte factors which affect electrochemical supercapacitor performance, requirements for designing better electrolytes and challenges of electrochemical supercapacitor electrolytes. Reaction chemistry of various types of electrolytes explored and reported in the literature is summarized and they are classified into liquid (aqueous, organic and ionic liquids), solid state and redox-active electrolytes. The effects of the electrolyte properties such as the ionic conductivity, mobility, radius of bare and hydrated ions, viscosity, dielectric constant of solvents, and thermal stability on the electrochemical supercapacitor performance such as the specific capacitance, energy and power densities, rate performance and cycling stability are reviewed in depth. Considerable achievements have been made in this field but still some significant challenges exist as discussed above. Several future research directions are given below to overcome these challenges:(1) The fundamental understanding of the reaction chemistry/compatibility between electrode–electrolyte can be improved by an in-depth study of theoretical and experimental investigations. It is essential to fundamentally understand the mechanisms of electrolyte ion dynamics and charge storage mechanism during the charging–discharging process. This can be accomplished by using both in situ characterization methods such as FTIR, NMR and Raman spectroscopy at the time of experimental approach and theoretical modelling (molecular/electronic level). Additionally, the fundamental understanding of the interaction between new electrolytes and hybrid electrode materials is also fundamental. Such a fundamental understanding will not only offer a direction for developing innovative electrolytes but also simplify the development of electrode materials to match with exact types of electrolytes. To reduce electrolyte degradation, it is also important to understand the mechanism of degradation of electrolytes, which can also be achieved by both experimental and theoretical modelling approaches. A variety of instrumental analysis methods (in situ characterization such as Raman microscopy and differential electrochemical mass spectrometry) can be used to characterize the electrolytes during lifetime tests.
(2) The charge capacity of the electrochemical supercapacitors can be enhanced by utilizing the pseudocapacitive contribution from both electrolytes (redox-active electrolytes) and pseudocapacitive electrodes materials.250,251 Hydrogel redox-active electrolytes can be highly promising candidates for high energy-density electrochemical supercapacitors because of their attractive advantages such as high safety, low cost and simplified fabrication procedures as compared to organic electrolytes.
(3) More fundamental experiments are required to fully understand the relationship between the pore size and surface-area-normalized capacitance regarding the specific capacitance. This would help optimize the matching between the pore size of carbonaceous materials and electrolyte ion size. Therefore, it is important to use more reliable methods to characterize the actual surface area of the electrode materials.
(4) The development of standard methods to evaluate the performance of different electrolytes is also necessary. At the time of characterizing the performance of electrochemical supercapacitors, it is also necessary to know the volume and mass of the electrolyte. Standard specifications and methods are required to evaluate the mechanical properties and the performance of flexible electrochemical supercapacitors using solid electrolytes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Bhupender Pal acknowledges the Research & Innovation Department of Universiti Malaysia Pahang (http://ump.edu.my) for awarding a Postdoctoral Fellowship. This project is funded under Flagship Strategic Leap 3 (RDU 172201) of Universiti Malaysia Pahang. V. T. also thanks the University of Calgary for the support of next generation electrochemical energy storage research.References
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- C. Zhou, S. Bag and V. Thangadurai, ACS Energy Lett., 2018, 3, 2181–2198 CrossRef CAS.
- S. Palakkathodi Kammampata and V. Thangadurai, Ionics, 2018, 24, 639–660 CrossRef CAS.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- V. Aravindan, W. Chuiling, M. V. Reddy, G. V. S. Rao, B. V. R. Chowdari and S. Madhavi, Phys. Chem. Chem. Phys., 2012, 14, 5808–5814 RSC.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- K. Naoi, S. Ishimoto, J. I. Miyamoto and W. Naoi, Energy Environ. Sci., 2012, 5, 9363–9373 RSC.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
- V. Aravindan, M. V. Reddy, S. Madhavi, S. G. Mhaisalkar, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2011, 196, 8850–8854 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816–1300859 CrossRef.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- M. Sathiya, A. S. Prakash, K. Ramesha, J. M. Tarascon and A. K. Shukla, J. Am. Chem. Soc., 2011, 133, 16291–16299 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P. L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- W. Shimizu, S. Makino, K. Takahashi, N. Imanishi and W. Sugimoto, J. Power Sources, 2013, 241, 572–577 CrossRef CAS.
- X. Wang and G. Shen, Nano Energy, 2015, 15, 104–115 CrossRef.
- W. Zuo, R. Li, C. Zhou, Y. Li, J. Xia and J. Liu, Adv. Sci., 2017, 4 CAS.
- J. W. Long, D. Bélanger, T. Brousse, W. Sugimoto, M. B. Sassin and O. Crosnier, MRS Bull., 2011, 36, 513–522 CrossRef CAS.
- J. R. Miller, R. A. Outlaw and B. C. Holloway, Science, 2010, 329, 1637–1639 CrossRef CAS PubMed.
- H. Wang, Y. Liang, M. Gong, Y. Li, W. Chang, T. Mefford, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, Nat. Commun., 2012, 3, 1–8 Search PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- B. Pal, B. L. Vijayan, S. G. Krishnan, M. Harilal, W. J. Basirun, A. Lowe, M. M. Yusoff and R. Jose, J. Alloys Compd., 2018, 740, 703–710 CrossRef CAS.
- M. Harilal, S. G. Krishnan, B. Pal, M. V. Reddy, M. H. Ab Rahim, M. M. Yusoff and R. Jose, Langmuir, 2018, 34, 1873–1882 CrossRef CAS PubMed.
- J. Deng, Y. Zhang, Y. Zhao, P. Chen, X. Cheng and H. Peng, Angew. Chem., Int. Ed., 2015, 54, 15419–15423 CrossRef CAS PubMed.
- Y. Huang, M. Zhu, Y. Huang, Z. Pei, H. Li, Z. Wang, Q. Xue and C. Zhi, Adv. Mater., 2016, 8344–8364 CrossRef CAS PubMed.
- V. Aravindan, M. V. Reddy, S. Madhavi, G. V. S. Rao and B. V. R. Chowdari, Nanosci. Nanotechnol. Lett., 2012, 4, 724–728 CrossRef CAS.
- M. V. Reddy, R. Jose, A. Le Viet, K. I. Ozoemena, B. V. R. Chowdari and S. Ramakrishna, Electrochim. Acta, 2014, 128, 198–202 CrossRef CAS.
- Y. Wu, R. Balakrishna, M. V. Reddy, A. S. Nair, B. V. R. Chowdari and S. Ramakrishna, J. Alloys Compd., 2012, 517, 69–74 CrossRef CAS.
- H. Li, Y. Hou, F. Wang, M. R. Lohe, X. Zhuang, L. Niu and X. Feng, Adv. Energy Mater., 2017, 7, 1601847–1601853 CrossRef.
- D. P. Dubal, N. R. Chodankar, D. H. Kim and P. Gomez-Romero, Chem. Soc. Rev., 2018, 47, 2065–2129 RSC.
- L. Liu, Z. Niu and J. Chen, Chin. Chem. Lett., 2018, 29, 571–581 CrossRef CAS.
- P. Yang and W. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- T. Xiong, T. L. Tan, L. Lu, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2018, 8, 1702630–1702640 CrossRef.
- B. K. Kim, S. Sy, A. Yu and J. Zhang, Electrochemical Supercapacitors for Energy Storage and Conversion, 2015 Search PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- A. S. Ulihin, Y. G. Mateyshina and N. F. Uvarov, Solid State Ionics, 2013, 251, 62–65 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- Q. Dou, S. Lei, D. W. Wang, Q. Zhang, D. Xiao, H. Guo, A. Wang, H. Yang, Y. Li, S. Shi and X. Yan, Energy Environ. Sci., 2018, 11, 3212–3219 RSC.
- L. Xia, L. Yu, D. Hu and G. Z. Chen, Mater. Chem. Front., 2017, 1, 584–618 RSC.
- X. Cheng, J. Pan, Y. Zhao, M. Liao and H. Peng, Adv. Energy Mater., 2018, 8, 1702184–1702200 CrossRef.
- B. Akinwolemiwa, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2015, 162, A5054–A5059 CrossRef CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- A. Ponrouch, E. Marchante, M. Courty, J. M. Tarascon and M. R. Palacín, Energy Environ. Sci., 2012, 5, 8572–8583 RSC.
- A. Bhide, J. Hofmann, A. Katharina Dürr, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2014, 16, 1987–1998 RSC.
- C. H. Wang, Y. W. Yeh, N. Wongittharom, Y. C. Wang, C. J. Tseng, S. W. Lee, W. S. Chang and J. K. Chang, J. Power Sources, 2015, 274, 1016–1023 CrossRef CAS.
- J. Feng, Z. Zhang, L. Li, J. Yang, S. Xiong and Y. Qian, J. Power Sources, 2015, 284, 222–226 CrossRef CAS.
- F. Wu, N. Zhu, Y. Bai, L. Liu, H. Zhou and C. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 21381–21386 CrossRef CAS PubMed.
- C. R. Pope, M. Kar, D. R. MacFarlane, M. Armand, M. Forsyth and L. A. O'Dell, ChemPhysChem, 2016, 17, 3187–3195 CrossRef CAS PubMed.
- J. Serra Moreno, G. Maresca, S. Panero, B. Scrosati and G. B. Appetecchi, Electrochem. Commun., 2014, 43, 1–4 CrossRef CAS.
- N. Wongittharom, T.-C. Lee, C.-H. Wang, Y.-C. Wang and J.-K. Chang, J. Mater. Chem. A, 2014, 2, 5655 RSC.
- G. Kamath, R. W. Cutler, S. A. Deshmukh, M. Shakourian-Fard, R. Parrish, J. Huether, D. P. Butt, H. Xiong and S. K. R. S. Sankaranarayanan, J. Phys. Chem. C, 2014, 118, 13406–13416 CrossRef CAS.
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine, X.-Z. Liao and Z.-F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC.
- Y. Cai, B. Zhao, J. Wang and Z. Shao, J. Power Sources, 2014, 253, 80–89 CrossRef CAS.
- S. Tian, L. Qi, M. Yoshio and H. Wang, J. Power Sources, 2014, 256, 1–11 CrossRef.
- W. H. Qu, F. Han, A. H. Lu, C. Xing, M. Qiao and W. C. Li, J. Mater. Chem. A, 2014, 2, 6549–6557 RSC.
- A. Janes, T. Thomberg, J. Eskusson and E. Lust, J. Electrochem. Soc., 2013, 160, A1025–A1030 CrossRef CAS.
- M. Y. Cho, M. H. Kim, H. K. Kim, K. B. Kim, J. R. Yoon and K. C. Roh, Electrochem. Commun., 2014, 47, 5–8 CrossRef CAS.
- S.-M. Oh, S.-T. Myung, C. S. Yoon, J. Lu, J. Hassoun, B. Scrosati, K. Amine and Y.-K. Sun, Nano Lett., 2014, 14, 1620–1626 CrossRef CAS PubMed.
- C. Ding, T. Nohira, K. Kuroda, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 238, 296–300 CrossRef CAS.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 1–5 Search PubMed.
- N. Tanibata, K. Noi, A. Hayashi and M. Tatsumisago, RSC Adv., 2014, 4, 17120–17123 RSC.
- J. Chen, Z. Huang, C. Wang, S. Porter, B. Wang, W. Lie and H. K. Liu, Chem. Commun., 2015, 51, 9809–9812 RSC.
- I. Hasa, S. Passerini and J. Hassoun, J. Power Sources, 2016, 303, 203–207 CrossRef CAS.
- L. Chancelier, A. O. Diallo, C. C. Santini, G. Marlair, T. Gutel, S. Mailley and C. Len, Phys. Chem. Chem. Phys., 2014, 16, 1967–1976 RSC.
- J. Feng, Y. An, L. Ci and S. Xiong, J. Mater. Chem. A, 2015, 3, 14539–14544 RSC.
- J. S. Gnanaraj, E. Zinigrad, L. Asraf, H. E. Gottlieb, M. Sprecher, M. Schmidt, W. Geissler and D. Aurbach, J. Electrochem. Soc., 2003, 150, A1533 CrossRef CAS.
- Y. Wang, J. Jiang and J. R. Dahn, Electrochem. Commun., 2007, 9, 2534–2540 CrossRef CAS.
- J. Jiang and J. R. Dahn, Electrochim. Acta, 2004, 49, 4599–4604 CrossRef CAS.
- L. Xia, Y. Xia and Z. Liu, J. Power Sources, 2015, 278, 190–196 CrossRef CAS.
- E. Zinigrad, L. Larush-Asraf, G. Salitra, M. Sprecher and D. Aurbach, Thermochim. Acta, 2007, 457, 64–69 CrossRef CAS.
- G.-Y. Kim and J. R. Dahn, J. Electrochem. Soc., 2014, 161, A1394–A1398 CrossRef CAS.
- B. E. Conway, J. Electroanal. Chem., 1981, 123, 81–94 CrossRef CAS.
- P. Debye and E. Hückel, Phys. Z., 1923, 24, 185–206 CAS.
- B. Pal, S. G. Krishnan, B. L. Vijayan, M. Harilal, C. C. Yang, F. I. Ezema, M. M. Yusoff and R. Jose, J. Electroanal. Chem., 2018, 817, 217–225 CrossRef CAS.
- M. Galiński, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- N. Jabeen, A. Hussain, Q. Xia, S. Sun, J. Zhu and H. Xia, Adv. Mater., 2017, 29, 1700804–1700813 CrossRef PubMed.
- J. Iqbal, A. Numan, S. Rafique, R. Jafer, S. Mohamad, K. Ramesh and S. Ramesh, Electrochim. Acta, 2018, 278, 72–82 CrossRef CAS.
- L. Zhao, Y. Qiu, J. Yu, X. Deng, C. Dai and X. Bai, Nanoscale, 2013, 5, 4902–4909 RSC.
- H. Xia, Y. Shirley Meng, G. Yuan, C. Cui and L. Lu, Electrochem. Solid-State Lett., 2012, 15, A60 CrossRef CAS.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Kang, J. Power Sources, 2013, 224, 86–92 CrossRef CAS.
- Y. Lin, X. Wang, G. Qian and J. J. Watkins, Chem. Mater., 2014, 26, 2128–2137 CrossRef CAS.
- S. Li, L. Qi, L. Lu and H. Wang, RSC Adv., 2012, 2, 3298–3308 RSC.
- S. Dhibar, P. Bhattacharya, G. Hatui, S. Sahoo and C. K. Das, ACS Sustainable Chem. Eng., 2014, 2, 1114–1127 CrossRef CAS.
- L. Demarconnay, E. Raymundo-Piñero and F. Béguin, Electrochem. Commun., 2010, 12, 1275–1278 CrossRef CAS.
- M. P. Bichat, E. Raymundo-Piñero and F. Béguin, Carbon, 2010, 48, 4351–4361 CrossRef CAS.
- C. Ramasamy, J. Palma Del Val and M. Anderson, J. Power Sources, 2014, 248, 370–377 CrossRef CAS.
- D. Jiménez-Cordero, F. Heras, M. A. Gilarranz and E. Raymundo-Piñero, Carbon, 2014, 71, 127–138 CrossRef.
- Q. Abbas, D. Pajak, E. Fr
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) ckowiak and F. Béguin, Electrochim. Acta, 2014, 140, 132–138 CrossRef CAS.
ckowiak and F. Béguin, Electrochim. Acta, 2014, 140, 132–138 CrossRef CAS. - Q. Wang, J. Yan, Y. Wang, T. Wei, M. Zhang, X. Jing and Z. Fan, Carbon, 2014, 67, 119–127 CrossRef CAS.
- Y. Mun, C. Jo, T. Hyeon, J. Lee, J. Lee, K. S. Ha, K. W. Jun, S. H. Lee, S. W. Hong, H. I. Lee and S. Yoon, Carbon, 2013, 64, 391–402 CrossRef CAS.
- Z. Jin, X. Yan, Y. Yu and G. Zhao, J. Mater. Chem. A, 2014, 2, 11706–11715 RSC.
- Q. Chen, Y. Hu, C. Hu, H. Cheng, Z. Zhang, H. Shao and L. Qu, Phys. Chem. Chem. Phys., 2014, 16, 19307–19313 RSC.
- L. F. Chen, Z. H. Huang, H. W. Liang, H. L. Gao and S. H. Yu, Adv. Funct. Mater., 2014, 24, 5104–5111 CrossRef CAS.
- H. J. Denisa, A. M. Puziy, O. I. Poddubnaya, S. G. Fabian, J. M. D. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026–5027 CrossRef PubMed.
- M. Jana, P. Khanra, N. C. Murmu, P. Samanta, J. H. Lee and T. Kuila, Phys. Chem. Chem. Phys., 2014, 16, 7618–7626 RSC.
- L. Deng, J. Wang, G. Zhu, L. Kang, Z. Hao, Z. Lei, Z. Yang and Z. H. Liu, J. Power Sources, 2014, 248, 407–415 CrossRef CAS.
- X. Liu, P. Shang, Y. Zhang, X. Wang, Z. Fan, B. Wang and Y. Zheng, J. Mater. Chem. A, 2014, 2, 15273–15278 RSC.
- S. S. Shinde, G. S. Gund, D. P. Dubal, S. B. Jambure and C. D. Lokhande, Electrochim. Acta, 2014, 119, 1–10 CrossRef CAS.
- Q. Wang, J. Yan, Z. Fan, T. Wei, M. Zhang and X. Jing, J. Power Sources, 2014, 247, 197–203 CrossRef CAS.
- Z. Zhang, Z. Zhou, H. Peng, Y. Qin and G. Li, Electrochim. Acta, 2014, 134, 471–477 CrossRef CAS.
- H. Wang, X. Sun, Z. Liu and Z. Lei, Nanoscale, 2014, 6, 6577–6584 RSC.
- B. S. Mao, Z. Wen, Z. Bo, J. Chang, X. Huang and J. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 9881–9889 CrossRef CAS PubMed.
- C. Feng, J. Zhang, Y. He, C. Zhong, W. Hu, L. Liu and Y. Deng, ACS Nano, 2015, 9, 1730–1739 CrossRef CAS PubMed.
- L. Li, S. Peng, Y. Cheah, P. Teh, J. Wang, G. Wee, Y. Ko, C. Wong and M. Srinivasan, Chem.–Eur. J., 2013, 19, 5892–5898 CrossRef CAS PubMed.
- I. I. Misnon, R. A. Aziz, N. K. M. Zain, B. Vidhyadharan, S. G. Krishnan and R. Jose, Mater. Res. Bull., 2014, 57, 221–230 CrossRef CAS.
- J. Zhu, Y. Xu, J. Wang, J. Lin, X. Sun and S. Mao, Phys. Chem. Chem. Phys., 2015, 17, 28666–28673 RSC.
- H. Wu, X. Wang, L. Jiang, C. Wu, Q. Zhao, X. Liu, B. Hu and L. Yi, J. Power Sources, 2013, 226, 202–209 CrossRef CAS.
- V. D. Nithya, R. K. Selvan, D. Kalpan, L. Vasylechko and C. Sanjeeviraj, Electrochim. Acta, 2013, 109, 720–731 CrossRef CAS.
- R. Wang, Q. Li, L. Cheng, H. Li, B. Wang, X. S. Zhao and P. Guo, Colloids Surf., A, 2014, 457, 94–99 CrossRef CAS.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 RSC.
- J. H. Chae and G. Z. Chen, Particuology, 2014, 15, 9–17 CrossRef CAS.
- H. Gao, A. Virya and K. Lian, Electrochim. Acta, 2014, 138, 240–246 CrossRef CAS.
- A. R. Koh, B. Hwang, K. Chul Roh and K. Kim, Phys. Chem. Chem. Phys., 2014, 16, 15146 RSC.
- J. K. McDonough, A. I. Frolov, V. Presser, J. Niu, C. H. Miller, T. Ubieto, M. V. Fedorov and Y. Gogotsi, Carbon, 2012, 50, 3298–3309 CrossRef CAS.
- M. Arulepp, L. Permann, J. Leis, A. Perkson, K. Rumma, A. Jänes and E. Lust, J. Power Sources, 2004, 133, 320–328 CrossRef CAS.
- S. D. Perera, M. Rudolph, R. G. Mariano, N. Nijem, J. P. Ferraris, Y. J. Chabal and K. J. Balkus, Nano Energy, 2013, 2, 966–975 CrossRef CAS.
- Y. Lei, Z. H. Huang, Y. Yang, W. Shen, Y. Zheng, H. Sun and F. Kang, Sci. Rep., 2013, 3, 2477–2483 CrossRef PubMed.
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623 RSC.
- E. Lim, H. Kim, C. Jo, J. Chun, K. Ku, S. Kim, H. I. Lee, I. S. Nam, S. Yoon, K. Kang and J. Lee, ACS Nano, 2014, 8, 8968–8978 CrossRef CAS PubMed.
- C. Zheng, M. Yoshio, L. Qi and H. Wang, J. Power Sources, 2014, 260, 19–26 CrossRef CAS.
- C. Zheng, J. Gao, M. Yoshio, L. Qi and H. Wang, J. Power Sources, 2013, 231, 29–33 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, ACS Nano, 2014, 8, 5069–5078 CrossRef CAS PubMed.
- N. Jung, S. Kwon, D. Lee, D. M. Yoon, Y. M. Park, A. Benayad, J. Y. Choi and J. S. Park, Adv. Mater., 2013, 25, 6854–6858 CrossRef CAS PubMed.
- R. Francke, D. Cericola, R. Kötz, D. Weingarth and S. R. Waldvogel, Electrochim. Acta, 2012, 62, 372–380 CrossRef CAS.
- A. Brandt, P. Isken, A. Lex-Balducci and A. Balducci, J. Power Sources, 2012, 204, 213–219 CrossRef CAS.
- Y. Lai, X. Chen, Z. Zhang, J. Li and Y. Liu, Electrochim. Acta, 2011, 56, 6426–6430 CrossRef CAS.
- A. Janes, J. Eskusson, T. Thomberg and E. Lust, J. Electrochem. Soc., 2014, 161, A1284–A1290 CrossRef.
- E. Perricone, M. Chamas, L. Cointeaux, J. C. Leprêtre, P. Judeinstein, P. Azais, F. Béguin and F. Alloin, Electrochim. Acta, 2013, 93, 1–7 CrossRef CAS.
- X. Yu, D. Ruan, C. Wu, J. Wang and Z. Shi, J. Power Sources, 2014, 265, 309–316 CrossRef CAS.
- W. Qian, F. Sun, Y. Xu, L. Qiu, C. Liu, S. Wang and F. Yan, Energy Environ. Sci., 2014, 7, 379–386 RSC.
- R. Väli, A. Laheäär, A. Jänes and E. Lust, Electrochim. Acta, 2014, 121, 294–300 CrossRef.
- X. Huang, B. Sun, S. Chen and G. Wang, Chem.–Asian J., 2014, 9, 206–211 CrossRef CAS.
- D. Hanlon, C. Backes, T. M. Higgins, M. Hughes, A. O'Neill, P. King, N. McEvoy, G. S. Duesberg, B. Mendoza Sanchez, H. Pettersson, V. Nicolosi and J. N. Coleman, Chem. Mater., 2014, 26, 1751–1763 CrossRef CAS.
- D. Yiǧit, M. Güllü, T. Yumak and A. Sinaǧ, J. Mater. Chem. A, 2014, 2, 6512–6524 RSC.
- H. Zhang, J. Wang, Y. Chen, Z. Wang and S. Wang, Electrochim. Acta, 2013, 105, 69–74 CrossRef CAS.
- S. G. Krishnan, M. V. Reddy, M. Harilal, B. Vidyadharan, I. I. Misnon, M. H. A. Rahim, J. Ismail and R. Jose, Electrochim. Acta, 2015, 161, 312–321 CrossRef CAS.
- S. G. Krishnan, M. Harilal, I. I. Misnon, M. V. Reddy, S. Adams and R. Jose, Ceram. Int., 2017, 43, 12270–12279 CrossRef CAS.
- M. Harilal, S. G. Krishnan, A. Yar, I. I. Misnon, M. V. Reddy, M. M. Yusoff, J. O. Dennis and R. Jose, J. Phys. Chem. C, 2017, 121, 21171–21183 CrossRef CAS.
- M. Harilal, S. G. Krishnan, B. L. Vijayan, M. Venkatashamy Reddy, S. Adams, A. R. Barron, M. M. Yusoff and R. Jose, Mater. Des., 2017, 122, 376–384 CrossRef CAS.
- V. Thangadurai, D. Pinzaru, S. Narayanan and A. K. Baral, J. Phys. Chem. Lett., 2015, 6, 292–299 CrossRef CAS PubMed.
- N. S. Nadiah, F. S. Omar, A. Numan, Y. K. Mahipal, S. Ramesh and K. Ramesh, Int. J. Hydrogen Energy, 2017, 42, 30683–30690 CrossRef CAS.
- Q. Zhang, J. Rong, D. Ma and B. Wei, Energy Environ. Sci., 2011, 4, 2152–2159 RSC.
- G. G. Amatucci, F. Badway, A. Du Pasquier and T. Zheng, J. Electrochem. Soc., 2001, 148, A930 CrossRef CAS.
- H. Maeshima, H. Moriwake, A. Kuwabara and C. A. J. Fisher, J. Electrochem. Soc., 2010, 157, A696 CrossRef CAS.
- R. D. Rogers and G. a. Voth, Glob. Health Action, 2010, 3, 5480 CrossRef PubMed.
- C. W. Liew, K. H. Arifin, J. Kawamura, Y. Iwai, S. Ramesh and A. K. Arof, J. Non-Cryst. Solids, 2017, 458, 97–106 CrossRef CAS.
- M. Shi, S. Kou and X. Yan, ChemSusChem, 2014, 7, 3053–3062 CrossRef CAS PubMed.
- Q. Dou, C. Lian, S. Lei, J. Chen, H. Liu and X. Yan, Energy Storage Mater., 2019, 18, 253–259 CrossRef.
- Q. Dou, L. Liu, B. Yang, J. Lang and X. Yan, Nat. Commun., 2017, 8, 2188–2197 CrossRef PubMed.
- B. Wu, K. Kuroda, K. Takahashi and E. W. Castner, J. Chem. Phys., 2018, 148, 193807-11 Search PubMed.
- L. Demarconnay, E. G. Calvo, L. Timperman, M. Anouti, D. Lemordant, E. Raymundo-Piñero, A. Arenillas, J. A. Menéndez and F. Béguin, Electrochim. Acta, 2013, 108, 361–368 CrossRef CAS.
- L. Timperman, P. Skowron, A. Boisset, H. Galiano, D. Lemordant, E. Frackowiak, F. Béguin and M. Anouti, Phys. Chem. Chem. Phys., 2012, 14, 8199–8207 RSC.
- L. Timperman, F. Beguin, E. Frackowiak and M. Anouti, J. Electrochem. Soc., 2013, 161, A228–A238 CrossRef.
- H. Kurig, M. Vestli, K. Tonurist, A. Janes and E. Lust, J. Electrochem. Soc., 2012, 159, A944–A951 CrossRef CAS.
- L. G. Bettini, M. Galluzzi, A. Podestà, P. Milani and P. Piseri, Carbon, 2013, 59, 212–220 CrossRef CAS.
- S. Pohlmann, T. Olyschläger, P. Goodrich, J. Alvarez Vicente, J. Jacquemin and A. Balducci, J. Power Sources, 2015, 273, 931–936 CrossRef CAS.
- T. Sato, G. Masuda and K. Takagi, Electrochim. Acta, 2004, 49, 3603–3611 CrossRef CAS.
- Y.-J. Kim, Y. Matsuzawa, S. Ozaki, K. C. Park, C. Kim, M. Endo, H. Yoshida, G. Masuda, T. Sato and M. S. Dresselhaus, J. Electrochem. Soc., 2005, 152, A710 CrossRef CAS.
- A. Senda, K. Matsumoto, T. Nohira and R. Hagiwara, J. Power Sources, 2010, 195, 4414–4417 CrossRef CAS.
- F. B. Sillars, S. I. Fletcher, M. Mirzaeian and P. J. Hall, Phys. Chem. Chem. Phys., 2012, 14, 6094–6100 RSC.
- A. J. R. Rennie, N. Sanchez-Ramirez, R. M. Torresi and P. J. Hall, J. Phys. Chem. Lett., 2013, 4, 2970–2974 CrossRef CAS PubMed.
- A. Orita, K. Kamijima and M. Yoshida, J. Power Sources, 2010, 195, 7471–7479 CrossRef CAS.
- R. Lin, P. Huang, J. Ségalini, C. Largeot, P. L. Taberna, J. Chmiola, Y. Gogotsi and P. Simon, Electrochim. Acta, 2009, 54, 7025–7032 CrossRef CAS.
- X. Zhang, D. Zhao, Y. Zhao, P. Tang, Y. Shen, C. Xu, H. Li and Y. Xiao, J. Mater. Chem. A, 2013, 1, 3706 RSC.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- V. Thangadurai and W. Weppner, Ionics, 2006, 12, 81–92 CrossRef CAS.
- M. Y. Chong, A. Numan, C. W. Liew, K. Ramesh and S. Ramesh, J. Appl. Polym. Sci., 2017, 134, 44636-11 CrossRef.
- M. Y. Chong, A. Numan, C. W. Liew, H. M. Ng, K. Ramesh and S. Ramesh, J. Phys. Chem. Solids, 2018, 117, 194–203 CrossRef CAS.
- A. Ramzy and V. Thangadurai, ACS Appl. Mater. Interfaces, 2010, 2, 385–390 CrossRef CAS PubMed.
- B. E. Francisco, C. M. Jones, S.-H. Lee and C. R. Stoldt, Appl. Phys. Lett., 2012, 100, 103902 CrossRef.
- A. A. Łatoszyńska, G. Z. Zukowska, I. A. Rutkowska, P. L. Taberna, P. Simon, P. J. Kulesza and W. Wieczorek, J. Power Sources, 2015, 274, 1147–1154 CrossRef.
- B. Pal, A. Yasin, R. Kunwar, S. Yang, M. M. Yusoff and R. Jose, Ind. Eng. Chem. Res., 2019, 58, 654–664 CrossRef CAS.
- M. L. Verma, M. Minakshi and N. K. Singh, Electrochim. Acta, 2014, 137, 497–503 CrossRef CAS.
- J. Duay, E. Gillette, R. Liu and S. B. Lee, Phys. Chem. Chem. Phys., 2012, 14, 3329–3337 RSC.
- C. W. Huang, C. A. Wu, S. S. Hou, P. L. Kuo, C. Te Hsieh and H. Teng, Adv. Funct. Mater., 2012, 22, 4677–4685 CrossRef CAS.
- Y. N. Sudhakar, M. Selvakumar and D. K. Bhat, Ionics, 2013, 19, 277–285 CrossRef CAS.
- C. Ramasamy, J. Palma Del Vel and M. Anderson, J. Solid State Electrochem., 2014, 18, 2217–2223 CrossRef CAS.
- X. Wang, B. Liu, Q. Wang, W. Song, X. Hou, D. Chen, Y. Cheng and G. Shen, Adv. Mater., 2013, 25, 1479–1486 CrossRef CAS PubMed.
- Z. Niu, H. Dong, B. Zhu, J. Li, H. H. Hng, W. Zhou, X. Chen and S. Xie, Adv. Mater., 2013, 25, 1058–1064 CrossRef CAS PubMed.
- G. Sun, J. An, C. K. Chua, H. Pang, J. Zhang and P. Chen, Electrochem. Commun., 2015, 51, 33–36 CrossRef CAS.
- L. Kou, T. Huang, B. Zheng, Y. Han, X. Zhao, K. Gopalsamy, H. Sun and C. Gao, Nat. Commun., 2014, 5, 3754-10 Search PubMed.
- G. Sun, J. Liu, X. Zhang, X. Wang, H. Li, Y. Yu, W. Huang, H. Zhang and P. Chen, Angew. Chem., Int. Ed., 2014, 53, 12576–12580 CAS.
- T. Chen, H. Peng, M. Durstock and L. Dai, Sci. Rep., 2014, 4, 3612-7 Search PubMed.
- L. Peng, X. Peng, B. Liu, C. Wu, Y. Xie and G. Yu, Nano Lett., 2013, 13, 2151–2157 CrossRef CAS PubMed.
- J. Ren, W. Bai, G. Guan, Y. Zhang and H. Peng, Adv. Mater., 2013, 25, 5965–5970 CrossRef CAS PubMed.
- L. Yuan, B. Yao, B. Hu, K. Huo, W. Chen and J. Zhou, Energy Environ. Sci., 2013, 6, 470–476 RSC.
- X. Wang, B. Liu, R. Liu, Q. Wang, X. Hou, D. Chen, R. Wang and G. Shen, Angew. Chem., Int. Ed., 2014, 53, 1849–1853 CrossRef CAS PubMed.
- S. Liu, J. Xie, H. Li, Y. Wang, H. Y. Yang, T. Zhu, S. Zhang, G. Cao and X. Zhao, J. Mater. Chem. A, 2014, 2, 18125–18131 RSC.
- W. Si, C. Yan, Y. Chen, S. Oswald, L. Han and O. G. Schmidt, Energy Environ. Sci., 2013, 6, 3218–3223 RSC.
- H. Fei, C. Yang, H. Bao and G. Wang, J. Power Sources, 2014, 266, 488–495 CrossRef CAS.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef CAS PubMed.
- G. Wang, H. Wang, X. Lu, Y. Ling, M. Yu, T. Zhai, Y. Tong and Y. Li, Adv. Mater., 2014, 26, 2676–2682 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed.
- G. Huang, C. Hou, Y. Shao, B. Zhu, B. Jia, H. Wang, Q. Zhang and Y. Li, Nano Energy, 2015, 12, 26–32 CrossRef CAS.
- H. F. Ju, W. L. Song and L. Z. Fan, J. Mater. Chem. A, 2014, 2, 10895–10903 RSC.
- Q. Chen, X. Li, X. Zang, Y. Cao, Y. He, P. Li, K. Wang, J. Wei, D. Wu and H. Zhu, RSC Adv., 2014, 4, 36253–36256 RSC.
- K. M. Kim, J. H. Nam, Y. G. Lee, W. Il Cho and J. M. Ko, Curr. Appl. Phys., 2013, 13, 1702–1706 CrossRef.
- J. M. Ko, J. H. Nam, J. H. Won and K. M. Kim, Synth. Met., 2014, 189, 152–156 CrossRef CAS.
- K. M. Kim, M. Latifatu, Y. G. Lee, J. M. Ko, J. H. Kim and W. Il Cho, J. Electroceramics, 2014, 32, 146–153 CrossRef CAS.
- C. Yang, J. Shen, C. Wang, H. Fei, H. Bao and G. Wang, J. Mater. Chem. A, 2014, 2, 1458–1464 RSC.
- H.-S. Nam, N.-L. Wu, K.-T. Lee, K. M. Kim, C. G. Yeom, L. R. Hepowit, J. M. Ko and J.-D. Kim, J. Electrochem. Soc., 2012, 159, A899 CrossRef CAS.
- R. Yuksel, Z. Sarioba, A. Cirpan, P. Hiralal and H. E. Unalan, ACS Appl. Mater. Interfaces, 2014, 6, 15434–15439 CrossRef CAS PubMed.
- P. M. Dicarmine, T. B. Schon, T. M. McCormick, P. P. Klein and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 8295–8307 CrossRef CAS.
- K. F. Chiu and S. H. Su, Thin Solid Films, 2013, 544, 144–147 CrossRef CAS.
- M. F. Hsueh, C. W. Huang, C. A. Wu, P. L. Kuo and H. Teng, J. Phys. Chem. C, 2013, 117, 16751–16758 CrossRef CAS.
- S. N. Syahidah and S. R. Majid, Electrochim. Acta, 2013, 112, 678–685 CrossRef CAS.
- Y. D. Chiou, D. S. Tsai, H. H. Lam, C. H. Chang, K. Y. Lee and Y. S. Huang, Nanoscale, 2013, 5, 8122–8129 RSC.
- C. Ramasamy, J. Palma and M. Anderson, J. Solid State Electrochem., 2014, 18, 2903–2911 CrossRef CAS.
- M. Schroeder, P. Isken, M. Winter, S. Passerini, A. Lex-Balducci and A. Balducci, J. Electrochem. Soc., 2013, 160, A1753–A1758 CrossRef CAS.
- S. A. Hashmi, J. Solid State Electrochem., 2014, 18, 465–475 CrossRef.
- P. Tamilarasan and S. Ramaprabhu, Mater. Chem. Phys., 2014, 148, 48–56 CrossRef CAS.
- Y. Lim, J. Yoon, J. Yun, D. Kim, S. Y. Hong, S. J. Lee, G. Zi and J. S. Ha, ACS Nano, 2014, 8, 11639–11650 CrossRef CAS PubMed.
- C. W. Liew, S. Ramesh and A. K. Arof, Int. J. Hydrogen Energy, 2014, 39, 2953–2963 CrossRef CAS.
- S. Ketabi and K. Lian, Electrochim. Acta, 2013, 103, 174–178 CrossRef CAS.
- M. Yamagata, K. Soeda, S. Ikebe, S. Yamazaki and M. Ishikawa, Electrochim. Acta, 2013, 100, 275–280 CrossRef CAS.
- X. Zhang, J. Xie, F. Shi, D. Lin, Y. Liu, W. Liu, A. Pei, Y. Gong, H. Wang, K. Liu, Y. Xiang and Y. Cui, Nano Lett., 2018, 18, 3829–3838 CrossRef CAS PubMed.
- C. Li, W. Wu, P. Wang, W. Zhou, J. Wang, Y. Chen, L. Fu, Y. Zhu, Y. Wu and W. Huang, Adv. Sci., 2019, 6, 1801665-8 Search PubMed.
- T. Zhai, S. Sun, X. Liu, C. Liang, G. Wang and H. Xia, Adv. Mater., 2018, 30, 1706640-9 Search PubMed.
- G. Lota and E. Frackowiak, Electrochem. Commun., 2009, 11, 87–90 CrossRef CAS.
- G. Lota, K. Fic and E. Frackowiak, Electrochem. Commun., 2011, 13, 38–41 CrossRef CAS.
- E. Frackowiak, K. Fic, M. Meller and G. Lota, ChemSusChem, 2012, 5, 1181–1185 CrossRef CAS PubMed.
- K. Lian and C. M. Li, Electrochem. Solid-State Lett., 2009, 12, A10 CrossRef CAS.
- Q. Tian and K. Lian, Electrochem. Solid-State Lett., 2010, 13, A4 CrossRef CAS.
- J. Suárez-Guevara, V. Ruiz and P. Gomez-Romero, J. Mater. Chem. A, 2014, 2, 1014–1021 RSC.
- L. Q. Mai, A. Minhas-Khan, X. Tian, K. M. Hercule, Y. L. Zhao, X. Lin and X. Xu, Nat. Commun., 2013, 4, 2923-7 Search PubMed.
- S. Roldán, M. Granda, R. Menéndez, R. Santamaría and C. Blanco, J. Phys. Chem. C, 2011, 115, 17606–17611 CrossRef.
- L. Chen, H. Bai, Z. Huang and L. Li, Energy Environ. Sci., 2014, 7, 1750–1759 RSC.
- S. Roldán, M. Granda, R. Menéndez, R. Santamaría and C. Blanco, Electrochim. Acta, 2012, 83, 241–246 CrossRef.
- J. Wu, H. Yu, L. Fan, G. Luo, J. Lin and M. Huang, J. Mater. Chem., 2012, 22, 19025–19030 RSC.
- H. Yu, L. Fan, J. Wu, Y. Lin, M. Huang, J. Lin and Z. Lan, RSC Adv., 2012, 2, 6736–6740 RSC.
- G. Lota and G. Milczarek, Electrochem. Commun., 2011, 13, 470–473 CrossRef CAS.
- L. Chen, Y. Chen, J. Wu, J. Wang, H. Bai and L. Li, J. Mater. Chem. A, 2014, 2, 10526–10531 RSC.
- K. Wasiński, M. Walkowiak and G. Lota, J. Power Sources, 2014, 255, 230–234 CrossRef.
- S. Roldán, Z. González, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Electrochim. Acta, 2011, 56, 3401–3405 CrossRef.
- T. Tooming, T. Thomberg, L. Siinor, K. Tonurist, A. Janes and E. Lust, J. Electrochem. Soc., 2013, 161, A222–A227 CrossRef.
- S. Yamazaki, T. Ito, M. Yamagata and M. Ishikawa, Electrochim. Acta, 2012, 86, 294–297 CrossRef CAS.
- C. M. Ionica-Bousquet, W. J. Casteel, R. M. Pearlstein, G. GirishKumar, G. P. Pez, P. Gómez-Romero, M. R. Palacín and D. Muñoz-Rojas, Electrochem. Commun., 2010, 12, 636–639 CrossRef CAS.
- R. Taniki, K. Matsumoto, T. Nohira and R. Hagiwara, J. Electrochem. Soc., 2013, 160, A734–A738 CrossRef CAS.
- V. Ozolinš, F. Zhou and M. Asta, Acc. Chem. Res., 2013, 46, 1084–1093 CrossRef PubMed.
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- T. A. Centeno, O. Sereda and F. Stoeckli, Phys. Chem. Chem. Phys., 2011, 13, 12403–12406 RSC.
- A. Numan, N. Duraisamy, F. Saiha Omar, D. Gopi, K. Ramesh and S. Ramesh, Prog. Nat. Sci. Mater. Int., 2017, 27, 416–423 CrossRef CAS.
- F. S. Omar, A. Numan, N. Duraisamy, S. Bashir, K. Ramesh and S. Ramesh, J. Alloys Compd., 2017, 716, 96–105 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |