
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of precursor pre-treatment on the vapor deposition of WS2 monolayers†
Mei Er
Pam
ab,
Yumeng
Shi
*acd,
Junping
Hu
a,
Xiaoxu
Zhao
 e,
Jiadong
Dan
e,
Xue
Gong
ac,
Shaozhuan
Huang
a,
Dechao
Geng
a,
Stephen
Pennycook
fg,
Lay Kee
Ang
ab and
Hui Ying
Yang
*a
e,
Jiadong
Dan
e,
Xue
Gong
ac,
Shaozhuan
Huang
a,
Dechao
Geng
a,
Stephen
Pennycook
fg,
Lay Kee
Ang
ab and
Hui Ying
Yang
*a
aPillar of Engineering Product Development, Singapore University of Technology and Design, 8 Somapah Road, Singapore 487372, Singapore. E-mail: yanghuiying@sutd.edu.sg
bScience and Math Cluster, Singapore University of Technology and Design, 8 Somapah Road, Singapore 487372, Singapore
cInternational Collaborative Laboratory of 2D Materials for Optoelectronic Science & Technology of Ministry of Education, Engineering Technology Research Center for 2D Material Information Function Devices and Systems of Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
dEngineering Technology Research Center for 2D Material Information Function Devices and Systems of Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
eDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore
fDepartment of Materials Science and Engineering, National University of Singapore, 9 Engineering Drive 1, Singapore 117575, Singapore
gNUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, 13 Centre for Life Science, #05-01, 28 Medical Drive, Singapore 117456, Singapore
First published on 5th November 2018
Abstract
Transition metal oxide powders have been widely used as the growth precursors for monolayer transition metal dichalcogenides (TMDCs) in chemical vapor deposition (CVD). It has been proposed that metal oxide precursors in the gas phase undergo a two-step reaction during CVD growth, where transition metal sub-oxides are likely formed first and then the sulfurization of these sub-oxides leads to the formation of TMDCs. However, the effects of stoichiometry of transition metal oxide precursors on the growth of TMDC monolayers have not been studied yet. In this contribution, we report the critical role of the WO3 precursor pre-annealing process on the growth of WS2 monolayers. Besides, several WO3 precursors with different types of oxygen vacancies have also been prepared and investigated by X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and density functional theory calculation. Among all the non-stoichiometric WO3 precursors, thermally annealed WO3 powder exhibits the highest oxygen vacancy concentration and produces WS2 monolayers with significantly improved quality in terms of lateral size, density, and crystallinity. Our comprehensive study suggests that the chemical composition of transition metal oxide precursors would be fundamentally critical for the growth of large-area and high-quality WS2 monolayers, which further pave the way for revealing their intrinsic properties and unique applications.
Introduction
The extraordinary electrical, optical, thermal and mechanical properties, which exist in monolayer transition metal dichalcogenides (TMDC) materials, offer various promising applications in future ultrathin logic devices, nanogenerators, flexible electronics and optoelectronic devices.1–4 Specifically, the tunable properties of band offset, carrier density,5 and band gap6–8 make TMDCs one of the most extensively researched subjects for practical and fundamental studies. Monolayer tungsten disulfide (WS2) has been intensively studied as a next generation nanoelectronic and optoelectronic material due to its indirect to direct band gap tunability,9,10 high on/off current ratio,11,12 high thermal stability,13 excellent electrostatic integrity14 and strong photoluminescence.15,16 Additionally, WS2 monolayers have shown an interesting spin orbit coupling effect resulting from the breaking of inversion symmetry of their crystal lattices, and the strong magnetic field interaction in WS2 monolayers favors their applications in optoelectronic and spintronic devices.17,18Various approaches have been employed to prepare WS2 monolayers. The layered structure of WS2 allows the mechanical or chemical exfoliation of monolayer crystals from their bulk counterparts. However, the exfoliated flakes show random thickness, size and shape that create great difficulties in large scale industrial applications.16 Chemical vapor deposition (CVD) has been widely adapted as one of the most effective strategies to grow wafer-scale two-dimensional (2D) materials including graphene, boron nitride, and TMDCs.19–23 In the pioneering studies of monolayer WS2 synthesis, tungsten trioxide (WO3) powder was chosen as the growth precursor and a low concentration of hydrogen (H2) gas was usually introduced to reduce WO3 to tungsten suboxide.22,24,25 Nevertheless, excessive hydrogen supply could also have an etching effect and induce various structural defects in monolayer WS2.16,25,26 Traditionally, tungsten oxides have been intensively studied previously for photocatalysis,27,28 electrochemistry,29 electrochromic application,30,31etc. According to the literature, WO3 powder is usually found in a non-stoichiometric composition and the W element may show various chemical states due to the spontaneous reduction of WO3 under ambient conditions.30 It has been demonstrated that the sulfurization process of transition-metal suboxides in the CVD process plays a critical role in the nucleation and crystallization of TMDC monolayers.22,32
In the CVD process, the generation and evolution of transition metal sub-oxide species can significantly affect the morphology and edge structure of TMDCs, which lead to dramatically different optical, electronical and magnetic properties.20 However, the study of the effects of chemical composition of WO3 on the CVD growth of WS2, in terms of quality, morphology and optical properties, is still lacking. Herein, we prepared various tungsten oxide precursors with different oxygen vacancy concentrations, that is, commercially available fresh WO3 (FWO3), thermally annealed WO3 (AWO3), air plasma treated WO3 (PTWO3) and thermally annealed PTWO3 powder (PTAWO3) to provide tungsten suboxide sources. The results reveal that AWO3 powder with the highest oxygen vacancy content produces WS2 monolayers with significantly improved domain size, higher coverage density, and higher crystallinity. Besides, we further demonstrate that the use of AWO3 powder instead of the introduction of hydrogen gas during the growth of monolayer WS2 to provide a tungsten suboxide source will enhance the growth of monolayer WS2 and produce monolayer WS2 with suppressed atomic defects. The control synthesis of WS2 monolayers by precursor pretreatment provides further insights into the growth mechanism and enables the production of WS2 monolayers with high quality for the development of a broad range of optoelectronic and electronic applications.
Experimental methods
Preparation of tungsten trioxide (WO3) precursors
The WO3 precursors were pretreated with heat, air plasma or both. Four WO3 precursors prepared with different methods were studied for comparison, including commercially available WO3 (FWO3) (Sigma-Aldrich), thermally annealed WO3 (AWO3), air plasma treated WO3 (PTWO3) and plasma treated annealed WO3 (PTAWO3) powder.For AWO3 powder, 500 mg of WO3 powder was loaded in an alumina boat, placed at the center of a furnace, and heated up to 500 °C in air for 30 minutes.
For PTWO3 powder, 500 mg of WO3 powder was loaded in an alumina boat, placed at the center of a plasma reactor center (Plasma Cleaner PDC-32G-2). The sample was directly exposed to a direct inductively coupled plasma. During the plasma treatment process, the chamber was evacuated to 1.5 mTorr with a mechanical pump and the radio frequency power was turned to ∼18 W, and at the same time air was introduced into the chamber and the process was continued for 5 minutes.
For PTAWO3 powder, 500 mg of WO3 powder was first treated using air plasma with the same preparation steps as the plasma treated WO3 powder, and the powder was then annealed using the same procedure that we employed to prepare the annealed WO3 powder sample.
Growth process of monolayer WS2
The employed CVD process for monolayer WS2 is based on our previous report.33,34 Prior to the growth process, single-side polished sapphire substrates were washed with the solution mixture of H2SO4 and H2O2 and blow-dried with nitrogen gas. An alumina boat filled with 500 mg pre-prepared WO3 powder (Sigma-Aldrich, 99.9%) was located at the center of a furnace, and the cleaned sapphire substrates were placed facing down on the boat with WO3 powder as illustrated in Fig. S1.† A separate boat with sulfur powder (Sigma-Aldrich, 99.5%) was located at the upstream of the furnace, which was heated up to the sulfur evaporation temperature at 120 °C with a heating belt during the growth of monolayer WS2. The CVD chamber was initially pumped down to a base pressure of ∼1 mTorr for 5 minutes and maintained at 15 torr with a 100 sccm flow of argon gas during the synthesis process. The furnace was then heated from room temperature to 200 °C and the temperature was kept constant for 10 minutes to evaporate the water vapor in the quartz tube. The temperature was then further heated up to 950 °C at a ramp rate of 25 °C per min and kept constant for 10 minutes. Finally, the growth chamber was cooled down to room temperature naturally. For the hydrogenated environmentally synthesized WS2 (HWS2) sample, the growth process was the same as that described above with commercially available WO3 powder except that 5% hydrogen gas is introduced into the tube furnace during the growth process.DFT calculation of the WO3 crystal structure
DFT calculations were performed on the crystal structure of WO3 with and without oxygen vacancies. Our calculations were based on density functional theory (DFT) using plane-wave pseudopotentials with the exchange-correlation of Perdew–Burke–Ernzerhof (PBE)35,36 form as implemented in the Vienna Ab initio Simulation Package (VASP).37 A cutoff energy of 500 eV was employed for the plane wave expansion of the wave functions. The Brillouin zone was sampled with a 9 × 5 × 5 Monkhorst–Pack k-point mesh38 for the structural optimization. The convergence criteria for the total energy and ionic forces were set to 10−5 eV and 0.005 eV Å−1 respectively. Spin polarization was carefully tested and no spin-polarization effect was observed in our calculations.Characterization
Photoluminescence (PL) and Raman measurements were obtained with a Witec Alpha300 Confocal Raman system with a laser excitation wavelength of 532 nm and grating density of 600 and 1800 mm−1 respectively. X-ray photoelectron spectroscopy (XPS, PHI Quantera II) with a monochromatic Al Kα source (1486.69 eV), operated at a base pressure of 10−7 Pa, was employed to obtain the chemical configuration spectra. X-ray diffraction (XRD, Bruker D8) with a Cu Kα source was employed to identify the crystalline phases and structures of the prepared WO3 powders. The annular dark field scanning transmission electron microscope (ADF-STEM) image was captured on an aberration-corrected JEOL ARM-200F equipped with a cold field emission gun, operating at 80 kV. The convergence semi-angle of the probe is ∼30 mrd. STEM-ADF images were captured for a half angle range from ∼85 to 280 mrd. A dwell time of 19 μs per pixel was set for a single-scan image.Transfer
The as-grown monolayer WS2 on sapphire was first spin coated with a thin layer of poly(methyl methacrylate) (PMMA) for one minute at a spin speed of 2000 rpm. Then, the edges of the sapphire substrate were removed and the substrate was immersed slowly in a hot solution of 3 mol L−1 NaOH (160 °C) to detach the PMMA layer from the sapphire substrate. The resulting sample was then delaminated in deionized water and washed a few times in deionized (DI) water. Finally, the sample was transferred onto a target substrate and carefully blow-dried with nitrogen gas. The PMMA coating was finally removed by using hot acetone vapor at 120 °C for 15 minutes. The samples for STEM study were prepared onto 200 mesh Quantifoil R2/1 micromachined copper grids from SPI Supplies, Structure Probe, Inc. using the same transfer method.Results and discussion
Generally, the synthesis of 2D TMDCs with the chemical vapor deposition method is based on the direct competition and multiple elementary processes in two reaction pathways between the transition metal suboxide and sulfur. The reaction pathways consist of: (1) transition metal suboxide diffuses and adsorbs on the substrate, and then reacts with S to form monolayer TMDC; (2) transition metal suboxide directly reacts with S in the gas phase to form a TMDC and the resulting WS2 clusters adsorb, nucleate and form on the substrate.22,32 Both of the two reaction routes strongly rely on the evaporation and stable delivery of the tungsten source. The metastable state tungsten suboxide can provide a much easier control over the supply of the tungsten source, and thus facilitate the growth process and enhance the growth of WS2 monolayers. Therefore, the formation of tungsten suboxide plays a critical role in the nucleation and crystallization of TMDC monolayers. In a previous report, in order to promote the growth of monolayer WS2, hydrogen gas was introduced during the synthesis, which assists the reduction of tungsten trioxide.16,24,25Fig. 1a illustrates the synthesis process of monolayer WS2 in an Ar/H2 atmosphere in which hydrogen gas acts as a tungsten trioxide reductant to reduce tungsten oxide to form volatile tungsten suboxides (WO3 + xH2 → WO3−x + xH2O).16,25,39 The resulting WO3−x are readily volatile, easily evaporate and release the tungsten source.40–42 The rapid evaporation of the tungsten source will eventually lead to an increase of nucleation sites and hence promote the growth of WS2.24,25 Unfortunately, the excessive supply of hydrogen has etching effects on WS2 (WS2 + 2H2 → W + 2H2S).16,25 The excess hydrogen in the growth atmosphere will react with the as-grown WS2 flakes and remove the sulfur elements, resulting in sulfur vacancies. Precursor pre-treatments such as thermal annealing and plasma treatment create oxygen vacancies in WO3, resulting in WO3−x (see Fig. 1b). The formation of volatile WO3−x suggests great potential in promoting the growth of monolayer WS2. Herein, to reveal the effects of oxygen vacancies in WO3 on the growth of WS2 monolayers, various tungsten trioxide powders with different oxygen vacancies were prepared, including thermally annealed WO3 (AWO3) powder, air plasma treated WO3 (PTWO3) powder, air plasma treated and annealed WO3 (PTAWO3) powder, and commercially available WO3 powder (FWO3). We first employed AWO3 powder to obtain the optimized WS2 growth temperature under the same pressure of 15 torr (see Fig. S1†). As illustrated in Fig. S1,† an optimized growth temperature of 950 °C was determined for WS2 monolayer synthesis.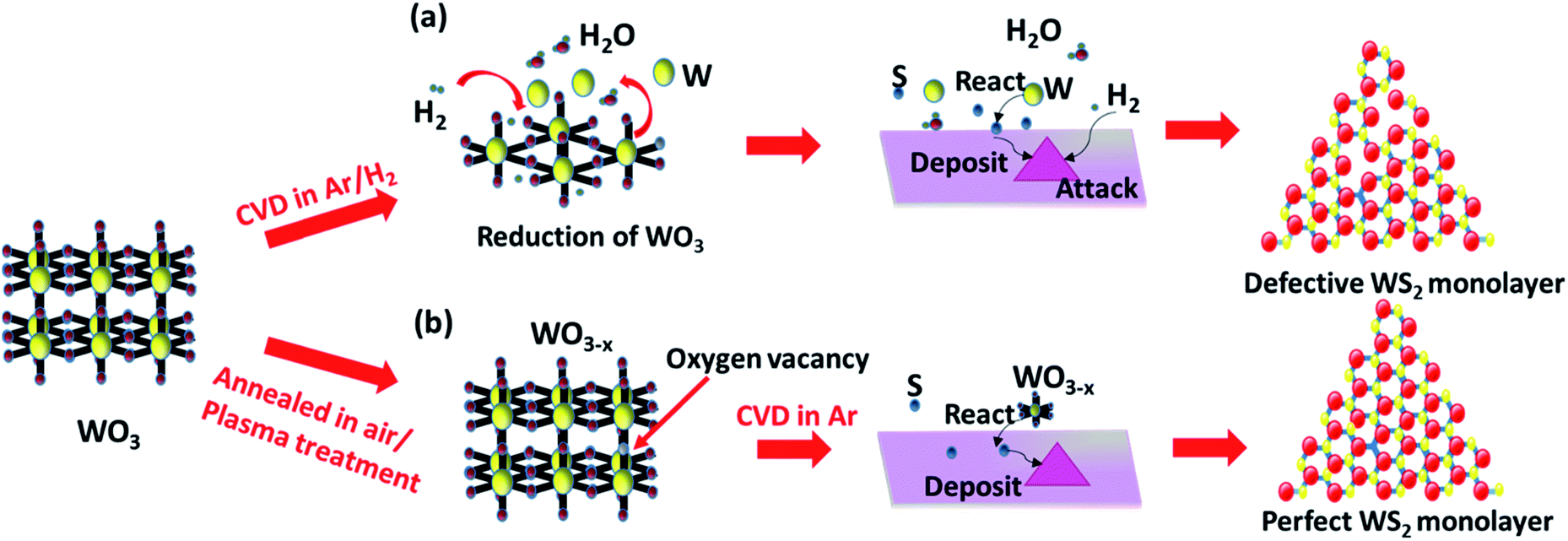 | ||
| Fig. 1 Reaction mechanism for CVD growth of (a) the commercial precursor in an Ar/H2 atmosphere; (b) pretreated precursors in an Ar only atmosphere. | ||
We further compared the as-grown WS2 monolayers produced from the four WO3 sources with different pre-treatment methods. The growth of monolayer WS2 strongly depends on the growth conditions, and in order to exclusively probe the effects of WO3 precursors on the CVD synthesis, regardless of the precursor pre-treatment methods used, all the growth processes were carried out under the same growth conditions with a growth temperature of 950 °C and a growth time of 10 minutes. AFM analysis and Raman characterization (see Fig. S2 and S3†) were carried out to confirm the monolayer nature of the WS2. The AFM line profiles at the edge of all the samples demonstrate that the thicknesses are in the 0.6 nm to 0.9 nm range, which fall in the thickness range of monolayer WS2. The thickness value differences in such a small range are commonly observed due to the sample's surface conditions. We further compare their Raman peaks, and the samples show two characteristic peaks of monolayer WS2: a sharp one at 355 cm−1 assigned to E2g mode and a broad one at 355 cm−1 assigned to A1g mode, matched well with previously reported Raman characterization of monolayer WS2.15,16,43 Therefore, all the samples studied in our work are monolayer WS2, and changing the precursor treatment conditions has no effects on the sample's thickness. According to the statistical analysis over an area of 1000 μm × 750 μm on the growth substrate (see Fig. S4†), the growth size of the monolayer WS2 is greatly influenced by the type of precursor. An average size of less than 10 μm for WS2 monolayers was obtained with the FWO3 precursor, while WS2 monolayers with a maximum size of ∼70 μm and an average size of ∼40 μm could be achieved with the AWO3 precursor. WS2 monolayers with reduced density and an average lateral size of ∼10 μm were obtained when the precursor was changed from FWO3 to PTWO3. Interestingly, a simple annealing process on the PTWO3 precursor resulted in WS2 monolayers with an average lateral size of ∼20 μm and improved surface coverage on the growth substrate. Besides, as shown in the optical images in Fig. 2a–d, truncated triangular shaped monolayer WS2 crystals were obtained with FWO3 and PTWO3 precursors, while WS2 monolayers in a regular triangular shape could be achieved with AWO3 and PTAWO3 precursors, revealing the morphology modification caused by the various precursor pre-treatment processes. The photoluminescence (PL) properties of WS2 monolayers in terms of the peak intensity and position can be dramatically changed due to the presence of various excitonic states such as trions, biexcitons and other charge bounded excitons, which strongly depend on the crystal quality and surface charge states of TMDC monolayers.26,44–47 As illustrated in Fig. 2e–l, the PL peak intensity mapping (Fig. 2e–h) and position mapping results (Fig. 2i–l) show that WS2 monolayers synthesized from the AWO3 precursor exhibit a more uniform PL peak intensity across the surface and have dominant PL peak with higher photon energy (∼2.0 eV), which is closer to the band gap of WS2 (2.1 eV),48 as compared with the other three synthesized samples.
 | ||
| Fig. 2 Optical characterization of the monolayer WS2 crystals that were obtained from various types of pretreated WO3 powder. (a–d) Optical images of the monolayer WS2 crystals that were grown from FWO3, PTWO3, AWO3, and PTAWO3 powders, respectively. (e–h) PL intensity mapping images of the monolayer WS2 crystals that were synthesized from FWO3, PTWO3, AWO3 and PTAWO3 powders, respectively. (i–l) PL position mapping images of the monolayer WS2 crystals that were synthesized from FWO3, PTWO3, AWO3 and PTAWO3 powders, respectively. | ||
To evaluate the oxygen vacancies in the precursors, we characterized the precursors with X-ray powder diffraction (XRD) as shown in Fig. 3, S5 and S6.† XRD patterns of all the four precursors consist of the monoclinic phase of WO3 diffraction peaks (see Fig. 3a), showing that the precursor treatment process did not change the crystal phase. However, the enlarged view of the peak ranging from 22 to 26° shows that all the precursors exhibit a peak position shift to a higher diffraction angle as compared to the standard XRD patterns for the monoclinic phase of WO3 (Fig. 3b). The XRD diffraction peaks from AWO3 powder exhibit the most significant peak shift (0.1°) to a higher diffraction angle with peak intensity reduction as compared to FWO3, followed by PTAWO3 (0.07°) and finally PTWO3 (0.03°). The peak position shifts to a higher diffraction angle are related to the contraction of lattice parameters, which results from the formation of more oxygen vacancies and the degradation of the crystallinity of the WO3 powder due to the thermal annealing process. As shown in Fig. S7,† WO3 powders prepared by different methods also show different colors from a greenish color to deep blue and these color changes have been attributed to the different oxygen vacancy contents in WO3 powders, which is in good consistency with the XRD results.30,49–51 To eliminate the effects of water content in the precursors on the growth of monolayer WS2, thermal gravimetric analysis (TGA) (see Fig. S8†) has been done and the results reveal that surface-absorbed water in the WO3 powders is less than 1%, implying negligible effects on the growth of monolayer WS2.
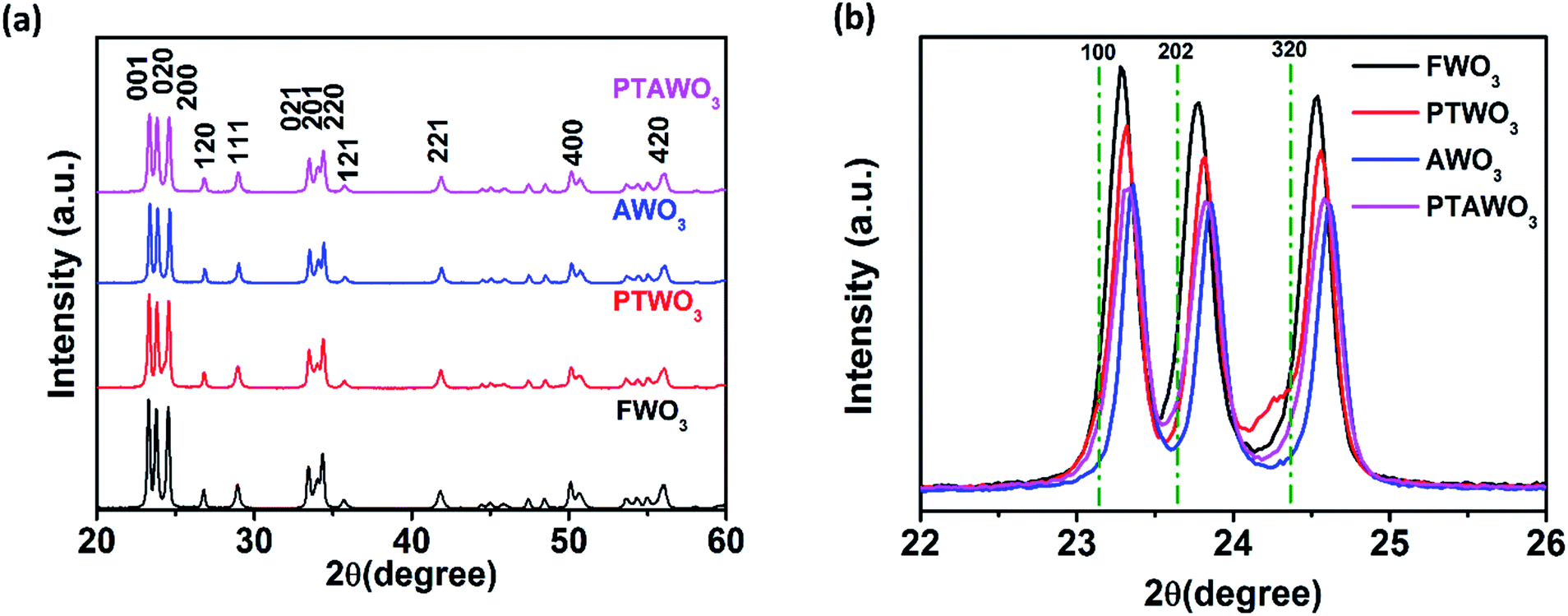 | ||
| Fig. 3 (a) XRD patterns of FWO3, PTWO3, AWO3 and PTAWO3 powders. (b) Enlarged view of XRD patterns at 001, 020 and 200 planes for FWO3, PTWO3, AWO3 and PTAWO3 powders. | ||
To further confirm the oxygen vacancy induced peak position and intensity change in XRD spectra, we further examined the optimized crystal structure of WO3 with and without oxygen vacancies by theoretical calculations (see Table 1). The lattice structures of WO3 have been studied using density functional theory (DFT) calculations with plane-wave pseudopotentials.52 The initial space group of stoichiometric WO3 is P21/a(#14), belonging to the monoclinic space group. After structural optimization (Fig. 4), the system's space group converts to Pbcm (#57). The optimized lattice parameters are a = 3.904 Å, b = 7.778 Å, and c = 7.558 Å. The unit cell volume is 229.54 Å3, and these four octahedrons in the system are equivalent. For the samples with oxygen vacancies, we remove one oxygen atom from the octahedron from three different directions, namely, a-, b-, and c-axis directions. For oxygen vacancies in the a-axis direction, the lattice parameter and volume decrease significantly. For oxygen vacancies in the b-axis direction, the bravais lattice shape changes, but the volume is substantially unchanged. For oxygen vacancies in the c-axis direction, the lattice constants of a and b decrease significantly, and the volume also decreases. In practice, oxygen vacancies may exist in any direction, and hence, from the above discussion, we can conclude that the presence of oxygen vacancies will cause the lattice constant of WO3 to shrink, which explains the peak position shift in XRD spectra for WO3 with different oxygen vacancy contents.
| Crystal structure of WO3 | Lattice parameters (Å) | Volume | Angle (Å) | ||||
|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | ||
| Without Oxygen vacancies | 3.904 | 7.778 | 7.558 | 229.54 | 90.00 | 90.00 | 90.00 |
| Oxygen vacancies in the a-axis direction | 3.796 | 7.799 | 7.598 | 224.95 | 90.00 | 90.00 | 90.02 |
| Oxygen vacancies in the b-axis direction | 3.808 | 7.795 | 7.573 | 229.48 | 90.00 | 90.00 | 90.03 |
| Oxygen vacancies in the c-axis direction | 3.824 | 7.651 | 7.721 | 225.85 | 90.84 | 90.00 | 90.00 |
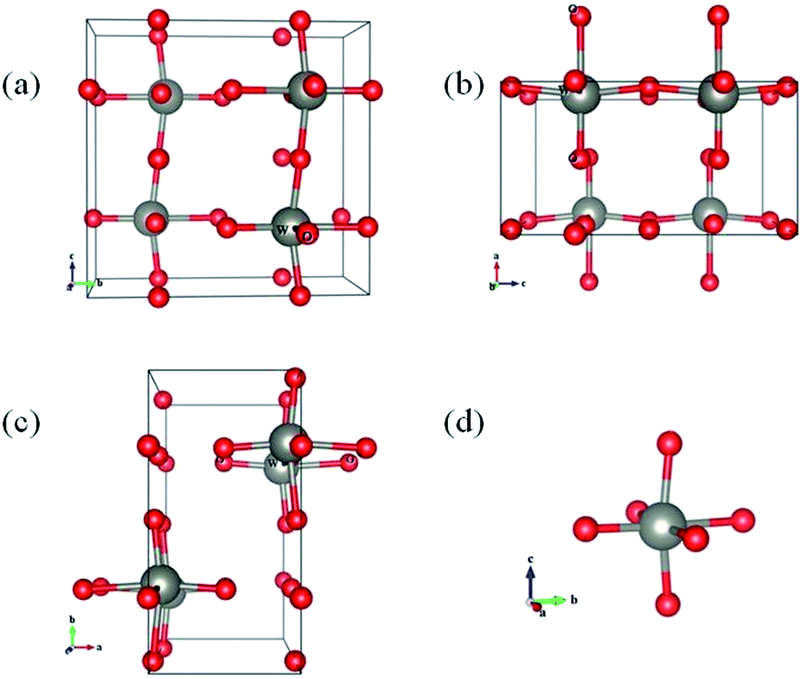 | ||
| Fig. 4 The view of the optimized structure of WO3 from the (a) a-axis direction, (b) b-axis direction, and (c) c-axis direction and (d) the octahedron in the WO3 system. There are 4 W atoms and 12 O atoms in this system. | ||
Moreover, X-ray photoelectron spectroscopy (XPS) analysis exhibits noticeable differences in the binding energy of tungsten and oxygen core levels as illustrated in Fig. 5, S9 and Table S1.† The XPS peak fitting results from all WO3 powders show that besides the dominating W4f(VI) valence state, a portion of the W4f(V) valence state also exists, confirming the presence of oxygen vacancies in all the WO3 powders. Yet, the XPS spectral fitting of W4f in AWO3 and PTWO3 powders suggests a higher contribution from the W4f(V) state as compared with that of FWO3 powder, indicating the higher oxygen vacancy concentration in both AWO3 and PTWO3 powders.
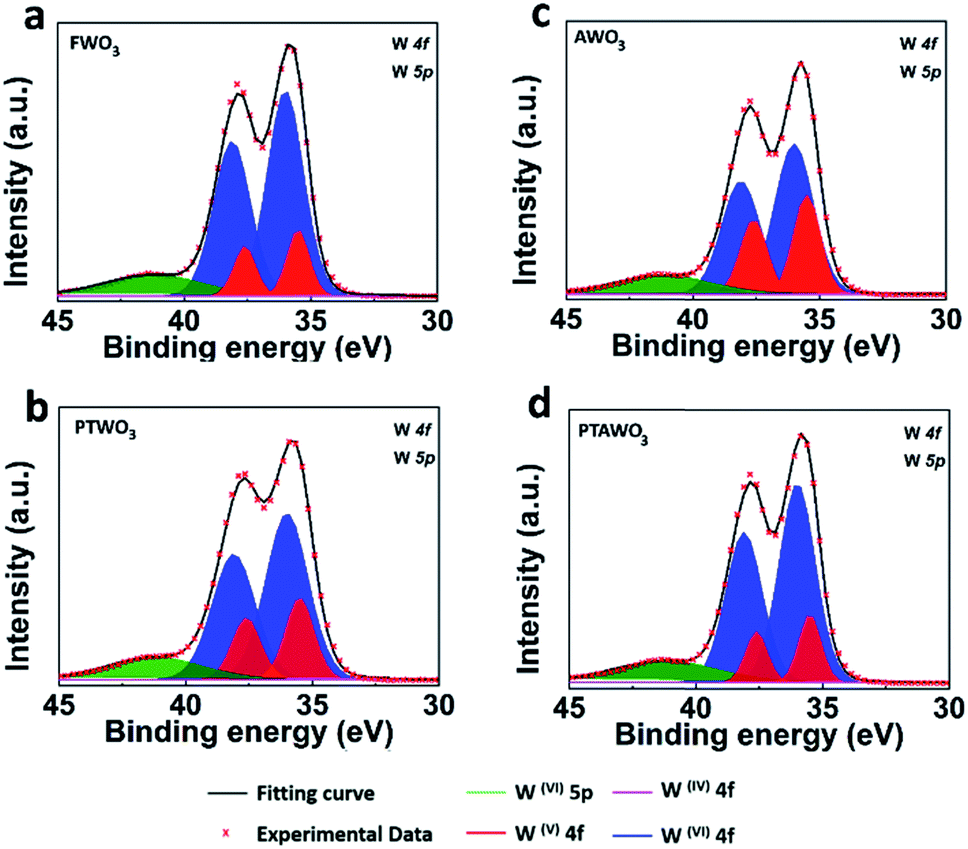 | ||
| Fig. 5 XPS spectra fitting of W4f and W5p peaks for WO3 powders. (a–d) XPS spectra of W4f for FWO3, PTWO3, AWO3 and PTAWO3 powders, respectively. | ||
Remarkably, compared with the other three control samples, a continuous dark oxide layer was formed after the plasma treatment of WO3 powder (Fig. S7b†). As reported by Wang et al., Raman spectroscopy is an effective way to characterize the crystallinity of WO3 powder.27 As illustrated in Fig. S10,† all the WO3 powders exhibit the two typical dominating Raman peaks of crystalline WO3 centered at 808 cm−1 and 715 cm−1, except the PTWO3 powder which shows a lower peak intensity and broader stretching mode peak at around 808 cm−1. The significant difference in Raman spectra suggests a strong plasma treatment effect on the surface crystallinity of WO3.
In fact, XPS analysis shows that the W4f(V) surface state is slightly reduced by thermal annealing for the PTWO3 samples, which could be due to the increased surface oxidation rate towards linear kinetics after plasma treatment.53 Therefore, the PTAWO3 powder possesses more W4f(VI) surface state compared with PTWO3. It is also noticed that XPS analysis and Raman characterization mainly reflect the surface information of chemical composition; meanwhile XRD presents the crystallinity of bulk sample powders. As shown in Fig. 3b, the XRD diffraction peaks from PTAWO3 powder exhibit a peak shift to a higher diffraction angle and peak intensity reduction, suggesting more oxygen vacancies formed by the annealing process. Combining the XRD, theoretical calculation and XPS analysis, we reach a conclusion that both thermal annealing and plasma treatment lead to the oxygen vacancy formation, while plasma treatment mainly affects the surface chemical states and the thermal annealing process can significantly change the metal oxide crystallinity and result in a smaller lattice spacing. Obviously, combining the precursor characterization and growth results, large-size triangular WS2 monolayer flakes could be produced with a higher oxygen vacancy concentration in the WO3 precursor source, while small-size truncated WS2 monolayers were generally obtained when the number of oxygen vacancies in the WO3 precursor reduced. The obtained morphology of the as-growth WS2 is largely related to the reaction conditions, such as the precursor concentration ratio, substrates used, gas flow rate. In pretreated precursors, the highly volatile tungsten suboxide will result in an increase of the amount of tungsten precursors, increase the rate of reaction toward sulfur and hence promote the growth of WS2. Overall, the thermal annealing process generates more oxygen vacancies in the WO3 precursor, which effectively promotes the growth of WS2 monolayers with larger size and better crystallinity. Besides, the PL results also suggest that the monolayer WS2 samples synthesized from thermal annealing pretreated precursors may have a lower lattice structure defect concentration.
To better reveal the superiority of the monolayer WS2 obtained from the AWO3 powder, we further explored the effect of the annealing atmosphere on the structure of the WS2. As aforementioned above, in previous studies, hydrogen gas (H2) was introduced to promotes the reduction of WO3 and hence promote the growth of WS2 monolayers.16,19,26,44 However, an excessive hydrogen supply could also induce defects. Therefore, we further study the quality of WS2 monolayers synthesized with a thermally annealed precursor. The thermally annealed precursor synthesized WS2 (AWS2) sample shows a uniform PL intensity feature on the whole surface and still retains its optical properties after being transferred onto a silicon dioxide substrate or more than one-month after the synthesis (see Fig. S11 and S12†). There is no aggregation/restacking effect observed for the AWS2 monolayers for prolonged periods (after one month). The optical image and optical characterization such as Raman and PL characterization (see Fig. S12†) show the good stability of the as-grown monolayer AWS2 after being exposed to ambient air for more than one month. The XPS spectra of W4f and S2p peak in AWS2 as shown in Fig. S13† confirm the presence of WS2.
Besides, atomic resolution annular dark field scanning transmission electron microscope (ADF-STEM) Z-contrast images for the AWS2 and hydrogenated environmentally synthesized WS2 (HWS2) samples were also obtained to study the atomic defect level in both samples (see Fig. 6). The obtained ADF-STEM image and the defect mapping analysis (see Fig. 6b and S14†) show that the AWS2 sample is almost defect-free, while there are a considerable number of lattice defects, including monosulfur vacancies (Vs), disulfur vacancies (VS2), and antisite defects where a W atom substituting a S2/S column (WS2, WS) or a S2 column substituting a W atom(S2w) is observed in the HWS2 sample (see Fig. 6d–j). These results suggest that the HWS2 sample contains more lattice defects than the AWS2 sample. This clearly illustrated that the AWS2 exhibits better crystallinity and fewer structural defects.
 | ||
| Fig. 6 Atomic structure of monolayer WS2. (a) STEMADF image of the as-transferred AWS2 monolayer onto a Quantifoil grid. (b) Atomic resolution ADF/STEM image of the AWS2 crystal. (c) SAED patterns of the WS2 crystal. (d) STEMADF image of the as-transferred HWS2 monolayer onto a Quantifoil grid. (e–f) Atomic resolution ADF/STEM image of the HWS2 crystal. (g) Defect mapping of the HWS2 monolayer. (h) Atomic resolution ADF/STEM images of various intrinsic point defects present in monolayer HWS2, including monosulfur vacancies (Vs), disulfur vacancies (VS2), and antisite defects where a W atom substituting a S2/S column (WS2, Ws) or a S2 column substituting a W atom (S2w) is observed. (i) Processed atomic resolution ADF/STEM image of various intrinsic point defects present in monolayer HWS2. (j) Relaxed structural models of the experimentally observed point defects of HWS2. | ||
Conclusions
In conclusion, we have comprehensively studied the effects of various stoichiometric WO3 precursors on the growth of WS2 monolayers. The morphology of WS2 monolayers transforms from a truncated triangle to a triangular shape when the oxygen vacancies in the WO3 precursor increase, resulting in the change of PL properties and crystallinity in the as-growth WS2 monolayers. The number of oxygen vacancies in WO3 induces a change in the growth atmosphere and hence significantly affects the lateral growth morphology as well as the optical properties of monolayer WS2. In our study, the annealed WO3 precursor with the highest oxygen vacancy concentration generates monolayer WS2 crystals with improved quality in terms of lateral size, surface coverage, and crystallinity, while PTWO3 and AWO3 powders with a lower oxygen vacancy concentration suppress the growth of monolayer WS2. Our results demonstrate the possibility of tailoring the morphology and crystal quality of WS2 monolayers by the tuning of oxygen vacancy content in the metal oxide precursor. The investigation on various non-stoichiometric WO3 precursors provides better understanding on the growth mechanism of large-area, high-quality WS2 monolayers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by A*STAR AME IRG (Grant no. A1783c0011). Y. S. acknowledges the support from the Science and Technology Project of Shenzhen (JCYJ20170817101100705), the Thousand Young Talents Program of China, the National Natural Science Foundation of China (Grant no. 51602200) and the (Key) Project of Department of Educational Commission of Guangdong Province (Grant no. 2016KZDXM008). This project was supported by the Shenzhen Peacock Plan (Grant no. KQTD2016053112042971).References
- Z. Sun, A. Martinez and F. Wang, Nat. Photonics, 2016, 10, 227–238 CrossRef CAS.
- G. Grosso, J. Graves, A. T. Hammack, A. A. High, L. V. Butov, M. Hanson and A. C. Gossard, Nat. Photonics, 2009, 3, 577–580 CrossRef CAS.
- C. Gong, H. Zhang, W. Wang, L. Colombo, R. M. Wallace and K. Cho, Appl. Phys. Lett., 2015, 107, 139904 CrossRef.
- H. Tan, Y. Fan, Y. Zhou, Q. Chen, W. Xu and J. H. Warner, ACS Nano, 2016, 10, 7866–7873 CrossRef CAS PubMed.
- A. P. Nayak, Z. Yuan, B. Cao, J. Liu, J. Wu, S. T. Moran, T. Li, D. Akinwande, C. Jin and J. F. Lin, ACS Nano, 2015, 9, 9117–9123 CrossRef CAS PubMed.
- T. Chu, H. Ilatikhameneh, G. Klimeck, R. Rahman and Z. Chen, Nano Lett., 2015, 15, 8000–8007 CrossRef CAS PubMed.
- W. X. Zhang, Z. S. Huang, W. L. Zhang and Y. R. Li, Nano Res., 2014, 7, 1731–1737 CrossRef CAS.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P. H. Tan and G. Eda, ACS Nano, 2013, 7, 791–797 CrossRef CAS PubMed.
- H. Shi, H. Pan, Y. W. Zhang and B. I. Yakobson, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 155304 CrossRef.
- X. Liu, J. Hu, C. Yue, N. Della Fera, Y. Ling, Z. Mao and J. Wei, ACS Nano, 2014, 8, 10396–10402 CrossRef CAS PubMed.
- M. W. Iqbal, M. Z. Iqbal, M. F. Khan, M. A. Shehzad, Y. Seo, J. H. Park, C. Hwang and J. Eom, Sci. Rep., 2015, 5, 10699 CrossRef CAS PubMed.
- L. Su, Y. Yu, L. Cao and Y. Zhang, Nano Res., 2015, 8, 2686–2697 CrossRef CAS.
- R. K. Ghosh and S. Mahapatra, IEEE J. Electron Devices Soc., 2013, 1, 175–180 Search PubMed.
- H. R. Gutiérrez, N. Perea-López, A. L. Elías, A. Berkdemir, B. Wang, R. Lv, F. López-Urías, V. H. Crespi, H. Terrones and M. Terrones, Nano Lett., 2013, 13, 3447–3454 CrossRef PubMed.
- K. M. McCreary, A. T. Hanbicki, G. G. Jernigan, J. C. Culbertson and B. T. Jonker, Sci. Rep., 2016, 6, 19159 CrossRef CAS PubMed.
- E. J. Sie, J. W. McIver, Y. H. Lee, L. Fu, J. Kong and N. Gedik, Nat. Mater., 2015, 14, 290–294 CrossRef CAS PubMed.
- H. Zeng, G. B. Liu, J. Dai, Y. Yan, B. Zhu, R. He, L. Xie, S. Xu, X. Chen, W. Yao and X. Cui, Sci. Rep., 2013, 3, 1608 CrossRef PubMed.
- H. Li, Y. Li, A. Aljarb, Y. Shi and L. J. Li, Chem. Rev., 2018, 118, 6134–6150 CrossRef CAS PubMed.
- Z. Cai, B. Liu, X. Zou and H. M. Cheng, Chem. Rev., 2018, 118, 6091–6133 CrossRef CAS PubMed.
- J. Chen, X. Zhao, S. J. R. Tan, H. Xu, B. Wu, B. Liu, D. Fu, W. Fu, D. Geng, Y. Liu, W. Liu, W. Tang, L. Li, W. Zhou, T. C. Sum and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 1073–1076 CrossRef CAS PubMed.
- Y. Shi, H. Li and L. J. Li, Chem. Soc. Rev., 2015, 44, 2744–2756 RSC.
- S. L. Wong, H. Liu and D. Chi, Prog. Cryst. Growth Charact., 2016, 62, 9–28 CrossRef CAS.
- Y. Sheng, H. Tan, X. Wang and J. H. Warner, Chem. Mater., 2017, 29, 4904–4911 CrossRef CAS.
- S. H. Choi, S. Boandoh, Y. H. Lee, J. S. Lee, J. H. Park, S. M. Kim, W. Yang and K. K. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 43021–43029 CrossRef CAS PubMed.
- K. N. Kang, K. Godin and E.-H. Yang, Sci. Rep., 2015, 5, 13205 CrossRef CAS PubMed.
- X. Zhao, J. Feng, S. Chen, Y. Huang, T. C. Sum and Z. Chen, Phys. Chem. Chem. Phys., 2017, 19, 1074–1082 RSC.
- Z. Lou and C. Xue, CrystEngComm, 2016, 18, 8406–8410 RSC.
- G. Wang, Y. Yang, Y. Ling, H. Wang, X. Lu, Y. C. Pu, J. Z. Zhang, Y. Tong and Y. Li, J. Mater. Chem. A, 2016, 4, 2849–2855 RSC.
- R. Chatten, A. V. Chadwick, A. Rougier and P. J. D. Lindan, J. Phys. Chem. B, 2005, 109, 3146–3156 CrossRef CAS PubMed.
- W. Wang, A. Janotti and C. G. Van de Walle, J. Mater. Chem. C, 2016, 4, 6641–6648 RSC.
- J. D. Cain, F. Shi, J. Wu and V. P. Dravid, ACS Nano, 2016, 10, 5440–5445 CrossRef CAS PubMed.
- Y. Shi, C. Hamsen, X. Jia, K. K. Kim, A. Reina, M. Hofmann, A. L. Hsu, K. Zhang, H. Li, Z. Y. Juang, M. S. Dresselhaus, L. J. Li and J. Kong, Nano Lett., 2010, 10, 4134–4139 CrossRef CAS PubMed.
- Y. Shi, H. Li, J. I. Wong, X. Zhang, Y. Wang, H. Song and H. Y. Yang, Sci. Rep., 2015, 5, 10378 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 77, 3865–3868 CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M. G. Charlton, Nature, 1954, 174, 703 CrossRef CAS.
- N. Birks, G. H. Meier and F. S. Pettit, Introduction to the High Temperature Oxidation of Metals, Cambridge University Press, Cambridge, 2 edn, 2006 Search PubMed.
- V. K. Sarin, J. Mater. Sci., 1975, 10, 593–598 CrossRef CAS.
- P. K. Sahoo, S. Memaran, Y. Xin, L. Balicas and H. R. Gutiérrez, Nature, 2018, 553, 63 CrossRef CAS PubMed.
- M. S. Kim, S. J. Yun, Y. Lee, C. Seo, G. H. Han, K. K. Kim, Y. H. Lee and J. Kim, ACS Nano, 2016, 10, 2399–2405 CrossRef CAS PubMed.
- S. Paradisanos, N. T. Germanis, C. Pelekanos, E. Fotakis, E. Kymakis, G. Kioseoglou and E. Stratakis, Appl. Phys. Lett., 2017, 110, 193102 CrossRef.
- Y. C. Lin, S. Li, H. P. Komsa, L. J. Chang, A. V. Krasheninnikov, G. Eda and K. Suenaga, Adv. Funct. Mater., 2018, 28, 1704210 CrossRef.
- J. H. Yun, J. Youngjo, Y. S. Joon, Z. Jiong, B. Jaeyoon, K. D. Hoon, L. H. Seok and L. Y. Hee, Adv. Mater., 2017, 29, 1605043 CrossRef PubMed.
- G. Plechinger, P. Nagler, J. Kraus, N. Paradiso, C. Strunk, C. Schüller and T. Korn, Phys. Status Solidi RRL, 2015, 9, 457–461 CrossRef CAS.
- X. H. Wang, J. Q. Ning, Z. C. Su, C. C. Zheng, B. R. Zhu, L. Xie, H. S. Wu and S. J. Xu, RSC Adv., 2016, 6, 27677–27681 RSC.
- C. G. Granqvist, Handbook of Inorganic Electrochromic Materials, Elsevier, 1995 Search PubMed.
- G. Wang, Y. Ling, H. Wang, X. Yang, C. Wang, J. Z. Zhang and Y. Li, Energy Environ. Sci., 2012, 5, 6180–6187 RSC.
- Y. Djaoued, S. Balaji and R. Brüning, J. Nanomater., 2012, 2012, 9 Search PubMed.
- J. Ihm, A. Zunger and M. L. Cohen, J. Phys. C: Solid State Phys., 1979, 12, 4409 CrossRef CAS.
- S. C. Cifuentes, M. A. Monge and P. Pérez, Corros. Sci., 2012, 57, 114–121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00212f |
| This journal is © The Royal Society of Chemistry 2019 |