
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMOF-templated cobalt nanoparticles embedded in nitrogen-doped porous carbon: a bifunctional electrocatalyst for overall water splitting†
Karabi
Nath
a,
Kousik
Bhunia
b,
Debabrata
Pradhan
 b and
Kumar
Biradha
*a
b and
Kumar
Biradha
*a
aDepartment of Chemistry, Indian Institute of Technology, Kharagpur-721302, India. E-mail: kbiradha@chem.iitkgp.ernet.in; Tel: +91-3222-283346
bMaterials Science Centre, Indian Institute of Technology, Kharagpur-721302, India
First published on 16th April 2019
Abstract
Development of cost-effective and efficient non noble metal electrocatalysts has immense importance towards sustainable energy technologies. Herein, a newly constructed porous Co(II)-metal organic framework (MOF) has been utilized for the synthesis of cobalt nanoparticles embedded in N-doped porous carbon, (Co@NPC), via a facile MOF-annealing strategy, at an optimum temperature of 800 °C under an argon atmosphere. DMF molecules present in the form of solvated guests and cations within the 3D-framework serve as a source for N-doping during the formation of the porous graphitic carbon upon carbonization. The nanocomposite was found to encapsulate homogeneously dispersed cobalt nanoparticles within the N-doped porous carbonaceous matrix. The synergistic effect of cobalt nanoparticles and the heteroatom-doped carbon framework makes Co@NPC electrochemically active towards both the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) under alkaline conditions. Furthermore, Co@NPC exhibits outstanding performance as a bifunctional electrocatalyst towards electrochemical water splitting with remarkable stability and durability. It achieves a current density of 10 mA cm−2 at a low cell voltage of 1.66 V in 1 M NaOH solution which is comparable with that of most of the self-templated ZIF-derived non-noble metal electrocatalysts.
Introduction
The quest for affordable and inexhaustible energy sources has stimulated much interest from the scientific community given the accelerated depletion of limited fossil fuels and deleterious environmental conditions.1,2 Hydrogen, is considered as the best eco-friendly source of energy that can be generated in the most viable way, through photo-/electrochemical water splitting.3–9 However, this “green” energy storage and conversion process entirely depends upon the efficiency of the water-splitting electro-catalytic systems. Due to the sluggish kinetics and high activation overpotentials associated with these reactions (hydrogen evolution reaction (HER) and oxygen evolution reaction (OER)) in each of the proton coupled electron transfer steps, the current challenge is to develop highly efficient and active electrocatalysts that can overcome the kinetic barriers for these processes.10,11 Moreover, to accomplish overall water splitting, the electrocatalysts should be efficient enough to drive both the half-cell reactions (HER and OER) in a two-electrode configuration in the same electrolyte with reduced overpotentials.12 According to the current state-of-the-art, noble metals such as platinum and its alloys are considered to be well-known electrocatalysts for the HER while ruthenium (Ru)/iridium (Ir) oxides for the OER.13 However, operation of a single metal-based electrocatalyst that could integrate high activities simultaneously for both these reactions (OER and HER) is rarely observed. In addition, certain intrinsic drawbacks, for instance, high cost, low abundance and poor stability further restricts the widespread applications of these noble-metal based electrocatalysts.Given these aspects, there is an essential necessity for the development of stable, cost-effective, durable and highly efficient bifunctional electrocatalysts for sustainable overall water splitting. A variety of alternatives including, heteroatom-doped nanostructured carbon materials14 and hybrid electrocatalysts based on transition metal oxides15/sulphides16/selenides17/phosphides,18,19 supported by carbon materials were developed that demonstrated promising electrocatalytic activity for the oxygen reduction reaction (ORR),20 OER21–26 and HER.27–29 Interestingly, among transition metals, Co-, Ni- and Fe-based hybrid electrocatalysts such as CoSe,30 NiCo2S4,31 FeNiOH/NF,32 iron phosphide nanotubes (IPNTs),33 Co-P,34 cobalt–cobalt oxide/N-doped carbon hybrids (CoOx@CN),35 porous Co phosphide/phosphate thin films (PCPTFs)36 and CoP NA/CC37 were explored as bifunctional electrocatalyst for overall water splitting. The carbon support with heteroatom doping, along with the active metallic sites (M–N–C), was responsible for enhanced electrocatalytic activity with favorable features including, high surface area, tailorable chemical composition, better conductivity, adjustable pore structures and morphologies. However, the synthetic strategy involved in the design of such an electrocatalyst with high-level performance still remains a matter of great challenge.
In this context, metal–organic frameworks (MOFs),38–42 on account of their intriguing architectures and well-defined porous structures, have recently evolved as ideal self-sacrificial templates for the tunable synthesis of metal/metal oxide decorated hierarchical nanostructured carbons via carbonization.43,44 Interestingly, these MOF-derived nanocomposites with embedded metal/metal-oxide nanoparticles have been widely used as electrode materials in a variety of potential applications.45–48 The metal-doped porous carbon materials further ensure strong coupling interactions between the metallic nanoparticles and the carbon support, leading to the favorable exposure of the active sites during electrocatalysis.49 A sub-class of MOFs, zeolitic-imidazolate frameworks (ZIFs), such as ZIF-8 and ZIF-67, are well-known precursors for the formation of metal-rich N-doped porous carbon frameworks that revealed prominent electrocatalytic activities.50–55 In contrast, carboxylate MOFs with occluded DMF guest molecules, as a source for C–N functionalities, have rarely been explored for the same.56 In this endeavour, we wish to present the synthesis of cobalt nanoparticles embedded in N-doped porous carbon, (Co@NPC), through one-step carbonization of a 3D-anionic MOF, [{Co6(μ3-OH)2(AOIA)3(H2O)6(DMF)2}·(NH2Me2)2·(solvent)x]n, (Co-AOIA), synthesized from a newly designed tetracarboxylate ligand, H4AOIA (Scheme 1). The MOF-derived Co@NPC was found to exhibit excellent bifunctional electrocatalytic activity towards the OER and HER under alkaline conditions, with its promising application in a two-electrode water electrolyzer.
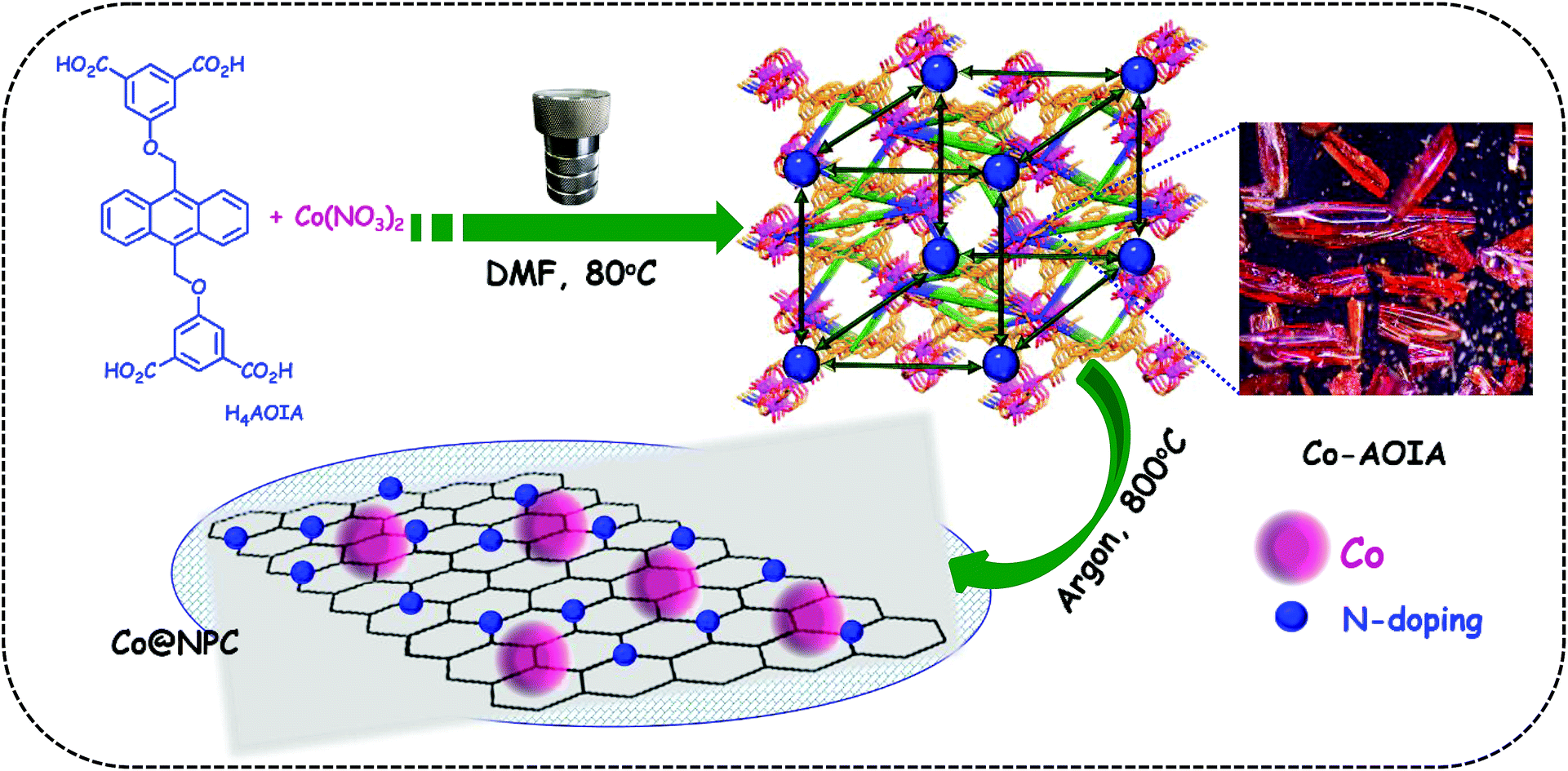 | ||
| Scheme 1 Molecular structure of H4AOIA and schematics for the formation of Co@NPC from Co-AOIA. | ||
Results and discussion
Tetracarboxylic acid H4AOIA, was prepared by nucleophilic substitution reaction of 9,10-bis-bromomethyl-anthracene with the dimethyl ester of 5-hydroxy isophthalic acid, followed by subsequent hydrolysis (Section S1, ESI†). Dark red plate-shaped crystals of Co-AOIA were synthesized by the reaction of H4AOIA with Co(NO3)2 in a DMF–H2O solvent system, under solvothermal conditions, at 80 °C for 24 h. Single crystal X-ray diffraction analysis revealed that the carboxylate based cobalt MOF (Co-AOIA) crystallized in the monoclinic P21/c space group and its asymmetric unit is composed of one and half units of AOIA, three Co(II) ions, one μ3-OH molecule, three water molecules (coordinated to the Co(II) center) and one coordinated DMF molecule. All three Co(II) ions were found to exhibit distorted octahedral geometry and were involved in the formation of a unique {Co3(μ3-OH)(COO)6} trigonal prismatic secondary building unit (SBU) that acts as a building block in the construction of the overall 3D framework.57–59 These three Co(II) centers resulted in the formation of a scalene triangle (Co⋯Co: 3.405, 3.459, 3.721 Å) where the centre of the triangle is being occupied by a bridging OH group (μ3-OH) that connects the three Co(II) ions (Co–O: 2.035, 2.050, 2.055 Å). Each trigonal prismatic SBU is linked to six carboxylate ligands (five in chelating mode and one in a monodentate fashion) and hence acts as a 6-connected node. Two crystallographically independent AOIA units act as 4-connected nodes leading to the formation of an unprecedented (4, 4, 6)-connected 3D-framework (Fig. 1). The SBU carries the charge of −1 which is neutralized by the inclusion of one dimethyl ammonium cation (HDMA) into the pores of the 3D-framework.60 Moreover, apart from the cation, the pores also entrap solvent molecules (DMF and water). The PLATON analysis reveals that the cation and solvents together occupy 33.2% (2115.9 Å3 out of 6376.2 Å3) of the unit cell volume.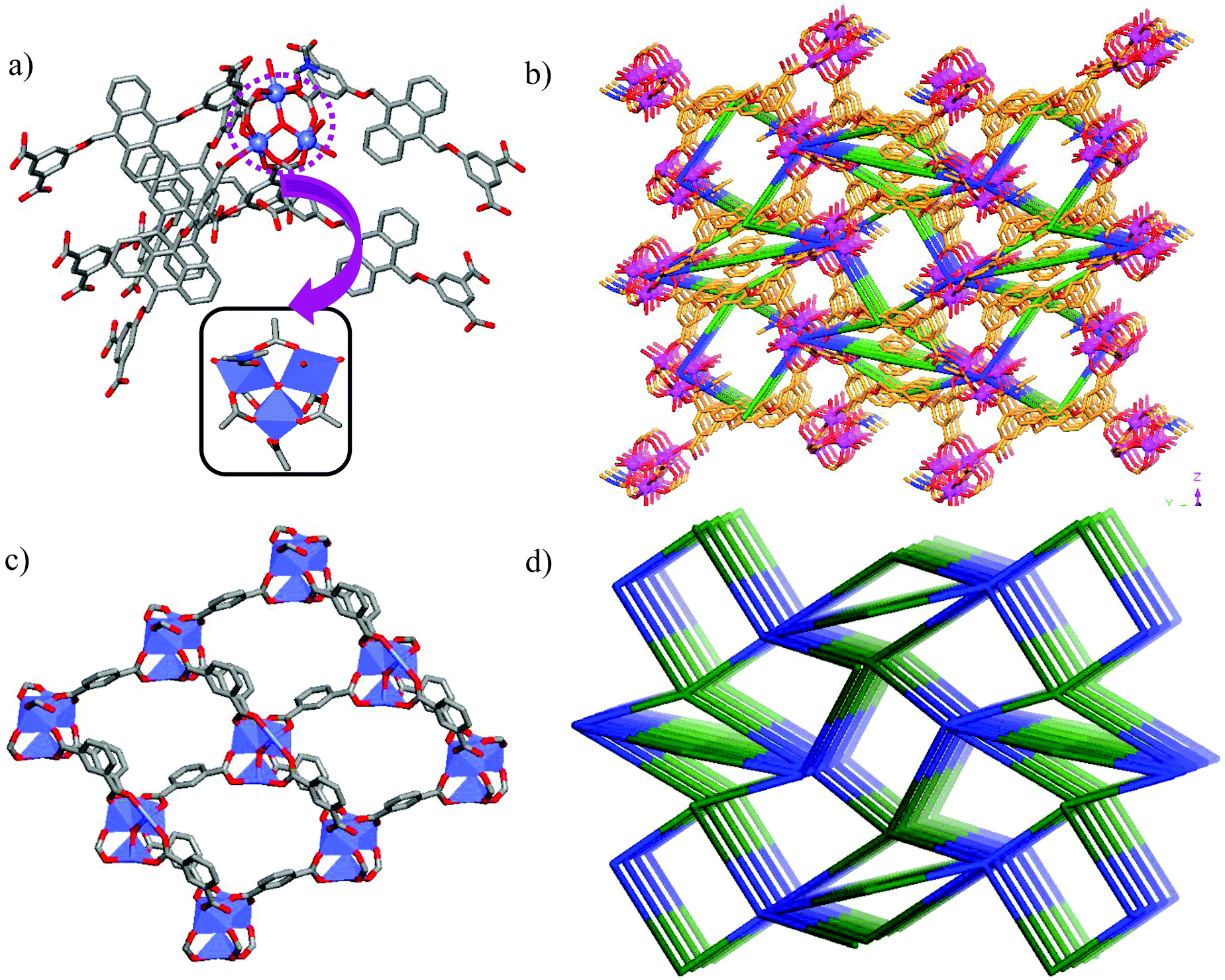 | ||
| Fig. 1 (a) Coordination of six AOIA units around each {Co3(μ3-OH)(COO)6} cluster; (b) molecular representation of the 3D porous framework of Co-AOIA, viewed along the x-axis direction; (c) two-dimensional layer formed by the isophthalate moieties; (d) simplified representation of the overall 3D-network. | ||
The bulk purity of Co-AOIA was verified by comparing the experimental powder patterns of the as-synthesized material to the simulated patterns, obtained from single crystal X-ray analysis (Fig. S1, ESI†). Thermogravimetric analysis (TGA) of Co-AOIA indicates that the framework is stable up to 370 °C. Furthermore, the permanent porosity of Co-AOIA was investigated through nitrogen sorption studies. Prior to gas adsorption analysis, the samples were activated by soaking them in a volatile solvent, such as methanol, to replace the high boiling DMF and H2O molecules in the pores, subsequently followed by degassing under high vacuum at 80 °C for 12 h. The N2 gas sorption isotherm revealed the typical microporous nature of Co-AOIA, as indicated by the reversible type-I behavior, and the BET (Brunauer–Emmett–Teller) surface area was calculated to be 703 m2 g−1, at 77 K and 1 bar pressure (Fig. S10a, ESI†).
In anticipation of MOF-based cost-effective and efficient electrocatalysts,61Co-AOIA was used as a precursor for the synthesis of Co@NPC, through controlled pyrolysis at an optimum temperature of 800 °C, under an inert atmosphere. It is to be noted that conventional synthetic procedures for the formation of an N-doped porous carbonaceous matrix often involve the use of additional precursors containing C–N functionalities like melamine and dicyandiamide.62 However, in the present study, the MOF, Co-AOIA, contains C–N functionalities in the form of entrapped DMF and HDMA which together can act as self-sacrificial templates for the suitable synthesis of N-doped carbon materials, without the use of any external reagent. The carbonized material, as obtained, was then characterized through X-ray powder diffraction (XRPD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) analysis.
The powder patterns of the carbonized material (Co@NPC) were found to be quite different from the parent MOF. The sharp and distinct peaks of the as-synthesized MOF material, Co-AOIA, were replaced by relatively broad peaks when pyrolysed. In XRPD of Co@NPC, the broad peak at 26° can be attributed to the (002) reflection of graphitic carbon, which is indicative of interlayer stacking (Fig. 2a). Furthermore, the Raman spectra of Co@NPC indicate the D and G bands for defective and graphitic carbons at 1342 cm−1 and 1598 cm−1, respectively (Fig. 2b). The ratio ID/IG, calculated from their peak intensities, was found to be 1.03, which revealed the presence of sufficient defect sites in the partially graphitized carbon framework, feasible for electrocatalysis.63 Moreover, carbonization of Co-AOIA at a lower annealing temperature of 600 °C leads to poor graphitization of the carbonaceous material.
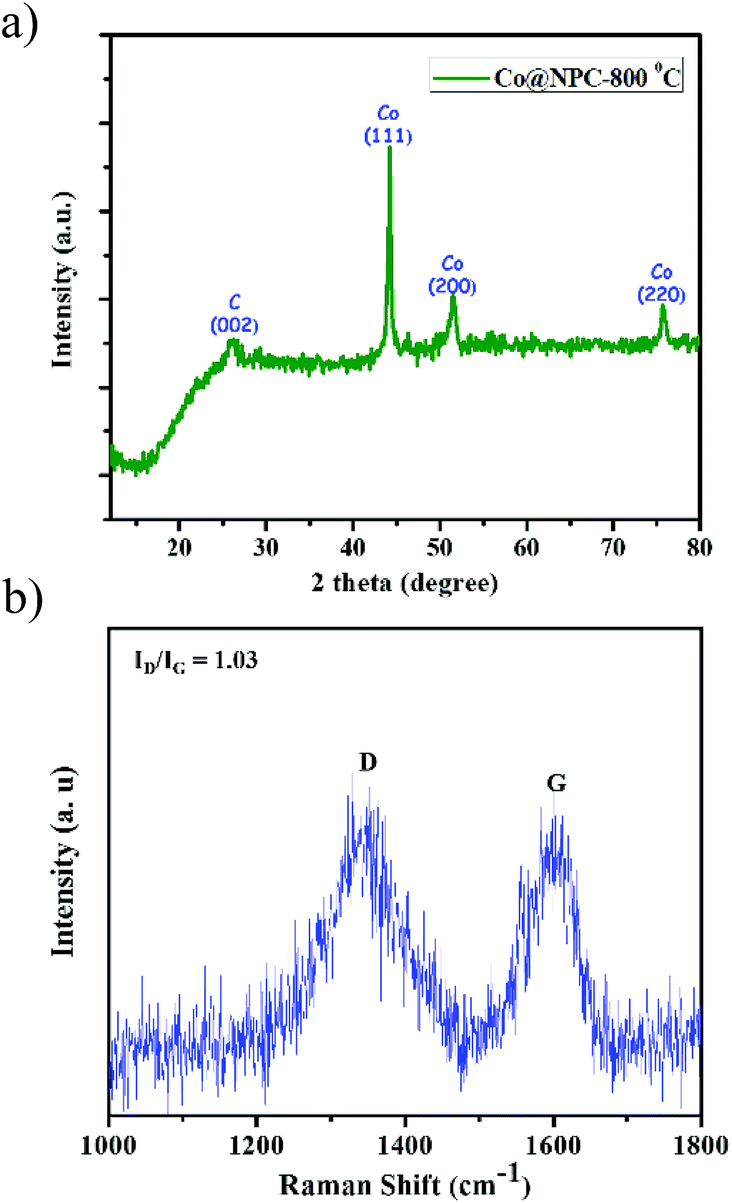 | ||
| Fig. 2 (a) Powder pattern of Co@NPC, formed after carbonization of Co-AOIA at 800 °C under an inert atmosphere; (b) Raman spectra of Co@NPC. | ||
In addition to this, sharp diffraction peaks in XRPD of Co@NPC at 2θ values of 44.1°, 51.5° and 75.7° could be indexed to the (111), (200) and (220) crystalline planes of face-centered cubic (fcc) Co (JCPDS file no. 15-0806), respectively (Fig. 2a). This indicates the complete transformation of the Co2+ ion in Co-AOIA into metallic cobalt nanoparticles by ligand derived carbon upon heat treatment. Fig. 3a and b display the optical microscopy image and typical SEM image of Co-AOIA, respectively. Formation of Co0 (cobalt nanoparticles) during the pyrolysis of Co-AOIA, under the inert atmosphere, was also evidenced through microscopic analysis. FESEM, TEM and high resolution TEM (HRTEM) images of Co@NPC revealed the uniform distribution of the cobalt nanospheres, with an average size of around 20–40 nm, within the graphitic carbon layers (Fig. 3c–e). The well-resolved lattice fringe for the encapsulating cobalt nanoparticle exhibited an interplanar spacing of 0.21 nm, corresponding to the (111) plane of cubic Co, while that for the surrounding carbon was 0.34 nm, attributable to the graphitic plane (002) (Fig. 3e). The energy dispersive X-ray spectroscopy (EDS) analysis showed the characteristic peaks for each of the elements (C, N, O and Co) within the matrix while elemental mapping indicated the homogeneous distribution of these elements (Fig. S9c and d, ESI†). The Co content in Co@NPC as determined from inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was found to be 35 wt%. Interestingly, the crystalline nature of these cobalt nanoparticles was also ascertained through the characteristic spot ring patterns in selected area electron diffraction (SAED) (Fig. 3f).
 | ||
| Fig. 3 (a) Optical microscopy image of Co-AOIA; (b) FESEM image of Co-AOIA; (c) FESEM image of Co@NPC; (d) TEM image of cobalt nanoparticles in Co@NPC; (e) HRTEM image of a single cobalt nanoparticle showing lattice fringes; (f) spot ring SAED pattern of cobalt nanoparticle in Co@NPC. | ||
Further detailed information regarding the chemical composition, valence state and the bonding configuration was attained through XPS analysis. The full survey spectrum of Co@NPC reveals the elemental composition as C = 87.87%, O = 10.11%, N = 0.68% and Co = 1.34% (Fig. S12a, ESI†). The corresponding high resolution C 1s spectrum showed a characteristic peak at 285.06 eV, which was further deconvoluted into three peaks at 284.4 eV (sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 285.2 eV (sp3 C–C/C–H) and 287.3 eV (C–O/C–N bonds) (Fig. S12b, ESI†).52,56 Along similar lines, the characteristic peak for the O 1s spectrum (532.40 eV) could be fitted into three peaks at 530.2 eV, 532.4 eV and 535.8 eV corresponding to O–Co, C–O and C–O–H bonds, respectively (Fig. S12c, ESI†).56,63 Deconvolution of the high resolution peak of N 1s at 399.43 eV resulted in four peaks at 398 eV, 399.9 eV, 401.4 eV and 405.5 eV which can be indicative of pyridinic N, Co–Nx, graphitic N and oxidized N, respectively (Fig. S12d, ESI†).52,55 Lastly, XPS analysis of the Co 2p spectrum indicated the presence of metallic Co0 (778.7 eV), the Co2+ state (780.4 eV), Co–Nx bonds (782.5 eV) and the corresponding satellite peak (786.4 eV) (Fig. S12e, ESI†).55,64 The presence of crystalline Co nanoparticles coupled with small fractions of amorphous Co–O and Co–Nx species could assist the electrocatalytic activities in the composite Co@NPC. In addition, involvement of electronegative N atoms within the graphitic carbon framework also influences its charge distribution and electronic properties, which in turn facilitates strong interaction with reactants and imparts better electrocatalytic activities. The electron-withdrawing nature of the pyridinic N eventually establishes a polarization effect amongst adjacent carbon skeletons, which enhances adsorption of various intermediates, formed during the OER and HER processes.
C), 285.2 eV (sp3 C–C/C–H) and 287.3 eV (C–O/C–N bonds) (Fig. S12b, ESI†).52,56 Along similar lines, the characteristic peak for the O 1s spectrum (532.40 eV) could be fitted into three peaks at 530.2 eV, 532.4 eV and 535.8 eV corresponding to O–Co, C–O and C–O–H bonds, respectively (Fig. S12c, ESI†).56,63 Deconvolution of the high resolution peak of N 1s at 399.43 eV resulted in four peaks at 398 eV, 399.9 eV, 401.4 eV and 405.5 eV which can be indicative of pyridinic N, Co–Nx, graphitic N and oxidized N, respectively (Fig. S12d, ESI†).52,55 Lastly, XPS analysis of the Co 2p spectrum indicated the presence of metallic Co0 (778.7 eV), the Co2+ state (780.4 eV), Co–Nx bonds (782.5 eV) and the corresponding satellite peak (786.4 eV) (Fig. S12e, ESI†).55,64 The presence of crystalline Co nanoparticles coupled with small fractions of amorphous Co–O and Co–Nx species could assist the electrocatalytic activities in the composite Co@NPC. In addition, involvement of electronegative N atoms within the graphitic carbon framework also influences its charge distribution and electronic properties, which in turn facilitates strong interaction with reactants and imparts better electrocatalytic activities. The electron-withdrawing nature of the pyridinic N eventually establishes a polarization effect amongst adjacent carbon skeletons, which enhances adsorption of various intermediates, formed during the OER and HER processes.
Furthermore, a better insight about the pore structure and surface area of the nanocomposite, Co@NPC, was obtained through N2-adsorption–desorption isotherms. Co@NPC exhibited an intermediate pattern of type-I and type-IV sorption isotherms, with sharp uptakes at relatively low pressure and prominent H3-type hysteresis loops within the pressure range from 0.4–1.0 (Fig. S11b†). These characteristic features indicate the existence of both microporous and mesoporous structures in Co@NPC. This is further evident from the pore size distribution plot in the range 2–25 nm and the cumulative pore volume, calculated to be 0.23 cm3 g−1 (Fig. S11c, ESI†). The BET surface area of Co@NPC was determined to be 179 m2 g−1, much lower than that of Co-AOIA (703 m2 g−1). Consequently, the above characterization clearly indicated that the encapsulation of cobalt nanoparticles in the N-doped porous matrix with a suitable electronic structure makes Co@NPC a potential material for various electrochemical applications.
Accordingly, the electrocatalytic OER activity of the nanocomposite Co@NPC was evaluated in an alkaline solution of 1 M NaOH by using a standard three-electrode system. 0.2 mg cm−2 of the as-synthesized material was homogeneously coated onto a polished rotating disk glassy carbon electrode (GCE) used as the working electrode, with Pt-wire as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The typical cyclic voltammetry (CV) curve of Co@NPC, revealed the presence of prominent redox peaks, in the anodic and cathodic regions at around 1.46 V and 1.30 V (vs. the reversible hydrogen electrode (RHE)), respectively, indicating the formation of the Co(III)/Co(IV) couple, preceding the onset of the OER (Fig. 4a). Interestingly, a small pair of peaks could also be identified at 1.05 V (anodic) and at 1.01 V (cathodic) vs. RHE, attributed to the Co(II)/Co(III) redox couple.65 The peak current densities for this quasi-reversible electron transfer process were found to be proportional to the square root of the scan rate, demonstrating a diffusion-controlled electrochemical phenomenon (Fig. S13b, ESI†).
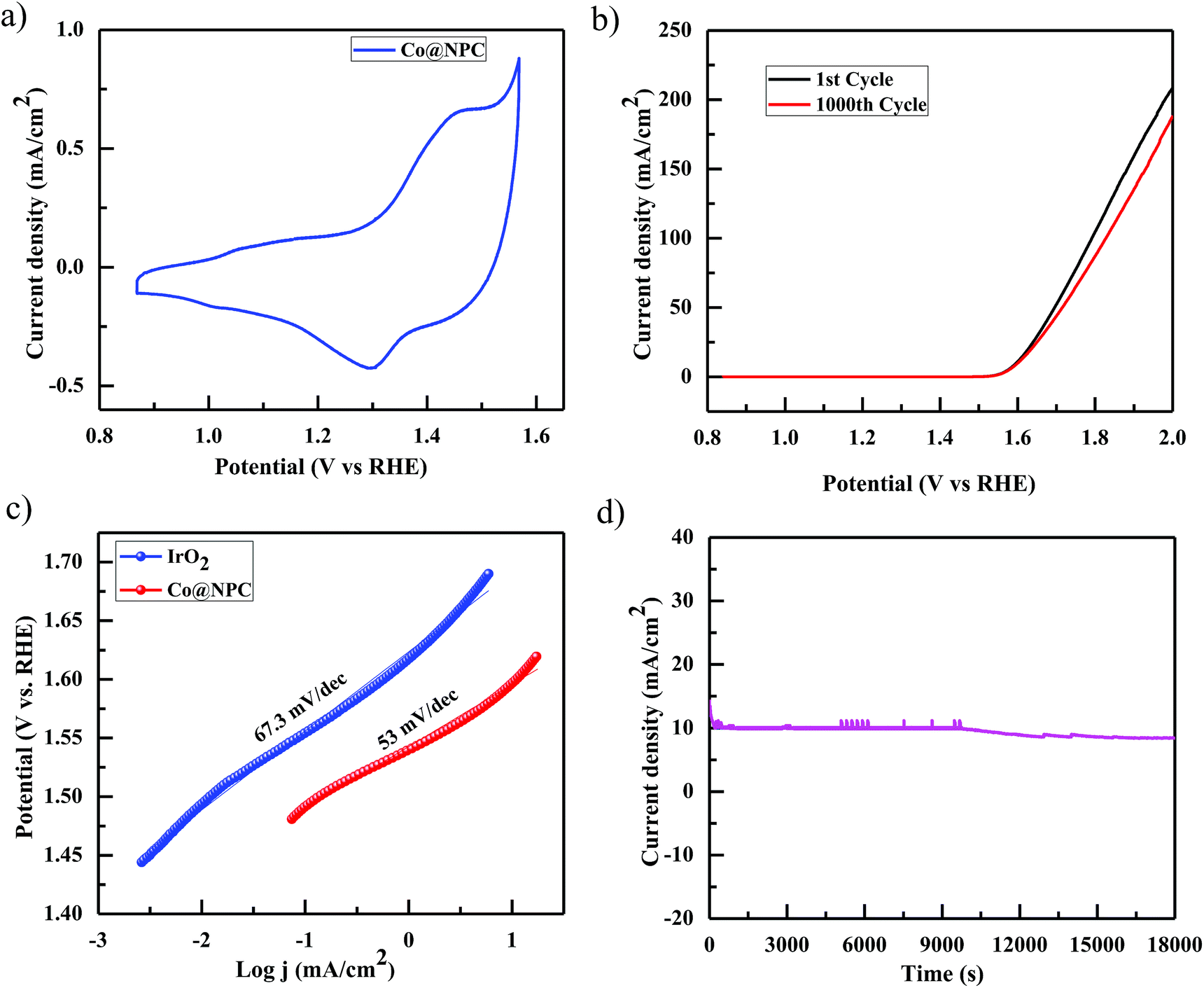 | ||
| Fig. 4 (a) Cyclic voltammogram of Co@NPC at a sweep rate of 100 mV s−1 in 1 M NaOH solution; (b) anodic polarization curve for Co@NPC before and after 1000 CV cycles, at a scan rate of 5 mV s−1; (c) Tafel plots for Co@NPC and IrO2; (d) chronoamperometric durability test of Co@NPC at a constant potential of 1.60 V (vs. RHE). | ||
Notably, Co@NPC experienced a pre-oxidation state, attributed to the formation of higher valent Co active sites from metallic Co, feasible for the OER. This activation is well recognized in most of the previously reported Co-based electrocatalysts.52,66,67 The anodic polarization curve (Fig. 4b) obtained from linear sweep voltammetry (LSV), performed at a scan rate of 5 mV s−1, indicated that Co@NPC requires an overpotential of 360 mV (1.59 V vs. RHE) to drive a current density of 10 mA cm−2 (Ej = 10), which is comparable to a recently reported MOF-derived Co@NC63 (C-MOF-C2-800 (Ej = 10; 1.65 V), C-MOF-C2-900 (Ej = 10; 1.58 V), and C-MOF-C2-1000 (Ej = 10; 1.73 V)) and several other Co-based OER electrocatalysts (Table S2†). The catalytic mechanism involved during the electrocatalytic water oxidation in the alkaline solution that initiates with the oxidation of the surface Co-atoms followed by the subsequent steps:68
| Co + 2OH− → Co(OH)2 + 2e− |
| 3Co(OH)2 + 2OH− → Co3O4 + 4H2O + 2e− |
| Co3O4 + H2O + OH− → 3CoOOH + e− |
| CoOOH + OH− → CoO2 + H2O + e− |
Summary of the OER:
| 4OH− → 2H2O + O2 + 4e− |
The kinetics for the electrocatalytic OER was also evaluated using the Tafel slope, which was determined from the correlation of the catalytic current density (i) with the overpotential (η) using the Tafel equation; η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logi + a (where, η is the overpotential, b is the Tafel slope, and i is the current density). The Tafel slope was calculated to be 53 mV dec−1, much lower than those of the highly efficient precious electrocatalyst IrO2 (67.3 mV dec−1) and several other Co-MOF derived electrocatalysts (Fig. 4c).52,63 Furthermore, the remarkable stability and long-term durability of the electrocatalyst, Co@NPC, was analyzed for its better OER performance. The LSV curve obtained after 1000 CV cycles was shown to exhibit almost similar overpotential to the initial one (η10 = 360 mV; 1.59 V), suggesting superior stability. In addition, the chronoamperometric i–t curve measured at 1.60 V (vs. RHE) indicated the potential of Co@NPC to maintain its catalytic activity for a period of at least 5 h (Fig. 4d). The structural integrity and morphology of the catalyst were found to retain even after the long-term OER cycling tests as indicated by SEM, TEM, HRTEM and XRPD analysis (Fig. S14a–d, ESI†). However, the crystalline nature of the encapsulating cobalt nanoparticles was partially converted to an amorphous form as confirmed by HRTEM analysis. To justify the contribution of various Co species in OER performance, Co@NPC was washed extensively with concentrated acids to prepare bare NPC (N-doped porous carbon). Compared to Co@NPC, bare NPC without Co nanoparticles, as verified by the XPS survey spectrum, demonstrated a weaker OER performance as revealed from its respective anodic polarization curve (Fig. S19a, ESI†).
logi + a (where, η is the overpotential, b is the Tafel slope, and i is the current density). The Tafel slope was calculated to be 53 mV dec−1, much lower than those of the highly efficient precious electrocatalyst IrO2 (67.3 mV dec−1) and several other Co-MOF derived electrocatalysts (Fig. 4c).52,63 Furthermore, the remarkable stability and long-term durability of the electrocatalyst, Co@NPC, was analyzed for its better OER performance. The LSV curve obtained after 1000 CV cycles was shown to exhibit almost similar overpotential to the initial one (η10 = 360 mV; 1.59 V), suggesting superior stability. In addition, the chronoamperometric i–t curve measured at 1.60 V (vs. RHE) indicated the potential of Co@NPC to maintain its catalytic activity for a period of at least 5 h (Fig. 4d). The structural integrity and morphology of the catalyst were found to retain even after the long-term OER cycling tests as indicated by SEM, TEM, HRTEM and XRPD analysis (Fig. S14a–d, ESI†). However, the crystalline nature of the encapsulating cobalt nanoparticles was partially converted to an amorphous form as confirmed by HRTEM analysis. To justify the contribution of various Co species in OER performance, Co@NPC was washed extensively with concentrated acids to prepare bare NPC (N-doped porous carbon). Compared to Co@NPC, bare NPC without Co nanoparticles, as verified by the XPS survey spectrum, demonstrated a weaker OER performance as revealed from its respective anodic polarization curve (Fig. S19a, ESI†).
These results confirmed the significant role of crystalline Co nanoparticles along with other amorphous oxidized Co species (Co–Nx and Co–O) embedded within the N-doped carbonaceous matrix in Co@NPC during the OER process. Furthermore, suitable control experiments were also performed, where only Co(NO3)2 was dissolved in DMF, dried and then the mixture was carbonized under conditions identical to those for the as-synthesized Co@NPC (without the contribution of Co-AOIA and the newly synthesized ligand H4AOIA). The powder patterns of this carbonized material were found to be different from Co@NPC and displayed well-defined peaks similar to that of pristine CoO nanoparticles (NPs) (JCPDS file no. 43-1004) (Fig. S18a, ESI†). Later on, CoO NPs were further annealed in a reducing atmosphere to produce pristine Co NPs (JCPDS file no. 15-0806). The catalytic effect of these resultant nanocomposites (pristine CoO NPs and Co NPs) towards the OER was investigated and found to be negligible as compared to Co@NPC (η10 = 540 mV, CoO NPs; 640 mV, Co NPs) (Fig. S19a, ESI†), confirming the contribution of the porous architecture, strong coupling interactions and the synergistic effects of the Co nanoparticles and the N-doped porous carbonaceous matrix in enhancing the OER activity in the case of Co@NPC. Interestingly, Co@NPC also exhibits an effectively larger electrochemical double layer capacitance (Cdl) of 366.8 μF cm−2, indicative of a larger electrochemical surface area (ECSA), essential for its electrochemical activity (Fig. S15b, ESI†).
The electrocatalytic HER performance of Co@NPC was also investigated in a similar alkaline environment (1 M NaOH) using the same electrode configuration. Fig. 5a shows the cathodic polarization curve of Pt/C and Co@NPC, obtained at a scan rate of 5 mV s−1, in 1 M NaOH solution. As evident, Pt/C was found to exhibit the best HER activity as compared to Co@NPC. To achieve a current density of 10 mA cm−2, Pt/C requires an overpotential of 98 mV; however, Co@NPC demands an overpotential of 325 mV. The corresponding Tafel slope of Pt/C and Co@NPC was determined to be 66 mV dec−1 and 117 mV dec−1, respectively (Fig. 5b). Similar HER contrast experiments were conducted in this case also with bare NPC and the as-prepared control samples from Co(NO3)2 and DMF (pristine CoO NPs and Co NPs), to elucidate the role of various Co species in the HER performance of Co@NPC. As evident from the cathodic polarization curve, Co@NPC exhibits a remarkably high activity as compared to bare NPC and other control samples (overpotential = 655 mV, CoO NPs; 475 mV, Co NPs and 815 mV, bare NPC) (Fig. S19b, ESI†). Furthermore, to determine the specific active site in the HER, the catalytic activity was investigated in the presence of SCN−. Addition of SCN− was found to inhibit the HER activity of Co@NPC to a certain extent owing to deactivation of some of the metallic Co sites (Fig. S16b, ESI†). This indicates that metallic Co acts as HER active centers in Co@NPC.52
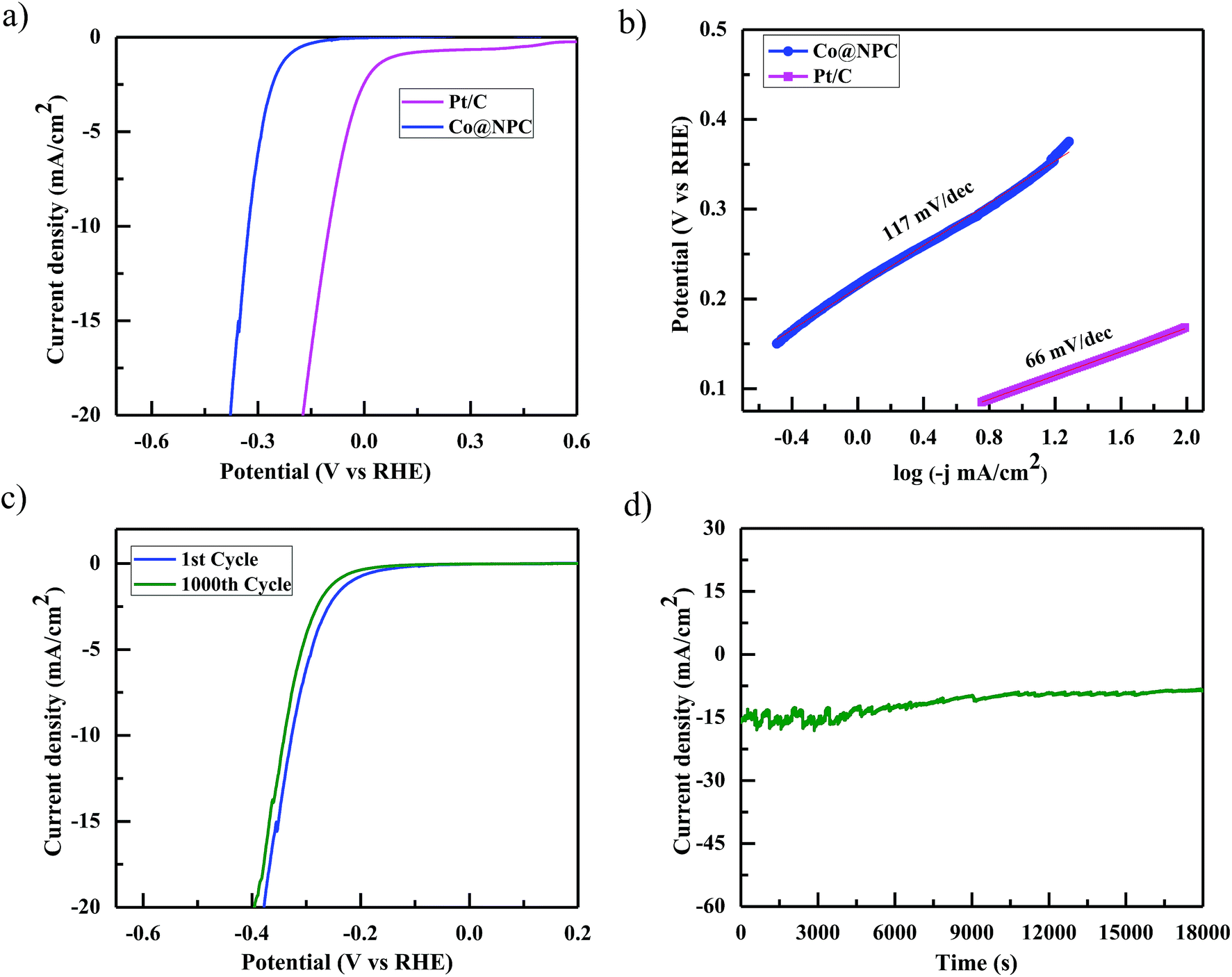 | ||
| Fig. 5 (a) Cathodic polarization curve of Co@NPC and a commercial Pt/C catalyst, performed at a scan rate of 5 mV s−1 in 1 M NaOH solution; (b) Tafel plots for Co@NPC and Pt/C; (c) durability test for Co@NPC through CV before and after the 1000th cycle; (d) chronoamperometric test for Co@NPC, performed at a constant potential. | ||
Interestingly, electrochemical impedance spectroscopy (EIS) conducted at varying potentials (50 and 300 mV) from 100000 Hz to 0.01 Hz provides a better insight about the electrode kinetics during the HER process (Fig. S16a, ESI†). The Nyquist plots revealed that the charge transfer resistance (Rct) decreased with the increased overpotential, indicating a favorable conductivity. As mentioned earlier, stability and durability are considered to be the most important parameters determining the performance of an electrocatalyst. Accordingly, the stability of Co@NPC was evaluated through long term 1000 CV cycles and time-dependent current density plots. The polarization curve obtained after 1000 CV cycles was found to retain its initial shape with the operational overpotential being slightly increased by 10 mV. The chronoamperometric measurements further revealed that the electrocatalytic activity could be maintained for a period of at least 5 h, in 1 M NaOH solution (Fig. 5d). XRPD analysis revealed the structural integrity; however, HRTEM analysis displayed partial amorphization of Co@NPC after the HER (Fig. S17, ESI†).
Therefore, based on the aforementioned results, it can be inferred that both N-doped carbon nanosheets and the entrapped cobalt nanoparticles synergistically participated in optimizing the electronic structure of the nanocomposite, Co@NPC, and thereafter enhanced electrochemical activity. The Co@NPC offers several advantages, in turn, for the development of efficient and cost-effective MOF-derived non-noble metal electrocatalysts. The N-doped carbon behaves as a supporting matrix and prevents the aggregation of the cobalt nanoparticles, thus exposing more active sites towards electrochemical activity. In addition, the high surface area and well developed porosity in Co@NPC regulates the easy diffusion of the electrolytes, thereby enabling faster charge transfer. The mechanistic processes proposed here are quite consistent with previously reported other Co-based electrocatalysts.35,55,69,70
Furthermore, inspired by the remarkable electrocatalytic activity of Co@NPC towards both the OER and HER, we designed and fabricated a bifunctional electrocatalyst using it. Accordingly, Co@NPC was deposited on a graphite sheet (1 cm2) and used both as anode and cathode in a two electrode configuration for practical water electrolysis. The overall water splitting was evaluated in the same alkaline electrolyte (1 M NaOH) and the linear sweep voltammogram revealed that the assembled Co@NPC/graphitic electrodes require a cell voltage of 1.66 V to drive a current density of 10 mA cm−2, which is comparable to most of the reported bifunctional electrocatalysts (Table S4†). In addition, the chronoamperometric curve obtained at a potential of 1.7 V supports the superior stability of the bifunctional electrocatalyst for a duration of at least 5 h (Fig. 6a and b). Subsequently, fabrication of Co@NPC highlights the cost-effectiveness and significance of the facile MOF-annealing strategy for synthesizing non-noble metal electrocatalysts.
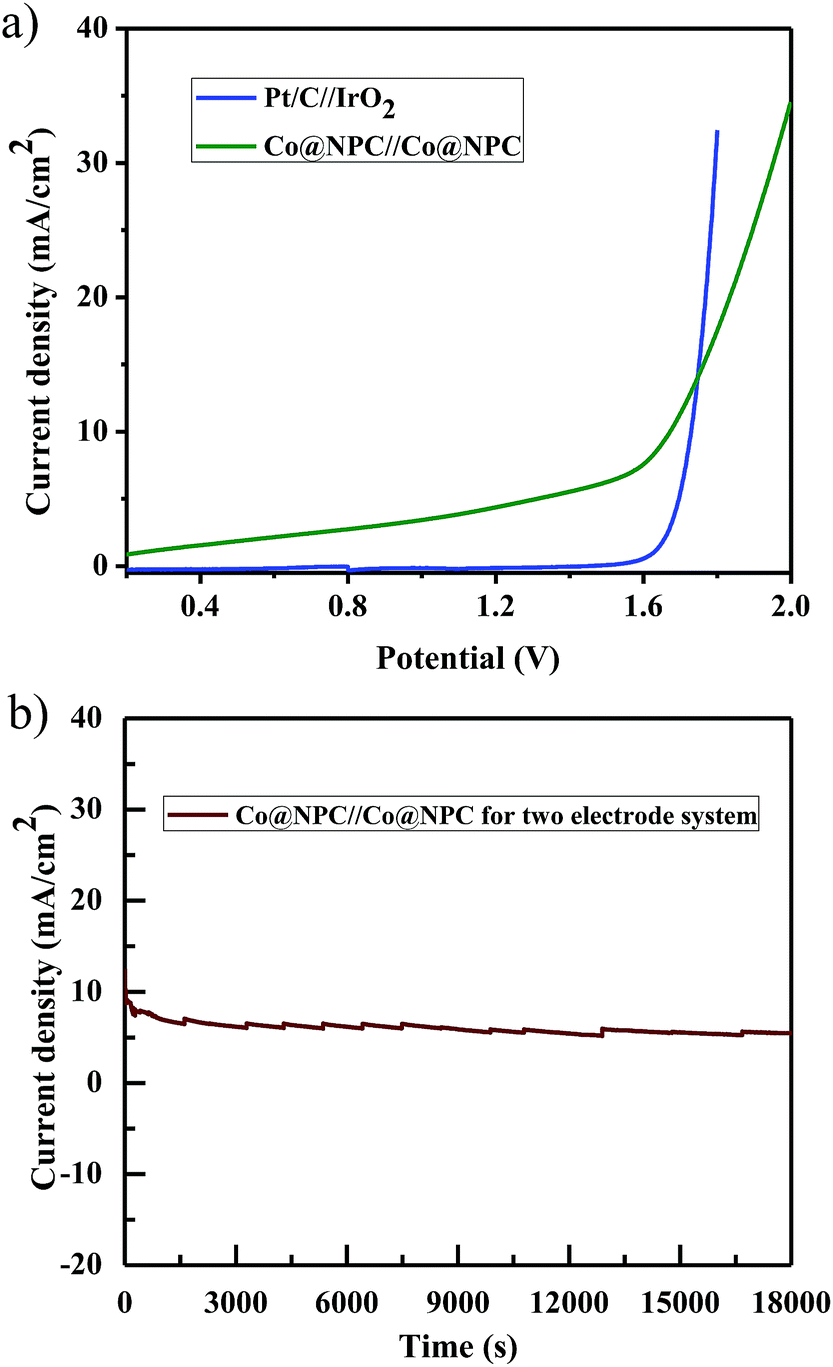 | ||
| Fig. 6 (a) LSV curves for water electrolysis by Co@NPC/graphitic electrodes, acting as both anode and cathode in 1 M NaOH solution; (b) durability test performed using Co@NPC/graphitic electrodes through chronoamperometric measurements. | ||
Conclusions
To summarize, a novel strategy has been demonstrated where solvated DMF molecules along with HDMA within a 3D-anionic MOF (Co-AOIA) have been utilized to generate porous N-doped carbon frameworks without the use of any N-containing external precursor. Introduction of DMF as a solvent serves two important purposes: (1) synthesis of crystalline Co-AOIA under solvothermal conditions, and (2) a source for N-doping into the graphitic carbon framework upon carbonization of Co-AOIA. The N-doped carbonaceous matrix was found to contain embedded Co-nanoparticles (Co@NPC), formed in situ from the cobalt cations in the MOF architecture, while carbonization under an inert atmosphere at a temperature of 800 °C. Co@NPC exhibits remarkable stability and durability as a bifunctional electrocatalyst towards both the HER and OER in an alkaline electrolyte (1 M NaOH). As for practical application in overall water splitting, Co@NPC requires a cell voltage of 1.66 V to achieve a current density of 10 mA cm−2 in a two-electrode cell configuration, comparable to the recently reported core–shell ZIF-8@ZIF-67 derived CoP/NCNHP hybrid bifunctional electrocatalyst.51 The study provides significant insights towards designing alternative high-performance electrocatalysts for its promising application in overall water splitting from carboxylate MOF-based materials.Experimental
Materials and methods
All chemicals including anthracene, paraformaldehyde, and dimethyl-5-hydroxyisophthalate were obtained from Sigma-Aldrich. All other solvents including glacial acetic acid and HBr were freshly distilled prior to use. Fourier transform IR (FTIR) spectra were recorded with a PerkinElmer instrument. Thermogravimetric analysis (TGA) data were recorded under a nitrogen atmosphere at a heating rate of 5 °C min−1 with a PerkinElmer instrument, Pyris Diamond TG/DTA. Powder X-ray diffraction (PXRD) data were recorded with a BRUKER-AXS-D8-ADVANCE diffractometer at room temperature. The surface morphology of the samples was characterized by using a field emission scanning electron microscope (ZEISS EVO 60 with Oxford EDS detector). The samples were also characterized through TEM (FEI-TECNAI G2 20S-TWIN, Type-5022/22) analysis.Synthesis of Co-AOIA
The as-synthesized ligand H4AOIA (28.3 mg, 0.05 mmol) was mixed with Co(NO3)2 (29 mg, 0.1 mmol) in the presence of DMF (3 ml), H2O (1 ml) and aqueous HBF4 (200 μl). The resulting mixture was taken in a 15 ml Pyrex tube and heated in an oven at 80 °C for 24 h. Dark red crystals of Co-AOIA were obtained after cooling down to room temperature. Yield: 70%.Synthesis of Co@NPC
For the preparation of Co@NPC, the cobalt MOF (Co-AOIA) was carbonized at an optimum temperature of 800 °C under an argon atmosphere for 4 h. The temperature inside the furnace was gradually increased from room temperature to the target temperature at a heating rate of 10 °C min−1. The resulting black powder obtained after cooling was then harvested in about 30% yield.Electrochemical measurements
Electrochemical measurements were carried out in a standard three-electrode system on an electrochemical CHI 760D workstation (CH Instruments, Inc., USA), in an electrolyte solution of 1 M NaOH. Platinum (Pt) wire was used as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The working electrode was a sample coated rotating disk glassy carbon electrode (GCE). For the fabrication of the working electrode, 2 mg of the as-synthesized catalyst (Co@NPC) was dispersed in a mixture of distilled water and ethanol (3:1 v/v, 1 ml). 10 μl of Nafion (5 wt%) was added to this solution. The resulting solution was sonicated for at least 40 minutes to prepare a homogeneous ink. 7 μl of this solution was then drop-cast onto a glassy carbon electrode (geometrical surface area: 0.07 cm2), and used as the working electrode for various electrochemical measurements. All potentials measured in this work were referenced to the reversible hydrogen electrode (RHE), according to the Nernst equation: ERHE = ESCE + 0.242 + 0.059pH.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the DST (SERB, EMR/2017/001499), New Delhi, India for financial support and DST-FIST for the single crystal X-ray diffractometer and KN thanks IIT KGP for a research fellowship. We are also thankful to Dr Manash R. Das, Scientist & Assistant Professor (AcSIR), CSIR-North East Institute of Science and Technology, Jorhat, for XPS measurements.References
- A. L. M. Reddy, S. R. Gowda, M. M. Shaijumon and P. M. Ajayan, Adv. Mater., 2012, 24, 5045–5064 CrossRef CAS PubMed.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Kampouri, T. N. Nguyen, C. Ireland, B. Valizadeh, F. Ebrahim, G. Capano, D. Ongari, A. Mace, N. Guijarro and K. Sivula, J. Mater. Chem. A, 2018, 6, 2476–2481 RSC.
- K. Nath, M. Chandra, D. Pradhan and K. Biradha, ACS Appl. Mater. Interfaces, 2018, 10, 29417–29424 CrossRef CAS PubMed.
- D. Shi, R. Zheng, M. J. Sun, X. Cao, C. X. Sun, C. J. Cui, C. S. Liu, J. Zhao and M. Du, Angew. Chem., Int. Ed., 2017, 56, 14637–14641 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, T. Wang, Y. Zhao, E. S. Smotkin and T. E. Mallouk, Energy Environ. Sci., 2012, 5, 7582–7589 RSC.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- T. Asefa, Acc. Chem. Res., 2016, 49, 1873–1883 CrossRef CAS PubMed.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS PubMed.
- C. C. Sun, Q. C. Dong, J. Yang, Z. Y. Dai, J. J. Lin, P. Chen, W. Huang and X. C. Dong, Nano Res., 2016, 8, 2234 CrossRef.
- R. Z. Zhang, C. M. Zhang and W. Chen, J. Mater. Chem. A, 2016, 4, 18723–18729 RSC.
- Y. Ji, L. Yang, X. Ren, G. Cui, X. Xiong and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 11186–11189 CrossRef CAS.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- S. Mukhopadhyay, J. Debgupta, C. Singh, A. Kar and S. K. Das, Angew. Chem., 2018, 130, 1936–1941 CrossRef.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- K. Maity, K. Bhunia, D. Pradhan and K. Biradha, ACS Appl. Mater. Interfaces, 2017, 9, 37548 CrossRef CAS PubMed.
- Z. Wang, X. Ren, L. Wang, G. Cui, H. Wang and X. Sun, Chem. Commun., 2018, 54, 10993–10996 RSC.
- X. Li, L. Yang, T. Su, X. L. Wang, C. Y. Sun and Z. M. Su, J. Mater. Chem. A, 2017, 5, 5000–5006 RSC.
- H. L. Fei, Y. Yang, Z. W. Peng, G. D. Ruan, Q. F. Zhong, L. Li, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 8083–8087 CrossRef CAS PubMed.
- C. Tang, R. Zhang, W. Lu, L. He, X. Jiang, A. M. Asiri and X. Sun, Adv. Mater., 2017, 29, 1602441 CrossRef PubMed.
- T. Liu, Q. Liu, A. M. Asiri, Y. Luo and X. Sun, Chem. Commun., 2015, 51, 16683–16686 RSC.
- D. Liu, Q. Lu, Y. Luo, X. Sun and A. M. Asiri, Nanoscale, 2015, 7, 15122–15126 RSC.
- J.-T. Ren, G.-G. Yuan, C.-C. Weng, L. Chen and Z.-Y. Yuan, Nanoscale, 2018, 10, 10620–10628 RSC.
- Y. Yan, B. Y. Xia, X. M. Ge, Z. L. Liu, A. Fisher and X. Wang, Chem.–Eur. J., 2015, 21, 18062–18067 CrossRef CAS PubMed.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- Y. Yang, H. Fei, G. Ruan and J. M. Tour, Adv. Mater., 2015, 27, 3175–3180 CrossRef CAS PubMed.
- T. Liu, L. Xie, J. Yang, R. Kong, G. Du, A. M. Asiri, X. Sun and L. Chen, ChemElectroChem, 2017, 4, 1840–1845 CrossRef CAS.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- M. Eddaoudi, D. B. Moler, H. L. Li, B. L. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- Z. Zhang and M. J. Zaworotko, Chem. Soc. Rev., 2014, 43, 5444–5455 RSC.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS PubMed.
- Y. V. Kaneti, J. Tang, R. R. Salunkhe, X. Jiang, A. Yu, K. C.-W. Wu and Y. Yamauchi, Adv. Mater., 2017, 29, 1604898 CrossRef PubMed.
- H. Yang, S. J. Bradley, A. Chan, G. I. N. Waterhouse, T. Nann, P. E. Kruger and S. G. Telfer, J. Am. Chem. Soc., 2016, 138, 11872–11881 CrossRef CAS PubMed.
- J. W. Jeon, R. Sharma, P. Meduri, B. W. Arey, H. T. Schaef, J. L. Lutkenhaus, J. P. Lemmon, P. K. Thallapally, M. I. Nandasiri, B. P. McGrail and S. K. Nune, ACS Appl. Mater. Interfaces, 2014, 6, 7214–7222 CrossRef CAS PubMed.
- W. Chaikittisilp, K. Ariga and Y. Yamauchi, J. Mater. Chem. A, 2013, 1, 14–19 RSC.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- H. B. Wu and X. W. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- Y. Xu, W. Tu, B. Zhang, S. Yin, Y. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 1605957 CrossRef PubMed.
- G. Zhong, D. Liu and J. Zhang, J. Mater. Chem. A, 2018, 6, 1887–1899 RSC.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS PubMed.
- X. Li, Z. Niu, J. Jiang and L. Ai, J. Mater. Chem. A, 2016, 4, 3204–3209 RSC.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- H. Yang, S. J. Bradley, X. Wu, A. Chan, G. I. N. Waterhouse, T. Nann, J. Zhang, P. E. Kruger, S. Q. Ma and S. G. Telfer, ACS Nano, 2018, 12, 4594–4604 CrossRef CAS PubMed.
- E. Zhang, Y. Xie, S. Ci, J. Jia, P. Cai, L. Yi and Z. Wen, J. Mater. Chem. A, 2016, 4, 17288–17298 RSC.
- S. Bhattacharyya, B. Konkena, K. Jayaramulu, W. Schuhmann and T. K. Maji, J. Mater. Chem. A, 2017, 5, 13573–13580 RSC.
- P. Chandrasekhar, G. Savitha and J. N. Moorthy, Chem.–Eur. J., 2017, 23, 7297–7305 CrossRef CAS PubMed.
- D.-M. Chen, J.-Y. Tian, C.-S. Liu, M. Chen and M. Du, Chem.–Eur. J., 2016, 22, 15035–15041 CrossRef CAS PubMed.
- X.-Y. Hou, X. Wang, S.-N. Li, Y.-C. Jiang, M.-C. Hu and Q.-G. Zhai, Cryst. Growth Des., 2017, 17, 3229–3235 CrossRef CAS.
- S. Seth, G. Savitha and J. N. Moorthy, J. Mater. Chem. A, 2015, 3, 22915–22922 RSC.
- K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef CAS.
- L. Ye, G. Chai and Z. Wen, Adv. Funct. Mater., 2017, 27, 1606190 CrossRef.
- M. D. Zhang, Q. B. Dai, H. G. Zheng, M. D. Chen and L. M. Dai, Adv. Mater., 2018, 30, 1705431 CrossRef PubMed.
- K. Zhang, C. Qu, Z. Liang, S. Gao, H. Zhang, B. Zhu, W. Meng, E. Fu and R. Zou, ACS Appl. Mater. Interfaces, 2018, 10, 30460–30469 CrossRef CAS PubMed.
- L. Wu, Q. Li, C. H. Wu, H. Zhu, A. Mendoza-Garcia, B. Shen, J. Guo and S. Sun, J. Am. Chem. Soc., 2015, 137, 7071–7074 CrossRef CAS PubMed.
- S. Han, S. Q. Liu, R. Wang, X. Liu, L. Bai and Z. He, ACS Appl. Mater. Interfaces, 2017, 9, 17186–17194 CrossRef CAS PubMed.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566–1569 CrossRef CAS PubMed.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- P. Li, R. Zhao, H. Chen, H. Wang, P. Wei, H. Huang, Q. Liu, T. Li, X. Shi, Y. Zhang, M. Liu and X. Sun, Small, 2019, 15, 1805103, DOI:10.1002/smll.201805103.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, synthesis, crystallographic description and details, powder patterns, graphs, tables, experimental spectra and references. See DOI: 10.1039/c9na00169g |
| This journal is © The Royal Society of Chemistry 2019 |