
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatial arrangement of block copolymer nanopatterns using a photoactive homopolymer substrate†
Zhen
Jiang
a,
Md Mahbub
Alam
a,
Han-Hao
Cheng
 c,
Idriss
Blakey
a and
Andrew K.
Whittaker
*ab
c,
Idriss
Blakey
a and
Andrew K.
Whittaker
*ab
aAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, 4072, Australia. E-mail: a.whittaker@uq.edu.au
bARC Centre of Excellence in Convergent Bio-Nano Science and Technology, The University of Queensland, St Lucia, 4072, Australia
cAustralian National Fabrication Facility-QLD Node, The University of Queensland, St Lucia, 4072, Australia
First published on 25th June 2019
Abstract
Spatial control of the orientation of block copolymers (BCPs) in thin films offers enormous opportunities for practical nanolithography applications. In this study, we demonstrate the use of a substrate comprised of poly(4-acetoxystyrene) to spatially control interfacial interactions and block copolymer orientation over different length scales. Upon UV irradiation poly(4-acetoxystyrene) undergoes a photo-Fries rearrangement yielding phenolic groups available for further functionalization. The wetting behaviour of PS-b-PMMA deposited on the poly(4-acetoxystyrene) films could be precisely controlled through controlling the UV irradiation dose. After exposure, and a mild post-exposure treatment, the substrate switches from asymmetric, to neutral and then to symmetric wetting. Upon exposure through photomasks, a range of high fidelity micro-patterns consisting of perpendicularly oriented lamellar microdomains were generated. Furthermore, the resolution of chemically patterned poly(4-acetoxystyrene) substrate could be further narrowed to submicrometer scale using electron beam lithography. When the BCP was annealed on an e-beam modified poly(4-acetoxystyrene) surface, the interface between domains of parallel and perpendicular orientation of the BCPs was well defined, especially when compared with the substrates patterned using the photomask.
Introduction
Block copolymers (BCPs) have the potential to self-assemble to form periodically-ordered, nanoscale morphologies with feature sizes on the order of several to hundreds of nanometers.1–5 Such self-assembled morphologies within thin films can be used as the template for functionalization to provide diverse properties,6 or to transfer patterns into an underlying substrate. The resulting self-assembled nanostructures have been applied in a range of areas including in nanolithography,2 electronic devices7 and biomaterials.6,8 In particular, owing to the ability to form sub-20 nm nanopatterns,9–15 block copolymer self-assembly has emerged as a viable alternative approach to conventional photolithography with high potential for gaining industrial relevance. Compared to other high resolution lithography techniques such as extreme ultraviolet16 and electron beam lithography,17 block copolymer lithography has advantages of potentially high throughput and low cost, and has become one of the most important alternative next-generation lithography techniques.A large body of work has been devoted to controlling alignment of BCPs to allow pattern transfer and hence device manufacture. The approaches adopted include modifying the surface energy of the substrate by coating with self-assembled monolayers,18,19 statistical copolymers,20–26 polymer blends27,28 or star copolymers.29 The purpose of these coatings is to provide a so-called neutral surface, in which the strengths of interactions between the substrate and the two blocks of the BCP are balanced. This is a necessary condition for achieving perpendicular alignment of the BCP domains with respect to the underlying substrate, and thus required for any subsequent pattern transfer step. The composition of the neutral surface cannot necessarily be determine a priori, so generally a range of candidate materials with a range of chemical compositions, e.g. copolymer composition, are synthesized and tested experimentally against the target block copolymer. The synthetic demands of such a process significantly lengthens the learning cycle and the time to identifying the optimal substrate composition. As a consequence it is highly desirable to have a single versatile material which can be used as a foundation for generation of candidate substrates. The use of homopolymers of para-substituted styrene derivatives, and copolymers of these monomers as neutral substrates was examined by Bates et al.30 Likewise, Pang and co-workers31 prepared a range of phenyl containing acrylates and methacrylates for application as surface-energy modifying polymer brushes, and examined the wetting behavior of a series of PSTY-containing block copolymers. In an approach similar to that reported here, Sun et al.32 irradiated polystyrene with UV light to induce oxidation reactions and changes in surface energy. They were able to control the surface energy through varying the exposure dose, and subsequently align poly(styrene-b-methyl methacrylate) into cylindrical or lamellar arrays.
In response to the need for matching the surface energies of the broad range of block copolymers being developed by many workers, including so-called high-chi BCPs, we introduce here a simple and highly versatile method based on the photolysis and functionalization of a homopolymer substrate. The homopolymer of 4-acetoxystyrene is capable of undergoing the photo-Fries reaction33–37 in which aryl esters are converted to reactive aromatic hydroxyl groups. The change in surface energy on UV irradiation, which depends on illumination dose, can be further manipulated by reaction with appropriate aliphatic or aromatic acids. This approach exploiting orthogonal methods for tuning surface energy lends great flexibility to the technology described here. As a proof-of-principle we demonstrate in this study the relationship between UV dose and surface wetting, and the achievement of preferential alignment of poly(styrene-b-methyl methacrylate) (PS-b-PMMA). Irradiation through simple contact masks demonstrates the potential for spatial control of alignment across a planar surface. The widely tunability of poly(4-acetoxystyrene) in terms of wetting behavior is demonstrated by switching the wetting behavior from asymmetric, to neutral and then to symmetric wetting, just by tuning the irradiation dose. To the best of our knowledge, such simple and versatile behavior has not been demonstrated in photopatternable underlayer polymer systems.11,32,38 Moreover, it is worthy to note that our polymer substrate was readily prepared from a commercially available monomer, avoiding the need to synthesize a series of multicomponent copolymers with different compositions for the purpose of tuning surface energies.11 We confirm that poly(4-acetoxystyrene) also undergoes a Fries rearrangement on exposure to high energy electron beam irradiation. Chemical patterns (300 nm line width) were fabricated using a high resolution e-beam writing technique, resulting in spatially control over the domain orientation of the BCP film at the submicron length scale. The interface between domains of parallel and perpendicular orientation of the BCPs are well defined.
Experimental
Materials
2-Cyano-2-propyl benzodithioate (CPDB, >97%), 2,2′-azobisisobutyronitrile (AIBN), glycidyl methacrylate (GMA), 4-acextoxystyrene, propylene glycol monomethyl ether acetate (PGMEA, 99%), were all obtained from Sigma-Aldrich. AIBN was purified by recrystallization from methanol. The monomers were purified by passing through a basic alumina column immediately before use. All other chemicals were used as received. PS-b-PMMA (38k–37k) block copolymers with molar mass dispersity of 1.08 were purchased from Polymer Source Inc. and used without further purification.Synthesis of crosslinkable poly(4-acetoxystyrene)
4-Acetoxystyrene (70 mmol), GMA (1.4 mmol), AIBN (6.7 × 10−2 mmol) and CPDB (0.23 mmol) were mixed in a 20 mL vial and sealed with a Suba-Seal. The solution was deoxygenated by bubbling with Ar for 20 min. The polymerization was allowed to proceed at 65 °C for at least 12 hours. The product was obtained by precipitation in n-hexane. A pink precipitate was obtained and this was dried under vacuum at 25 °C.Coating of Si wafers with crosslinkable poly(4-acetoxystyrene)
The uncoated silicon wafers used in this study were initially cleaned via sonication in methanol for 3 min, acetone for 3 min, and finally in deionized water for 3 min, followed by drying on a hotplate for 10 min at 100 °C. The surface of each silicon wafer was then treated with an O2 plasma in a reactive ion etcher with a gas flow rate of 10 sccm, a chamber pressure of 50 mTorr, a power of 150 W, and an exposure time of 60 s. Following the cleaning process, all wafers were stored in a class P10000 clean room prior to use. Solutions of the crosslinkable poly(4-acetoxystyrene) in toluene (0.8% w/w) were spin-coated at 3000 rpm onto silicon wafers and then annealed under vacuum at 160 °C for 2 days to effect crosslinking. Uncrosslinked polymer was removed by sonication of the coated substrate immersed in toluene for 1 min to leave an approximately 9 nm film of poly(4-acetoxystyrene), as determined by ellipsometry.UV irradiation
UV irradiation experiments were carried out with an unfiltered high-pressure Hg lamp in air. For comparison, UV irradiation experiments were also conducted by irradiating coated silicon wafers inside quarts cells that were purged with nitrogen. For these experiments, the light intensity (power density) at the sample surface was measured with a spectroradiometer. The integrated power density at 254 nm was measured to be 20 mW cm−2. Patterned structures were obtained by placing a photomask (Cr pattern on quartz) directly onto the polymer film prior to illumination.e-Beam irradiation
Electron-beam irradiation was performed on a Raith eLINE system. The chamber pressure was 9.0 × 10−5 Pa and the working distance was 10 mm. The beam current was 25.6 pA and the accelerating voltage was 10 kV.Post-exposure modification with carboxylic acid chlorides
After UV illumination of crosslinked poly(4-acetoxystyrene) thin films, the wafers were immersed in a solution of 200 μL acid chloride and 50 μL trimethylamine in 2 mL DCM for 14 h. The wafers were then rinsed with DCM and water, followed by drying in a vacuum oven at a pressure of (600 mmHg) at 70 °C for 4 h.Preparation of films of PS-b-PMMA
Thin films of PS-b-PMMA were spin coated from PGMEA solutions onto a series of wafers coated with post-exposure modified poly(4-acetoxystyrene). The thickness of the films was controlled by modifying the concentration of the block copolymer solutions and by adjusting the spin speed of the spin coater. Samples were annealed at 220 °C on a hotplate for 300 s, and where specified for a further 24 h at 190 °C under a nitrogen atmosphere.Characterization
The chemical composition of the polymers was confirmed by 1H NMR spectroscopy in solution (CDCl3) on a Bruker Avance 300 high resolution NMR spectrometer (Fig. S1†). Size exclusion chromatography (SEC) was performed with a Waters Alliance 2690 Separations Module equipped with Waters 2414 Refractive Index (RI) Detector, Waters 2489 UV/Visible Detector, Waters 717 Plus Autosampler and Waters 1515 Isocratic HPLC Pump. THF was used as an eluent, and monodisperse polystyrene standards were used for calibration. The thickness of the films was measured by a Woollam VUV-VASE32 variable angle spectroscopic ellipsometer. FTIR spectra of the thin films on silicon wafers were obtained using a Nicolet Nexus 5700 FTIR spectrometer (Thermo Electron Corp., Waltham, MA) equipped with a Harrick grazing angle attenuated total reflectance (GATR) accessory (Harrick Scientific Products, Pleasantville, NY) fitted with a KRS-5 MIR polarizer (Harrick Scientific Products, Pleasantville, NY). The morphologies of selected films were analyzed using a JEOL JSM-7800F scanning electron microscope (SEM). Films were mounted onto appropriate stubs using carbon tape, and stored in a vacuum oven at 50 °C for at least 12 hours prior to analysis. In a typical setup, SEM micrographs were collected with a working distance of 2.7 mm while operating at 2 kV. XPS measurements were performed using a Kratos Axis ULTRA spectrometer (Kratos Analytical, Manchester, U.K.) with a 165 mm hemispherical electron-energy analyzer and monochromatic AlKα X-ray source (1486.6 eV) operating at 300 W (15 kV, 20 mA). Static contact angles were measured using a Data Physics Instruments Optical Contact Angle Series 5 (OCA 5) goniometer. The contact angles reported here were measured by the addition of a 5 μL drop of either water or diiodomethane at five different locations for each thin film. Surface energies were calculated using the harmonic mean method.39 Atomic force microscopy (AFM) was performed using a stand-alone MFP-3D instrument in tapping mode in air. The AFM was mounted on an antivibration table (Herzan LLC) and operated within an acoustic isolation enclosure (TMC). Height images were captured at 512 points per line at 0.8 Hz. Multi75DLC-50 cantilevers (75 ± 15 kHz, 3 N m−1, and radius of curvature < 15 nm) were obtained from Budget Sensors and were used for the characterization of the surface topography. The surface morphology of the thin films was observed by optical microscopy using 50× or 100× objectives (Olympus BX 60). At least ten AFM or optical microscope images were taken in different areas of a given sample to explore the film homogeneity.Results and discussion
Design and preparation of polymer substrate
Previous studies have confirmed that on irradiation with UV light, poly(4-acetoxystyrene) undergoes a partial photo-Fries rearrangement to form the 2-acetyl phenol product (Fig. 1A). The maximum yield of the photo-Fries reaction reported by Frechet et al.40 was 40–50%, whilst Höfler et al.35 reported a yield of 32% photo-Fries product. This latter group also reported through IR spectroscopic measurement a loss of over 80% of the ester group, indicating other unspecified photo-products. These are most likely 4-hydroxystyrene formed by direct decarboxylation of the acetyl ester. Despite the apparent non-quantitative conversion of the ester to the photo-product, poly(4-acetoxystyrene) was chosen as the substrate material due to the extensive experience within the lithography community of this polymer and the copolymers of 4-acetoxystyrene, the excellent film forming properties and high glass transition temperature which will affords high stability of the films. The stability of the films was further enhanced by copolymerization with 1.8 mol% glycidyl methacrylate (GMA). The polymers were prepared through reversible addition–fragmentation chain-transfer (RAFT) polymerization (Mn = 40 kDa and ĐM = 1.10) and details of the synthesis and characterization are provided in the experiment section and the ESI.† | ||
| Fig. 1 (A) Schematic illustration of photo-Fries rearrangement and post-exposure modification. (B) GATR FTIR spectra of thin films of poly(4-acetoxystyrene) thin films after exposure to UV light at doses of 0, 0.4, 0.9, 1.5, 2.6 and 3.5 J cm−2. (C) The yield of hydroxyketone and ester group content as a function of UV irradiation dose. (D) Contact angles of water and diiodomethane on exposed poly(4-acetoxystyrene) thin film as a function of UV irradiation doses. (E) AFM height images of PS-b-PMMA films after annealing at 190 °C for 24 h in vacuum placed on substrates prior to irradiation. | ||
This films of poly(4-acetoxystyrene), with thickness of 9 nm, were prepared by spin-coating onto silicon wafers. To allow crosslinking to occur the films were heated to 160° for different lengths of times and then quenched to room temperature. Uncrosslinked polymer was removed by sonication in toluene. The thicknesses of the poly(4-acetoxystyrene) film (Fig. S2†) remaining after sonication rapidly increased with increasing crosslinking time, reaching a maximum after 5 h of heating. A similar approach was reported for poly(styrene-stat-methyl methacrylate) containing a higher content of the crosslinkable GMA monomer.41
Photo-induced changes in surface properties
The photochemical changes occurring on UV irradiation of the film were followed using GATR FTIR, which permits analysis of thin films when coated on high refractive index substrates such as silicon. Fig. S3† shows the GATR FTIR spectra of poly(4-acetoxystyrene) before and after UV irradiation to a dose of 1.5 J cm−2. In the spectrum of the unirradiated film, the peaks at 1764 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) and 1197 cm−1 (asym. C–O–C stretch) are assigned to the ester units. Significant changes were observed after UV irradiation, the signals of the phenyl ester group have significantly decreased in intensity. New bands are observable at 3400 cm−1, 1640 cm−1 and 1612 cm−1, which can be ascribed to O–H stretching vibration of hydroxyl groups and aromatic–aliphatic ketones. These signals are evidence of the formation of the photo-Fries product.
O stretch) and 1197 cm−1 (asym. C–O–C stretch) are assigned to the ester units. Significant changes were observed after UV irradiation, the signals of the phenyl ester group have significantly decreased in intensity. New bands are observable at 3400 cm−1, 1640 cm−1 and 1612 cm−1, which can be ascribed to O–H stretching vibration of hydroxyl groups and aromatic–aliphatic ketones. These signals are evidence of the formation of the photo-Fries product.
The yield of hydroxyketone groups as a function of dose was determined from the GATR FTIR spectra (Fig. 1B). The ester CO stretching band was observed to decrease significantly with increasing irradiation dose, while the signals from photo-Fries product at 1640 cm−1 and 1612 cm−1 increased. The photochemical yield of hydroxyketone units was estimated using IR absorbance coefficients of model compounds acetoxybenzene and 2-hydroxyacetophenone as previously reported.35Fig. 1C shows the percentage of hydroxyketone units with increasing exposure dose. The maximum hydroxyketone yield was 42% at an irradiation dose of 3.5 J cm−2. Previously Frechet et al.40 reported a conversion to the photo-Fries product of around 40% at a dose of 3 J cm−2, while Höfler et al.35 reported 30–35% yield at much higher dose of 27 J cm−2. Higher doses did not generate additional hydroxyketone units, consistent with previous reports.40 The extent of loss of the acetyl ester group was estimated from the decrease in the peak at 1764 cm−1 and is shown in Fig. 1C. The maximum loss in ester groups was approximately 65%, slightly less than the value of 83% reported by Höfler at a higher exposure dose (27 J cm−2).
Changes in the surface chemistry of the photoirradiated substrates were determined by measurements of contact angles using water and diiodomethane as test liquids. Fig. 1D illustrates the reduction in water contact angle with increasing light dose from 75° to 63° after exposure of 3.2 J cm−2, while the contact angle of diiodomethane increased strongly over the same dose range. The results indicate a significant increase in hydrophilicity as a result of the photo-Fries reaction.
Controlling BCP nanodomain orientation by light
The overall objective of this study is to control the alignment of block copolymers on the photo-irradiated poly(4-acetoxystyrene). Initially we examined the behaviour of a test BCP, poly(styrene-block-methyl methacrylate) (PS-b-PMMA), a well-studied system in block copolymer lithography,2 on the virgin substrate. PS-b-PMMA was spin-coated onto the poly(4-acetoxystyrene) surface and annealed at 190 °C for 24 h. An “island” morphology was observed by AFM. It was reported that when the thickness of P(S-b-MMA) film is not within commensurate conditions ((n + 1/2)L0, where n is an integer number and L0 is the domain spacing of the lamellae), holes (for 0 < ε < 1/2L0) or islands (for 1/2L0 < ε < L0) will form on the free surface for asymmetric wetting by the BCP.19 We conclude therefore that the observed “island” morphology indicates that the PMMA block of the BCP preferentially wets the poly(4-acetoxystyrene) (Fig. 1E). The PMMA block is more hydrophilic than the PS block. After irradiation with UV light the surface became increasingly hydrophilic (Table 1). Therefore to achieve a so-called neutral substrate, in which the PMMA and PS block interact equally with the substrate, a hydrophobic functional group should be attached to the irradiated material.| θ H20 | θ CH2I2 | γ (mJ m−2) | γ D (mJ m−2) | γ P (mJ m−2) | |
|---|---|---|---|---|---|
| Unirradiated | 75.1 ± 0.6° | 31.2 ± 0.5° | 51.36 | 40.94 | 8.42 |
| Exposed (3.5 J cm−2) | 63.1 ± 0.7° | 43.2 ± 0.8° | 48.68 | 31.42 | 17.26 |
| +Benzoyl chloride | 82.2 ± 1.1° | 48.6 ± 0.7° | 42.19 | 33.47 | 8.75 |
To achieve a more hydrophobic surface, the irradiated poly(4-acetoxystyrene) was treated with benzoyl chloride.34 GATR FTIR spectra of poly(4-acetoxystyrene) prior to reaction, the UV exposed film and the post-exposure modified film are shown in Fig. 2A. After treatment with benzoyl chloride, peaks at 1736 cm−1 and 1190 cm−1 are evident, supporting the formation of the carboxylic acid ester poly(4-vinylphenyl benzoate).
 | ||
| Fig. 2 (A) GATR spectra of poly(4-acetoxystyrene) before and after illumination with UV-light, followed by post treatment with benzoyl chloride. (B–D) XPS O1s spectra of poly(4-acetoxystyrene) thin films before (B) and after illumination with UV-light at dose of 3.0 J cm−2 (C), followed by post treatment with benzoyl chloride (D). | ||
X-ray photoelectron spectroscopy (XPS) was performed to further investigate the changes in surface chemistry in the modified poly(4-acetoxystyrene) films. The XPS survey scans (Fig. S4†) confirm the purity of the films. The high resolution O1s spectra of unexposed, exposed and post-exposure modified poly(4-acetoxystyrene) films are shown in Fig. 2D–F. For the unexposed material, the peaks at 532.0 eV and 533.4 are due to CO and OC–O, respectively. After irradiation, the O1s spectrum can be fitted with three peaks located at 531.8, 532.6, 533.6 eV associated with CO, C–OH and OC–O, respectively. It is worth noting that the peak due to C–OH in the spectrum of the exposed sample was absent after post-exposure modification (Fig. 2D), while the peak ascribed to hydroxyl groups was found to be almost absent in the GATR IR spectrum after post-exposure modification (Fig. S5†). These results indicate the quantitative conversion of hydroxyl groups into carboxylic acid esters, consistent with GATR FTIR results.
Table 1 lists the results from contact angle measurements before and after illumination of poly(4-acetoxystyrene) to a dose of 3.5 J cm−2, as well as after the post-exposure reaction with benzoyl chloride. After post-treatment with benzoyl chloride, the water contact angle increased to 82.2° and the contact angle of diiodomethane slightly increased from 43.2° to 48.6°, which is consistent with conversion of hydroxyl groups to aromatic ester units. Using the static contact angles of water and diiodomethane, we have calculated the surface energy parameters of the surfaces based on the method reported in the literature.42 Moreover, the overall surface energy (γ) was reduced upon irradiation, due to the significantly reduced dispersive component, γD. After treatment with benzoyl chloride, the overall surface energy decreased further because of the reduced polar components, γP.
Using the calculated dispersive, polar, and total surface energies of exposed and treated substrate, and of the PS and PMMA blocks, the interfacial energies of two blocks39 can be calculated as a function of UV dose (Fig. 3A). The interfacial energy of the PMMA block with the substrate (γPMMA–Sub) was at a minimum before irradiation and functionalization. On irradiation and treatment with benzoyl chloride, the interfacial energy γPMMA–Sub became larger. The opposite trend was observed for the energy at the PS block–substrate interface (γPS–Sub). Most importantly, the interfacial energies of the PS and PMMA blocks were close to identical when the irradiation dose is between 2.3 and 2.7 J cm−2 (Fig. 3A). Therefore, poly(4-acetoxystyrene) films irradiated to these doses and treated with benzoyl chloride are potentially neutral substrates to drive perpendicular orientation in thin films of PS-b-PMMA.
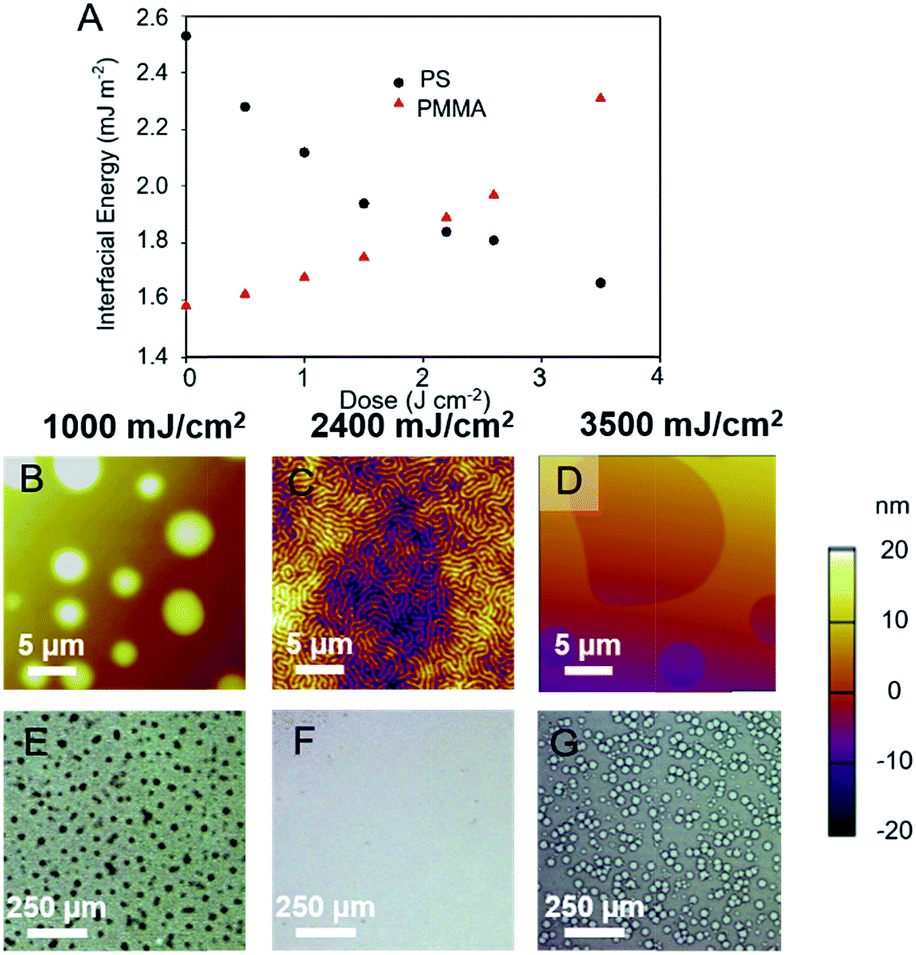 | ||
| Fig. 3 (A) Interfacial energies of PS (black circles) and PMMA (red triangles) blocks against the post-exposure modified poly(4-acetoxystyrene) exposed to different UV doses. AFM height images (B–D) and optical microscope images (E–G) of PS-b-PMMA films annealed at 190 °C for 24 h in vacuum placed on substrates at different irradiation doses. The film thickness of PS-b-PMMA thin film was 1.7L0. | ||
The block copolymer, PS-b-PMMA, was deposited on the post-exposure modified poly(4-acetoxystyrene) films and the morphology examined by AFM and optical microscopy. For copolymers irradiated to 2.4 J cm−2, the deposited BCP displayed characteristic fingerprint patterns indicative of perpendicular alignment of the BCP lamellae with respect to the substrate. At least ten AFM images were taken at different areas of the wafer, and a highly uniform morphology was observed in each location. Optical microscope images show that PS-b-PMMA film appeared featureless at that resolution on this substrate (Fig. 3F), confirming that the perpendicular orientation of the nanodomains on the substrate is homogeneous over a large sample area. At a lower dose (1 J cm−2Fig. 3B) an island morphology was observed, and at higher doses (3.5 J cm−2Fig. 3D) holes were observed. At low dose the PMMA block preferentially wet the substrate whilst at higher doses the substrate was PS wetting. Furthermore, we have demonstrated that the surface energy of the substrate as well as the wetting behaviour of PS-b-PMMA could be well tuneable by controlling the composition of the mixture of acid chlorides used in the subsequent functionalization step (Fig. S6†).
Patterning substrate through diverse photomasks
The combination of the photo-Fries rearrangement of poly(4-acetoxystyrene) with lithographic methods can potentially allow spatial control of the orientation of BCP domains. To demonstrate this, the films poly(4-acetoxystyrene) were exposed with UV light through multiple micron-scale photomasks and treated with benzoyl chloride to form benzyl esters. Fig. 4 shows optical microscopic images of thermally annealed PS-b-PMMA thin films deposited on a range of patterned substrates. Customized micro-patterns with perpendicularly oriented lamella nanodomains in the exposed area (Fig. 4D–F) were formed. In contrast, an “island” morphology was observed on the unexposed areas which is evidence of parallel BCP domains on the strongly PMMA-preferential substrate. Higher magnification SEM images of the boundary between the perpendicular and parallel nanodomain orientations shows relatively small extension of the fingerprint patterns into the unexposed regions (Fig. 4B). The extensions of the fingerprint patterns could be ascribed to the low-resolution contact printing method used, as was previously reported for patterning of a grafted polymer film.34 In summary, these results demonstrate manipulation of BCP lamellar nanodomains to form various customized micro-patterns in high fidelity even when using a low resolution photomask. Experiments using higher resolution patterning are currently ongoing. | ||
| Fig. 4 (A, D, E and F) Optical microscopy images of self-assembled PS-b-PMMA films showing a range of micropatterns; (B) SEM image of the area corresponding to the red rectangle on the optical image in (A). (C) SEM image of the area corresponding to the black square on the optical image in (A). | ||
Patterning with the aid of electron beam writing
In the preceding section we demonstrated spatial control over BCP nanodomain orientation using a low resolution photomask. However, more sophisticated high resolution lithography tools may be needed to reduce the roughness of the boundary between the domains of perpendicular and parallel orientation. A suitable technology for high resolution patterning is electron beam lithography.43,44 A number of years ago the JAERI group demonstrated that the Fries rearrangement in sulfonic acid esters and amides can be initiated by electron beam irradiation.45,46 It may not be unexpected that the Fries rearrangement plays a role in the high energy radiation chemistry of aryl esters since the photochemical Fries rearrangement proceeds via the excited singlet state resulting in homolytic scission of the O–CO bond.47 High energy irradiation is expected to result in cleavage of this bond as a major reaction pathway, although the energy of the photons and secondary electrons, and hence the product distribution may differ considerably from the photochemical process. Accordingly we have examined the effect of electron beam irradiation on films of poly(4-acetoxystyrene). To confirm that a Fries mechanism is contributing to the chemical transformations on irradiation, we initially exposed an area of 1 × 1 mm2 of the film of the polymer to 10 kV electrons using a Raith eLINE system. The results of XPS measurements of the exposed area are shown in Fig. 5A. The high resolution O1s spectrum in Fig. 5A–C demonstrates the formation of C–OH groups after electron beam irradiation with peak intensity increasing with e-beam dose. The phenolic product may result from either direct ester bond cleavage or indeed from a Fries mechanism. The presence of the ketone product was tested by reaction with pentafluorophenylhydrazine, which reacts rapidly with ketones.48 As shown in Fig. 5D, the XPS survey scan shows the presence of fluorine atoms in the exposed samples after treatment with pentafluorophenylhydrazine. We also confirmed the absence of fluorine in the unexposed sample (Fig. S7†). The high resolution F1s spectrum of the exposed sample showed one component at around 689.0 eV due to –CF groups (Fig. 5E). The results indicate the presence of rearranged hydroxy ketone groups upon high energy electron beam irradiation.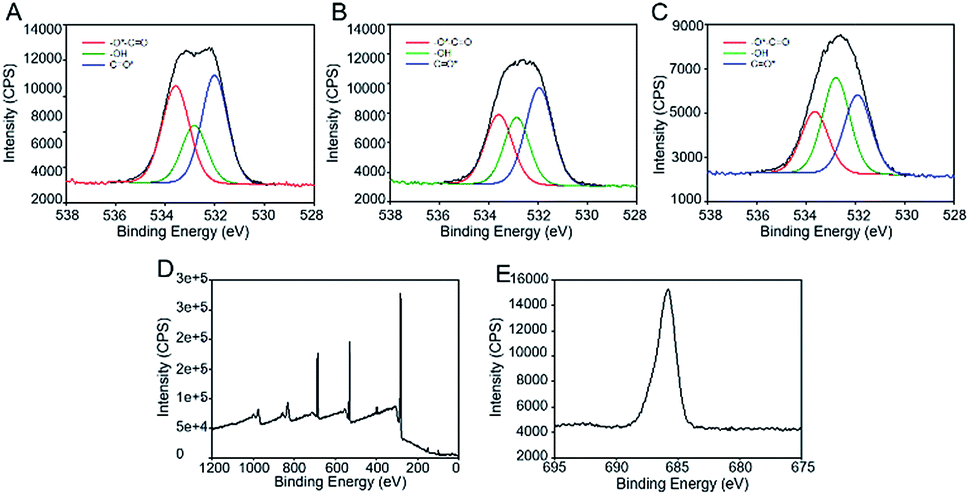 | ||
| Fig. 5 (A–C) High resolution XPS O1s spectra of poly(4-acetoxystyrene) films irradiated by electron beam to doses of 100 μC cm−2 (A), 250 μC cm−2 (B) and 400 μC cm−2 (C). XPS survey scan (D) and high resolution XPS F1s (E) of e-beam irradiated poly(4-acetoxystyrene) film after treatment with pentafluorophenylhydrazine. | ||
Exposure of an area of 1 × 1 mm2 of the film of poly(4-acetoxystyrene) is sufficient for XPS measurements, but does not allow measurement of contact angles. Therefore, the changes in surface energy after exposure and treatment with benzoyl chloride have been inferred from observations of the morphology of films of PS-b-PMMA coated onto the substrate. Fig. 6A–C show SEM images of the morphology of self-assembled lamellar PS-b-PMMA at different e-beam doses. At low exposure dose of 100 μC cm2, perpendicular lamellae were observed over very small areas, suggesting the modified substrate is still preferentially wet by the PMMA block. Fully perpendicular alignment and the characteristic fingerprint pattern was observed at a higher e-beam dose of 250 μC cm2, indicating we had achieved a neutral substrate. At a higher e-beam dose 400 μC cm−2 the orientation of the BCP domains switched from perpendicular to parallel.
 | ||
| Fig. 6 (A–C) SEM images of PS-b-PMMA assembled on e-beam modified poly(4-acetoxystyrene) substrate with irradiated region of 1 × 1 mm2 at e-beam doses of 100 μC cm−2 (A), 250 μC cm−2 (B) and 400 μC cm−2 (C). Top view SEM image (D) and high magnification SEM image (E) of the lamella forming PS-b-PMMA, assembled on the electron beam patterned poly(4-acetoxystyrene) layer at dose of 250 μC cm−2 with line width of 300 nm. | ||
To achieve spatially-resolved BCP alignment, the poly(4-acetoxystyrene) substrate was patterned by electron beam with 300 nm lines and spaces at a dose of 250 μC cm2, followed by treatment with benzoyl chloride. Assembly of PS-b-PMMA on the e-beam patterned substrate shows clear switching of wetting behaviour toward the BCP from neutral to preferential as shown in Fig. 6D and E, indicating that the resulting nanoscale chemical pattern provides controlled polymer surface interactions for perpendicular assembly of PS-b-PMMA domains on the exposed area. Importantly, the boundary between the domains of perpendicular and parallel domains are significantly better resolved than the micro-patterns produced by UV illumination through the photomask.
It is necessary finally to comment on the sensitivity of the chemical processes described here to the dose of incident photons. The UV exposure dose of 2.4 mJ used to achieve an optimal yield of hydroxyketone units will likely prevent direct implementation of this approach into commercial processes for directed self-assembly. However, it is likely that an optimised process will be able to exploit significantly lower yields of phenol units, by, for e.g. functionalisation with different, highly-surface active acid chloride derivatives. Alternatively, it has been reported that the naphthyl-esters and their derivatives37,49 also undergo the photo-Fries rearrangement; these will have stronger absorbance at UV wavelengths and hence greater yields compared with the phenyl derivatives. Finally we have shown for the first time that high energy electrons also initiate the Fries rearrangement with high sensitivity, indicating the potential for e-beam writing to be used in high throughput to pattern substrates.
Conclusions
A novel strategy for achieving spatial control of orientation of BCP nanodomain over different length scales, based on irradiation and functionalization of a homopolymer, is reported. The approach is simple and versatile, and potentially suitable for inducing preferential alignment of a large range of block copolymers. A photoresponsive homopolymer, poly(4-acetoxystyrene) is exposed to an appropriate dose of UV radiation and treated with acyl acid chlorides, allowing control of surface energy and for perpendicularly orientated BCP nanodomains to be formed. Exposure through a photomask resulted in neutral wetting behaviour of PS-b-PMMA only in the exposed areas, enabling the fabrication in high fidelity of microscale patterns with perpendicularly oriented lamella nanodomains. It was shown for the first time that the Fries-rearrangement process could by induced in poly(4-acetoxystyrene) by high resolution e-beam writing, resulting in local control over surface interactions and orientation of BCPs at the submicron length scale. Importantly, the use of electron beam lithography results in a sharper interface between domains of parallel and perpendicular orientation, a result which is encouraging for device applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in part at the Queensland node of the Australian National Fabrication Facility (ANFF), at the University of Queensland node of the Australian Microscopy and Microanalysis Research Facility (AMMRF) within the Centre of Microscopy and Microanalysis (CMM). Financial support from the Australian Research Council (LE110100028, LE110100033, LP120100737, DP130103774, LE140100087, CE140100036, DP180101221) is also acknowledged. ZJ would like to thank The University of Queensland for an International Postgraduate Research Scholarship.References
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146 CrossRef CAS PubMed.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Macromolecules, 2014, 47, 2 CrossRef CAS.
- C. J. Hawker and T. P. Russell, MRS Bull., 2005, 30, 952 CrossRef CAS.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401 CrossRef CAS.
- C. Cummins, T. Ghoshal, J. D. Holmes and M. A. Morris, Adv. Mater., 2016, 28, 5586–5618 CrossRef CAS PubMed.
- C. Zhou, T. Segal-Peretz, M. E. Oruc, H. S. Suh, G. Wu and P. F. Nealey, Adv. Funct. Mater., 2017, 27, 1701756 CrossRef.
- H. G. Yoo, M. Byun, C. K. Jeong and K. J. Lee, Adv. Mater., 2015, 27, 3982 CrossRef CAS PubMed.
- J. M. Kim, Y. Kim, W. I. Park, Y. H. Hur, J. W. Jeong, D. M. Sim, K. M. Baek, J. H. Lee, M.-J. Kim and Y. S. Jung, Adv. Funct. Mater., 2015, 25, 306–315 CrossRef CAS.
- X. Gu, Z. Liu, I. Gunkel, S. Chourou, S. W. Hong, D. L. Olynick and T. P. Russell, Adv. Mater., 2012, 24, 5688–5694 CrossRef CAS PubMed.
- I. Keen, A. Yu, H. H. Cheng, K. S. Jack, T. M. Nicholson, A. K. Whittaker and I. Blakey, Langmuir, 2012, 28, 15876 CrossRef CAS PubMed.
- M. J. Maher, C. M. Bates, G. Blachut, M. C. Carlson, J. L. Self, D. W. Janes, W. J. Durand, A. P. Lane, C. J. Ellison and C. G. Willson, ACS Macro Lett., 2014, 3, 824 CrossRef CAS.
- A. Vora, K. Schmidt, G. Alva, N. Arellano, T. Magbitang, A. Chunder, L. E. Thompson, E. Lofano, J. W. Pitera, J. Y. Cheng and D. P. Sanders, ACS Appl. Mater. Interfaces, 2016, 8, 29808 CrossRef CAS PubMed.
- J. Zhang, M. B. Clark, C. Wu, M. Li, P. Trefonas III and P. D. Hustad, Nano Lett., 2016, 16, 728 CrossRef CAS PubMed.
- A. P. Lane, X. Yang, M. J. Maher, G. Blachut, Y. Asano, Y. Someya, A. Mallavarapu, S. M. Sirard, C. J. Ellison and C. G. Willson, ACS Nano, 2017, 11, 7656 CrossRef CAS PubMed.
- Y. Liao, W. C. Chen and R. Borsali, Adv. Mater., 2017, 29, 1701645 CrossRef PubMed.
- D. Bratton, D. Yang, J. Dai and C. K. Ober, Polym. Adv. Technol., 2006, 17, 94 CrossRef CAS.
- A. S. Gangnaik, Y. M. Georgiev and J. D. Holmes, Chem. Mater., 2017, 29, 1898 CrossRef CAS.
- S. O. Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. de Pablo and P. F. Nealey, Nature, 2003, 424, 411–414 CrossRef CAS PubMed.
- R. D. Peters, X. M. Yang, T. K. Kim, B. Sohn and P. F. Nealey, Langmuir, 2000, 16, 4625–4631 CrossRef CAS.
- J. Bang, U. Jeong, Y. Ryu du, T. P. Russell and C. J. Hawker, Adv. Mater., 2009, 21, 4769–4792 CrossRef CAS PubMed.
- P. Mansky, Y. Liu, E. Huang, T. Russell and C. Hawker, Science, 1997, 275, 1458–1460 CrossRef CAS.
- D. Y. Ryu, K. Shin, E. Drockenmuller, C. J. Hawker and T. P. Russell, Science, 2005, 308, 236–239 CrossRef CAS PubMed.
- E. Han, I. In, S. M. Park, Y. H. La, Y. Wang, P. F. Nealey and P. Gopalan, Adv. Mater., 2007, 19, 4448–4452 CrossRef CAS.
- G. W. Yang, G. P. Wu, X. Chen, S. Xiong, C. G. Arges, S. Ji, P. F. Nealey, X. B. Lu, D. J. Darensbourg and Z. K. Xu, Nano Lett., 2017, 17, 1233–1239 CrossRef CAS PubMed.
- W. Lee, S. Park, Y. Kim, V. Sethuraman, N. Rebello, V. Ganesan and D. Y. Ryu, Macromolecules, 2017, 50, 5858–5866 CrossRef CAS.
- J. Bang, J. Bae, P. Löwenhielm, C. Spiessberger, S. A. Given-Beck, T. P. Russell and C. J. Hawker, Adv. Mater., 2007, 19, 4552–4557 CrossRef CAS.
- S. Ji, G. Liu, F. Zheng, G. S. W. Craig, F. J. Himpsel and P. F. Nealey, Adv. Mater., 2008, 20, 3054–3060 CrossRef CAS.
- Z. Qiang, S. A. Akolawala and M. Wang, ACS Macro Lett., 2018, 7, 566–571 CrossRef CAS.
- S. Jang, K. Lee, H. C. Moon, J. Kwak, J. Park, G. Jeon, W. B. Lee and J. K. Kim, Adv. Funct. Mater., 2015, 25, 5414–5419 CrossRef CAS.
- C. M. Bates, J. R. Strahan, L. J. Santos, B. K. Mueller, B. O. Bamgbade, J. A. Lee, J. M. Katzenstein, C. J. Ellison and C. G. Willson, Langmuir, 2011, 27, 2000–2006 CrossRef CAS PubMed.
- Y. Pang, L. Wan, G. Huang, X. Zhang, X. Jin, P. Xu, Y. Liu, M. Han, G.-P. Wu and S. Ji, Macromolecules, 2017, 50, 6733 CrossRef CAS.
- Y.-S. Sun, C.-T. Wang and J.-Y. Liou, Soft Matter, 2016, 12, 2923–2931 RSC.
- S.-K. Li and J. Guillet, Macromolecules, 1977, 10, 840–844 CrossRef CAS.
- T. Griesser, T. Höfler, S. Temmel, W. Kern and G. Trimmel, Chem. Mater., 2007, 19, 3011 CrossRef CAS.
- T. Höfler, T. Grießer, X. Gstrein, G. Trimmel, G. Jakopic and W. Kern, Polymer, 2007, 48, 1930 CrossRef.
- T. Griesser, J. Adams, J. Wappel, W. Kern, G. J. Leggett and G. Trimmel, Langmuir, 2008, 24, 12420 CrossRef CAS PubMed.
- T. Höfler, T. Grießer, M. Gruber, G. Jakopic, G. Trimmel and W. Kern, Macromol. Chem. Phys., 2008, 209, 488 CrossRef.
- C. He and M. P. Stoykovich, Small, 2015, 11, 2407–2416 CrossRef CAS PubMed.
- D. K. Owens and R. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- J. M. Frechet, T. G. Tessier, C. G. Willson and H. Ito, Macromolecules, 1985, 18, 317 CrossRef CAS.
- E. Han and P. Gopalan, Langmuir, 2010, 26, 1311 CrossRef CAS PubMed.
- D. K. Owens and R. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- M. L. Wu, D. Wang and L. J. Wan, Nat. Commun., 2016, 7, 10752 CrossRef PubMed.
- C. K. Hohle, R. Gronheid, H. Yamamoto, G. Dawson, T. Kozawa and A. P. G. Robinson, Proc. SPIE, 2017, 10146, 1014613 CrossRef.
- K. Yuasa, K. Enomoto, Y. Maekawa, J. Kato, T. Yamashita and M. Yoshida, J. Photopolym. Sci. Technol., 2004, 17, 21–27 CrossRef CAS.
- J. Kato, H. Kakehata, Y. Maekawa and T. Yamashita, Chem. Commun., 2006, 43, 4498–4500 RSC.
- V. Ramamurthy and K. S. Schanze, Photochemistry of organic molecules in isotropic and anisotropic media, CRC Press, 2003 Search PubMed.
- D. S. Everhart and C. N. Reilley, Anal. Chem., 1981, 53, 665 CrossRef CAS.
- L. Merle-Aubry, D. A. Holden, Y. Merle and J. E. Guillet, Macromolecules, 1980, 13, 1138–1143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00095j |
| This journal is © The Royal Society of Chemistry 2019 |