
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular assembly of plasmonic core–satellite structures as highly brilliant SERS-encoded nanoparticles†
Nicolas
Pazos-Perez
 *a,
Jamie M.
Fitzgerald
b,
Vincenzo
Giannini
bc,
Luca
Guerrini
a and
Ramon A.
Alvarez-Puebla
*ad
*a,
Jamie M.
Fitzgerald
b,
Vincenzo
Giannini
bc,
Luca
Guerrini
a and
Ramon A.
Alvarez-Puebla
*ad
aDepartamento de Quimica Fisica e Inorganica, EMaS, Universitat Rovira i Virgili, Carrer de Marcel·lí Domingo s/n, 43007 Tarragona, Spain. E-mail: nicolas.pazos@urv.cat; ramon.alvarez@urv.cat
bDepartment of Physics Condensed Matter Theory, Imperial College London, England, UK
cInstituto de Estructura de la Materia (IEM-CSIC), Consejo Superior de Investigaciones Cientificas, Serrano 121, 28006 Madrid, Spain
dICREA, Passeig Lluís Companys 23, 08010 Barcelona, Spain
First published on 29th October 2018
Abstract
Herein, we present a fabrication approach that produces homogeneous core–satellite SERS encoded particles with minimal interparticle gaps (<2–3 nm) and maximum particle loading, while positioning the encoding agents at the gaps. Integration of plasmonic building blocks of different sizes, shapes, compositions, surface chemistries or encoding agents is achieved in a modular fashion with minimal modification of the general synthetic protocol. These materials present an outstanding optical performance with homogeneous enhancement factors over 4 orders of magnitude as compared with classical SERS encoded particles, which allows their use as single particle labels.
1 Introduction
The use of encoded nanoparticles is becoming a powerful method to solve analytical problems in complex media such as biological fluids,1,2 or to perform basic research in combinatorial chemistry3 and drug discovery.4 Such nanoparticles address some of the limitations posed by other conventional substrates. Thus, encoded nanoparticles are a very suited material for high-throughput screening and multiplexing,5 as they present a large surface area for receptor conjugation or solid phase synthesis, better accessibility of the analytes to the entire sample volume for interaction with the bead-conjugated receptors and greater versatility in sample analysis and data acquisition.Encoded particles can be achieved by using a myriad of strategies, ranging from the combinatorial tuning of the shape of the particle6 to the addition of external labels such as dyes, photonic crystals,7 semiconductor quantum dots8 or molecules with large SERS cross-sections.9 The use of the latter as encoding agents is gaining prominence as these SERS encoded particles (SEPs) provide materials with larger photostabilities and increased multiplexing and quantitative abilities.10
In general, SEPs comprise a plasmonic core (mostly Au or Ag), a SERS encoding agent, a protective layer, and a surface recognition element, selective toward a given target analyte. The brightness of a given SEP depends on the molecular Raman encoder but primarily on the optical properties of the plasmonic core. Generally, single plasmonic particles are preferred over aggregates because of their relatively small size and consistent SERS signals. However, although SEPs composed of a single particle plasmonic nucleus may have an extraordinary application in static experiments such as bioimaging,11 the fact that, usually, they cannot form electromagnetic hot spots12,13 hinders their applicability to other more demanding applications in which acquisition time or spatial resolution are of paramount importance. Thus, methodologies have been developed for the generation of plasmonic cores comprising several particles capable of generating hot spots to improve the overall SERS performance of the SEP. For example, one of the first approaches consisted in the codification of randomly formed colloidal aggregates.14 These SEPs remarkably increased the SERS signal, as compared with those comprising single particle cores, but at the cost of sacrificing signal intensity constancy in between particles while substantially increasing the SEP size. Thus, to produce a complex plasmonic nucleus for SEPs, controlled in terms of size (suitable for bioapplications) and optical response (suitable for quantitative analysis), core–satellite structures (i.e. a large particle surrounded other smaller ones) are particularly appropriate as they concentrate a dense collection of symmetrically arranged hot-spots in a small volume.15,16
Core–satellite structures are typically produced via wet chemical methods where the nanoparticle assembly is chemically mediated by either (i) complementary ligands with selective recognition capabilities such as DNA, antibodies/antigens, etc.,17–22 or (ii) molecular linkers that directly bridge the plasmonic units via covalent or electrostatic interactions of their functional groups (usually located at opposite ends of the molecular structure).23–28 However, the intrinsic DNA length required to ensure stable hybridization precludes the possibility of tuning the interparticle separation at short distances, which results in rather moderate enhancing capabilities.29 Similar limitations are encountered with the use of other bulky macromolecules, such as proteins,25 or branched polymers.24 On the other hand, the simplest method for encoding the plasmonic nucleus of the SEP is the addition of the encoding agent to the pre-formed plasmonic superstructures. However, the efficient diffusion of the SERS label to the volume of maximum enhancements (the hot spots) can be severely hampered, especially when the metallic surfaces at the hot spots are passivated with covalently bonded linkers and, also, when the accessibility is limited by very narrow gap distances. Also, the addition of large amounts of SERS labels irreversibly perturbs the colloidal stability and integrity of the core–satellite assembly. Thus, the preferable choice is represented by the use of small SERS active bifunctional linkers which operate as subnanometric interparticle bridges and, therefore, automatically position themselves at the hot spots.30 However, this approach raises some important practical issues. This class of aromatic linkers is structurally restricted to molecules with terminal mercapto or amino groups, which drastically diminishes the number of commercially available species. On the other hand, the chemical nature of these reporters normally decreases the colloidal stability to the point of exposing the plasmonic building blocks to uncontrolled agglomeration. These are likely the reasons why most of the examples reported in the literature repeatedly exploit 4-aminobenzenethiol (4-ABT) as the SERS active linker.31–35
Thus, despite extensive efforts, current fabrication strategies are still far from meeting the necessary criteria for the efficient production of core–satellite SEPs. Herein, we present an alternative fabrication approach that produces core–satellite SEPs with minimal interparticle distances (<2–3 nm) and maximum satellite loading (i.e., maximum number of hot spots per assembly), while positioning the encoding agents at the gaps. Integration of plasmonic building blocks of different sizes, shapes, compositions and surface chemistries is achieved in a modular fashion with minimal modification of the synthetic protocol. Similar considerations apply to the choice of encoding agent which is not restricted to a small group of molecules. In addition to such versatility, the fabrication method is simple, cheap, scalable and robust, yielding stable plasmonic constructs in high yields.
2 Results and discussion
Fig. 1 outlines the assembling strategy for the fabrication of silica-coated SERS-encoded core–satellites. In this scheme, citrate-stabilized spherical gold or silver nanoparticles were synthesized as the core unit (core@Cit). SERS codification is then performed following the MUA-mediated protocol as previously described.36 Mercaptoundecanoic acid (MUA) is firstly added to the colloidal suspension at a basic pH as the stabilizing agent. MUA firmly binds the metallic surface via the thiol group while its long aliphatic chain and the terminal COOH moiety impart steric and electrostatic repulsions, respectively (Fig. 1B). SERS codes (SCs) can then diffuse within the aliphatic pockets and attach to the metallic surface without perturbing the colloidal stability, regardless of the chemical nature of the molecular reporter (core@MUA/SC). Importantly, the MUA concentration is optimized to safely guarantee the colloidal stability at minimal surface coverages (i.e., sub-monolayer regime) to maximize the final number of SCs per particle. Specifically, the ratio between the number of MUA molecules and the available metallic surface was fixed at 1.25 molecules per nm2 range, which approximately corresponds to ca. 30% of a full monolayer of MUA.37 Negatively charged encoded particles were then wrapped with a single layer of positively charged branched polyethylenimine (PEI) to yield the corresponding positively charged core@MUA/SC@PEI nanoparticles. TEM images reveal a thin layer of approximately 1.5–2 nm thickness on top of these particles (Fig. 1C and S1†). It is worth noting that removal of unbound or loosely bound PEI is critical for preventing the formation of chain-like structures protruding from the core particles (Fig. S2†). These core units are subsequently exposed to a large excess of negatively charged satellites (satellites![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :core ∼ 400:1), which, via electrostatic interactions, saturates the core particles yielding the plasmonic assemblies. Finally, silica encapsulation is performed using a modification of the Stöber method directly on the mixture containing the core–satellite assemblies (Fig. 1E) and the residual unbound satellites. The silica shell protects both plasmonic particles and SCs from the environment, while providing high colloidal stability and an external surface which can be easily functionalized based on the well-known silica surface chemistry. Additionally, in this specific synthetic approach, the silica coating allows efficient separation of the light satellites from the clustered particles via post-centrifugation cycling with no risks of perturbing the aggregation state of the assemblies (Fig. S3†).
:core ∼ 400:1), which, via electrostatic interactions, saturates the core particles yielding the plasmonic assemblies. Finally, silica encapsulation is performed using a modification of the Stöber method directly on the mixture containing the core–satellite assemblies (Fig. 1E) and the residual unbound satellites. The silica shell protects both plasmonic particles and SCs from the environment, while providing high colloidal stability and an external surface which can be easily functionalized based on the well-known silica surface chemistry. Additionally, in this specific synthetic approach, the silica coating allows efficient separation of the light satellites from the clustered particles via post-centrifugation cycling with no risks of perturbing the aggregation state of the assemblies (Fig. S3†).
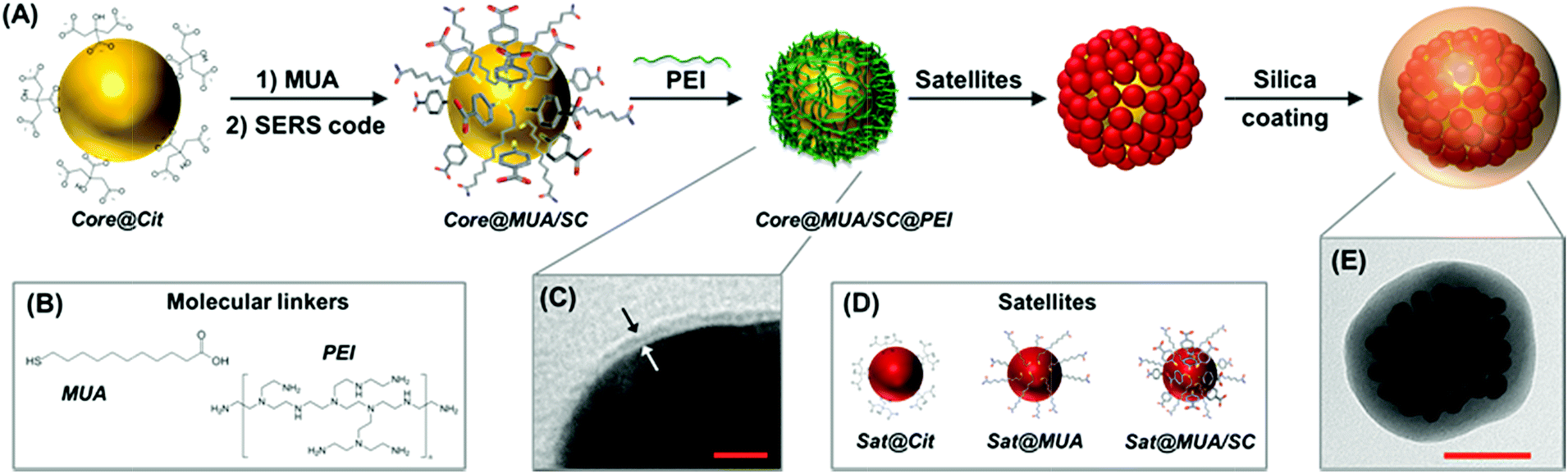 | ||
| Fig. 1 Illustrative description of the MUA/PEI-mediated assembling approach. (A) Schematic of the progressive functionalization of the citrate-stabilized plasmonic core (core@Cit) with molecular linkers (B) and SERS codes (SC), generating a thin layer of approximately 2 nm thickness (core@MUA/SC@PEI). (C) Representative TEM image of a ca. 61 nm diameter gold nanoparticle functionalized with a mixed layer of MUA and SERS code (SC; in this case, 4-mercaptobenzoic acid – MBA) and subsequently wrapped with PEI. Scale bar = 5 nm. (D) Schematic of small gold nanoparticle satellites with different surface elements: original citrate-stabilized nanoparticles, and upon functionalization with either a full monolayer of MUA or a mixed bilayer of MUA and SC. (E) Representative TEM image of a silica-encapsulated core–satellite assembly comprising a spherical gold core of ca. 63 nm diameter and a dense collection of ca. 12 nm size gold satellites. Scale bar = 50 nm. | ||
Obviously, the sizes of the core and satellite building blocks play a central role in determining the final features of the nanoassemblies. In this regard, the fabrication of homogenous and ultrabright SERS clusters in the ≤100–150 nm range remains a major synthetic challenge. On the other hand, this also severely impacts the exploitation of such structures in, most notably, biological applications. For instance, it has been shown that the circulation and extravasation of particles from the bloodstream into cancer tissues efficiently occurs for nanomaterials in the 100 nm size range and below.38,39 Thus, we initially targeted the fabrication of core–satellite clusters in the ca. 100 nm size range, using spherical Au or Ag nanoparticle cores of ca. 60–65 nm diameter and small Au satellites of ca. 12 nm (Fig. S4†). In particular, it has been shown that satellites in that size range offer a good compromise between their SERS enhancing capabilities, which increase with the particle size up to ca. 100 nm diameter,36,40 and number of satellites that can accumulate at the core surfaces (i.e., the number of interparticle gaps is larger for smaller satellites for a given core particle).32 On the other hand, theoretical studies indicated that a minimum core diameter of ca. 50 nm is preferable to avoid an excessive reduction of the surface area that would significantly lower the satellite coverage.41
Small gold satellites were used as such, with citrate-capping ligands as stabilizers, or after surface modification with either a full monolayer of MUA or a mixed bilayer of MUA and SC (Fig. 1D), as similarly performed for larger core particles. This allows us to gain a better insight into the impact of the MUA coating on the extent of satellite accumulation at the core particle as well as the relative contribution of core–satellite vs. satellite–satellite plasmonic coupling to the final SERS signal. In this regard, 4-mercaptobenzoic acid (MBA) was selected as the SERS code in this foundational comparative study. Functionalization of plasmonic particles with MUA and MBA leads to a slight red-shift of the LSPRs due to an increase in the refractive index of the medium surrounding the nanoparticles, with negligible perturbation of the colloidal stability.36 In contrast, a drastic reshaping of the extinction profile is observed upon agglomeration of satellites at single particle cores (Fig. 2A and B) promoted by the plasmon coupling of the closely spaced nanoparticles. Oxidative dissolution of silver nanoparticles (e.g., via ammonia addition) enables the selective removal of the Ag-core from the core–satellite nanoassemblies. As a result, hollow structures comprising a shell of satellites enclosed in the silica matrix are generated, exposing the red-shifted resonances of gold satellites ascribed to the satellite–satellite plasmon coupling (Fig. 2C). The remarkable red-shift observed in the extinction profile of the satellite-corona compared to the isolated particles is proof of the closely spaced organization of Au satellites. For instance, Ross et al.41 have shown that, for a similar nanoassembly configuration, a dense shell of 2 nm-spaced gold satellites only results in a modest red-shift of the LSPRs up to ca. 550 nm, far smaller than the one displayed by our satellite-corona.
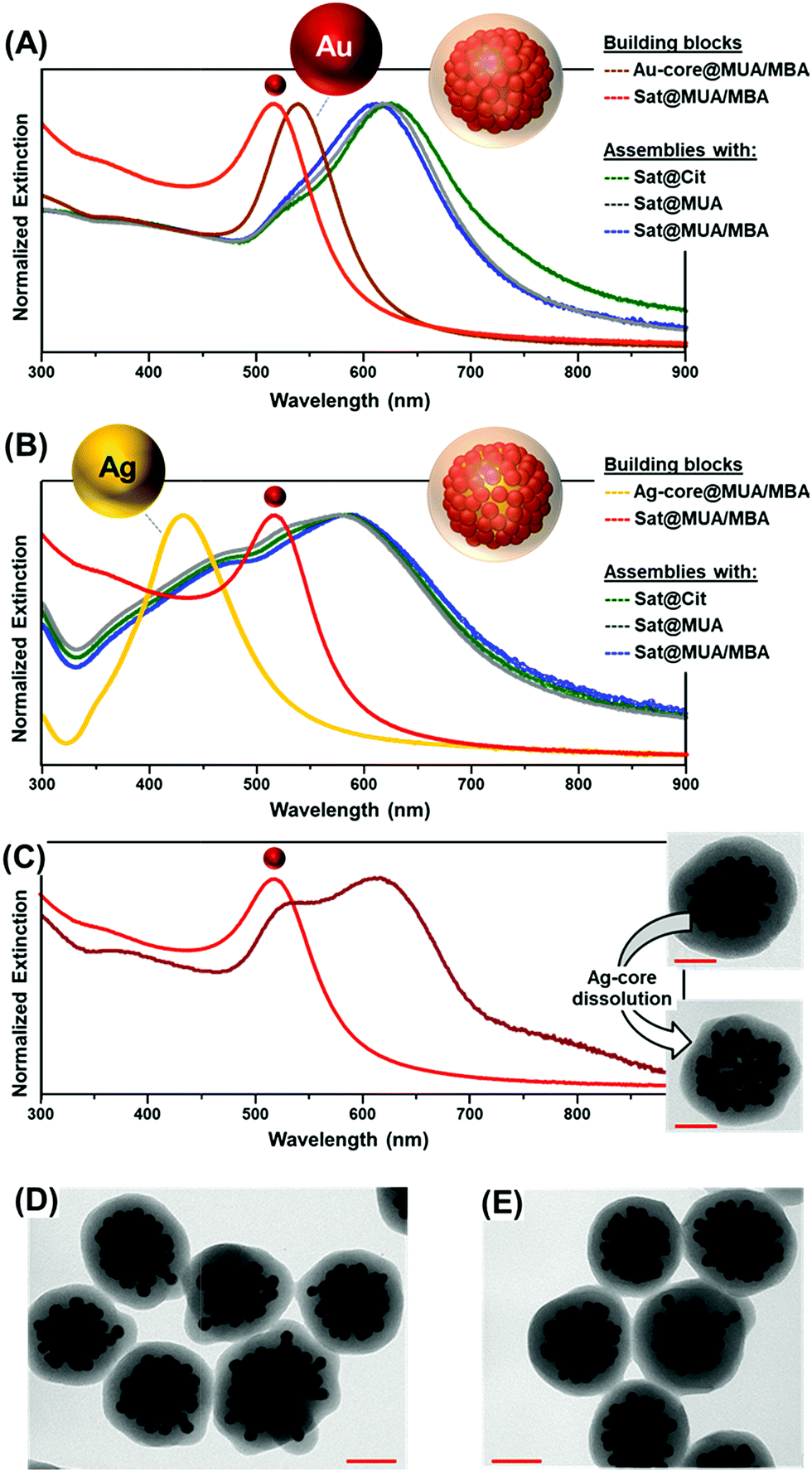 | ||
| Fig. 2 (A) Normalized extinction spectra of MUA/MBA functionalized Au-core and Au-satellite colloids (Au-core@MUA/MBA and Sat@MUA/MBA), and core–satellite assemblies comprising satellites with different surface functionalities (Sat@Cit, Sat@MUA, and Sat@MUA/MBA). (B) Normalized extinction spectra of MUA/MBA functionalized Ag-core and Au-satellite colloids (Ag-core@MUA/MBA and Sat@MUA/MBA), and core–satellite assemblies comprising satellites with different surface functionalities (as above). (C) Extinction spectrum of Ag–Au core satellites upon removal of the silver particle, as well as representative TEM images illustrating the silver dissolution process. Scale bars = 40 nm. (D and E) Representative TEM images of core–satellites comprising a Au-core and Sat@Cit (C) and a Ag-core and Sat@Cit (D). Scale bars = 50 nm. | ||
Random packing of closely spaced satellites cannot exceed a density limit of 63.4% of the available core particle surface area,42 which, for a 63 nm Au-core and 12 nm Au-satellites, corresponds approximately to 70 satellites per particle. Remarkably, the average number of satellites per cluster estimated from TEM images falls in the same range (ca. 66 ± 10 satellites per particle, N = 30, see Fig. S5 and S6† for details about satellite counting), demonstrating the extremely efficient packing of the small gold nanoparticles at the central core. This result far exceeds those previously reported in the literature, which typically display satellite coverage in the 20–35% range of maximum random packing.24,25,32,35,43–45 This outstanding result can be interpreted as follows. When branched PEI is replaced by poly-allylamine hydrochloride (PAH), a linear positively charged polymer, a drastic reduction of the satellite surface coverage is noted (Fig. 3A). This piece of evidence suggests that, for a PAH coating, a thin shell of positive charges is deposited over the particle core, therefore promoting the satellite adhesion via a relatively small area of anchoring points. Thus, lateral electrostatic repulsions between negatively charged ligands on satellites hamper an efficient packing on the core surface (Fig. 3A). In contrast, the branched PEI coating generates a dense cloud of positive amino groups which increases the extent of binding points with the small gold nanoparticles, in turn, reducing the satellite-to-satellite repulsions on the outer shell (Fig. 3B).
 | ||
| Fig. 3 Schematics of satellite adhesion onto (A) PAH coated and (B) branched PEI coated core particles. Representative TEM images of the corresponding nanoassemblies are also shown. | ||
Overall, the extinction spectra and TEM images (Fig. 2D and E and S7†) show that neither the composition of the metal core (Au vs. Ag), nor the diverse surface chemistries of the Au satellites, nor the encoding agent has any apparent repercussion on the final structural features of the assemblies. These pieces of evidence demonstrate that the assembling process is solely driven by the electrostatic interactions between the molecular linkers (MUA and PEI), while the composition of the spherical core particle (for a given size) and the chemical nature of the SCs do not play any appreciable role.
The exceptional universality of the MUA/PEI assembling approach is further validated by the straightforward fabrication of MBA encoded core-assemblies using core particles with diverse sizes (Fig. S8†), shapes and surface chemistries, such as PVP-stabilized gold nanostars and CTAB-stabilized gold nanorods (Fig. 4A–D). Analogously, the codification with other SERS active molecules (Fig. 4E) can be achieved by simply replacing MBA with other reporters, without altering the experimental protocol (Fig. 4E).
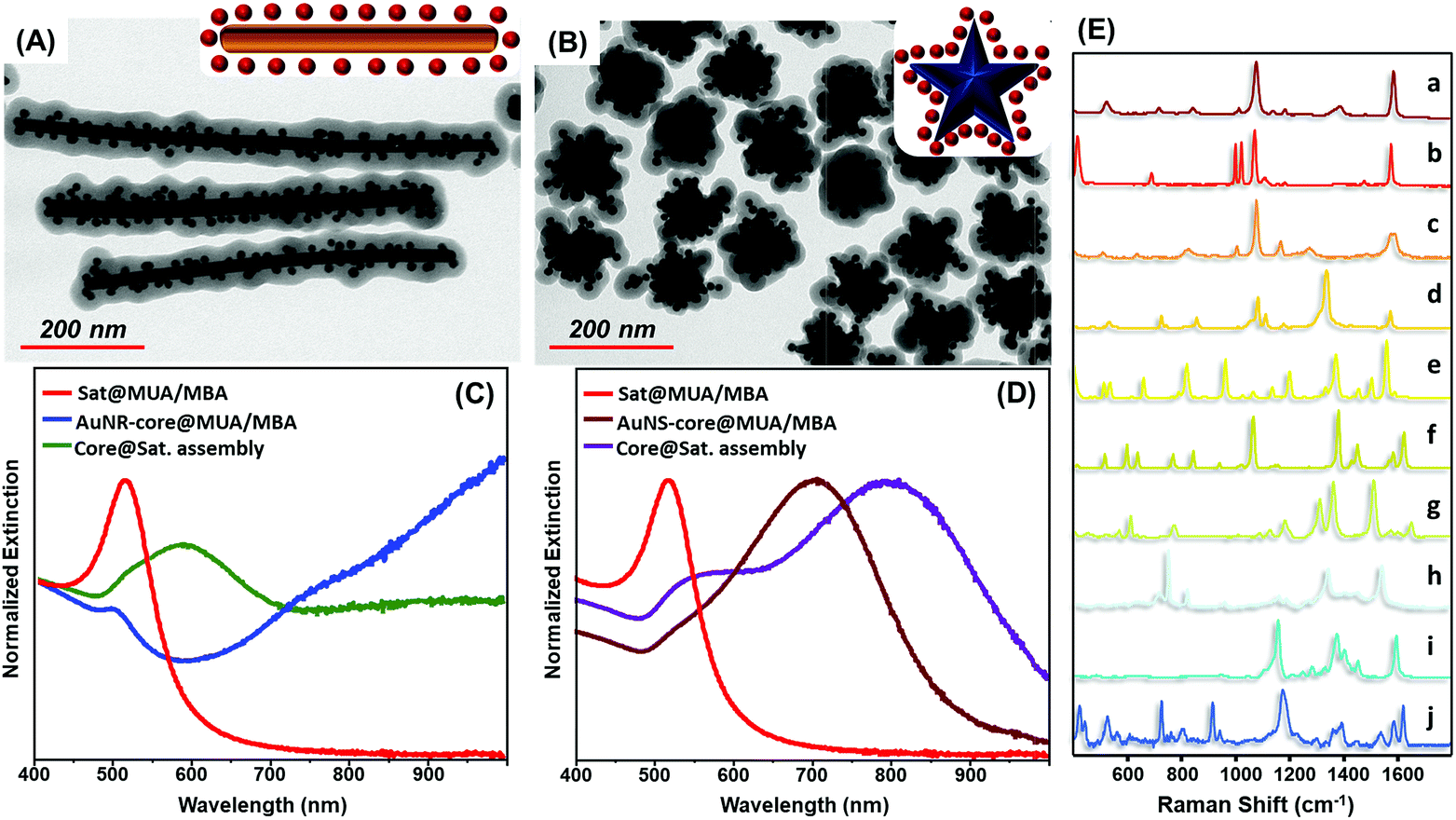 | ||
| Fig. 4 (A and B) Schematics and representative TEM images of core–satellite assemblies comprising gold satellites (12 nm diameter) and, as the core particle, (A) long gold nanorods (length 610 ± 130 nm, width: 20 ± 2 nm) or (B) gold nanostars (tip-to-tip length = 77 ± 13 nm). Core particles were modified with a MUA/MBA mixed layer prior to wrapping with PEI. (C and D) Normalized extinction spectra of MUA/MBA functionalized Au-cores (either gold nanorods or gold nanostars) and Au satellites. The normalized extinction spectra of the corresponding core–satellite assemblies are also included. (E) SERS spectra of different core–satellites encoded with (a) MBA; (b) thiophenol; (c) 4-mercaptothiophenol; (d) 4-nitrothiophenol; (e) naphthalene-1-thiol; (f) naphthalene-2-thiol; (g) rhodamine 6G; (h) astra blue; (i) congo red; and (j) crystal violet. | ||
To test the optical enhancing properties of the materials produced and the possibility of single particle detection, diluted solutions of the colloidal preparations (all encoded with MBA, Fig. 5A, and coated with silica) were spin-coated on silicon slides to reach concentrations below one particle per μm2 (Fig. 5B), the spatial resolution of a laser focused through a 50× objective. Then, the samples were imaged on a Raman microscope with 633 and 785 nm laser lines (Fig. 5C), in the case of gold and gold–gold composites and also a 532 nm laser line for silver and silver–gold composites. It is important to note that although all the composite materials yielded strong SERS signals for all lasers in the single particle regime (particle density below 0.2 particles per μm2), in the case of the silica isolated single silver and gold nanoparticles, more concentrated films were required to provide a measurable signal, specifically, 18 and 5 particles per μm2 for gold and silver cores, respectively, and 45 particles per μm2 for gold satellites.
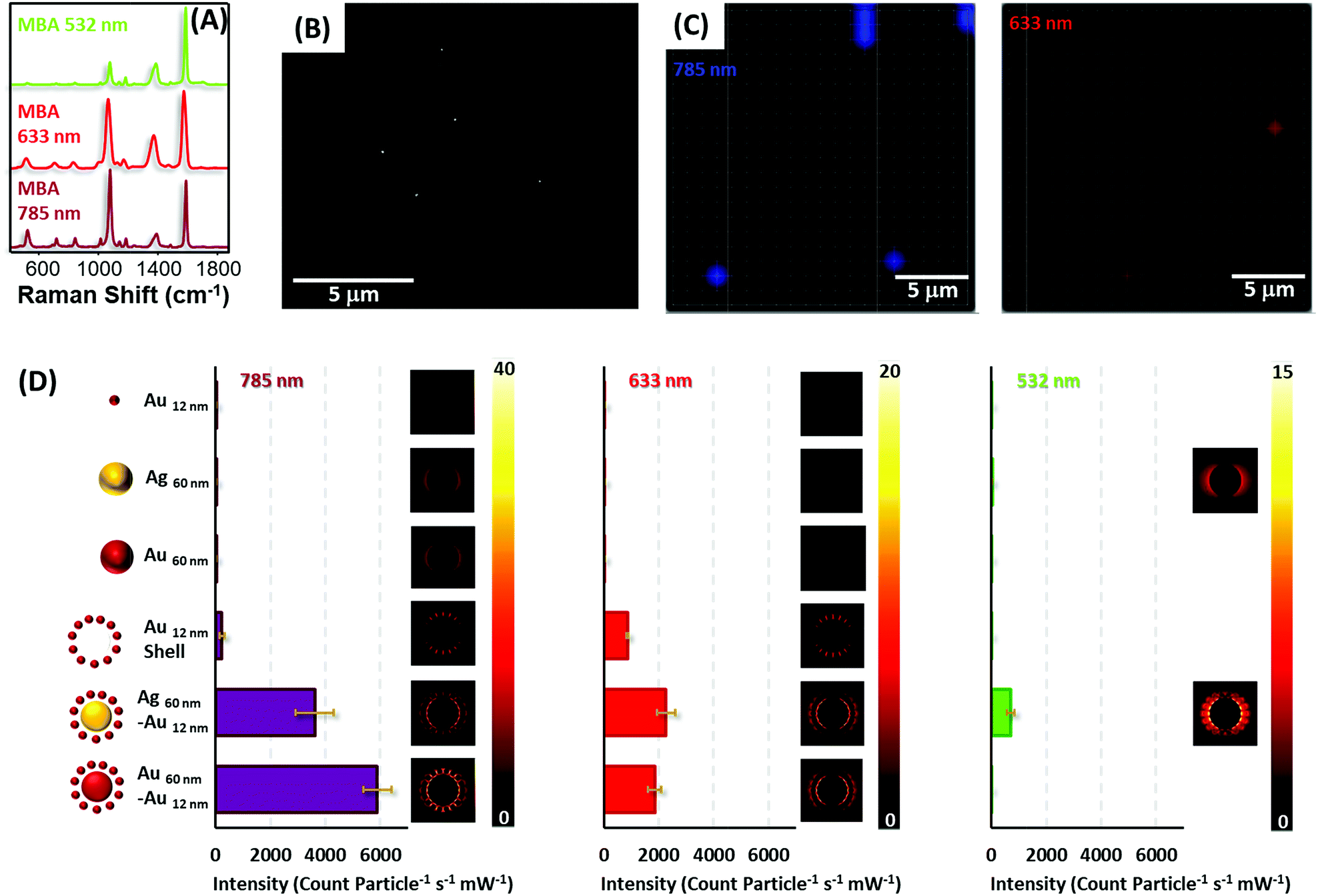 | ||
| Fig. 5 (A) SERS spectra of MBA at different laser lines on Aucore@Ausatellites. (B) Example of a SEM image of a sample spin-coated with a diluted solution of Aucore@Ausatellites. (C) SERS imaging of the sample with 785 and 633 nm laser lines. Acquisition time 0.5 s. (D) SERS intensity, normalized for a single particle, provided by each material at three laser lines: 785, 633 and 532 nm. Simulation of the electric field maps provided by each of the materials. | ||
Fig. 5D shows the normalized SERS intensity of the ring breathing vibrational mode of MBA, normalized for time and power at the sample and particle for the three laser lines. Experiments with the 785 nm laser show extremely high intensities for both core–shell materials. In fact, the enhancement factors for these composites as compared with those of the constituent gold or silver cores surpass 4 orders of magnitude, in full agreement with the theoretical calculations shown in the same graph. This fact can be explained by the generation of multiple hot spots46 formed between the satellite particles and between these satellites and the core. Notably, the abrupt decrease in SERS intensity for the satellite shells, upon removal of the cores, indicates that the larger electromagnetic contribution is due to the core. Thus, although satellite shells can provide SERS signals in the single nanoparticle regime, the contribution of a core increases that signal dramatically over 20-fold. Another interesting detail lies in the fact that standard deviations for all composite materials are always below 10% of the average intensity. This indicates a good homogeneous SERS response between particles, subsequently allowing their use for quantitative ultrasensitive determinations.
In the case of the red line, the same trends are demonstrated. However, in contrast to the NIR laser, silver–gold composites offer a slightly larger signal than gold due to a better electromagnetic coupling between the laser and LSPR.47 Notably, for the green laser, only silver containing materials yielded SERS due to the presence of strongly damped plasmons in gold when excited with green or more energetic lasers because of coupling to interband transitions.48 Yet, even at this energetic excitation, silver–gold composites are able to provided single particle SERS albeit at a more modest level than with less energetic lines.
3 Experimental
Materials
Trisodium citrate dihydrate (≥99.5%, C6H5Na3O7·2H2O), absolute ethanol (≥99.9%, EtOH), L-ascorbic acid (≥99.0%, AA), 4-mercaptobenzoic acid (≥99%, 4MBA), thiophenol, 4-mercaptothiophenol, 4-nitrothiophenol, naphthalene-1-thiol, naphthalene-2-thiol, rhodamine 6G, astra blue, congo red, crystal violet, 11-mercaptoundecanoic acid (≥95%, MUA), silver nitrate (≥99.9999%, AgNO3), gold(III) chloride trihydrate (≥99.9%, HAuCl4·3H2O), sodium hydroxide (≥98%, NaOH), ammonia solution (29%, NH4OH), tetraethoxysilane (≥99.999%, TEOS), polyvinylpyrrolidone (average MW = 8000, PVPk8), N,N′-dimethylformamide (≥99%, DMF), branched polyethylenimine (≥99.5%, PEI), cetyltrimethylammonium bromide (99.72%, CTAB), sodium borohydride (99%, NaBH4), poly-allylamine hydrochloride (average MW = 17500, PAH), and magnesium sulfate (≥98%, MgSO4) were purchased from Sigma-Aldrich Gmbh (Munich, Germany). All reactants were used without further purification. Milli-Q water (18 MΩ cm−1) was used in all aqueous solutions, and all glassware was cleaned with aqua regia before the experiments.
Synthesis of citrate-stabilized spherical gold nanoparticles (ca. 63 nm diameter)
Spherical gold nanoparticles of approximately 63 nm diameter were produced by a modification of the kinetically controlled seeded growth method.49 Briefly, 1.54 mL of an aqueous solution of HAuCl4 (0.081 M) was added to a boiling aqueous solution of C6H5Na3O7·2H2O (500 mL, 0.27 mM) under vigorous stirring. A condenser was utilized to prevent the evaporation of the solvent. During this time, the color of the solution gradually changed from colorless to purple to finally become deep red. 30 min after the HAuCl4 addition, the reaction was cooled until the temperature of the solution reached 90 °C. Then 4.0 mL of C6H5Na3O7·2H2O (0.1 M) were injected. After 5 min, 2 mL of C6H5Na3O7·2H2O (0.1 M) and 808.5 μL of HAuCl4 (0.1 M) were sequentially injected (time delay ∼ 2 min). This process (sequential addition of C6H5Na3O7·2H2O and HAuCl4) was repeated 3 times to finally obtain Au NPs of the desired diameter (in each step, gold particles were progressively grown to larger sizes). After 30 min of the third addition, one aliquot of 2 mL was extracted for further characterization by transmission electron microscopy (TEM) and UV-vis spectroscopy. The concentration of NPs was approximately the same as that of the original seed particles (5.7 × 1010 NPs mL−1, [Au] = 7.28 × 10−4 M). Finally, the NPs were cleaned by centrifugation at 3000 rpm for 8 minutes to eliminate the excess of citrate. The nanoparticle size was estimated to be 62.9 ± 4.7 nm (Fig. S4†).Spherical gold nanoparticle (ca. 63 nm) codification and PEI wrapping
To provide colloidal stability to the Au nanoparticles during the encoding process, 30 mL of the as-produced Au NPs were functionalized with a small amount of MUA (1.25 molecules per nm2). To this end, a solution containing NH4OH (120 μL, 2.9% aqueous solution) and MUA (440.8 μL, 10−4 M in EtOH) was prepared. This solution was then rapidly added under vigorous stirring to the gold nanoparticle solution. Agitation was continued for 30 min to ensure MUA functionalization of the Au surface. Then, a solution containing 3.0 MBA molecules per nm2 (1058.0 μL, 10−4 M in EtOH) was also rapidly added under vigorous stirring to the MUA-stabilized gold nanoparticles. 1 hour later, the NPs were cleaned by centrifugation at 2800 rpm for 8 minutes to remove carefully the C6H5Na3O7·2H2O and redispersed in 43.7 mL of Milli-Q water to achieve [Au] ∼ 5 × 10−4 M. Finally, the Au NPs were wrapped with a polyelectrolyte monolayer. To this end, a solution of MBA encoded Au NPs (43.7 mL, [Au] ∼ 5 × 10−4 M) was added drop by drop to a solution of PEI (43.7 mL, 2 g L−1, previously sonicated for 30 min) under vigorous stirring. Stirring was continued for 3 hours and then the particles were washed twice with 43.7 mL of Milli-Q water (3800 rpm, 8 min). No sonication was performed in any of the cleaning steps to avoid any detachment of the polyelectrolyte layer.Synthesis of citrate-stabilized spherical silver nanoparticles (ca. 65 nm diameter)
Spherical silver nanoparticles of approximately 65 nm diameter were produced by heating 500 mL of H2O until boiling under strong magnetic stirring. When it boils energetically, a mixture containing AA (500 μL, 0.1 M) and C6H5Na3O7·2H2O (6.818 mL, 0.1 M) is added. 1 minute after the addition, a solution previously incubated for 5 min containing AgNO3 (1488 μL, 0.1 M) and MgSO4 (1.118 μL, 0.1 M) is also added. Boiling and stirring were continued for 1 h. Finally, the NPs were cleaned by centrifugation (5400 rpm, 20 min) and redispersed in Milli-Q water. The nanoparticle size was estimated to be 64.6 ± 5.7 nm (Fig. S4†).Spherical silver nanoparticle codification and PEI wrapping
A solution containing MUA (189 μL, 1.0 × 10−4 M in EtOH) and MBA (189 μL, 1.0 × 10−4 M in EtOH) was prepared. This solution was then rapidly added to 30 mL of silver nanospheres (Ø ∼ 65 nm, [Ag] = 2.5 × 10−4 M, in Milli-Q water) and sonicated for 30 seconds (the number of molecules per nm2 was 1.25 for MUA and 3.0 for the SERS code). Consequently, 150.0 μL of a NaOH solution (0.23 M, in Milli-Q water) was added to the mixture under vigorous stirring and sonicated again for 30 seconds. 1 hour later, the NPs were cleaned by centrifugation (4200 rpm, 12 min) and redispersed in 15 mL of Milli-Q water to achieve [Ag] ∼ 5 × 10−4 M. Finally, the Ag NPs were wrapped with a polyelectrolyte monolayer. To this end, a solution of MBA encoded Ag NPs (15 mL, [Ag] ∼ 5 × 10−4 M) was added drop by drop to a solution of PEI (15 mL, 2 g L−1, previously sonicated for 30 min) under vigorous stirring. Stirring was continued for 1.5 hours and then the particles were washed twice with 10 mL Milli-Q water (4200 rpm, 12 min). No sonication was performed in any of the cleaning steps to avoid any detachment of the polyelectrolyte layer.Synthesis of citrate-stabilized spherical gold nanoparticles (ca. 12 nm diameter)
Spherical gold nanoparticles of approximately 12 nm diameter were produced by a modification of the well-known Turkevich method.50 Briefly, 833.0 μL of an aqueous solution of HAuCl4 (0.1 M) was added to a boiling aqueous solution of sodium citrate (500 mL, 2.2 mM) under vigorous stirring. A condenser was utilized to prevent the evaporation of the solvent. During this time, the color of the solution gradually changed from colorless to purple to finally become deep red. Finally, the NPs were cleaned by centrifugation at 9000 rpm for 28 minutes to eliminate the excess of citrate. The nanoparticle size was estimated to be 11.5 ± 0.8 nm (Fig. S4†).Spherical gold nanoparticle (ca. 12 nm diameter) codification
To provide colloidal stability to the Au nanoparticles during the encoding process, 60 mL of the as-produced satellite gold nanoparticles were functionalized with a small amount of MUA (1.25 molecules per nm2). To this end, a solution containing MUA (106 μL, 10−3 M in EtOH) was prepared. This solution was then rapidly added under vigorous stirring to the gold nanoparticle solution. Agitation was continued for 2 hours to ensure MUA functionalization of the Au surface. Then, a solution containing 3.0 MBA molecules per nm2 (254.4 μL, 10−3 M in EtOH) was also rapidly added under vigorous stirring to the MUA-stabilized gold nanoparticles. 1 hour later, a solution of NH4OH (120 μL, 2.9% aqueous solution) was added and the stirring was continued for 2 hours. Finally, the NPs were cleaned by centrifugation at 9000 rpm for 28 minutes to remove carefully the unreacted chemicals and redispersed in 20 mL of Milli-Q water to achieve [Au] ∼ 5 × 10−4 M.MUA-stabilized spherical gold nanoparticles (ca. 12 nm diameter)
60 mL of the as-produced satellite gold nanoparticles were functionalized with full surface coverage of MUA (5.0 molecules per nm2). To this end, a solution containing MUA (424.0 μL, 10−3 M in EtOH) was prepared. This solution was then rapidly added under vigorous stirring to the gold nanoparticle solution. Agitation was continued for 2 hours to ensure MUA functionalization of the Au surface. 3 hours later, a solution of NH4OH (120 μL, 2.9% aqueous solution) was added and the stirring was continued for 2 hours. Finally, the NPs were cleaned by centrifugation at 9000 rpm for 28 minutes to remove carefully the unreacted chemicals and redispersed in 20 mL of Milli-Q water to achieve [Au] ∼ 5 × 10−4 M.Synthesis of PVP-stabilized gold nanostars
The synthesis of gold nanostars (GNSs) was performed using a modified PVP-based method. In the first step, spherical gold seeds of approximately 10 nm diameter were produced by a modification of the well-known Turkevich method. Briefly, a solution of sodium citrate in Milli-Q water (150 mL, 1.93 mM) was heated with a heating mantle in a 250 mL three-necked round-bottom flask for 15 min under vigorous stirring. A condenser was utilized to prevent the evaporation of the solvent. After boiling had commenced, 308 μL of HAuCl4 (0.081 M) was injected. The color of the solution changed from yellow to bluish gray and then to soft pink in 10 min and to wine red after 30 min. The resulting particles were coated with negatively charged citrate ions and, hence, were well suspended in H2O. Then, the seeds were concentrated in a 250 mL beaker to a Au concentration of 9.45 × 10−4 M with a constant flow of N2 for 120 min. In the second step, a solution of PVPk8 (15 mM) in dimethylformamide (35 mL) was sonicated for 15 min. Then, 216 μL of HAuCl4 (0.081 M) was added to the mixture under rapid stirring at room temperature, followed by the concentrated seeds (1.108 mL, [Au] = 9.45 × 10−4 M). Within 5 min, the color of the solution changed from pink to blue, indicating the formation of gold nanostars. The solution was left stirring overnight. Then, the excess PVPk8 was removed by a 6-fold centrifugation (7000 rpm, 15 min) and redispersed in ethanol (the particle solution was adjusted to yield a final concentration of [Au] ∼ 4.5 × 10−4 M).Gold nanostar codification and PEI wrapping
First of all, 10 mL of the as-produced GNSs was functionalized with a small amount of MBA (0.5 molecules per nm2). To this end, a solution containing NH4OH (40 μL, 2.9% aqueous solution) and MBA (38.2 μL, 10−4 M in EtOH) was prepared. This solution was then rapidly added under vigorous stirring to the gold nanoparticle solution. Agitation was continued for 4 hours to ensure MBA adsorption on the GNS tips. Then, a solution containing 1.25 MUA molecules per nm2 (95.4 μL, 10−4 M in EtOH) was also rapidly added under vigorous stirring. Agitation was continued for 12 hours to guarantee MUA adsorption on the metallic surface. Again, and to further ensure the presence of the MBA molecules on the metallic surface, a solution containing 2.5 MBA molecules per nm2 (57.3 μL, 10−4 M in EtOH) was added under vigorous stirring. 24 hours later, the NPs were cleaned by centrifugation at 4500 rpm for 15 minutes to remove carefully the excess of reactants and redispersed in 10 mL of Milli-Q water to achieve [Au] ∼ 4.5 × 10−4 M. Finally, GNSs were wrapped with a polyelectrolyte monolayer. To this end, a solution of MBA-encoded GNSs (10 mL) was added drop by drop to a solution of PEI (10 mL, 2 g L−1, previously sonicated for 30 min) under vigorous stirring. Stirring was continued for 3.0 hours and then the particles were washed twice with 10 mL Milli-Q water (4200 rpm, 12 min). No sonication was performed in any of the cleaning steps to avoid any detachment of the polyelectrolyte layer.Synthesis of high aspect ratio CTAB-stabilized gold nanorods
Gold nanorods were produced by changing the seed crystallography of a previously reported seed-mediated procedure.51 Briefly, seed particles were prepared by dissolving 0.0015 g of sodium citrate in 20 mL of Milli-Q water. An aliquot of HAuCl4 solution (46.28 μL, 0.108 M) was added to yield a final HAuCl4 concentration of 2.5 × 10−4 M. Next, a freshly prepared NaBH4 solution (600 μL, 0.1 M) was quickly injected; meanwhile, the solution was energetically stirred (1200 rpm). After addition, the color immediately changed from light yellow to red. Stirring was continued for 1 h in an open atmosphere to allow the NaBH4 to decompose, avoiding overpressure. The growth solution was prepared by mixing a CTAB solution (0.1 M, 250 mL, thermostated at 32 °C) with an aqueous HAuCl4 solution (1213 μL, 1.08 M) to give a final concentration of 2.5 × 10−4 M. After that, ascorbic acid (1837 μL, 0.1 M) was added, followed by the careful addition of 500 μL seeds to the growth solution foam. After the addition, the mixture was energetically shaken and left undisturbed at 32 °C for 48 h to allow the high aspect ratio rods to sediment. Finally, the supernatant containing spheres was removed and the sediment containing the rods was redispersed in 40 mL H2O. This solution was again allowed to sediment for 2 days and the supernatant still containing spheres was removed. The collected Au rods were dispersed in CTAB (30 mL, 1 mM). This process was repeated until the supernatant did not contain any spheres.Gold nanorod codification and PEI wrapping
To provide colloidal stability to the Au rods during the encoding process, the produced rods were first functionalized with a small amount of MUA (1.25 molecules per nm2). To do this, 20 mL of the as-synthesized Au rods ([Au] = 0.5 mM, [CTAB] = 1 mM) were diluted with H2O to achieve 0.5 mM CTAB and 0.25 mM Au. Next, a solution containing NH4OH (120 μL, 2.9% aqueous solution) and MUA (898 μL, 10−4 M in EtOH) was rapidly added under vigorous stirring to the CTAB stabilized nanoparticle solution (40 mL). Agitation was continued for 30 min to ensure MUA functionalization of the Au surface. The MUA modified particles were centrifuged twice to remove the remaining CTAB and redispersed in ethanol (40 mL). For the encoding of the Au nanoparticles, a solution containing 3.0 MBA molecules per nm2 (2155 μL, 10−4 M in EtOH) was also rapidly added under vigorous stirring to the MUA-stabilized gold nanoparticles and left under magnetic stirring for 2 h. The NPs were cleaned by centrifugation at 2500 rpm for 5 minutes and redispersed in 40 mL of Milli-Q water to achieve a Au concentration of 2.5 × 10−4 M. Finally, the Au rods were wrapped with PEI. To this end, the solution of MBA encoded rods was added drop by drop to 40 mL of a PEI solution (2 g L−1, previously sonicated for 30 min) under vigorous stirring. After 3 hours, the particles were washed twice (2500 rpm, 5 min) to finally redisperse them in 40 mL Milli-Q water.Formation of core–satellite assemblies
2 mL of the PEI wrapped Au encoded NPs ([Au] = 5 × 10−4 M) or Ag encoded NPs ([Ag] = 5 × 10−4 M) were added respectively, drop by drop, to three different solutions: MUA/MBA encoded Au satellites (5.52 mL, ∼[Au] = 5 × 10−4 M), MUA stabilized Au satellites (5.52 mL, [Au] = 5 × 10−4 M) and citrate capped Au satellites (5.52 mL, [Au] = 5 × 10−4 M) under vigorous stirring. The stoichiometric ratio of Au cores:Au satellites was 1:400. To ensure an isotropic 3D coverage of the core NPs, the solutions were stirred for 12 hours. Finally, each of the assembled nanostructure solutions was added respectively to a solution containing 48.8 mL of EtOH and 995.2 μL of NH4OH (29%, aqueous solution).
Silica encapsulation and purification
The silica encapsulation of the assembled nanostructures was achieved through a modified Stöber method. The appropriate concentrations of H2O, NH4OH and EtOH for silica growth on the SERS-encoded planet–satellite nanostructures were previously adjusted, during the assembly step, to yield final concentrations of 7.94, 0.128 and 14.60 M, respectively (EtOH/H2O molar ratio of 1.84). Next TEOS (72 μL, 10% v/v in EtOH) was added, and the solution was energetically shaken and left undisturbed at room temperature for 14 hours, and then it was left undisturbed at 60 °C for 12 hours for the silica hardening. Finally, the resulting nanostructures were cleaned by centrifugation (5 × 3000 rpm, 6 min) to remove excess reactants and redispersed in 1 mL of Milli-Q water. Finally, the few non-assembled Au satellites that remained in solution were cleaned by precipitation (leaving the solution undisturbed for 96 hours in a 15 mL conical centrifuge tube and then removing the supernatant).Dissolution of Ag cores in core–satellite assemblies
After the formation and encapsulation of the core–satellite assemblies, the Ag cores were dissolved inducing the formation of the diamine-silver(I) complex [Ag(NH3)2]+ using NH4OH. To this end, 0.4 mL of the silica encapsulated Ag–Au core–satellite assemblies (59.3 pM, in Milli-Q water) was added to a solution containing 0.6 mL of EtOH and 33.0 μL of NH4OH (29%, aqueous solution). The solution was energetically shaken and left undisturbed at room temperature for 12 hours. Then, the resulting nanostructures were cleaned by centrifugation (4000 rpm, 10 min) to remove excess reactants and redispersed in 0.4 mL of Milli-Q water. This procedure was repeated 3 times to ensure that Ag cores were completely dissolved by formation of [Ag(NH3)2]+. Finally, the resulting nanostructures were cleaned by centrifugation (5000 rpm, 10 min) and redispersed in 0.4 mL of EtOH to achieve a final concentration of 59.3 pM.Characterization
UV-vis spectra were recorded using a Thermo Scientific Evolution 201 UV-vis spectrophotometer. Silver concentration of Ag NPs was calculated using the Lambert–Beer law with an extinction coefficient of 6.61 × 1010 M−1 cm−1. Electron micrographs were recorded with a transmission electron microscope (JEOL JEM-1011 operating at 80 kV) and an environmental scanning electron microscope (JEOL 6400) for the structural characterization of the samples and their distribution on the silicon wafers.Substrate deposition of core–satellite assemblies and SERS characterization
Silicon wafers (1 cm × 1 cm) were cleaned with a standard RCA-1 solution to remove any possible organic residue. Solutions of all types of NP building blocks and their corresponding assemblies were prepared at different Au concentrations ranging from 10−3 M to 10−5 M. Next, 50 μL of each concentration were spin coated ((1st ramp) 500 rpm, 10 s; (2nd ramp) 3000 rpm, 30 s, with an acceleration rate of 500 rpm s−1 for both ramps) on the silicon wafers to achieve particle densities ranging from approx. 0.1 to 2 particles per μm2. Samples were characterized by SEM and the number of particles per μm2 enumerated. SERS spectra were collected in backscattering geometry with a Renishaw inVia Reflex system equipped with a 2D-CCD detector and a Leica confocal microscope. Excitation of the sample was carried out with either 785, 633 or 532 nm laser lines with acquisition times ranging from 0.5 to 2 s and power at the sample of about 0.15 and 3 mW. The laser was focused onto the sample with a ×50 objective, providing a spatial resolution of ca. 1 μm.Near field enhancement calculations
The near field intensity normalized by the incident intensity was obtained by using a finite-difference time-domain method for the Maxwell equation problem.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was funded by the Spanish Ministerio de Economia y Competitividad (CTQ2017-88648R, RYC-2016-20331, and RYC-2015-19107), the Generalitat de Cataluñya (2017SGR883), the Universitat Rovira i Virgili (2017PFR-URV-B2-02), and the Universitat Rovira i Virgili and Banco Santander (2017EXIT-08).Notes and references
- N. Pazos-Perez, E. Pazos, C. Catala, B. Mir-Simon, S. Gómez-de Pedro, J. Sagales, C. Villanueva, J. Vila, A. Soriano, F. J. García de Abajo and R. A. Alvarez-Puebla, Sci. Rep., 2016, 6, 29014 CrossRef CAS PubMed.
- M. Y. Sha, H. Xu, M. J. Natan and R. Cromer, J. Am. Chem. Soc., 2008, 130, 17214–17215 CrossRef CAS PubMed.
- A. Prasanna de Silva, M. R. James, B. O. F. McKinney, D. A. Pears and S. M. Weir, Nat. Mater., 2006, 5, 787 CrossRef CAS PubMed.
- R. Pérez-Pineiro, M. A. Correa-Duarte, V. Salgueirino and R. A. Alvarez-Puebla, Nanoscale, 2012, 4, 113–116 RSC.
- H. Fenniri and R. Alvarez-Puebla, Nat. Chem. Biol., 2007, 3, 247 CrossRef CAS PubMed.
- D. C. Pregibon, M. Toner and P. S. Doyle, Science, 2007, 315, 1393–1396 CrossRef CAS PubMed.
- F. Ramiro-Manzano, R. Fenollosa, E. Xifré-Pérez, M. Garín and F. Meseguer, Adv. Mater., 2011, 23, 3022–3025 CrossRef CAS PubMed.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2002, 21, 41 CrossRef PubMed.
- L. Guerrini, N. Pazos-Perez, E. Garcia-Rico and R. Alvarez-Puebla, Cancer Nanotechnol., 2017, 8, 5 CrossRef.
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed.
- A.-I. Henry, B. Sharma, M. F. Cardinal, D. Kurouski and R. P. Van Duyne, Anal. Chem., 2016, 88, 6638–6647 CrossRef CAS PubMed.
- V. Giannini, A. I. Fernandez-Dominguez, S. C. Heck and S. A. Maier, Chem. Rev., 2011, 111, 3888–3912 CrossRef CAS PubMed.
- N. J. Halas, S. Lal, W. S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- L. O. Brown and S. K. Doorn, Langmuir, 2008, 24, 2178–2185 CrossRef CAS PubMed.
- J. M. Romo-Herrera, R. A. Alvarez-Puebla and L. M. Liz-Marzan, Nanoscale, 2011, 3, 1304–1315 RSC.
- E. C. Cho, L. Au, Q. Zhang and Y. Xia, Small, 2010, 6, 517–522 CrossRef CAS PubMed.
- I. Choi, H. D. Song, S. Lee, Y. I. Yang, T. Kang and J. Yi, J. Am. Chem. Soc., 2012, 134, 12083–12090 CrossRef CAS PubMed.
- R. C. Mucic, J. J. Storhoff, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 12674–12675 CrossRef CAS.
- X. Xu, N. L. Rosi, Y. Wang, F. Huo and C. A. Mirkin, J. Am. Chem. Soc., 2006, 128, 9286–9287 CrossRef CAS PubMed.
- L. Y. T. Chou, F. Song and W. C. W. Chan, J. Am. Chem. Soc., 2016, 138, 4565–4572 CrossRef CAS PubMed.
- L. Xu, H. Kuang, C. Xu, W. Ma, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 1699–1709 CrossRef CAS PubMed.
- K. Li, K. Wang, W. W. Qin, S. H. Deng, D. Li, J. Y. Shi, Q. Huang and C. H. Fan, J. Am. Chem. Soc., 2015, 137, 4292–4295 CrossRef CAS PubMed.
- C. Rossner and P. Vana, Angew. Chem., Int. Ed., 2014, 53, 12639–12642 CAS.
- P. Dey, S. Zhu, K. J. Thurecht, P. M. Fredericks and I. Blakey, J. Mater. Chem. B, 2014, 2, 2827–2837 RSC.
- R. P. M. Höller, M. Dulle, S. Thomä, M. Mayer, A. M. Steiner, S. Förster, A. Fery, C. Kuttner and M. Chanana, ACS Nano, 2016, 10, 5740–5750 CrossRef PubMed.
- S. Borsley, S. Flook and E. R. Kay, Chem. Commun., 2015, 51, 7812–7815 RSC.
- M. Schutz and S. Schlucker, Phys. Chem. Chem. Phys., 2015, 17, 24356–24360 RSC.
- W. Xie, B. Walkenfort and S. Schlücker, J. Am. Chem. Soc., 2013, 135, 1657–1660 CrossRef CAS PubMed.
- R. Alvarez-Puebla, L. M. Liz-Marzán and F. J. García de Abajo, J. Phys. Chem. Lett., 2010, 1, 2428–2434 CrossRef CAS.
- L. Guerrini and D. Graham, Chem. Soc. Rev., 2012, 41, 7085–7107 RSC.
- G. B. Braun, S. J. Lee, T. Laurence, N. Fera, L. Fabris, G. C. Bazan, M. Moskovits and N. O. Reich, J. Phys. Chem. C, 2009, 113, 13622–13629 CrossRef CAS.
- W. Xiong, D. Sikdar, L. W. Yap, M. Premaratne, X. Li and W. Cheng, Nanoscale, 2015, 7, 3445–3452 RSC.
- A. Shiohara, S. M. Novikov, D. M. Solís, J. M. Taboada, F. Obelleiro and L. M. Liz-Marzán, J. Phys. Chem. C, 2015, 119, 10836–10843 CrossRef CAS.
- N. Gandra, A. Abbas, L. Tian and S. Singamaneni, Nano Lett., 2012, 12, 2645–2651 CrossRef CAS PubMed.
- Q. Ruan, L. Shao, Y. Shu, J. Wang and H. Wu, Adv. Opt. Mater., 2014, 2, 65–73 CrossRef.
- B. Mir-Simon, I. Reche-Perez, L. Guerrini, N. Pazos-Perez and R. A. Alvarez-Puebla, Chem. Mater., 2015, 27, 950–958 CrossRef CAS.
- H. Hiramatsu and F. E. Osterloh, Langmuir, 2003, 19, 7003–7011 CrossRef CAS.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815 CrossRef CAS PubMed.
- F. Benz, R. Chikkaraddy, A. Salmon, H. Ohadi, B. de Nijs, J. Mertens, C. Carnegie, R. W. Bowman and J. J. Baumberg, J. Phys. Chem. Lett., 2016, 7, 2264–2269 CrossRef CAS PubMed.
- B. M. Ross, J. R. Waldeisen, T. Wang and L. P. Lee, Appl. Phys. Lett., 2009, 95, 193112 CrossRef PubMed.
- C. Song, P. Wang and H. A. Makse, Nature, 2008, 453, 629–632 CrossRef CAS PubMed.
- Z. Rong, R. Xiao, C. Wang, D. Wang and S. Wang, Langmuir, 2015, 31, 8129–8137 CrossRef CAS PubMed.
- J. H. Yoon, J. Lim and S. Yoon, ACS Nano, 2012, 6, 7199–7208 CrossRef CAS PubMed.
- J. H. Yoon, Y. Zhou, M. G. Blaber, G. C. Schatz and S. Yoon, J. Phys. Chem. Lett., 2013, 4, 1371–1378 CrossRef CAS PubMed.
- N. Pazos-Perez, C. S. Wagner, J. M. Romo-Herrera, L. M. Liz-Marzan, F. J. G. de Abajo, A. Wittemann, A. Fery and R. A. Alvarez-Puebla, Angew. Chem., Int. Ed., 2012, 51, 12688–12693 CrossRef CAS PubMed.
- A. D. McFarland, M. A. Young, J. A. Dieringer and R. P. Van Duyne, J. Phys. Chem. B, 2005, 109, 11279–11285 CrossRef CAS PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- N. G. Bastus, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef CAS PubMed.
- B. V. Enustun and J. Turkevich, J. Am. Chem. Soc., 1963, 85, 3317–3328 CrossRef CAS.
- S. Jessl, M. Tebbe, L. Guerrini, A. Fery, R. A. Alvarez-Puebla and N. Pazos-Perez, Small, 2018, 14, 1703879 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00257f |
| This journal is © The Royal Society of Chemistry 2019 |