
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultistage rocket: integrational design of a prodrug-based siRNA delivery system with sequential release for enhanced antitumor efficacy†
Qian
Jiang‡
ab,
Xiaobing
Chen‡
a,
Hong
Liang
a,
Yu
Nie
 *a,
Rongrong
Jin
a,
Matthias
Barz
c,
Dong
Yue
a and
Zhongwei
Gu
*d
*a,
Rongrong
Jin
a,
Matthias
Barz
c,
Dong
Yue
a and
Zhongwei
Gu
*d
aNational Engineering Research Center for Biomaterials, Sichuan University, Chengdu, 610064, P. R. China. E-mail: nie_yu@scu.edu.cn
bDepartment of Pharmacy, The Second People’s Hospital of Chengdu, Chengdu 610017, P. R. China
cInstitute of Organic Chemistry, Johannes Gutenberg-University Mainz Duesbergweg 10-14, 55099 Mainz, Germany
dCollege of Materials Science and Engineering, Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, P. R. China. E-mail: zwgu1006@hotmail.com
First published on 20th October 2018
Abstract
An integrated peptide-camptothecin prodrug (RSC) system was designed as a nano-sized multistage rocket for the efficient complexation and controlled sequential release of siRNA and anticancer drug under tumor-relevant reductive and esterase-enriched conditions, which facilitated the avoidance of negative interactions and maximized the synergistic effect.
Emergence, development, deterioration and metastasis constitute the extremely complex physiological process of cancer, which enables tumor cells to be disencumbered by normal death/apoptosis in their cellular cycle and to elude being killed by the immune system. Individual therapies including surgery, chemotherapy, radiotherapy, and gene therapy against cancer are inevitably sub-optimal and highly ineffective.1–3 Among these, chemotherapy has been an important treatment in clinics for decades using low-molecular weight drugs to directly damage genetic DNA or proteins.4–6 Gene therapy (such as RNA interference by small interfering RNA, siRNA) works using hereditary material from oneself to induce sequence-specific mRNA down-regulation, and results in the knocking down of a target gene protein on a mRNA transcriptional level.7,8 Due to different molecular mechanisms, the combination of drug and gene therapy may exhibit excellent complementary and synergistic effects to maximize therapeutic efficacy and narrow down their respective limitations. For instance, siRNA could down-regulate drug efflux pumps (e.g., Pgp, MRP1, ABCG2) and reduce the administration dosage of doxorubicin,9,10 or serve as anti-angiogenic factors (e.g., VEGFR/VEGF, HIF-1α, integrin αvβ3)11,12 to show a synergistic effect with paclitaxel.
However, these combinations could increase the complexity of the delivery system. Several key factors should be considered for efficient carrier design, including loading capacity, biological activity and release kinetics.1,13 Evidence shows that antecedently released doxorubicin would implant into the double helix of loaded DNA and impact its activity and transfection efficiency,1,4 which only shows the tip of the iceberg of the evident dilemma in drug and gene co-delivery systems. On the other hand, molecular biologists found that treatment of breast cancer cells with siRNAs targeting PLK1 improved the sensitivity toward paclitaxel and Herceptin in a synergistic manner.14 Hence, control of the timing, duration and order of drug and gene release remains vital for the design of co-delivery systems.1 Interestingly, spatiotemporally or temporally controlled co-delivery systems have been rarely reported so far. Some studies utilized exogenous cues, e.g., electric field, magnetic field, and ultrasound, in more situations, but these methods fail to guide with precision and the releasing process remains largely empirical.15,16 Could we utilize the inherent conditions of the tumor microenvironment (including pH, redox, enzyme, etc.) as triggers to realize this precise stepwise control? Herein, our experience in the design of various biological stimuli-sensitive systems17–20 might be beneficial for actualizing such specifically controlled release, like in a multistage rocket.
Another challenge of loading drugs and genes together in the same carrier is to ensure that the presence of one therapeutic agent does not affect the loading capacity of the other. Some researchers found that co-encapsulating genes and doxorubicin within microspheres led to a much lower encapsulation efficiency than when encapsulating one species alone.21 Similarly, Han et al. observed that doxorubicin loading in conjugated cyclodextrins–PEI resulted in reduced pDNA binding ability.22 In general, drug content cannot exceed 10% by physical encapsulation in nanoparticles or liposomes, which requires administering a high dose of carriers or repeated therapeutic interventions to guarantee the anti-tumor effect. However, this compromise may cause severe toxicity and create a burden for the patient to absorb or excrete the drug carrier materials. Covalent linkage of the drug to the carrier system provides a straightforward method for prodrug fabrication to enhance encapsulation efficiency and minimize the initial burst release of the drug into the blood stream. Prof. Shen’s group conjugated a dimeric camptothecin with a short oligomer chain of ethylene glycol to form an amphiphilic phospholipid-mimicking prodrug with exceptionally high drug loading (40–58%).23 Prof. Cheng’s group utilized cytotoxic drugs as the hydrophobic part in micelles, reaching 17% drug loading.24,25 It was found that conjugating a drug to the carrier system does not only enhance encapsulation efficiency, but also postpones the drug effect to some extent via steric hindrance and inactivation.15,26,27 Following these designs, the present study aims to explore whether this approach is suitable for the construction of vehicles for the co-delivery of genes and drug.
As a proof-of-concept, we designed a bioinspired co-delivery system (R2/3SC) consisting of an integrated dendritic peptide–prodrug conjugate with high loading efficiency, in which the anticancer drug camptothecin (CPT) acts as the hydrophobic segment of an amphiphile. Besides, dendritic arginine was chosen as the hydrophilic part to provide enhanced membrane-penetrating ability and thus ensure high transfection efficiency.17,20,28,29 These two parts were linked through a disulfide linkage that can be cleaved inside cells while providing high stability outside.18,30 In the presence of siRNA, R2/3SC is expected to assemble into nano-sized particles with a high loading efficiency of therapeutic agents (CPT and siRNA targeting the polo-like kinase 1 (PLK1) gene). It is speculated that siPLK1 condensed by the arginine-rich periphery would release earlier and faster in the delivery process due to the configuration damage to the self-assemblies by disulfide bond cleavage. Next, the inactive prodrug would revert back to the pharmacologically active form through interaction with esterase, which is abundant inside various cells.31 Hence, CPT has a delayed release with two activation steps, allowing the siPLK1 to sufficiently down-regulate the PLK1 in advance to improve the sensitivity of cancer cells to CPT.32–34 Finally, CPT can efficiently exert the desired therapeutic effect (Scheme 1).
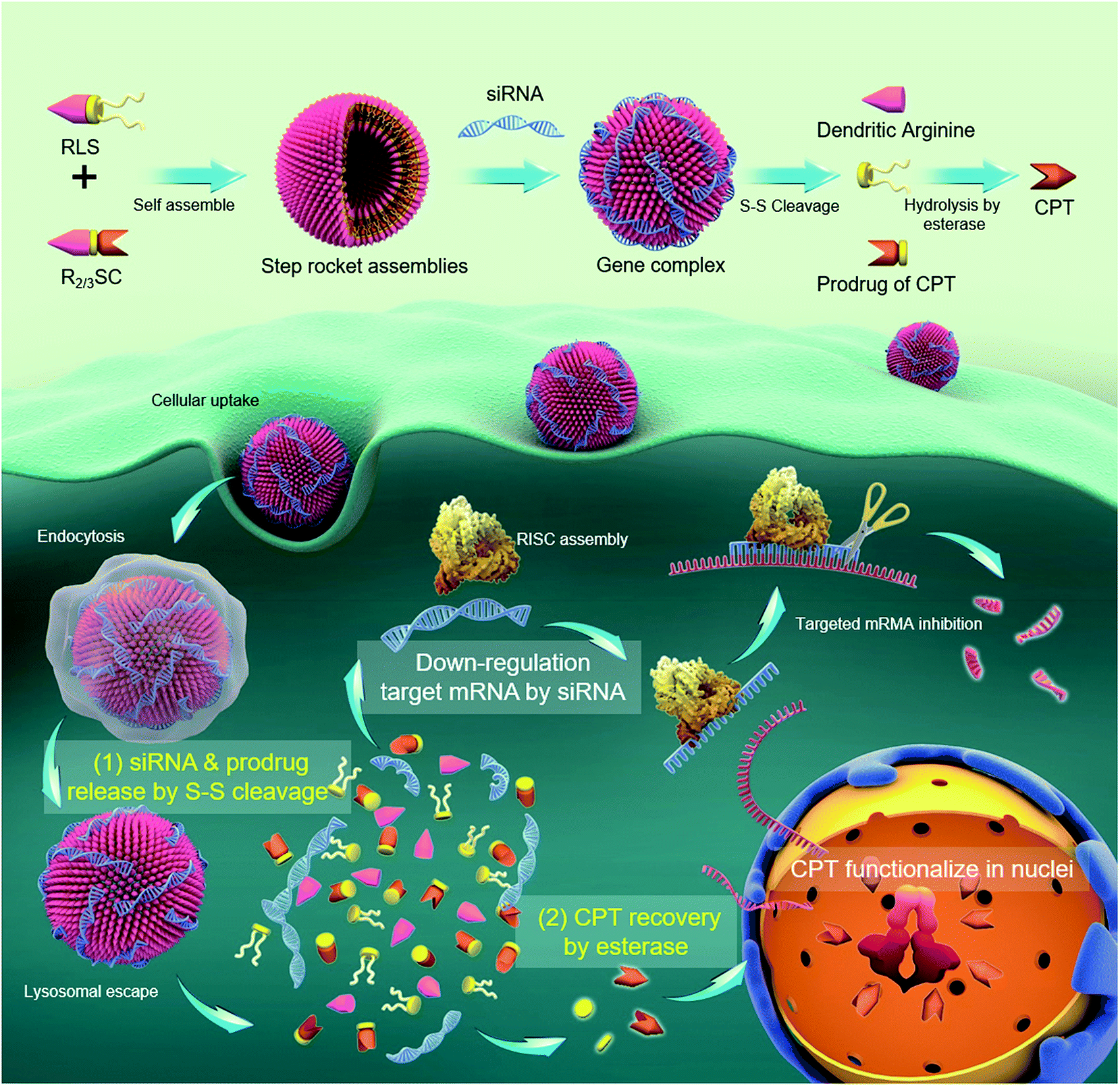 | ||
| Scheme 1 Schematic representation of a “multistage rocket” (R2/3SC): R2/3SC assembles into nano-sized vesicles with a high loading efficiency of therapeutic agents (CPT and siRNA targeting the polo-like kinase 1 (PLK1) gene). (1) Earlier and faster release of siPLK1 condensed by the arginine-rich periphery in the delivery process due to the configuration damage to the self-assemblies by redox cleavage. (2) The inactive prodrug reverts back to the pharmacologically active moiety triggered by the unique biological stimulation of esterase, which is abundant inside the cells. Finally, CPT is released via esterase cleavage and accumulated in nuclei to exert a therapeutic effect. Hence, the release of CPT is delayed by two activation steps, allowing the siPLK1 to sufficiently down-regulate the PLK1 in advance to improve the sensitivity of cancer cells to CPT. | ||
The structures of R2/3SC (Fig. 1) were designed as a “multistage rocket”, synthesized from an inactive CPT derivative and dendrimers of different generations using a straightforward three-step reaction (Fig. S1 and S2†), and purified using silica gel column chromatography with an overall yield of ∼34%. The synthetic intermediates and the final chemical structure (Fig. 1) of R2/3SC have been confirmed by nuclear magnetic resonance 1H-NMR spectroscopy, electrospray ionization mass spectrometry (ESI-MS) and matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) (Fig. S3 and S4†). Based on the NMR data, the lactone ring structure of CPT remained intact in all intermediates and the final product. Structural differences between R2SC and R3SC can be distinguished by the number of characteristic signals and their intensity, including the –CH– of the amino acid residue at δ = 4.10 ppm (Fig. S5A and S6A†). Based on the molecular weight of both systems, the drug loading efficiency of R2SC and R3SC is 34.0% and 21.8%, respectively (Fig. S5B and S6B†). When assembled together with RLS,20 R2SC showed uniform spherical nanoparticles with a size of about 200 nm under a transmission electron microscope (TEM) (Fig. S7 and S8†). The size of R2SC vesicles as measured using dynamic light scattering (DLS) was about 195 nm by intensity distribution (PDI 0.16) with a zeta-potential of +20.1 mV, while those of R3SC were 251.1 nm (PDI 0.26, +30.2 mV). It was found that the assembly of R3SC was somehow inferior to that of R2SC, either from the particle dispersion index (PDI) or the TEM images.
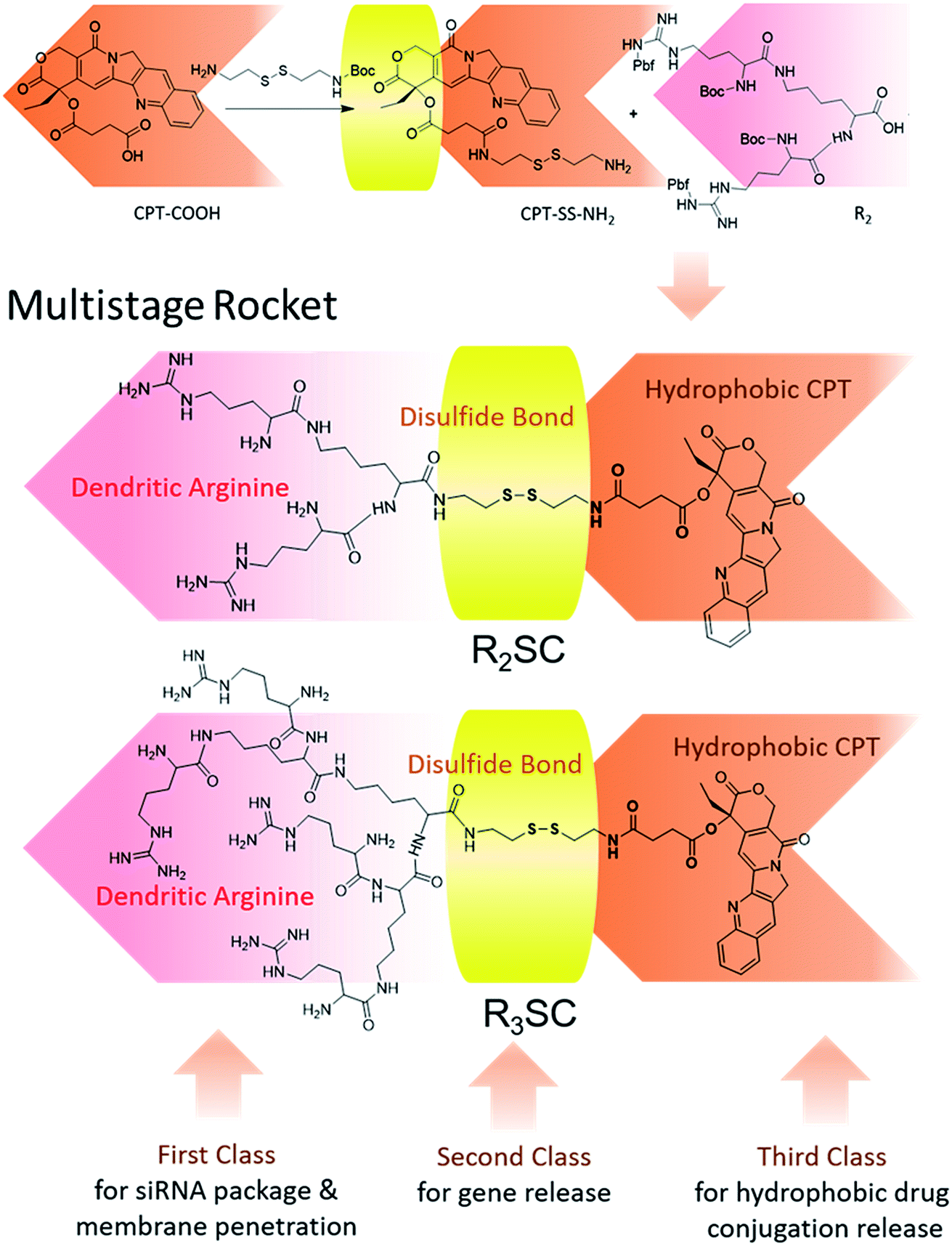 | ||
| Fig. 1 Chemical structure of the “multistage rocket” (R2/3SC): first class for the siRNA package and bio-membrane penetration, second class for reduction-responsive disassembly and gene release, and third class for hydrophobic camptothecin (CPT) conjugation release. | ||
Next, the assemblies were gently mixed with siRNA to construct gene complexes (R2/3SC/siRNA) and characterized by size and surface charge (Fig. S9†). As the N/P ratio increased from 10 to 40, the size of the complexes followed a decreasing trend, and the zeta-potential increased. For instance, the average size of R2SC/siRNA complexes was about 400 nm with a 14.2 mV zeta-potential at a N/P ratio of 10, and the particle size reduced below 270 nm (zeta-potential of 45 mV) at higher N/P ratios (20–40). Both cationic R2SC and R3SC assemblies were able to condense siRNA at appropriate N/P ratios, preventing the migration of genes in agarose gel (Fig. S10†). Moreover, the siRNA binding ability of R3SC (completely condensing siRNA at N/P 5) was clearly stronger than in the case of R2SC (completely condensing siRNA at N/P 8), arising from the higher surface charge induced by the third generation-arginine dendrimer.35,36
Since the goal was to develop a co-delivery system for CPT and siRNA that releases the two payloads in a temporal manner, we investigated these properties mimicking the intracellular redox potential and esterase microenvironment of tumor cells ex vivo. In a reductive solution of 5 mM GSH, fast changes to the R2SC assemblies occurred. The particle size of the R2SC assemblies dramatically increased from 200 nm (PDI = 0.15) to 850 nm (PDI = 0.56) in 1 h (Fig. 2A), and the morphologies became non-homogeneous, loose and irregular with many small pieces appearing in the TEM image (Fig. S11A†). After that, the disintegrated pieces formed some huge aggregations in 2 h (Fig. S11B†), with the particle zeta-average size expanded to around 1200 nm (PDI = 0.62, Fig. 2A). The size of the R3SC assemblies even became up to 4-fold larger after 2 h incubation. The zeta potentials of the R2SC and R3SC assemblies decreased from about +22–28 mV to neutral potentials (−1.2 mV) due to size changes and poor positive segment assembly. R2/3SC/siRNA complexes displayed a similar tendency with gene free R2/3SC assemblies in a reductive solution of 5 mM GSH (Fig. S11C†). Whether the disintegration of the co-delivery systems would result in the liberation of RNA was verified using a gel retardation assay (Fig. 2B). The released siRNA bands were clearly visible at N/P ratios of 10, where complete condensation was observed before, which indicates that in the presence of 5 mM GSH after 2 h, the complexes release siRNAs. After incubation with GSH, the disulfide bonds of the R2/3SC/siRNA complexes could be cleaved and the complexes soon became unstable and disintegrated. The dendritic cationic arginine would distribute in the solution as small pieces (with low charge density) that showed poor interaction with siRNA, further continuously resulting in some aggregation.
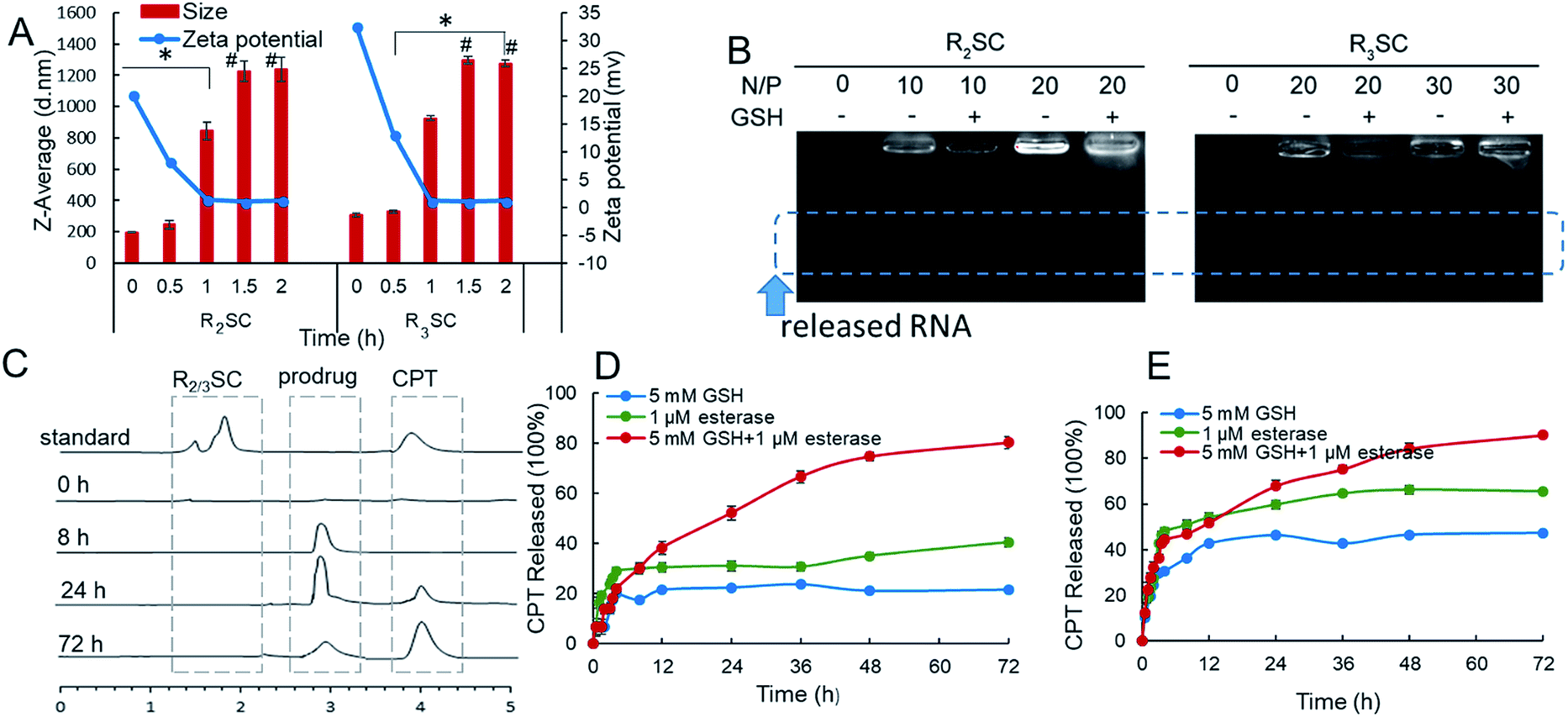 | ||
| Fig. 2 (A) Representative size distribution and zeta potential of R2/3SC assemblies with thiol trigger (5 mM GSH) from DLS (# poor distribution). (B) Release behavior of siRNA from R2SC and R3SC complexes with or without 5 mM GSH (after 2 h-incubation). (C) Release chromatogram of CPT from assemblies detected using HPLC. Quantitative release profiles of CPT from R2SC (D) and R3SC (E) complexes under different conditions at 37 °C. | ||
In addition, the release profile of camptothecin from the R2SC assemblies was investigated under reductive conditions and with high esterase content using reverse-phase high-performance liquid chromatography (HPLC). R2SC assemblies and CPT were first injected into the HPLC to confirm the peak position (Fig. 2C), showing that the free CPT molecule exhibited a sharp retention time peak at 3.9 min, while the assemblies appeared at 1.8 min. Due to the molecular weight cut-off of the applied dialysis bag (MWCO 1000), only small molecules, such as CPT and the CPT prodrug, could permeate the membrane. There was no detectable peak after exposure to 5 mM GSH and esterase (1 μM) at the very beginning. Only after 8 h, a peak appeared at 2.9 min, which could be assigned to the inactive prodrug molecule using MS. Over time the intensity of this peak decreases while the intensity of the peak of free CPT increases, indicating the formation of the active drug. The in vitro quantitative release profile of CPT in the presence of 5 mM GSH, esterase or a mixture of the two is displayed in Fig. 2D and E. We found that in the first 2 h, namely the time interval for complete siRNA release, only a minimal amount of drug was released (∼13%) from the R2SC assemblies (in the presence of GSH or esterase). Moreover, less than 25% of CPT was released in the presence of 5 mM GSH and ≤40% in the presence of 1 μM esterase from the R2SC assemblies even after 72 h incubation. It is supposed that the 25% release might be from general hydrolysis, while the 40% free CPT resulted from enzymatic degradation. At that time, the kinetic profile almost reached a plateau, due to the difficult entry of esterase into non-disassociated assemblies due to steric hindrance. On the contrary, in the simultaneous presence of GSH and esterase, the release of CPT from the R2SC assemblies occurred in a sustained manner over time, reaching 14% after 2 h and 80% after 72 h, indicating that the release of CPT was first triggered by GSH breaking the internal structure of the aggregates and providing accessibility to the esterase. The release profile of the R3SC assemblies indicated an accelerated release (27–32% at 2 h) at the beginning along with 1 mM esterase or stimulation by GSH and esterase. These findings may relate to the lower stability of R3SC based assemblies, which facilitates contact between the enzyme and the prodrug molecule. In conclusion, our data points out that both the R2SC and R3SC based systems provide the desired sequential release mechanism leading to a dual-stage release profile. However, the R2SC based formulation was able to leave more time for siRNA to silence the mRNA in comparison to the R3SC one.
Whilst the R2/3SC/siRNA complexes performed different release order/profile of siRNA and camptothecin, the influence of such kinetics on therapeutic effects remains to be addressed. To address this point, we first investigated the in vitro gene silencing efficiency of camptothecin prodrug-based siRNA delivery systems. Therefore, the fluorescence degree of GFP-HeLa cells treated with the R2/3SC/siGFP complexes at various N/P ratios was determined in the presence of serum (Fig. S12†). The GFP expression in the R2SC/siGFP complexes at a N/P of 20 showed a clear reduction by approximately 75% compared to that in the untreated cells, while the knockdown by R3SC/siGFP complexes at the highest N/P ratio of 40 exhibited a maximum reduction down to ∼55% fluorescence intensity. Besides, cells treated with RLS showed a similar down-regulation effect to those with R2SC (Fig. S12C†), and R2/3SC/siGFP complexes caused no distinct damage to cells during the transfection process (in 24 h, Fig. S13†). This phenomenon implied that the conjugated CPT did not exert a therapeutic effect during the knockdown of GFP expression. Based on the knockdown efficiency of GFP expression, the appropriate N/P ratios of R2SC and R3SC were set to be 20 and 40 for further experiments, respectively.
Following this, polo-like kinase 1 (PLK1) was selected as the oncogenic target since it is a well-known key regulator of the mitotic progression of mammalian cells and its activity is elevated in many cancer cells.37 Down-regulation of PLK1 expression could disturb the integrity of the mitotic apparatus that enables SN-38 (active metabolite of CPT-11) to exert cytotoxicity as an enzyme topoisomerase I inhibitor.33,34 Therefore, based on the molecular mechanism, the therapeutic gene needs to functionalize in advance in order to improve the cellular sensitivity to CPT and achieve a better co-effect. Real time quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and western blotting were used for timing and tracing the down-regulation at the mRNA and protein levels, as well as some synergistic effect for the sequential release. As shown in the histogram (Fig. 3A), the PLK1 mRNA of HeLa cells treated with R2SC/siPLK1, RLS/siPLK1 + CPT, R3SC/siPLK1 and Lipo/siPLK1 complexes clearly decreased. More importantly, the R2SC/siPLK1 group showed the highest knock down effect of 75.1% compared with 26.7% reduction for RLS/siPLK1+CPT, confirming that R2SC/siPLK1 complexes that release genes and drugs in a sequential release manner were more efficient than those that use a simultaneous release manner. Meanwhile, direct usage of free CPT had no evident effect on the down-regulation of PLK1, which was also verified by the similar down-regulation efficiency (∼25%) of the RLS/siPLK1 complexes and RLS/siPLK1 + CPT. However, it is interesting that the reduction effect of the R2SC/siPLK1 complexes was stronger than that of the RLS/siPLK1 complexes, suggesting that the prodrug of CPT may synergize with siPLK1 to enhance the down-regulation of PLK1.38 Besides, the R2SC/siPLK1 complexes showed better mPLK1 reduction than its analogous R3SC/siPLK1 complexes (46.8%) and positive control Lipo/siPLK1 complexes (53.1%). Similar tendency of PLK1 protein expression inhibition between various groups was observed by western blot analyses (Fig. 3B–D). The band of R2SC/siPLK1 group were the dimmest compared to the other groups (Fig. 3B), resulting in the best inhibition of PLK1 protein expression (only 9.8%). The down-regulation caused by Lipofectamine2000/siPLK1 and RLS/siPLK1 + CPT was 19.8% and 67.5%, respectively (Fig. 3C). It was exciting to find that the sequential design showed the expected result that the delayed release of CPT made enough time for siPLK1 expression, which would enhance the cellular CPT-sensitivity to accelerate the apoptosis.39,40
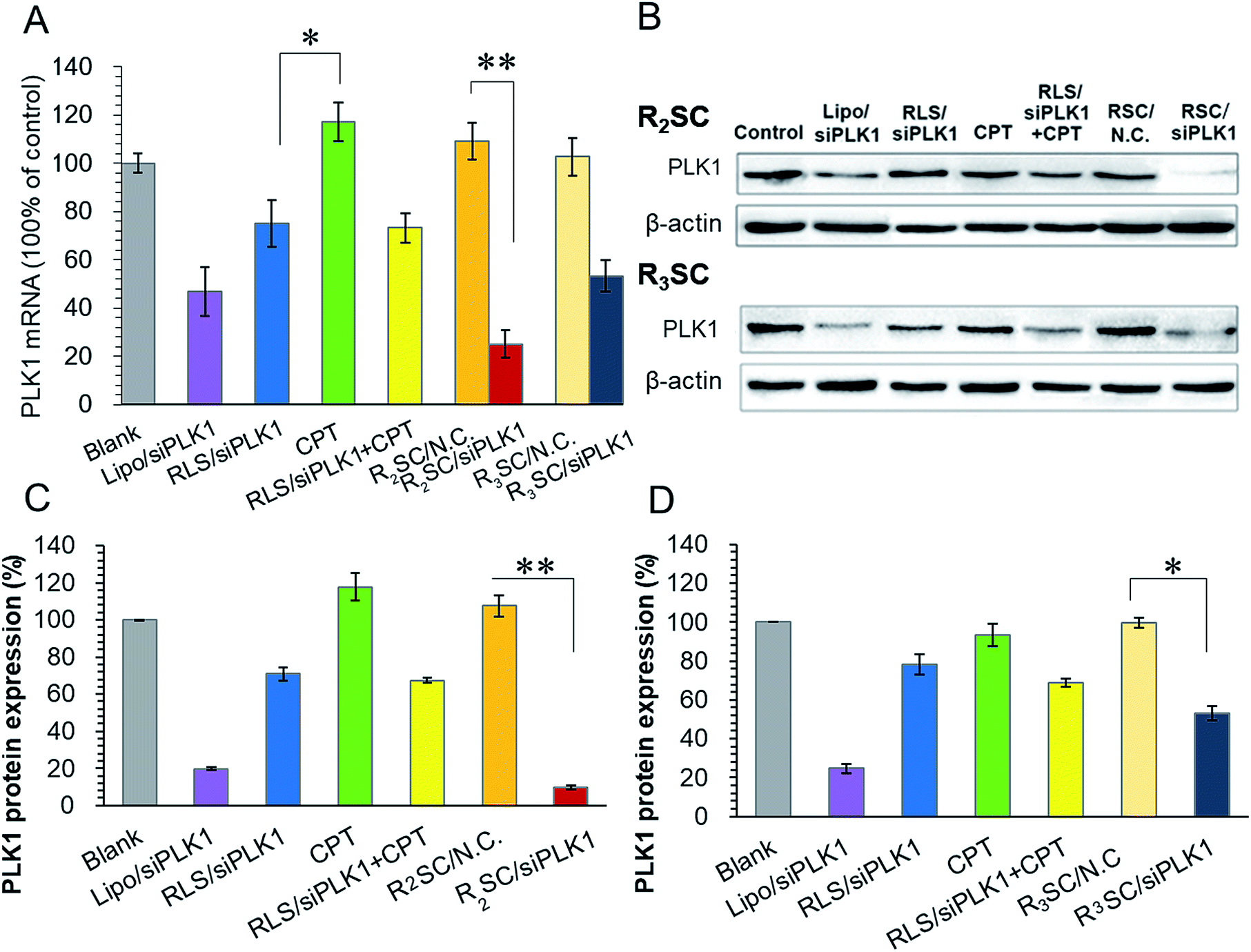 | ||
| Fig. 3 Down-regulation of PLK1 mRNA and protein levels in HeLa cells after treatment with various formulations. (A) mRNA levels of PLK1 in HeLa cells treated with different complexes and determined using qRT-PCR at 24 h. Data were referenced to the PLK1 mRNA of untreated cells. (B) Western blotting assay of PLK1 protein expression in HeLa cells incubated with different complexes transfected by R2SC (top) and R3SC (bottom) complexes for 48 h. Analysis of light intensities of PLK1 protein expression as the ratio of PLK1 to β-actin from western blot results from R2SC (C) and R3SC (D) complexes. Data represent the mean ± S.D. *p < 0.05, **p < 0.01. | ||
During the evaluation, it was found that there was a difference between R2SC/ and R3SC/siRNA complexes where lower generation dendrimers displayed better gene silencing efficiency. Previous studies have shown that the positively charged head groups of the assemblies were responsible for the interaction between the carriers and the genes, complexes and cell membranes, or other components of the cell, which plays a crucial role in successful gene delivery.17,41 Thus, the transport process, including cellular internalization and endosomal/lysosomal escape, was explored using confocal laser scanning microscopy (CLSM, Fig. S14A†). Red fluorescence in the cytoplasm originating from Cy5-labelled siN.C. particles indicated their cellular distribution (including uptake & endo/lysosomal escape, Fig. S14A†), and semi-quantitative evaluation of the cellular uptake was assessed using mean fluorescent intensity (MFI) comparisons of red fluorescence (Fig. S14B†). HeLa cells treated with R2SC complexes showed much higher Cy5-labelled siN.C. signals within the cells than those with R3SC complexes after 2 and 4 h incubation. The amount of internalized R2SC complexes was up to 3.3 times higher than that of R3SC complexes after 4 h. Mander’s overlap coefficients (R) were acquired from CLSM images to calculate the fraction of co-localized red intensity divided by the total red intensity (Fig. S14C†)42 using Image-Pro Plus 6.0 software, which indicated co-localization of the red Cy5-labelled siRNA and green fluorescent acidic endo/lysosomes (labelled with Lysotracker Green). At the start of the incubation, the R2SC complexes clearly showed much brighter orange fluorescent dots in the cytoplasm compared to those of its analogue R3SC. As the time extended, the orange changed to red fluorescence with a strongly decreased R value for the R2SC group, while that of R3SC showed only a slight change (Fig. S14C†). This phenomenon indicates that the R2SC with a 2nd generation dendritic arginine head is more efficient concerning cellular internalization and endosomal escape than R3SC containing a 3rd generation dendritic arginine. We suspect that the difference might result from the steric hindrance of higher generation dendrons,43 relatively irregular spherical-morphology assemblies, and the stronger hydrophilic properties of R3SC.44
PLK1 regulates the assembly of mitotic spindles and its overexpression contributes to oncogenic transformation. Silencing PLK1 may disturb the integrity of the mitotic spindle apparatus and may increase the activity of caspase 3, which enables camptothecin analogues to exert higher cytotoxicity in order to jointly upregulate the expression of caspase 3.33 HeLa cells were treated with various formulations containing 2 μg siRNA/mL, and then stained with annexin-V-FITC and propidine iodide (PI) for cell apoptosis determination (Fig. 4A). Not surprisingly, temporal/sequential delivery of siRNA and camptothecin in the R2SC/siPLK1 complexes led to the highest rate of cell apoptosis (20.0%, including the early apoptotic cells and fully apoptotic cells), which was obviously superior to that of RLS/siPLK1 (7.0%) and the simultaneous administration of siPLK1 and the drug (RLS/siPLK1 + CPT, 11.9%). In addition, the cell apoptosis induced by R2SC/siPLK1 was almost two-fold higher than that of free CPT (13.1%) and R2SC/siN.C. A TUNEL assay also confirmed similar results for cell apoptosis, which labels DNA at the 3′-hydroxyl termini in the double-strand DNA breaks generated during apoptosis (Fig. 4B and S15†). R2SC/siPLK1 complex treated HeLa cells showed the brightest green fluorescent dots, indicating the highest number of TUNEL positive cells. The results indicate that R2SC/siPLK1 enables the efficient knockdown of PLK1 expression as well as the induction of synergistic cell apoptosis in this in vitro study. Furthermore, the superiority of the sequential release of siRNA and anti-cancer drugs for inducing anti-proliferative effects was verified using a Cell Counter Kit-8 (CCK-8), using RLS/siPLK1 + CPT, CPT and Lipofectamine 2000/siPLK1 groups as controls (Fig. 4C). As expected, R2SC/siPLK1 complexes significantly reduced cell viability to approximately 40% with the highest amount of cytotoxicity, while the values for non-sequential release (RLS/siPLK1 + CPT) and single CPT were 50% and 57%, respectively. It should be pointed out that cells treated with Lipofectamine 2000/siPLK1 did not show satisfying anti-proliferative effects on tumor cells, although these complexes have performed well in the down-regulation of the mPLK1 (53.1%). The moderate decline of cell viability after pLK1 knockdown is probably caused by residual pLK1, which was in line with the findings of Zhang et al.45 Thus, we have deduced that the cytotoxicity of R2SC/siPLK1 complexes is attributed to their excellent siPLK1 gene silencing efficiency by improving the sensitivity of HeLa cells to CPT and the temporally controlled release behavior, which facilitated the synergistic effect of CPT and siPLK1.39,40 The findings mentioned above indicate that the timing of drug and gene release contributes to enhanced cell apoptosis and in vitro cytotoxicity.
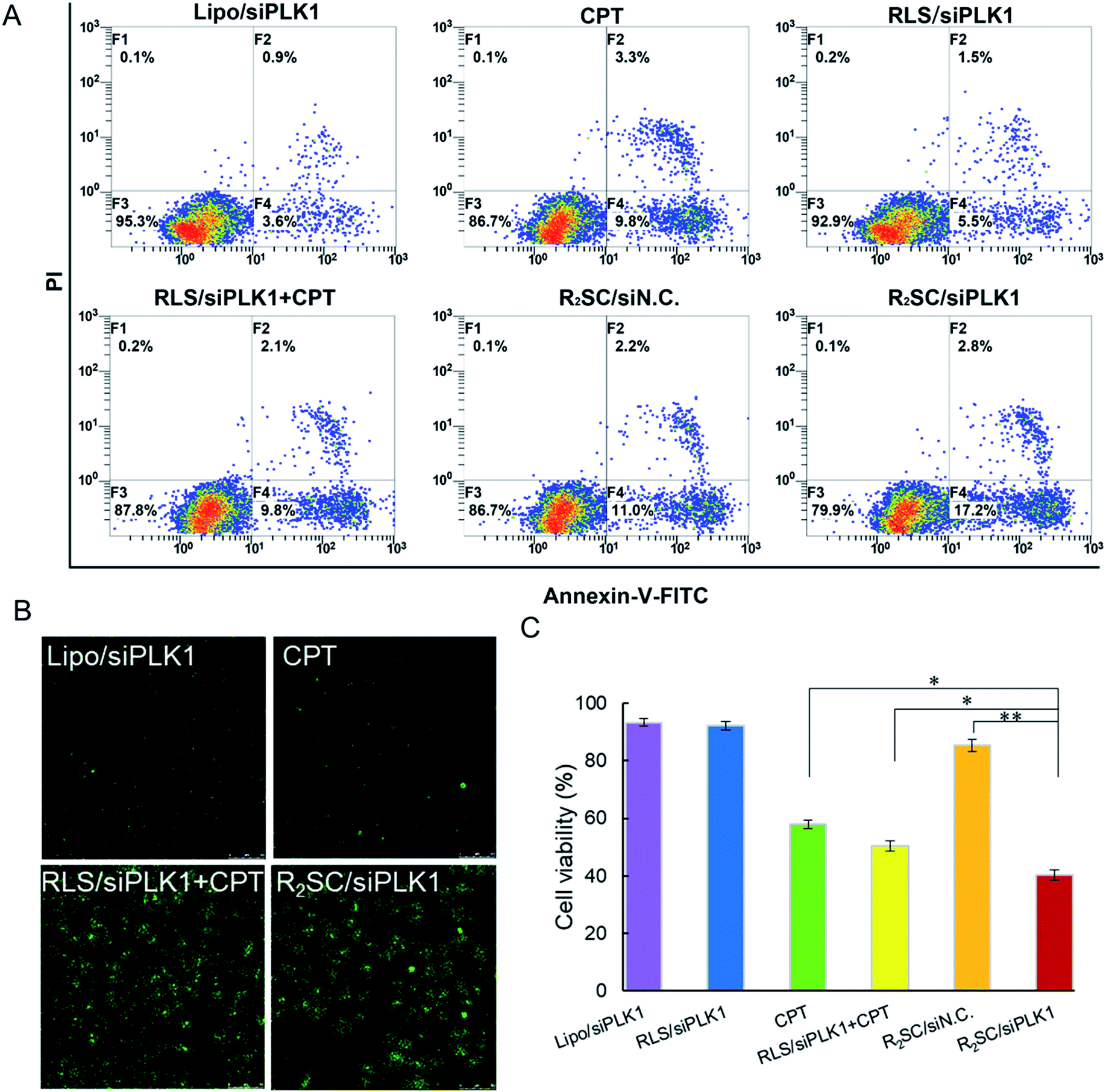 | ||
| Fig. 4 In vitro antitumor activity of various formulations. (A) Induction of apoptosis in HeLa cells by various formulations and free CPT after 48 h incubation. The early apoptotic cells are presented in the lower right quadrant, and fully apoptotic cells are presented in the upper right quadrant. (B) The representative images of apoptotic cells stained using a TUNEL cell apoptosis detection kit. (C) In vitro cytotoxicity of different complexes on HeLa cells after 72 h incubation. The concentration of free CPT was equal to the concentration of CPT from prodrug-based complexes corresponding to siRNA concentration. Data represent the mean ± S.D. (n = 5). *p < 0.05, **p < 0.01. | ||
Finally, the synergistic effect of R2SC/siPLK1 on tumor growth inhibition was assessed in tumor-bearing nude mice in vivo by intratumoral administration, using CPT and a mixture of RLS/siPLK1 and CPT as a comparison. It was obvious that the tumors in mice treated with saline grew rapidly and reached a relative final tumor volume ∼8-fold larger, whereas those with CPT as well as the mixture (RLS/siPLK1 + CPT) administered demonstrated moderately inhibited tumor growth, with final relative tumor volumes of 450% and 350% that of the initial volume, respectively (Fig. 5A). Satisfactorily, R2SC/siPLK1 was the most effective for tumor growth inhibition, showing only 2.5-fold increase in relative tumor volume. On the other hand, groups with free CPT (CPT and RLS/siRNA + CPT) showed a striking 15% reduction in body weight during the 14 day treatment (Fig. 5B), which indicates the serious systemic toxicity of the free drug even when using intra-tumoral injection. Importantly, the R2SC/siPLK1 group showed no distinct body weight loss, resulting from minimal side effects. After six treatments, the PLK1 protein level of each tumor mass showed a more obvious reduction in the R2SC/siPLK1 group than that with CPT or RLS/siPLK1 + CPT (Fig. 5C and D). The PLK1 protein expression was about 46.6% for RLS/siPLK1 + CPT, whereas the R2SC/siPLK1 group would significantly knockdown PLK1 protein expression to a lower level of 28.8%. There was no significant decrease in the PLK1 protein level after treatment with CPT when compared to that after treatment with saline. The H&E stained sections of tumor tissue from the saline group showed more chromatin and binucleolates, while different degrees of nucleus atypia and tumor necrosis were observed in the CPT, RLS/siPLK1 + CPT and R2SC/siPLK1 groups. Furthermore, a TUNEL assay was employed to detect apoptotic programmed cell death and Ki-67 antigen staining was performed to assess the antitumor efficacy on tumor cell proliferation. From the TUNEL assay, the R2SC/siPLK1 group showed the largest area of TUNEL-positive cells compared to the other groups, indicating the most serious tumor apoptosis (∼5-fold higher than the group without therapy, in TUNEL staining) (Fig. 5E, S16A†). The Ki-67 level (a proliferation marker) of the R2SC/siPLK1 treatment group was much lower than those in other groups (Fig. S16B†), implying that less cellular proliferation but higher antitumor activity was induced by R2SC/siPLK1. These data demonstrate that R2SC/siPLK1 with excellent siPLK1 gene silencing and temporally controlled drug release behavior can efficiently suppress tumor growth by inhibiting proliferation and by inducing apoptosis of tumor cells, contributing to a synergistic effect of CPT and siPLK1.
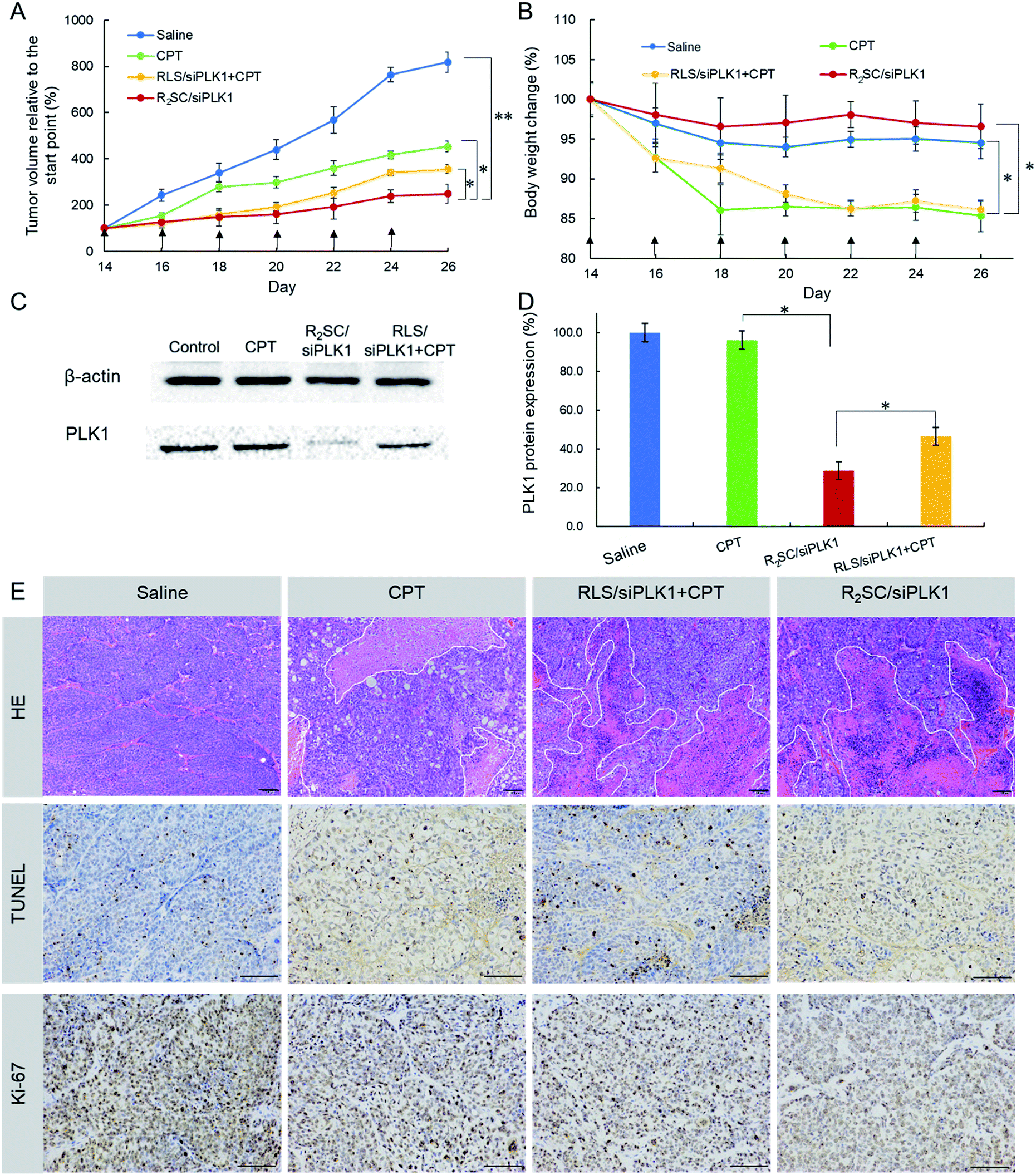 | ||
| Fig. 5 Inhibition of tumor growth in BALB/c nude mice with HeLa xenografts by intra-tumoral administration. (A) Tumor growth curve of mice after six injections of the indicated formulation. (B) Body weight change of mice during the treatment. The arrows indicate the injection times. Data represent the mean ± S.D. (n = 5). * p < 0.05, ** p < 0.01. (C) Representative PLK1 protein expression in tumors determined using western blotting analysis. (D) Analysis of the light intensities of the PLK1 protein expression as the ratio of PLK1 to β-actin from western blot results. (E) H&E, TUNEL, and Ki-67 analyses of tumor tissues after the various treatments. Scale bar = 100 μm. | ||
Conclusions
In summary, we have successfully developed a camptothecin prodrug-based siRNA delivery system (RSC), in which CPT was simultaneously used as the hydrophobic segment of the amphiphilic carrier with high loading efficiency. After assembly with siRNA into nano-sized vesicles, the loaded therapeutic agents (CPT and siRNA) were located in two separate compartments. R2SC/siRNA complexes were shown to perform earlier and faster siRNA release due to redox cleavage, and then the inactive CPT-prodrug slowly reverted back to the pharmacologically active moiety, triggered by the unique biological stimulation of esterase in a temporally controlled release manner. The delayed release of CPT in two steps provides a sufficient period of time for siPLK1 down-regulation in advance, and sequentially improves the sensitivity of cancer cells to CPT. As a result, the camptothecin prodrug-based siRNA delivery system synergistically promoted excellent apoptotic and anti-proliferative effects in vitro and in vivo, thus providing a promising approach for effective combination therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key Research and Development Plan of China (2017YFC1104601), the National Natural Science Foundation of China (NSFC, No. 81571794, No. 81873921), The Fundamental Research Funds for the Central Universities (2012017yjsy222) and the Spark innovation Project of Sichuan University (2018SCUH0045). Dr Q. Jiang and X. Chen contributed equally to this work.Notes and references
- P. Y. Teo, W. Cheng, J. L. Hedrick and Y. Y. Yang, Adv. Drug Delivery Rev., 2016, 98, 41–63 CrossRef CAS PubMed.
- Q. Zheng, D. Lin, L. Lei, X. Li and S. Shi, J. Biomed. Nanotechnol., 2017, 13, 1565–1580 CrossRef CAS PubMed.
- X. Li, L. Hong, T. Song, A. Rodríguezpatón, C. Chen, H. Zhao and X. Shi, J. Biomed. Nanotechnol., 2017, 13, 747–757 CrossRef CAS.
- Z. Yang, D. Gao, Z. Cao, C. Zhang, D. Cheng, J. Liu and X. Shuai, Biomater. Sci., 2015, 3, 1035–1049 RSC.
- K. Lin, Z. Gao, W. Huang, M. Jin and Q. Wang, Acta Pharm. Sin. B, 2015, 5, 169–175 CrossRef PubMed.
- J. Li, Y. Chen, L. Zeng, G. Lian, S. Chen, Y. Li, K. Yang and K. Huang, J. Biomed. Nanotechnol., 2016, 12, 1654–1666 CrossRef CAS PubMed.
- S. Hao, Y. Yan, X. Ren, Y. Xu, L. Chen and H. Zhang, Biotechnol. Bioprocess Eng., 2015, 20, 550–560 CrossRef CAS.
- N. S. Gandhi, R. K. Tekade and M. B. Chougule, J. Controlled Release, 2014, 194, 238–256 CrossRef CAS PubMed.
- H. Yang, L. Deng, T. Li, X. Shen, J. Yan, L. Zuo, C. Wu and Y. Liu, J. Biomed. Nanotechnol., 2015, 11, 2124–2136 CrossRef CAS PubMed.
- H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji and J. I. Zink, ACS Nano, 2013, 7, 994–1005 CrossRef CAS PubMed.
- C. Zhu, S. Jung, S. Luo, F. Meng, X. Zhu, T. G. Park and Z. Zhong, Biomaterials, 2010, 31, 2408–2416 CrossRef CAS PubMed.
- P. Ofek, G. Tiram and R. Satchi-Fainaro, Adv. Drug Delivery Rev., 2017, 119, 3–19 CrossRef CAS PubMed.
- X. Dai and C. Tan, Adv. Drug Delivery Rev., 2015, 81, 184–197 CrossRef CAS PubMed.
- B. Spänkuch, E. Kuruncicsacsko, M. Kaufmann and K. Strebhardt, Oncogene, 2007, 26, 5793–5807 CrossRef PubMed.
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani and M. Bozorgomid, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- S.-J. Seo, S.-Y. Lee, S.-J. Choi and H.-W. Kim, Macromol. Biosci., 2015, 15, 1198–1204 CrossRef CAS PubMed.
- Q. Jiang, D. Yue, Y. Nie, X. Xu, Y. He, S. Zhang, E. Wagner and Z. Gu, Mol. Pharm., 2016, 13, 1809–1821 CrossRef CAS PubMed.
- Y. He, Y. Nie, G. Cheng, L. Xie, Y. Shen and Z. Gu, Adv. Mater., 2013, 26, 1534–1540 CrossRef.
- Y. He, G. Cheng, L. Xie, Y. Nie, B. He and Z. Gu, Biomaterials, 2013, 34, 1235–1245 CrossRef CAS PubMed.
- X. Chen, J. Yang, H. Liang, Q. Jiang, B. Ke and Y. Nie, J. Mater. Chem. B, 2017, 5, 1482–1497 RSC.
- Q. Xu, Y. Xia, C. H. Wang and D. W. Pack, J. Controlled Release, 2012, 163, 130–135 CrossRef CAS PubMed.
- L. Han, R. Huang, J. Li, S. Liu, S. Huang and C. Jiang, Biomaterials, 2011, 32, 1242–1252 CrossRef CAS PubMed.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- H. Hu, W. Yuan, F.-S. Liu, G. Cheng, F.-J. Xu and J. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 8942–8951 CrossRef CAS PubMed.
- Q. Tang, B. Cao and G. Cheng, Chem. Commun., 2014, 50, 1323–1325 RSC.
- P. Zhong, J. Zhang, C. Deng, R. Cheng, F. Meng and Z. Zhong, J. Controlled Release, 2017, 259, e104 CrossRef.
- H. Deng, X. Zhao, J. Liu, J. Zhang, L. Deng, J. Liu and A. Dong, Nanoscale, 2016, 8, 1437–1450 RSC.
- Q. Jiang, Y. Nie, X. Chen, Y. He, D. Yue and Z. Gu, Adv. Funct. Mater., 2017, 1701571 CrossRef.
- X. Xu, Q. Jiang, X. Zhang, Y. Nie, Z. Zhang, Y. Li, G. Cheng and Z. Gu, J. Mater. Chem. B, 2015, 3, 7006–7010 RSC.
- D. Yue, G. Cheng, Y. He, Y. Nie, Q. Jiang, X. Cai and Z. Gu, J. Mater. Chem. B, 2014, 2, 7210–7221 RSC.
- Y. Talukdar, J. T. Rashkow, G. Lalwani, S. Kanakia and B. Sitharaman, Biomaterials, 2014, 35, 4863–4877 CrossRef CAS PubMed.
- Y. Li, R. Liu, J. Yang, G. Ma, Z. Zhang and X. Zhang, Biomaterials, 2014, 35, 9731–9745 CrossRef CAS PubMed.
- P. Fan, S. Zhang, H. Tian, N. Yan, L. Dai, X. Zhang, L. Cheng, C. Li, Y. Li and X. Chen, Neoplasma, 2012, 59, 676–684 CrossRef CAS PubMed.
- N. Kreis, K. Sommer, M. Sanhaji, A. Kramer, Y. Matthess, M. Kaufmann, K. Strebhardt and J. Yuan, Cell Cycle, 2009, 8, 460–472 CrossRef CAS PubMed.
- D. Luong, P. Kesharwani, R. Deshmukh, M. C. Mohd Amin, U. Gupta, K. Greish and A. K. Iyer, Acta Biomater., 2016, 43, 14–29 CrossRef CAS PubMed.
- R. J. Bose, Y. Arai, J. C. Ahn, H. Park and S. H. Lee, Int. J. Nanomed., 2015, 10, 5367–5382 CAS.
- X. Z. Yang, S. Dou, T. M. Sun, C. Q. Mao, H. X. Wang and J. Wang, J. Controlled Release, 2011, 156, 203–211 CrossRef CAS PubMed.
- X. Li, Y. Zhu, J. Shao, C. Min, Y. Hao, G. Li, Z. Yi, Z. Xu, Y. Bo and P. Luo, Br. J. Cancer, 2017, 116, 1027–1036 CrossRef PubMed.
- B. Qin, B. Gao, J. Yu, J. Yuan and Z. Lou, J. Biol. Chem., 2013, 288, 16139–16144 CrossRef CAS PubMed.
- R. W. Gunawardena, H. Siddiqui, D. A. Solomon, C. N. Mayhew, J. Held, S. P. Angus and E. S. Knudsen, J. Biol. Chem., 2004, 279, 29278–29285 CrossRef CAS PubMed.
- J. Gao, P. Chen, Y. Singh, X. Zhang, Z. Szekely, S. Stein and P. J. Sinko, Bioconjugate Chem., 2013, 24, 1332–1344 CrossRef CAS PubMed.
- V. Zinchuk, O. Zinchuk and T. Okada, Acta Histochem. Cytochem., 2007, 40, 101–111 CrossRef CAS PubMed.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed.
- J. C. Sunshine, M. I. Akanda, D. Li, K. L. Kozielski and J. J. Green, Biomacromolecules, 2011, 12, 3592–3600 CrossRef CAS PubMed.
- L. Zhang, W. Zheng, R. Tang, N. Wang, W. Zhang and X. Jiang, Biomaterials, 2016, 104, 269–278 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00191j |
| ‡ Equal contribution to the manuscript as co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |